

Abstract
Disruptions of dopamine (DA) signaling contribute to a broad spectrum of neuropsychiatric disorders, including attention-deficit hyperactivity disorder (ADHD), addiction, bipolar disorder, and schizophrenia. Despite evidence that risk for these disorders derives from heritable variation in DA-linked genes, a better understanding is needed of the molecular and circuit context through which gene variation drives distinct disease traits. Previously, we identified the DA transporter (DAT) variant Val559 in subjects with ADHD and established that the mutation supports anomalous DAT-mediated DA efflux (ADE). Here, we demonstrate that region-specific contributions of D2 autoreceptors (D2AR) to presynaptic DA homeostasis dictate the consequences of Val559 expression in adolescent male mice. We show that activation of D2ARs in the WT dorsal striatum (DS), but not ventral striatum (VS), increases DAT phosphorylation and surface trafficking. In contrast, the activity of tyrosine hydroxylase (TH) is D2AR-dependent in both regions. In the DS but not VS of Val559 mice, tonic activation of D2ARs drives a positive feedback loop that promotes surface expression of efflux-prone DATs, raising extracellular DA levels and overwhelming DAT-mediated DA clearance capacity. Whereas D2ARs that regulate DAT are tonically activated in the Val559 DS, D2ARs that regulate TH become desensitized, allowing maintenance of cytosolic DA needed to sustain ADE. Together with prior findings, our results argue for distinct D2AR pools that regulate DA synthesis versus DA release and inactivation and offer a clear example of how the penetrance of gene variation can be limited to a subset of expression sites based on differences in intersecting regulatory networks.
SIGNIFICANCE STATEMENT Altered dopamine (DA) signaling has been linked to multiple neuropsychiatric disorders. In an effort to understand and model disease-associated DAergic disturbances, we previously screened the DA transporter (DAT) in subjects with attention-deficit hyperactivity disorder (ADHD) and identified multiple, functionally impactful, coding variants. One of these variants, Val559, supports anomalous DA efflux (ADE) and in transgenic mice leads to changes in locomotor patterns, psychostimulant sensitivity, and impulsivity. Here, we show that the penetrance of Val559 ADE is dictated by region-specific differences in how presynaptic D2-type autoreceptors (D2ARs) constrain DA signaling, biasing phenotypic effects to dorsal striatal projections. The Val559 model illustrates how the impact of genetic variation underlying neuropsychiatric disorders can be shaped by the differential engagement of synaptic regulatory mechanisms.
Keywords: dopamine, dopamine D2 autoreceptor, dopamine transporter, neuropsychiatric disorders
Introduction
Dopamine (DA) neurotransmission provides critical modulatory support of multiple behaviors including movement (Salamone, 1992), reward, and motivation (Wise, 2004) and attention (Robbins, 2003). The DA transporter (DAT) plays an important role in terminating DA signaling, clearing extracellular DA through reuptake (Gowrishankar et al., 2014), a process that also augments de novo DA synthesis, ensuring adequate vesicular stores (Jones et al., 1998). DAT is a target for abused and therapeutic psychostimulants including cocaine, methylphenidate and amphetamine (AMPH) that produce rapid elevations in extracellular DA by blocking reuptake (Vaughan and Foster, 2013) and/or DAT-mediated DA efflux (Sulzer et al., 1995). DAT surface density and activity can be regulated by multiple receptors, associated proteins and kinase pathways (Bermingham and Blakely, 2016). Relevant to this study, presynaptic D2-type autoreceptor (D2AR) activation can elevate DAT surface expression, thereby enhancing DA uptake (Chen et al., 2013), in addition to decreasing cytosolic DA levels via tyrosine hydroxylase (TH) inhibition (Lindgren et al., 2001), and limiting vesicular DA release (Benoit-Marand et al., 2011).
Longstanding evidence implicates aberrant DA signaling in the pathophysiology of multiple neuropsychiatric disorders including Parkinson's disease (PD; Hornykiewicz, 1962), attention-deficit/hyperactivity disorder (ADHD; Mazei-Robinson and Blakely, 2006; Volkow et al., 2007), autism spectrum disorders (ASD; Pavăl, 2017), schizophrenia (Grace, 2016), bipolar disorder (BPD; Ashok et al., 2017), and addiction (Everitt and Robbins, 2016). We and others have identified and characterized multiple rare, functional DAT coding variants from patients with ADHD (Mazei-Robison et al., 2005; Sakrikar et al., 2012), ASD (Hamilton et al., 2013), PD (Kurian et al., 2009; Hansen et al., 2014; Ng et al., 2014), and BPD (Grünhage et al., 2000). One of the variants we identified in ADHD, the Ala559Val (Val559), has also been detected in BPD and ASD (Bowton et al., 2014). We determined in vitro that although Val559 did not alter DAT surface expression, DA affinity or uptake capacity, it imposed a state of constitutive anomalous DA efflux (ADE; Mazei-Robison et al., 2008), a process normally triggered by AMPH. Further in vitro studies revealed that Val559 imparts elevated N-terminal DAT phosphorylation and that D2-type receptor signaling sustains ADE (Bowton et al., 2010). Subsequently, we developed Val559 knock-in mice (Mergy et al., 2014a,b) and, as predicted by in vitro findings, detected elevated striatal levels of extracellular DA that leads to tonic, D2AR-mediated suppression of DA release. Additionally, Val559 mice exhibited alterations in basal (rearing) and approach-induced locomotion (darting), as well as a reduction in responsiveness to psychostimulants. Recently, we documented that the Val559 mice display impulsivity and increased motivation (Davis et al., 2018). Together, these studies provide compelling evidence that Val559 mice are a construct-valid model for disorders linked to tonic striatal hyperdopaminergia, with behavioral alterations aligning reasonably well with ADHD, or disorders that often feature ADHD traits, such as ASD.
In considering the in vivo impact of ADE, it is important to recognize that DA neurons, despite sharing a core molecular program to synthesize, release and inactivate DA, are increasingly understood to be heterogeneous (Roeper, 2013; Chuhma et al., 2017). The majority of CNS DA neurons are located in the substantia nigra pars compacta (SNpc) and the ventral tegmental area (VTA), with major projections to the dorsal (DS) or ventral striatum (VS), respectively. Recent studies have revealed multiple molecular contributors to DA neuron signaling differing by origin and projection field, and that this heterogeneity supports distinct behavioral states (Matsumoto and Hikosaka, 2009; Howe and Dombeck, 2016; Parker et al., 2016). However, whether genetic variation in shared molecular features of DA projections can exhibit pathway-biased phenotypes is unclear. Here, we provide evidence for previously unknown, region-specific D2AR-linked regulatory complexity in how distinct DA terminals insure homeostatic control of DA signaling that contribute to unexpected, region-dependent bias in penetrance of the Val559. More broadly, our studies provide an example of how pathway-specific differences in neurotransmitter signaling regulation can shape the impact of gene variants on distinct clinical features of neuropsychiatric disorders.
Materials and Methods
Animals.
All experiments were performed under protocols approved by the Institutional Animal Care and Use Committee of Florida Atlantic University and Vanderbilt University. Unless noted otherwise, 4- to 6-week-old mice homozygous for DAT Val559 (Val559) or DAT Ala559 (WT) were bred from homozygous dams and sires derived from heterozygous breeders, no more than two generations removed. Genetic background for all animals was 75% 129/6: 25% C57 as with prior studies (Mergy et al., 2014b; Davis et al., 2018). Only age-matched juvenile males were used because of the male bias reported for ADHD diagnoses and the reported age of symptom onset. Animals were housed on a 12 h reverse light cycle with water and food available ad libitum. All experiments were performed during the animal's active cycle.
Brain slice preparation, drug treatments, and immunoblotting.
All chemical reagents used in experiments were obtained from Sigma-Aldrich unless otherwise specified. Brain slice preparation and experiments were performed under constant oxygenation (95%O2:5%CO2). Animals were killed by rapid decapitation and excised brains were moved to ice-cold sucrose-artificial CSF (S-ACSF; in mm: sucrose 250, KCl 2.5, NaH2PO4 1.2, NaHCO3 26, d-glucose 11, MgCl2.6H2O 1.2, CaCl2.2H2O 2.4, pH 7.4, 300–310 mOsm). A vibratome (Leica VT100 S) was used to prepare 250 μm slices that contained the DS and/or VS (matching coordinates of 0.2–0.5 mm A/P for DS, and 1.0–1.5 mm A/P for VS in the Allen Mouse Brain Atlas) in S-ACSF following which, slices were allowed to recover at 30–32°C for 1 h in ACSF, substituting 92 mm NaCl for sucrose (pH 7.4, 300–310 mOsm). Before drug treatments, slices were washed with ACSF at 37°C and then treated with vehicle, 1 μm (−)−quinpirole hydrochloride (quinpirole), a D2/D3 agonist for 5 min (biotinylation) or 10 min (immunoprecipitation), 10 μm (±)-sulpiride hydrochloride (sulpiride), a D2/D3 antagonist for 20 min (biotinylation and immunoprecipitation) or 100 μm 3-hydroxybenzylhydrazine dihydrochloride (NSD1015), an amino acid decarboxylase inhibitor for 10 min [l-3,4-dihydroxyphenylalanine (l-DOPA) accumulation]. With the exception of biotinylation and l-DOPA accumulation, slices were washed in ice-cold ACSF, followed by rapid dissection of DS and VS, and tissue solubilization in lysis buffer (in mm: NaCl 150, KCl 2.5 m, Tris 50, and 1% Triton X-100). Protein concentrations were determined using the BCA protein assay (ThermoFisher) with bovine serum albumin (BSA) as standard, with a FLUOstar Omega microplate reader (BMG Labtech). Following SDS-PAGE (10% gel), proteins were transferred to PVDF membranes (Millipore) at 4°C overnight and then blocked with 5% dry milk (for DAT) or 5% BSA (for TH) in room temp wash buffer (in mm: NaCl 150, 2.5 KCl, 50 Tris, and 0.1% Tween 20). Blocked membranes were incubated with rat anti-DAT (MAB369, Millipore; RRID:AB_2190413) and rabbit anti-TH (2025-THRAB, PhosphoSolutions; RRID:AB_2492276) at a dilution of 1:1000 in wash buffer overnight at 4°C. Membranes were then rinsed 4× in wash buffer before incubation with goat anti-rat or goat anti-rabbit HRP-conjugated secondary antibody (Jackson ImmunoResearch) at a 1:10,000 dilution for 1 h. Following multiple washes using wash buffer, membranes were subjected to chemiluminescent visualization (Bio-Rad Clarity ECL) and bands were visualized using an ImageQuant LAS 4000 imager (GE Healthcare Life Sciences). Blot images were minimally processed during quantitation and cropped for the presentation of representative figures.
Slice biotinylation.
Methods to assess DAT surface expression in acute brain slices were adapted from those described previously (Gabriel et al., 2014). Briefly, following washes in ice-cold ACSF, slices were treated with sulfo-NHS-SS-biotin (1 mg/ml, ThermoFisher) on ice for 30 min. Reactions were quenched by treatment with 0.1 m glycine 2× for 10 min, followed by rapid washes with ice-cold ACSF. DS and VS were rapidly dissected, solubilized in lysis buffer as noted above, and protein concentration determined. Detergent lysates were added to streptavidin agarose beads (ThermoFisher) at a ratio of 20 μg protein per 50 μl bead slurry and mixed overnight at 4°C. Following washes with lysis buffer, protein was eluted/denatured at room temp for 30 min, subjected to SDS-PAGE, PVDF transfer, and immunoblotting for DAT and TH.
[3H]DA slice uptake.
[3H]DA uptake in acute slices was performed as described previously (Wu et al., 2015). In brief, brain slices containing DS and VS were washed in 37°C were incubated in 50 nm [3H]DA [dihdroxyphenylethylamine, 3, 4-(Ring-2, 5, 6-3H), PerkinElmer] for 10 min, which lies within the linear portion of the time dependence of uptake in our assays (r2 = 0.86), in the presence of GBR12909, a DAT inhibitor. Slices were then rapidly washed in ice-cold ACSF, DS and VS dissected, and proteins extracted using lysis buffer. Lysates were then added to Ecoscint H (National Diagnostics), followed by gentle shaking overnight. Accumulated radioactivity was determined using a TriCarb 2900TR scintillation counter (PerkinElmer). Counts were normalized to DAT total levels in each condition.
High-speed chronoamperometry.
For in vivo chronoamperometric assessment of DA clearance, 8- to 10-week-old mice were used. High-speed chronoamperometry (HSC) for DA was performed as described previously (Owens et al., 2012). Animals were anesthetized by intraperitoneal injection of α-chloralose (400 mg/kg) and urethane (4 mg/kg), fitted with an endotracheal tube to ensure steady breathing, and placed on a heating pad at 37°C with head mounted in a stereotaxic frame (Kopf Instruments). Electrode-micropipette assemblies were made before the experiment using a single, 20-μm-diameter carbon fiber (Specialty Materials) and 7-barrel micropipettes (FHC), ensuring a distance of 200 μm between the electrode tip and micropipette. The electrode was coated with 5% Nafion to enhance selectivity for DA and calibrated in vitro using 2 μm serial additions of DA to PBS. Each barrel of the micropipette was filled with either 200 μm DA (in PBS) or 400 μm sulpiride (in ACSF+10% DMSO). The electrode-micropipette assembly was lowered into the DS (1.5 mm M/L, 0.5 mm A/P, 2.5 mm D/V) or VS (1.0 mm M/L, 1.5 mm A/P, 3.5 mm D/V). DA pressure ejection was achieved using a Picrospritzer (Parker Hannifin) with an ejection volume of 8–125 nl (10–30 psi, 0.05–5 s). Drugs were applied at a volume of 100–125 nl (0.5 psi). Recordings of DA clearance were conducted using the FAST12 system (Quanteon), following 100 ms of 550 mV step potentials to the carbon fiber, separated by a 0.9 ms return to 0 mV, relative to a reference electrode mounted under the neck. DA was identified using its current oxidation/reduction ratio, 0.5:0.8. Signal parameters used in quantitation were the T80(s), the time for amperometric signal to decay to 80% signal amplitude, and the clearance rate (TC in nm/s), estimated as the slope of the linear portion of current decay from 20 to 60% signal amplitude.
Phospho-Thr53 DAT immunoprecipitation.
Quantification of DAT phosphorylation at Thr53 was assessed as previously described (Foster et al., 2012). Briefly, rabbit Thr53 DAT antibody (Roxanne A. Vaughan; RRID:AB_2492078) was crosslinked to protein A magnetic beads (Dynabeads, ThermoFisher) at a ratio of 1 mg Thr53 antibody or rabbit IgG (Antibodies) to 10 μl bead slurry using 25 mm dimethyl pimelimidate in 0.2 m triethylamine 3× for 30 min each, quenched with 50 mm ethanolamine then treated with 5% BSA in PBS + 0.1% Triton X-100 at 4°C for 1 h to reduce nonspecific binding. Detergent lysates of brain slices were added to cross-linked DAT Thr53 antibody-conjugated beads at a ratio of 250 μg protein to 25 μl of bead slurry, rocked at 4°C for 4 h, washed with lysis buffer, treated with 2× Laemmli sample buffer (Bio-Rad) to elute/denature protein before SDS-PAGE and immunoblotting for DAT.
Slice l-DOPA accumulation.
Following slice preparation and recovery, slices were treated with NSD1015 (100 μm) alone, or quinpirole (1 μm) and NSD1015 (100 μm) for 10 min at 37°C, then rapidly washed in ice-cold ACSF and sonicated in 250 μl of 0.2 m HClO4. Isoproterenol (10 ng) was added as an internal standard, the protein denatured and samples centrifuged at 20,000 × g for 15 min at 4°C, with 1 m sodium acetate addition to modify the supernatant to pH 3.0. The supernatant was then filtered with a spin column and analyzed by HPLC. The HPLC system consisted of an Eicom Insight Autosampler (AS-700), EiCOM Stand-Alone HPLC-Electrochemical Detection System (HTEC-510), Eicompak SC-30DS (ID 3.0 × 100 mm) reverse-phase column with a graphite working electrode (WE-3G, EiCOM). Assays used an applied potential of +750 mV versus Ag/AgCl. A mobile phase consisting of 85% 0.1 m citrate-acetate buffer, pH 3.5, 15% methanol, 220 mg/L sodium octane sulfonate and 5 mg/L EDTA-2Na was used to separate l-DOPA and its metabolites. Signals were quantitated relative to known concentrations of standards using Envision Data System software. The final oxidation current values were converted to ng and adjusted to milligrams protein amounts determined by BCA protein assay as described above.
Experimental design and statistical analyses.
Statistical analyses and graphical presentations were pursued using GraphPad Prism 6.0, with p < 0.05 taken as statistically significant. For all experiments, each N value represents data obtained from an individual animal. For all slice experiments, hemisections obtained from the same slice were treated with either vehicle (Veh) or drug. DS or VS slices from WT and Val559 mice for slice biotinylation and p-Thr53 immunoprecipitation experiments were paired and samples run on the same gel. Data are presented normalized to WT Veh and a two-way ANOVA was used to compare all groups. Post hoc Bonferroni tests were performed if there was a significant drug and genotype interaction. For slice [3H]DA uptake, each paired hemi-slice was treated with Veh or GBR12909 before [3H]DA application. Data were normalized to milligrams of protein obtained from lysates, and lysates were subjected to SDS-PAGE and immunoblotting for DAT to ensure comparable levels of DAT in each hemi-slice. For in vivo HSC, peak DA signal amplitudes were generated in triplicate for each animal. Basal clearance at 1 μm DA signal amplitude was compared between genotypes using a two-tailed Student's t test. TC curves were analyzed using a two-way ANOVA and post hoc Bonferroni test comparing DA signal amplitudes and genotypes, and subjected to Michaelis-Menten analyses to obtain fit (r2) and KM values. For sulpiride injection studies, a stable DA signal amplitude of 1 μm was obtained in triplicate before Veh or sulpiride injection. Veh injection always preceded sulpiride injection; data are presented normalized to Veh at 20 min and analyzed using a two-tailed Student's t test, comparing sulpiride effects on TC and T80 in WT or Val559. For slice l-DOPA accumulation experiments, DS or VS slices were collected from WT or Val559 mice and analyzed for l-DOPA levels independently. Data are presented normalized to the NSD-only condition and analyzed using two-tailed Student's t test, comparing effects of quinpirole on NSD-stimulated l-DOPA accumulation in WT or Val559 mice. Specific statistical tests and exact p values for experiments are provided in the Figure Legends, along with the sample size (N).
Results
Val559 induces elevated DAT surface expression in DS, but not VS, consistent with tonic, region-specific presynaptic D2AR activation
Our prior studies using transfected cells demonstrated that whereas Val559-driven ADE in transfected cells and cultured neurons is supported by DA stimulation of endogenous D2Rs (Bowton et al., 2010), this receptor engagement did not appear to enhance DAT surface trafficking (Mazei-Robison and Blakely, 2005). In contrast, synaptosomal D2AR activation with an exogenous agonist has been reported to increases DAT surface expression (Chen et al., 2013), suggesting differences between culture and native preparations that could limit detection of Val559 phenotypes. Therefore, we evaluated the effects of in vivo D2AR activation on transporter surface density using the D2-type DA receptor agonist quinpirole (1 μm, 5 min) by ex vivo biotinylation approaches (Gabriel et al., 2014). Recognizing the aforementioned molecular and functional diversity of different DA projections, we assessed transporter trafficking in WT and Val559 mice using slices containing either the DS or VS. Whereas total levels of DAT protein were not impacted by Val559 in either DS or VS (Fig. 1A,B), surface levels of DAT were significantly elevated in the DS of Val559 mice relative to WT animals (Fig. 1C). This genotype effect was absent in slices prepared from the VS (Fig. 1D). Additionally, whereas quinpirole treatment elevated cell-surface transporter levels in WT DS slices, no effect of quinpirole was detected in Val559 DS slices (Fig. 1C). Remarkably, quinpirole was without effect on surface DAT levels in VS slices regardless of genotype (Fig. 1D). We hypothesized that maximal, ongoing activation of D2ARs might be responsible for the elevated transporter surface expression detected under basal conditions in Val559 DS slices, as well as the inability of quinpirole to effect membrane trafficking. This hypothesis was supported by biotinylation experiments conducted with the D2-type receptor antagonist sulpiride (10 μm, 20 min), which eliminated the Val559 genotype effect on basal surface DAT levels (Fig. 1E).
Figure 1.
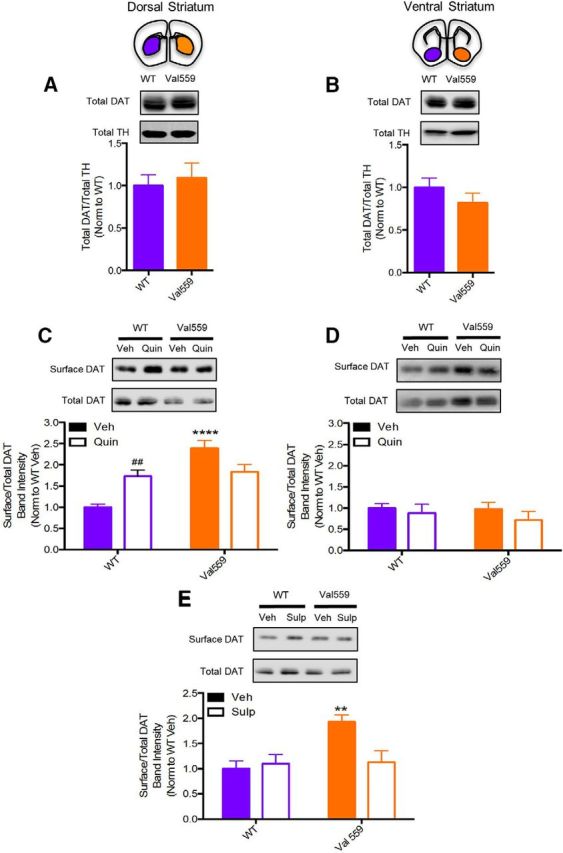
Val559 induces elevated DAT surface expression in DS, but not VS, consistent with tonic, region-specific presynaptic D2AR activation. Total DAT levels, normalized to total TH, are unchanged across genotypes in both (A) DS (two-tailed Student's t test, t(14) = 0.4344, p = 0.671, N = 8) and (B) VS (two-tailed Student's t test, t(10) = 1.164 p = 0.271, N = 6). In the DS (C) surface DAT in Val559 is higher relative to WT and quinpirole (Quin; 1 μm, 5 min) elevates WT surface DAT, not Val559 DAT (two-way ANOVA, genotype: F(1,24) = 25.64, p < 0.0001; Quin: F(1,24) = 0.3471, p = 0.561; interaction: F(1,24) = 19.23, p = 0.0001. Bonferroni's multiple-comparisons test: p < 0.0001 for WT Veh vs Val559 Veh, p = 0.009 for WT Veh vs WT Quin, p = 0.072 for Val559 Veh vs Val559 Quin, N = 8). In the VS, (D) membrane DAT is unchanged across genotypes and surface levels are unaffected in response to Quin (1 μm, 5 min; two-way ANOVA, genotype: F(1,16) = 0.306, p = 0.5873; Quin F(1,16) = 1.145, p = 0.300; interaction F(1,16) = 0.159, p = 0.696, N = 5). E, Sulpiride (Sulp; 10 μm, 20 min) treatment abolishes genotype effects in Val559 (two-way ANOVA, genotype: F(1,20) = 7.117, p = 0.015; Sulp: F(1,20) = 3.855, p = 0.064; interaction: F(1,20) = 6.402, p = 0.0199. Bonferroni's multiple-comparisons test: p = 0.009 for WT Veh vs Val559 Veh, p > 0.999 for WT Veh vs Val559 Sulp, N = 6). * denotes comparisons across genotypes (WT vs. Val559); **** for p < 0.0001 in (C) and ** for p < 0.01 in (E). # denotes comparisons across drug (Veh vs. Quin); ## for p < 0.01 in (C).
Val559 impacts in vivo basal and D2AR-mediated clearance of exogenous DA, highlighting impact of ADE in the DS, not VS
Our findings of elevated surface DAT in Val559 DS were surprising, as previously we had reported no changes in DAT-mediated [3H]DA uptake using transfected cells (Mazei-Robison and Blakely, 2005) or striatal synaptosomes (Mergy et al., 2014b). We reasoned that a failure to detect elevations in DA transport in Val559 synaptosomes could arise from either the disruptive nature of synaptosomal preparation or to the free diffusion of DA that arises from ADE in synaptosomal incubations, lessening both D2AR activation and subsequent D2AR-dependent changes in transporter surface expression and DA uptake. Our attempt to assess genotype effects on [3H]DA uptake assays in brain slices, however, revealed further complexities, as here we detected a significant reduction in [3H]DA uptake in Val559 versus WT DS slices (Fig. 2A,B). One possible explanation for the latter findings is that, in brain slices, DA released by ADE could remain trapped in the tissue and result in a reduction of [3H]DA-specific activity, thereby appearing to diminish levels of [3H]DA uptake. If this is the case, we should detect a decrease in apparent DA affinity, or an increase in KM in DA dose–response studies. To explore this idea, and to move our functional evaluations in vivo, we pursued assessments of DA clearance in anesthetized WT and Val559 mice using HSC. Pressure ejection of DA resulting in a signal amplitude of 1 μm corroborated our ex vivo findings of reduced DAT function, showing a delay in the time to clear 80% of the DA signal (T80) and reduction in DA clearance rate (TC) in the Val559 DS (Fig. 3A). Remarkably, VS DA clearance was unchanged across genotypes (Fig. 3B). To perform dose–response studies, we pressure-ejected increasing concentrations of DA and determined DA clearance rates (TC) at each concentration. As shown in Figure 3, C and D, the concentration dependence of TC profiles conformed well to Michaelis–Menten kinetics, regardless of genotype or region (see legend for r2 values). Consistent with our hypothesis, we obtained a significant increase in the exogenous DA KM from fits of DS recordings in Val559 versus WT mice (Fig. 3C; see legend for KM values). In contrast, no genotype effect in DA KM was evident in recordings from the VS (Fig. 3D).
Figure 2.

Val559 expression causes a reduction in [3H]DA uptake in DS slices. Val559 DS slices show a profound reduction in (A) total (two-way ANOVA, genotype: F(1,12) = 25.56, p = 0.0003; GBR: F(1,12) = 42.11, p < 0.0001; interaction: F(1,12) = 18.98, p = 0.0009. Bonferroni's multiple-comparisons test: p = 0.0001 for WT Veh vs Val559 Veh, p < 0.0001for WT Veh vs WT GBR and p = 0.944 for Val559 Veh vs Val559 GBR, N = 4) and (B) specific [3H]DA (50 nm, 10 min) uptake (two-tailed Student's t test, t(6) = 9.188, p < 0.003, N = 4). * denotes comparisons across genotypes (WT vs. Val559); *** for p < 0.001 in (A) and ** for p < 0.01 in (B). # denotes comparisons across drug (Veh vs. GBR); #### for p < 0.0001 in (A).
Figure 3.
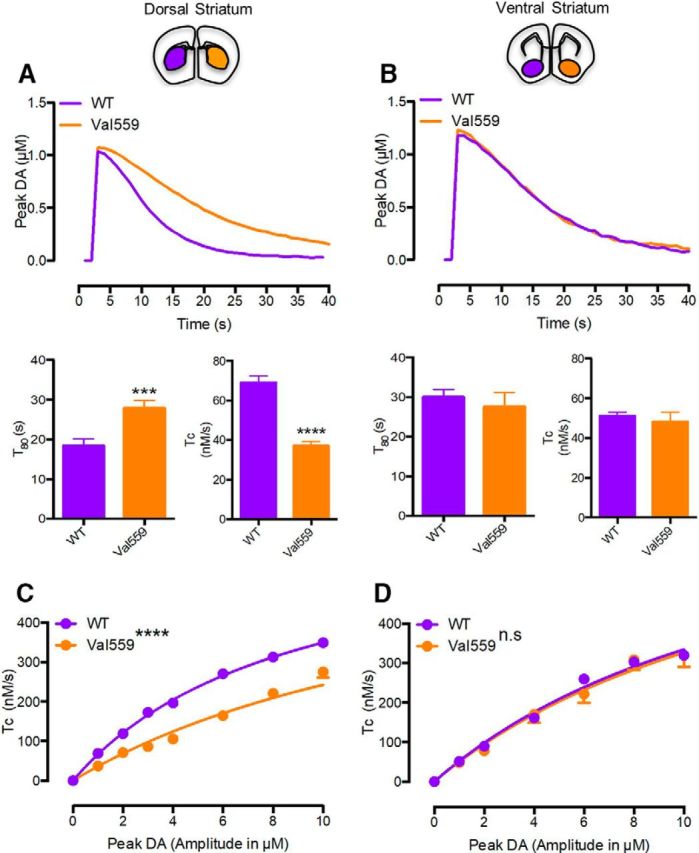
Val559 reduces in vivo clearance of exogenous DA, highlighting impact of ADE in the DS, not VS. In the DS, (A) Val559 expression results in decreased TC (two-tailed Student's t test, t(30) = 8.499, p < 0.0001, N = 5–6) and a delay in T80 (two-tailed Student's t test, t(30) = 3.661, p = 0.001, N = 5–6), also apparent in the representative clearance trace at 1 μm, but in the VS (B) causes no change in TC (two-tailed Student's t test, t(22) = 0.549, p = 0.568, N = 4) or T80 (two-tailed Student's t test, t(22) = 0.400, p = 0.693, N = 4). In dose-dependent studies in the DS, (C) clearance rates (TC) are attenuated in the Val559 compared with the WT (two-way ANOVA, genotype: F(1,218) = 401.8, p < 0.0001; peak DA: F(7,218) = 506.7, p < 0.0001; interaction: F(7,218) = 18.33, p < 0.0001. Bonferroni's multiple-comparisons test shows p = 0.002 for WT vs Val559 at 1 μm and p < 0.0001 at 2, 3, 4, 6, 8, and 10 μm peak DA, N = 5–6). TC profiles conformed to Michaelis–Menten kinetics in both WT and Val559 (WT-r2 = 0.969, Val559-r2 = 0.873), but show apparent KM for DA being significantly higher in Val559 (WT KM = 8.9 ± 0.14 μm, Val559 KM = 17.3 ± 0.50 μm, two-tailed Student's t test, t(9) = 14.49, p < 0.0001). In the VS, (D) Val559 TC is comparable to WT over all DA signal amplitudes (two-way ANOVA, genotype: F(1,154) = 0.561, p = 0.478; peak DA: F(6,154) = 162.2, p < 0.0001; interaction: F(6,154) = 0.619, p = 0.715, N = 4) and conformed well to Michaelis–Menten kinetics for WT and Val559 (WT-r2 = 0.971, Val559-r2 = 0.861), with no change in apparent KM (WT KM = 14.99 ± 0.24 μm, Val559 KM = 15.59 ± 0.82 μm, two-tailed Student's t test, t(6) = 0.7, p =0.528). * denotes comparisons across genotypes (WT vs. Val559); denotes comparisons across genotypes (WT vs. Val559); ***p < 0.001 for T80 comparisons and ****p < 0.0001 for TC comparisons in (A), and ****p < 0.0001 for TC comparisons in (C).
Previous studies using HSC have shown that local injection of D2AR antagonists can delay DA clearance and enhance DA signal amplitude, likely due to the inhibition of D2AR-mediated elevations in DAT surface expression that are triggered by the exogenous DA pulse (Cass and Gerhardt, 1994; Dickinson et al., 1999). We reasoned that ADE induced by Val559 in the DS would diminish D2AR antagonist effects, because of the greater amount of DA that must be blocked to constrain D2AR activation. In WT DS, sulpiride (50 pmol, 20 min) injections significantly elevated DA signal amplitude and increased T80 compared with vehicle (Fig. 4A; 1 μm DA signal amplitude presented). Under the same conditions, in the DS of Val559 mice, sulpiride lacked the ability to elevate DA signal amplitude (actually a small but significant decrease) or to delay T80 (Fig. 4B). Together with our slice DAT trafficking and [3H]DA uptake studies, our HSC dose–response and D2AR antagonist studies support a model where the Val559 DS exhibits elevated surface levels of efflux-prone DATs, driven by ADE in the context of D2AR coupling to DAT. In the VS, Val559 ADE fails to modify DAT surface trafficking and extracellular DA levels, due to a lack of D2AR–DAT coupling.
Figure 4.

Val559 attenuates the effects of in vivo D2AR-antagonism on clearance of exogenous DA, highlighting impact of ADE in the DS, not VS. In the WT DS, (A) Sulp injection (50pmol, 20 min) enhanced signal amplitude (two-tailed Student's t test, t(12) = 2.519, #p = 0.027, N = 7) and delayed T80 (two-tailed Student's t test, t(12) = 2.998, #p = 0.011, N = 7), but (B) reduced signal amplitude (two-tailed Student's t test, t(6) = 2.608, #p < 0.04, N = 4) and had no effect on T80 (two-tailed Student's t test, t(6) = 0.431, p = 0.681, N = 4) in the Val559 DS. # denotes comparisons across drug (Veh vs. Sulp); # for p < 0.05 for Signal Amplitude and T80 comparisons in (A) and for Signal Amplitude comparisons in (B).
Val559 causes enhanced DAT phosphorylation in DS, but not VS, arising from tonic, region-specific presynaptic D2AR activity
Multiple DAT N-terminal Ser/Thr residues are phosphorylated under basal conditions (Foster et al., 2002; Foster et al., 2012), and although elevated DAT phosphorylation has been demonstrated following drug treatments (Foster et al., 2006; Challasivakanaka et al., 2017), the role of endogenous receptors (such as D2AR) in the phosphorylation-dependent control of native DAT trafficking and function, and how transporter phosphorylation is impacted by disease-associated mutations, remains an active area of investigation (Ramamoorthy et al., 2011; Bermingham and Blakely, 2016; Foster and Vaughan, 2017). Recently, Vaughan and colleagues detected basal phosphorylation of DAT at the juxtamembrane residue Thr53 (p-Thr53) in both transfected cells and striatal synaptosomes (Foster et al., 2012). These investigators also associated Thr53 phosphorylation with AMPH-induced DA efflux, an important finding in the context of the ADE induced spontaneously by Val559. Moreover, Thr53 lies within a MAPK consensus phosphorylation site, of relevance to our studies in that D2AR regulation of DAT trafficking has been found to be dependent on signaling by the MAPK ERK1/2 (Bolan et al., 2007). Paralleling our observations of region-dependent changes in DAT trafficking, we found a significant elevation in p-Thr53 DAT with DS, but not VS slices of Val559 mice (Fig. 5A,B). To determine whether elevated Thr53 phosphorylation might derive from ADE-driven D2AR stimulation, we first treated slices with quinpirole (1 μm, 10 min) and detected elevated p-Thr53 levels in WT DS but not in Val559 DS slices (Fig. 5A), consistent with tonic D2AR activation in the DS that cannot be further elevated by the D2AR agonist. Consistent with this hypothesis, the basal elevations in DS p-Thr53 levels were normalized by treatment of slices with sulpiride (10 μm, 20 min; Fig. 5C). In keeping with a lack of D2AR–DAT coupling in the VS, quinpirole treatment failed to elevate p-Thr53 levels in VS slices (Fig. 5B).
Figure 5.
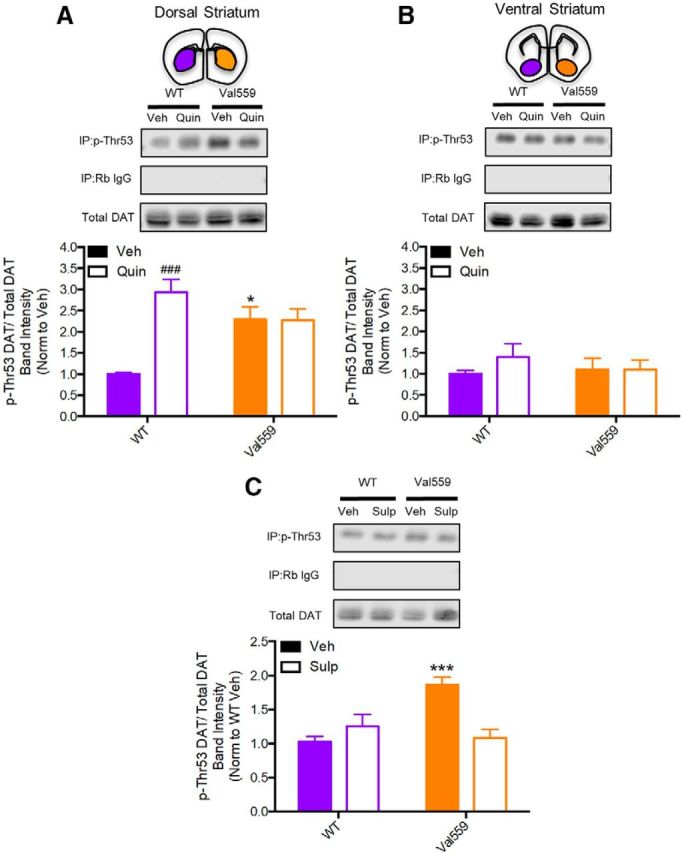
Val559 causes enhanced DAT phosphorylation in DS, but not VS, arising from tonic, region-specific presynaptic D2AR activity. In the DS (A) basal Val559 p-Thr53 is higher relative to WT and Quin (1 μm, 10 min) increases WT p-Thr53, whereas Val559 p-Thr53 is unchanged (two-way ANOVA, genotype: F(1,16) = 1.553, p = 0.231; Quin: F(1,16) = 14.47, p = 0.001; interaction: F(1,16) = 14.89, p = 0.002. Bonferroni's multiple-comparisons test: *p = 0.014 for WT Veh vs Val559 Veh, p = 0.0003 for WT Veh vs WT Quin, p > 0.9999 for Val559 Veh vs Val559 Quin, N = 5). In the VS (B) Val559 and WT p-Thr53 are comparable and Quin (1 μm, 10 min) has no effect on WT or Val559 p-Thr53 DAT (two-way ANOVA, genotype: F(1,16) = 0.566, p = 0.699; Quin: F(1,16) = 0.155, p = 0.430; interaction: F(1,16) = 0.670, p = 0.425, N = 5). C, Sulp (10 μm, 20 min) normalized Val559 p-Thr53 to WT levels (two-way ANOVA, genotype: F(1,24) = 7.132, p = 0.013; Sulp effect: F(1,24) = 4.998, p = 0.035; interaction: F(1,24) = 16.05, p = 0.0005. Bonferroni's multiple-comparisons test: *p = 0.0003 for WT Veh vs Val559 Veh, p > 0.999 for WT Veh vs Val559 Sulp, N = 7). * denotes comparisons across genotypes (WT vs. Val559); * for p < 0.05 in (A) and *** for p < 0.001 in (C). # denotes comparisons across drug (Veh vs. Quin); ### for p < 0.001 in (A).
Val559 attenuates D2AR-induced inhibition of TH activity in the DS, but not VS
Our studies of DAT trafficking, clearance and phosphorylation demonstrate a region-dependent perturbation by Val559, with genotype effects seen for both basal and D2AR activation in the DS but not VS. Because VS DA projections express functional D2ARs (Sesack et al., 1994), we explored whether the impact of the Val559 allele extends from the control of DAT to other aspects of D2AR-mediated presynaptic regulation, specifically to the regulation of TH, the rate-limiting determinant of DA synthesis. Prior studies have shown that striatal D2AR stimulation via quinpirole can inhibit TH activity, as measured by l-DOPA accumulation in the presence of the amino acid decarboxylase inhibitor, NSD1015 (Lindgren et al., 2001; Bello et al., 2011). We quantified quinpirole-modulation of NSD-stimulated l-DOPA accumulation in DS and VS slices prepared from WT and Val559 mice. Although we found no genotype impact on TH activity in either DS or VS following NSD treatment (Fig. 6A,B), or total TH protein levels (Fig. 6C,D), we observed an inability for DS slices to translate quinpirole application to an attenuation of TH activity in the Val559 DS, whereas WT DS demonstrated the expected quinpirole sensitivity. Additionally, D2AR stimulation reduced TH activity in the VS of both WT and Val559 slices (Fig. 6E,F). These effects broaden the region-dependent effects of Val559 to include perturbations of control of DA synthesis as well as DAT phosphorylation and trafficking.
Figure 6.
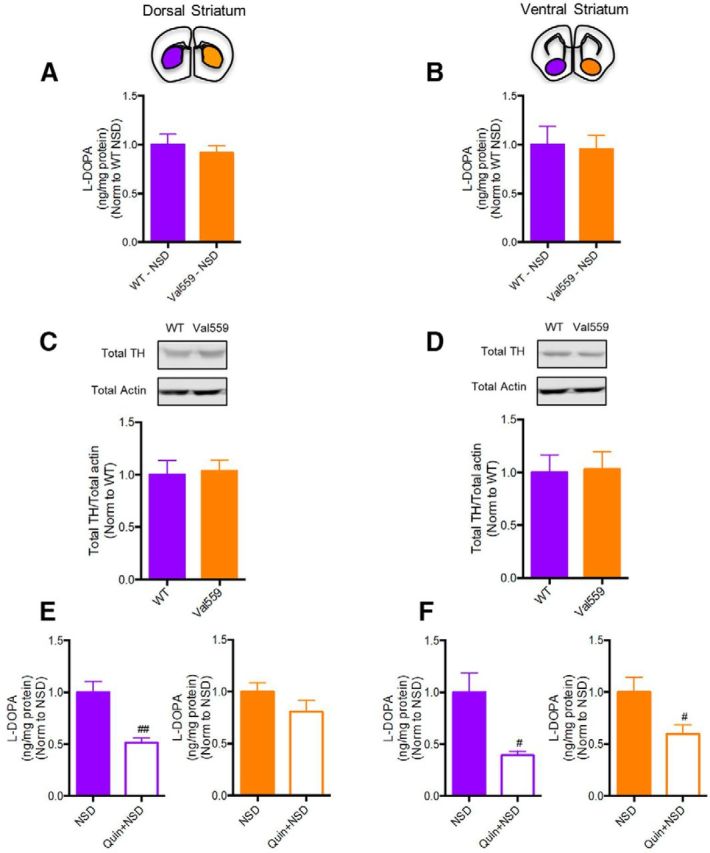
Val559 diminishes the effects of D2AR-induced inhibition of TH activity in the DS, but not VS. A, B, l-DOPA levels post-NSD1015 only across genotypes was unchanged in both DS (two-tailed Student's t test, t(12) = 0.659, p = 0.524, N = 7) and VS (two-tailed Student's t test, t(8) = 0.191, p = 0.854, N = 5). C, D, Total TH levels are unchanged across genotypes in both DS (two-tailed Student's t test, t(10) = 0.216, p = 0.834, N = 6) and VS (two-tailed Student's t test, t(14) = 0.126, p = 0.904, N = 8). In the DS, (E) Quin (1 μm, 10 min) along with NSD (100 μm, 10 min) treatment reduced WT l-DOPA accumulation compared with NSD (100 μm, 10 min) alone (two-tailed Student's t test, t(12) = 4.155, #p = 0.001, N = 7), with no effect in Val559 (two-tailed Student's t test, t(12) = 1.406, #p = 0.187, N = 7). In the VS (F) Quin (1 μm, 10 min) decreased l-DOPA levels comparably in both WT (two-tailed Student's t test, t(8) = 3.18, #p = 0.013, N = 5) and Val559 (two-tailed Student's t test, t(8) = 2.411, #p = 0.041, N = 5). # denotes comparisons across drug (NSD vs. Quin+NSD); #p < 0.05 in (E) and ##p < 0.01 in (F).
Discussion
Genetic, pharmacological, and brain imaging studies indicate that synaptic DA availability and signaling is disrupted in neuropsychiatric disorders, including addiction (Volkow and Morales, 2015), ADHD (Cook et al., 1995; Volkow et al., 2007), schizophrenia (Slifstein et al., 2015; Howes et al., 2017), and ASD (Jeste and Geschwind, 2014). Past efforts to model DAergic contributions to these disorders, although important, have used lesion and pharmacological approaches, or knock-out models that perturb DA synthesis, release, or inactivation (Zhou and Palmiter, 1995; Giros et al., 1996; Wang et al., 1997). Thus, we pursued the identification of functionally penetrant, disease-associated variants in DAT (Mazei-Robison et al., 2005; Mergy et al., 2014a) with which animal models with improved construct validity could be constructed. The current report arises from an effort to model the impact of Val559, a variant we identified in two male siblings with ADHD, and that others identified in a girl with bipolar disorder (Grünhage et al., 2000) and two unrelated boys with ASD (Bowton et al., 2014).
Following our initial demonstration that Val559 exhibited ADE in transfected cells (Mazei-Robison et al., 2008), studies established that endogenous D2 receptors expressed in cells and cultured DA neurons were required to sustain ADE (Bowton et al., 2010). Although D2ARs had been reported to elevate DAT surface expression and function (Bolan et al., 2007), presumably matching demands from elevated vesicular release, we detected no effects of Val559 on these measures in transfected cells (Mazei-Robison and Blakely, 2005) or Val559 synaptosomes (Mergy et al., 2014b). We reasoned that the lack of an effect of ADE on trafficking might be from the use of in vitro assays, where its impact might be minimized through rapid diffusion of extracellular DA.
To pursue this idea, we used striatal slices with which we previously demonstrated D2AR-dependent, tonic inhibition of DA vesicular release as a consequence of Val559 ADE (Mergy et al., 2014b). Acknowledging that DS and VS receive distinct DA projections, we assessed DAT trafficking in DS and VS slices separately. In WT DS slices, we demonstrated that D2AR activation elevates DAT surface expression. Moreover, Val559 slices displayed basal elevation in surface DAT that is normalized by D2AR antagonism (Fig. 1). Together, these results suggest that Val559 ADE in the DS drives tonic elevations in surface DAT via D2AR-induction of DAT trafficking. In the VS, where D2AR-DAT regulation is absent, Val559 ADE and subsequent D2AR effects are greatly diminished or absent.
To complement our trafficking studies, we performed [3H]DA uptake in DS slices, and observed a striking reduction in Val559 [3H]DA uptake compared with WT (Fig. 2). To assess the impact of Val559 on in vivo DA clearance we chose HSC, where DA is applied exogenously, over other approaches (e.g., fast-scan cyclic voltammetry) due to studies indicating rapid DAT trafficking following electrical stimulation (Richardson et al., 2016). Our recordings demonstrated Michaelis–Menten kinetics for DA clearance at concentrations expected to reflect the physiological range of DAT function. Consistent with slice uptake, we found decreased DA clearance in Val559 DS compared with WT DS (Fig. 3). We could reconcile these seemingly contradictory findings if these measures are impacted by competition of exogenous DA with endogenous, ADE-generated DA. The detection of an increase in DA KM, as well as the inability of D2AR inhibition to affect DA clearance in DS (Fig. 4), supports this hypothesis. As with our surface DAT results, we failed to detect a genotype effect on DA clearance in the VS. Together, these findings suggest that an intrinsic lack of D2AR–DAT coupling in the VS precludes an ability of Val559 to establish ADE. Additionally, they indicate that any released DA in the DS is inefficiently cleared by DAT, despite elevated surface levels.
In vitro studies indicate that the conformational changes biasing Val559 to ADE rely on N-terminal phosphorylation at a cluster of distal Ser residues (Ramamoorthy et al., 2011) and Thr53, located proximal to the membrane within a MAPK consensus site (Gorentla et al., 2009). As ERK1/2 signaling participates in D2AR-DAT trafficking (Bolan et al., 2007) and DA efflux (Foster et al., 2012), we assessed whether changes in p-Thr53 are linked to D2AR-DAT regulation. Similar to our previous observations, D2AR activation elevated p-Thr53 levels. Moreover, these levels were significantly elevated in Val559 DS relative to WT and could be reversed by D2AR inhibition, whereas the VS displayed no genotype differences (Fig. 5). These findings compel further studies to assess an in vivo requirement for elevated p-Thr53 in Val559-ADE and/or D2AR-modulation of DAT trafficking. They also suggest that a lack of regulation of DAT by D2AR in the VS compared with the DS, may arise from differences in D2AR signaling to DAT via ERK1/2. Interestingly, a recent study (Calipari et al., 2017) found estrus cycle-dependent elevations in p-Thr53 in the VS associated with enhanced reward, warranting further studies into sex-dependent mechanisms that elevate DAT p-Thr53 and their convergence with D2AR-linked pathways. In this regard, DAT can also be regulated by other presynaptic G-protein-coupled receptors (Opazo et al., 2010; Kivell et al., 2014). Future studies should consider whether DAT regulation by heteroreceptors in the DS is impacted by tonic D2AR activation.
The absence in D2AR control of DAT regulation does not necessarily indicate an altogether lack of functional D2ARs. Prior studies have shown that D2ARs on VS DA terminals control DA synthesis (Onali and Olianas, 1989) and release (Holroyd et al., 2015). Notably, we found that D2AR inhibits TH activity comparably in WT DS and VS slices, though regional differences emerged in genotype comparisons. Thus, TH activity in Val559 DS, but not VS, was insensitive to D2AR activation (Fig. 6). D2AR can downregulate TH activity through a reduction in PKA-dependent TH phosphorylation (Dunkley et al., 2004). Compromised D2AR signaling through this pathway could involve D2AR desensitization by ADE-derived DA. In this regard, Jones et al. (1999) demonstrated a similar lack of TH control by D2AR in the DAT KO, suggesting a loss of D2AR function. In this case, our ability to reverse changes in Val559-mediated DAT phosphorylation, surface trafficking and DA clearance (this study), and DA release via acute D2AR antagonism (Mergy et al., 2014b) provides evidence, the first to our knowledge, that D2ARs regulating DA transport are distinct from those regulating synthesis. Distinct D2AR populations could derive from differential receptor post-translational modifications, localization with proteins, or pools of D2AR-accessible G-protein subunits, some of which have been shown to support DAT-mediated DA efflux (Garcia-Olivares et al., 2017). D2AR associates with DAT (Lee et al., 2007), with potential disease relevance (Lee et al., 2009); however, reports suggest that this interaction is static and not influenced by D2AR stimulation. Whether such associations preclude D2AR engagement of G-protein signaling mechanisms that regulate TH are unclear, but an important area for future studies. Importantly, evidence that D2ARs in the Val559 DS fail to provide negative feedback control of DA synthesis indicates that cytosolic DA, unabated by feedback inhibition, can continue to sustain ADE.
Our report adds to a growing appreciation of the inherent diversity of midbrain DA neurons and their projections. Approaches to assess in vivo DAT function in DA projections without reliance on membrane stimulation have yet to be demonstrated. Nonetheless, striatum-projecting DA neurons that populate the SNpc and VTA, preferentially project to the DS and VS respectively and exhibit differences in afferent inputs (Watabe-Uchida et al., 2012), modulation by neurotransmitters and peptides (Zhang et al., 2009; Xiao et al., 2017), and intrinsic electrophysiological properties (Gantz et al., 2018). Further microheterogeneity has emerged from studies showing diverse properties of DA projections to dorsomedial versus dorsolateral striatum (Lerner et al., 2015), nucleus accumbens core versus shell (Shin et al., 2017), and striatal striosome versus patch matrices (Salinas et al., 2016). Here, we demonstrate region-specific differences in presynaptic D2AR regulation of DAT, which, in the Val559 DS, leads to tonic activation of presynaptic D2AR, elevated DAT trafficking, DA availability unabated by feedback inhibition, increased ADE, and inefficient DA clearance. Functionally, these changes are predicted to shift DA terminals in the DS away from reliance on tightly controlled excitation-secretion mechanisms (Fig. 7).
Figure 7.

D2AR and DAT are functionally coupled selectively in the DS, biasing Val559 phenotypic penetrance. A, In WT DS, D2AR control over DAT plays an important role in the termination of DA signaling, in addition to D2AR-induced inhibition of DA synthesis and release. B, Functional D2AR–DAT coupling selectively in the DS results in the upregulation of Val559 DAT regulation desensitization of D2AR coupled to TH, biasing the enhancement of efflux-prone transporters that amplify ADE unabated by D2AR feedback synthesis inhibition, ultimately compromising presynaptic DA signaling. However, at (C) VS DA terminals, DAT lacks D2AR regulation. Hence, (D) despite Val559 expression, the lack of D2AR–DAT coupling in the VS leaves DAT regulation, DA clearance and TH activity unperturbed.
A desire to understand underlying molecular and circuit-level mechanisms that drive specific behavioral traits has inspired creation of the Research Domain Criteria framework (Insel et al., 2010). To pursue its goals, targeted studies aimed at delineating potentially different pathways that ultimately drive behavioral heterogeneity in disease are needed, but difficult to pursue in humans. At a circuit level, DA projections to the DS support movement and habit formation whereas those to the VS underlie motivation and response to reward. We have found that Val559 mice exhibit altered spontaneous and psychostimulant-induced locomotion and impulsivity, behavioral abnormalities that could arise through DS-specific ADE, though we have not analyzed the contribution of Val559 in the cortex. We have also found changes in motivation for reward, which we speculate may derive from both a lack of effect of Val559 on novelty and value-sensitive VS circuitry, and/or an inability for behavior to shift from responses to the positive salience of the reward to habit-based performance.
Footnotes
This work was supported by the Vanderbilt International Scholars Program and Vanderbilt Brain Institute (R.G.), an Elaine Sanders-Bush Scholar's Award from the Vanderbilt Silvio O. Conte Center for Neuroscience Research (G.L.D.) and NIH awards MH107132 (G.L.D.) and MH105094 (R.D.B). We thank Christina Svitek, Jane Wright, Tracey Moore-Jarrett, Sarah Sturgeon, Qiao Han, and Angela Steele at Vanderbilt, and Matthew Gross, Peter Rodriguez, Rania Katamish, and Erika Castriz at Florida Atlantic University for expert laboratory support; and Nicole Baganz and Matthew Robson for training and advice on chronoamperometry methods. R.G. is a Ph.D. candidate of the Vanderbilt Neuroscience Graduate Program.
The authors declare no competing financial interests.
References
- Ashok AH, Marques TR, Jauhar S, Nour MM, Goodwin GM, Young AH, Howes OD (2017) The dopamine hypothesis of bipolar affective disorder: the state of the art and implications for treatment. Mol Psychiatry 22:666–679. 10.1038/mp.2017.16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bello EP, Mateo Y, Gelman DM, Noaín D, Shin JH, Low MJ, Alvarez VA, Lovinger DM, Rubinstein M (2011) Cocaine supersensitivity and enhanced motivation for reward in mice lacking dopamine D2 autoreceptors. Nat Neurosci 14:1033–1038. 10.1038/nn.2862 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benoit-Marand M, Ballion B, Borrelli E, Boraud T, Gonon F (2011) Inhibition of dopamine uptake by D2 antagonists: an in vivo study. J Neurochem 116:449–458. 10.1111/j.1471-4159.2010.07125.x [DOI] [PubMed] [Google Scholar]
- Bermingham DP, Blakely RD (2016) Kinase-dependent regulation of monoamine neurotransmitter transporters. Pharmacol Rev 68:888–953. 10.1124/pr.115.012260 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolan EA, Kivell B, Jaligam V, Oz M, Jayanthi LD, Han Y, Sen N, Urizar E, Gomes I, Devi LA, Ramamoorthy S, Javitch JA, Zapata A, Shippenberg TS (2007) D2 receptors regulate dopamine transporter function via an extracellular signal-regulated kinases 1 and 2-dependent and phosphoinositide 3 kinase-independent mechanism. Mol Pharmacol 71:1222–1232. 10.1124/mol.106.027763 [DOI] [PubMed] [Google Scholar]
- Bowton E, Saunders C, Erreger K, Sakrikar D, Matthies HJ, Sen N, Jessen T, Colbran RJ, Caron MG, Javitch JA, Blakely RD, Galli A (2010) Dysregulation of dopamine transporters via dopamine D2 autoreceptors triggers anomalous dopamine efflux associated with attention-deficit hyperactivity disorder. J Neurosci 30:6048–6057. 10.1523/JNEUROSCI.5094-09.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowton E, Saunders C, Reddy IA, Campbell NG, Hamilton PJ, Henry LK, Coon H, Sakrikar D, Veenstra-VanderWeele JM, Blakely RD, Sutcliffe J, Matthies HJ, Erreger K, Galli A (2014) SLC6A3 coding variant Ala559Val found in two autism probands alters dopamine transporter function and trafficking. Transl Psychiatry 4:e464. 10.1038/tp.2014.90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calipari ES, Juarez B, Morel C, Walker DM, Cahill ME, Ribeiro E, Roman-Ortiz C, Ramakrishnan C, Deisseroth K, Han MH, Nestler EJ (2017) Dopaminergic dynamics underlying sex-specific cocaine reward. Nat Commun 8:13877. 10.1038/ncomms13877 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cass WA, Gerhardt GA (1994) Direct in vivo evidence that D2 dopamine receptors can modulate dopamine uptake. Neurosci Lett 176:259–263. 10.1016/0304-3940(94)90096-5 [DOI] [PubMed] [Google Scholar]
- Challasivakanaka S, Zhen J, Smith ME, Reith MEA, Foster JD, Vaughan RA (2017) Dopamine transporter phosphorylation site threonine 53 is stimulated by amphetamines and regulates dopamine transport, efflux, and cocaine analog binding. J Biol Chem 292:19066–19075. 10.1074/jbc.M117.787002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen R, Daining CP, Sun H, Fraser R, Stokes SL, Leitges M, Gnegy ME (2013) Protein kinase Cβ is a modulator of the dopamine D2 autoreceptor-activated trafficking of the dopamine transporter. J Neurochem 125:663–672. 10.1111/jnc.12229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chuhma N, Mingote S, Kalmbach A, Yetnikoff L, Rayport S (2017) Heterogeneity in dopamine neuron synaptic actions across the striatum and its relevance for schizophrenia. Biol Psychiatry 81:43–51. 10.1016/j.biopsych.2016.07.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cook EH Jr, Stein MA, Krasowski MD, Cox NJ, Olkon DM, Kieffer JE, Leventhal BL (1995) Association of attention-deficit disorder and the dopamine transporter gene. Am J Hum Genet 56:993–998. [PMC free article] [PubMed] [Google Scholar]
- Davis GL, Stewart A, Stanwood GD, Gowrishankar R, Hahn MK, Blakely RD (2018) Functional coding variation in the presynaptic dopamine transporter associated with neuropsychiatric disorders drives enhanced motivation and context-dependent impulsivity in mice. Behav Brain Res 337:61–69. 10.1016/j.bbr.2017.09.043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dickinson SD, Sabeti J, Larson GA, Giardina K, Rubinstein M, Kelly MA, Grandy DK, Low MJ, Gerhardt GA, Zahniser NR (1999) Dopamine D2 receptor-deficient mice exhibit decreased dopamine transporter function but no changes in dopamine release in dorsal striatum. J Neurochem 72:148–156. 10.1046/j.1471-4159.1999.0720148.x [DOI] [PubMed] [Google Scholar]
- Dunkley PR, Bobrovskaya L, Graham ME, von Nagy-Felsobuki EI, Dickson PW (2004) Tyrosine hydroxylase phosphorylation: regulation and consequences. J Neurochem 91:1025–1043. 10.1111/j.1471-4159.2004.02797.x [DOI] [PubMed] [Google Scholar]
- Everitt BJ, Robbins TW (2016) Drug addiction: updating actions to habits to compulsions ten years on. Annu Rev Psychol 67:23–50. 10.1146/annurev-psych-122414-033457 [DOI] [PubMed] [Google Scholar]
- Foster JD, Vaughan RA (2017) Phosphorylation mechanisms in dopamine transporter regulation. J Chem Neuroanat 83–84:10–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foster JD, Pananusorn B, Vaughan RA (2002) Dopamine transporters are phosphorylated on N-terminal serines in rat striatum. J Biol Chem 277:25178–25186. 10.1074/jbc.M200294200 [DOI] [PubMed] [Google Scholar]
- Foster JD, Cervinski MA, Gorentla BK, Vaughan RA (2006) Regulation of the dopamine transporter by phosphorylation. Handb Exp Pharmacol 175:197–214. 10.1007/3-540-29784-7_10 [DOI] [PubMed] [Google Scholar]
- Foster JD, Yang JW, Moritz AE, Challasivakanaka S, Smith MA, Holy M, Wilebski K, Sitte HH, Vaughan RA (2012) Dopamine transporter phosphorylation site threonine 53 regulates substrate reuptake and amphetamine-stimulated efflux. J Biol Chem 287:29702–29712. 10.1074/jbc.M112.367706 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gabriel LR, Wu S, Melikian HE (2014) Brain slice biotinylation: an ex vivo approach to measure region-specific plasma membrane protein trafficking in adult neurons. J Vis Exp 86:e51240. 10.3791/51240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gantz SC, Ford CP, Morikawa H, Williams JT (2018) The evolving understanding of dopamine neurons in the substantia nigra and ventral tegmental area. Annu Rev Physiol 80:219–241. 10.1146/annurev-physiol-021317-121615 [DOI] [PubMed] [Google Scholar]
- Garcia-Olivares J, Baust T, Harris S, Hamilton P, Galli A, Amara SG, Torres GE (2017) Gbetagamma subunit activation promotes dopamine efflux through the dopamine transporter. Mol Psychiatry 22:1673–1679. 10.1038/mp.2017.176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giros B, Jaber M, Jones SR, Wightman RM, Caron MG (1996) Hyperlocomotion and indifference to cocaine and amphetamine in mice lacking the dopamine transporter. Nature 379:606–612. 10.1038/379606a0 [DOI] [PubMed] [Google Scholar]
- Gorentla BK, Moritz AE, Foster JD, Vaughan RA (2009) Proline-directed phosphorylation of the dopamine transporter N-terminal domain. Biochemistry 48:1067–1076. 10.1021/bi801696n [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gowrishankar R, Hahn MK, Blakely RD (2014) Good riddance to dopamine: roles for the dopamine transporter in synaptic function and dopamine-associated brain disorders. Neurochem Int 73:42–48. 10.1016/j.neuint.2013.10.016 [DOI] [PubMed] [Google Scholar]
- Grace AA. (2016) Dysregulation of the dopamine system in the pathophysiology of schizophrenia and depression. Nat Rev Neurosci 17:524–532. 10.1038/nrn.2016.57 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grünhage F, Schulze TG, Müller DJ, Lanczik M, Franzek E, Albus M, Borrmann-Hassenbach M, Knapp M, Cichon S, Maier W, Rietschel M, Propping P, Nöthen MM (2000) Systematic screening for DNA sequence variation in the coding region of the human dopamine transporter gene (DAT1). Mol Psychiatry 5:275–282. 10.1038/sj.mp.4000711 [DOI] [PubMed] [Google Scholar]
- Hamilton PJ, Campbell NG, Sharma S, Erreger K, Herborg Hansen F, Saunders C, Belovich AN, Sahai MA, Cook EH, Gether U, McHaourab HS, Matthies HJ, Sutcliffe JS, Galli A (2013) De novo mutation in the dopamine transporter gene associates dopamine dysfunction with autism spectrum disorder. Mol Psychiatry 18:1315–1323. 10.1038/mp.2013.102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen FH, Skjørringe T, Yasmeen S, Arends NV, Sahai MA, Erreger K, Andreassen TF, Holy M, Hamilton PJ, Neergheen V, Karlsborg M, Newman AH, Pope S, Heales SJ, Friberg L, Law I, Pinborg LH, Sitte HH, Loland C, Shi L, et al. (2014) Missense dopamine transporter mutations associate with adult parkinsonism and ADHD. J Clin Invest 124:3107–3120. 10.1172/JCI73778 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holroyd KB, Adrover MF, Fuino RL, Bock R, Kaplan AR, Gremel CM, Rubinstein M, Alvarez VA (2015) Loss of feedback inhibition via D2 autoreceptors enhances acquisition of cocaine taking and reactivity to drug-paired cues. Neuropsychopharmacology 40:1495–1509. 10.1038/npp.2014.336 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hornykiewicz O. (1962) Dopamine (3-hydroxytyramine) in the central nervous system and its relation to the parkinson syndrome in man [in German]. Dtsch Med Wochenschr 87:1807–1810. 10.1055/s-0028-1114024 [DOI] [PubMed] [Google Scholar]
- Howe MW, Dombeck DA (2016) Rapid signalling in distinct dopaminergic axons during locomotion and reward. Nature 535:505–510. 10.1038/nature18942 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howes OD, McCutcheon R, Owen MJ, Murray RM (2017) The role of genes, stress, and dopamine in the development of schizophrenia. Biol Psychiatry 81:9–20. 10.1016/j.biopsych.2016.07.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Insel T, Cuthbert B, Garvey M, Heinssen R, Pine DS, Quinn K, Sanislow C, Wang P (2010) Research domain criteria (RDoC): toward a new classification framework for research on mental disorders. Am J Psychiatry 167:748–751. 10.1176/appi.ajp.2010.09091379 [DOI] [PubMed] [Google Scholar]
- Jeste SS, Geschwind DH (2014) Disentangling the heterogeneity of autism spectrum disorder through genetic findings. Nat Rev Neurol 10:74–81. 10.1038/nrneurol.2013.278 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones SR, Gainetdinov RR, Jaber M, Giros B, Wightman RM, Caron MG (1998) Profound neuronal plasticity in response to inactivation of the dopamine transporter. Proc Natl Acad Sci U S A 95:4029–4034. 10.1073/pnas.95.7.4029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones SR, Gainetdinov RR, Hu XT, Cooper DC, Wightman RM, White FJ, Caron MG (1999) Loss of autoreceptor functions in mice lacking the dopamine transporter. Nat Neurosci 2:649–655. 10.1038/10204 [DOI] [PubMed] [Google Scholar]
- Kivell B, Uzelac Z, Sundaramurthy S, Rajamanickam J, Ewald A, Chefer V, Jaligam V, Bolan E, Simonson B, Annamalai B, Mannangatti P, Prisinzano TE, Gomes I, Devi LA, Jayanthi LD, Sitte HH, Ramamoorthy S, Shippenberg TS (2014) Salvinorin A regulates dopamine transporter function via a kappa opioid receptor and ERK1/2-dependent mechanism. Neuropharmacology 86:228–240. 10.1016/j.neuropharm.2014.07.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurian MA, Zhen J, Cheng SY, Li Y, Mordekar SR, Jardine P, Morgan NV, Meyer E, Tee L, Pasha S, Wassmer E, Heales SJ, Gissen P, Reith ME, Maher ER (2009) Homozygous loss-of-function mutations in the gene encoding the dopamine transporter are associated with infantile parkinsonism-dystonia. J Clin Invest 119:1595–1603. 10.1172/JCI39060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee FJ, Pei L, Moszczynska A, Vukusic B, Fletcher PJ, Liu F (2007) Dopamine transporter cell surface localization facilitated by a direct interaction with the dopamine D2 receptor. EMBO J 26:2127–2136. 10.1038/sj.emboj.7601656 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee FJ, Pei L, Liu F (2009) Disruption of the dopamine transporter-dopamine D2 receptor interaction in schizophrenia. Synapse 63:710–712. 10.1002/syn.20648 [DOI] [PubMed] [Google Scholar]
- Lerner TN, Shilyansky C, Davidson TJ, Evans KE, Beier KT, Zalocusky KA, Crow AK, Malenka RC, Luo L, Tomer R, Deisseroth K (2015) Intact-brain analyses reveal distinct information carried by SNc dopamine subcircuits. Cell 162:635–647. 10.1016/j.cell.2015.07.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindgren N, Xu ZQ, Herrera-Marschitz M, Haycock J, Hökfelt T, Fisone G (2001) Dopamine D2 receptors regulate tyrosine hydroxylase activity and phosphorylation at Ser40 in rat striatum. Eur J Neurosci 13:773–780. 10.1046/j.0953-816x.2000.01443.x [DOI] [PubMed] [Google Scholar]
- Matsumoto M, Hikosaka O (2009) Two types of dopamine neuron distinctly convey positive and negative motivational signals. Nature 459:837–841. 10.1038/nature08028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazei-Robison MS, Blakely RD (2005) Expression studies of naturally occurring human dopamine transporter variants identifies a novel state of transporter inactivation associated with Val382Ala. Neuropharmacology 49:737–749. 10.1016/j.neuropharm.2005.08.012 [DOI] [PubMed] [Google Scholar]
- Mazei-Robinson MS, Blakely RD (2006) ADHD and the dopamine transporter: are there reasons to pay attention? Handb Exp Pharmacol 175:373–415. 10.1007/3-540-29784-7_17 [DOI] [PubMed] [Google Scholar]
- Mazei-Robison MS, Couch RS, Shelton RC, Stein MA, Blakely RD (2005) Sequence variation in the human dopamine transporter gene in children with attention deficit hyperactivity disorder. Neuropharmacology 49:724–736. 10.1016/j.neuropharm.2005.08.003 [DOI] [PubMed] [Google Scholar]
- Mazei-Robison MS, Bowton E, Holy M, Schmudermaier M, Freissmuth M, Sitte HH, Galli A, Blakely RD (2008) Anomalous dopamine release associated with a human dopamine transporter coding variant. J Neurosci 28:7040–7046. 10.1523/JNEUROSCI.0473-08.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mergy MA, Gowrishankar R, Davis GL, Jessen TN, Wright J, Stanwood GD, Hahn MK, Blakely RD (2014a) Genetic targeting of the amphetamine and methylphenidate-sensitive dopamine transporter: on the path to an animal model of attention-deficit hyperactivity disorder. Neurochem Int 73:56–70. 10.1016/j.neuint.2013.11.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mergy MA, Gowrishankar R, Gresch PJ, Gantz SC, Williams J, Davis GL, Wheeler CA, Stanwood GD, Hahn MK, Blakely RD (2014b) The rare DAT coding variant Val559 perturbs DA neuron function, changes behavior, and alters in vivo responses to psychostimulants. Proc Natl Acad Sci U S A 111:E4779–4788. 10.1073/pnas.1417294111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng J, Zhen J, Meyer E, Erreger K, Li Y, Kakar N, Ahmad J, Thiele H, Kubisch C, Rider NL, Morton DH, Strauss KA, Puffenberger EG, D'Agnano D, Anikster Y, Carducci C, Hyland K, Rotstein M, Leuzzi V, Borck G, et al. (2014) Dopamine transporter deficiency syndrome: phenotypic spectrum from infancy to adulthood. Brain 137:1107–1119. 10.1093/brain/awu022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Onali P, Olianas MC (1989) Involvement of adenylate cyclase inhibition in dopamine autoreceptor regulation of tyrosine hydroxylase in rat nucleus accumbens. Neurosci Lett 102:91–96. 10.1016/0304-3940(89)90313-3 [DOI] [PubMed] [Google Scholar]
- Opazo F, Schulz JB, Falkenburger BH (2010) PKC links Gq-coupled receptors to DAT-mediated dopamine release. J Neurochem 114:587–596. 10.1111/j.1471-4159.2010.06788.x [DOI] [PubMed] [Google Scholar]
- Owens WA, Williams JM, Saunders C, Avison MJ, Galli A, Daws LC (2012) Rescue of dopamine transporter function in hypoinsulinemic rats by a D2 receptor–ERK-dependent mechanism. J Neurosci 32:2637–2647. 10.1523/JNEUROSCI.3759-11.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parker NF, Cameron CM, Taliaferro JP, Lee J, Choi JY, Davidson TJ, Daw ND, Witten IB (2016) Reward and choice encoding in terminals of midbrain dopamine neurons depends on striatal target. Nat Neurosci 19:845–854. 10.1038/nn.4287 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pavăl D. (2017) A dopamine hypothesis of autism spectrum disorder. Dev Neurosci 39:355–360. 10.1159/000478725 [DOI] [PubMed] [Google Scholar]
- Ramamoorthy S, Shippenberg TS, Jayanthi LD (2011) Regulation of monoamine transporters: role of transporter phosphorylation. Pharmacol Ther 129:220–238. 10.1016/j.pharmthera.2010.09.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richardson BD, Saha K, Krout D, Cabrera E, Felts B, Henry LK, Swant J, Zou MF, Newman AH, Khoshbouei H (2016) Membrane potential shapes regulation of dopamine transporter trafficking at the plasma membrane. Nat Commun 7:10423. 10.1038/ncomms10423 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robbins TW. (2003) Dopamine and cognition. Curr Opin Neurol 16:S1–2. 10.1097/00019052-200312002-00001 [DOI] [PubMed] [Google Scholar]
- Roeper J. (2013) Dissecting the diversity of midbrain dopamine neurons. Trends Neurosci 36:336–342. 10.1016/j.tins.2013.03.003 [DOI] [PubMed] [Google Scholar]
- Sakrikar D, Mazei-Robison MS, Mergy MA, Richtand NW, Han Q, Hamilton PJ, Bowton E, Galli A, Veenstra-Vanderweele J, Gill M, Blakely RD (2012) Attention deficit/hyperactivity disorder-derived coding variation in the dopamine transporter disrupts microdomain targeting and trafficking regulation. J Neurosci 32:5385–5397. 10.1523/JNEUROSCI.6033-11.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salamone JD. (1992) Complex motor and sensorimotor functions of striatal and accumbens dopamine: involvement in instrumental behavior processes. Psychopharmacology (Berl) 107:160–174. 10.1007/BF02245133 [DOI] [PubMed] [Google Scholar]
- Salinas AG, Davis MI, Lovinger DM, Mateo Y (2016) Dopamine dynamics and cocaine sensitivity differ between striosome and matrix compartments of the striatum. Neuropharmacology 108:275–283. 10.1016/j.neuropharm.2016.03.049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sesack SR, Aoki C, Pickel VM (1994) Ultrastructural localization of D2 receptor-like immunoreactivity in midbrain dopamine neurons and their striatal targets. J Neurosci 14:88–106. 10.1523/JNEUROSCI.14-01-00088.1994 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin JH, Adrover MF, Alvarez VA (2017) Distinctive modulation of dopamine release in the nucleus accumbens shell mediated by dopamine and acetylcholine receptors. J Neurosci 37:11166–11180. 10.1523/JNEUROSCI.0596-17.2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slifstein M, van de Giessen E, Van Snellenberg J, Thompson JL, Narendran R, Gil R, Hackett E, Girgis R, Ojeil N, Moore H, D'Souza D, Malison RT, Huang Y, Lim K, Nabulsi N, Carson RE, Lieberman JA, Abi-Dargham A (2015) Deficits in prefrontal cortical and extrastriatal dopamine release in schizophrenia: a positron emission tomographic functional magnetic resonance imaging study. JAMA Psychiatry 72:316–324. 10.1001/jamapsychiatry.2014.2414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sulzer D, Chen TK, Lau YY, Kristensen H, Rayport S, Ewing A (1995) Amphetamine redistributes dopamine from synaptic vesicles to the cytosol and promotes reverse transport. J Neurosci 15:4102–4108. 10.1523/JNEUROSCI.15-05-04102.1995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaughan RA, Foster JD (2013) Mechanisms of dopamine transporter regulation in normal and disease states. Trends Pharmacol Sci 34:489–496. 10.1016/j.tips.2013.07.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volkow ND, Morales M (2015) The brain on drugs: from reward to addiction. Cell 162:712–725. 10.1016/j.cell.2015.07.046 [DOI] [PubMed] [Google Scholar]
- Volkow ND, Wang GJ, Newcorn J, Fowler JS, Telang F, Solanto MV, Logan J, Wong C, Ma Y, Swanson JM, Schulz K, Pradhan K (2007) Brain dopamine transporter levels in treatment and drug naive adults with ADHD. Neuroimage 34:1182–1190. 10.1016/j.neuroimage.2006.10.014 [DOI] [PubMed] [Google Scholar]
- Wang YM, Gainetdinov RR, Fumagalli F, Xu F, Jones SR, Bock CB, Miller GW, Wightman RM, Caron MG (1997) Knock-out of the vesicular monoamine transporter 2 gene results in neonatal death and supersensitivity to cocaine and amphetamine. Neuron 19:1285–1296. 10.1016/S0896-6273(00)80419-5 [DOI] [PubMed] [Google Scholar]
- Watabe-Uchida M, Zhu L, Ogawa SK, Vamanrao A, Uchida N (2012) Whole-brain mapping of direct inputs to midbrain dopamine neurons. Neuron 74:858–873. 10.1016/j.neuron.2012.03.017 [DOI] [PubMed] [Google Scholar]
- Wise RA. (2004) Dopamine, learning and motivation. Nat Rev Neurosci 5:483–494. 10.1038/nrn1406 [DOI] [PubMed] [Google Scholar]
- Wu S, Bellve KD, Fogarty KE, Melikian HE (2015) Ack1 is a dopamine transporter endocytic brake that rescues a trafficking-dysregulated ADHD coding variant. Proc Natl Acad Sci U S A 112:15480–15485. 10.1073/pnas.1512957112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao L, Priest MF, Nasenbeny J, Lu T, Kozorovitskiy Y (2017) Biased oxytocinergic modulation of midbrain dopamine systems. Neuron 95:368–384.e365. 10.1016/j.neuron.2017.06.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang T, Zhang L, Liang Y, Siapas AG, Zhou FM, Dani JA (2009) Dopamine signaling differences in the nucleus accumbens and dorsal striatum exploited by nicotine. J Neurosci 29:4035–4043. 10.1523/JNEUROSCI.0261-09.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou QY, Palmiter RD (1995) Dopamine-deficient mice are severely hypoactive, adipsic, and aphagic. Cell 83:1197–1209. 10.1016/0092-8674(95)90145-0 [DOI] [PubMed] [Google Scholar]