

Abstract
The present study demonstrates HIV-1 Tat-mediated epigenetic downregulation of microglial miR-124 and its association with microglial activation. Exposure of mouse primary microglia isolated from newborn pups of either sex to HIV-1 Tat resulted in decreased expression of primary miR-124-1, primary miR-124-2 as well as the mature miR-124. In parallel, HIV-1 Tat exposure to mouse primary microglial cells resulted in increased expression of DNA methylation enzymes, such as DNMT1, DNMT3A, and DNMT3B, which were also accompanied by increased global DNA methylation. Bisulfite-converted genomic DNA sequencing in the HIV-1 Tat-exposed mouse primary microglial cells further confirmed increased DNA methylation of the primary miR-124-1 and primary miR-124-2 promoters. Bioinformatic analyses identified MECP2 as a novel 3′-UTR target of miR-124. This was further validated in mouse primary microglial cells wherein HIV-1 Tat-mediated downregulation of miR-124 resulted in increased expression of MECP2, leading in turn to further repression of miR-124 via the feedback loop. In addition to MECP2, miR-124 also modulated the levels of STAT3 through its binding to the 3′-UTR, leading to microglial activation. Luciferase assays and Ago2 immunoprecipitation determined the direct binding between miR-124 and 3′-UTR of both MECP2 and STAT3. Gene silencing of MECP2 and DNMT1 and overexpression of miR-124 blocked HIV-1 Tat-mediated downregulation of miR-124 and microglial activation. In vitro findings were also confirmed in the basal ganglia of SIV-infected rhesus macaques (both sexes). In summary, our findings demonstrate a novel mechanism of HIV-1 Tat-mediated activation of microglia via downregulation of miR-124, leading ultimately to increased MECP2 and STAT3 signaling.
SIGNIFICANCE STATEMENT Despite the effectiveness of combination antiretroviral therapy in controlling viremia, the CNS continues to harbor viral reservoirs. The persistence of low-level virus replication leads to the accumulation of early viral proteins, including HIV-1 Tat protein. Understanding the epigenetic/molecular mechanism(s) by which viral proteins, such as HIV-1 Tat, can activate microglia is thus of paramount importance. This study demonstrated that HIV-1 Tat-mediated DNA methylation of the miR-124 promoter leads to its downregulation with a concomitant upregulation of the MECP2-STAT3-IL6, resulting in microglial activation. These findings reveal an unexplored epigenetic/molecular mechanism(s) underlying HIV-1 Tat-mediated microglial activation, thereby providing a potential target for the development of therapeutics aimed at ameliorating microglial activation and neuroinflammation in the context of HIV-1 infection.
Keywords: DNA methylation, epigenetics, MECP2, microglia, miR-124, neuroinflammation
Introduction
Microglia, the primary brain-resident immune cells, are recognized as versatile effectors and regulators of the CNS in the context of executing both protective as well as pathogenic roles in various neurodegenerative diseases (Grubman et al., 2016; Hambardzumyan et al., 2016; Le et al., 2016; Song and Suk, 2017; Wolf et al., 2017). Under physiological conditions, microglia are dynamically surveying and providing vigilance of the surrounding environment to ensure proper CNS homeostasis (Michell-Robinson et al., 2015; Han et al., 2017; Mosser et al., 2017). Microglia can, however, be quickly activated in response to pathogenic stimuli by interactions with cell debris or microbial products. This activation leads to altered microglia functioning via phagocytosis and/or secretion of a plethora of cytokines and chemokines (Han et al., 2017). Interestingly, microglia are also the key target cells for HIV-1 infection in the CNS. HIV-1 infection leads to microglial activation and the concomitant release of several toxic viral and cellular proteins (Bansal et al., 2000).
HIV-1 Transactivator of transcription (Tat) protein is one of nine HIV-1 proteins actively secreted by HIV-1-infected cells and is an early viral protein expressed following viral entry into the cells. HIV-1 Tat released from the infected cells can be taken up by neighboring noninfected cells, such as the neurons (Bagashev and Sawaya, 2013), thereby affecting both immunological and neurological functioning within the CNS (Falkensammer et al., 2007; Mediouni et al., 2012; Maubert et al., 2015). Moreover, because combination antiretroviral therapy (cART) does not impact levels of HIV-1 Tat and the CNS is often inaccessible to the cART regimens, HIV-1 Tat has been implicated as an underlying mediator of HIV-1-associated neurocognitive disorders. Accumulating evidence indicates that HIV-1 Tat alters microglial functional dynamics by controlling the intracellular signaling cascades that regulate the levels of cAMP, intracellular Ca2+, thereby leading to the production of various neurotoxic mediators, such as ROS and proinflammatory cytokines (Minghetti et al., 2004; Bagashev and Sawaya, 2013). Additionally, HIV-1 Tat also elicits other detrimental effects in the CNS, such as neurotoxicity, aberrant cellular activation, and endothelial dysfunction (Maubert et al., 2015).
miRs belong to a class of small noncoding RNAs and function as key regulators of the post-transcriptional expression of various genes. Increasing evidence also points to the role of brain-enriched miRs in regulating gene expression, microglial quiescence, and neuronal activities (Davis et al., 2015; Yu et al., 2015). miR-124 is one such brain-enriched miR that plays crucial roles in neurogenesis, synaptic signal transmission, and glial–neuronal interactions to maintain brain homeostasis. Under basal conditions, miR-124 is abundantly expressed in both microglia and neurons (Sun et al., 2015). Indeed, Ponomarev et al. (2011) showed that increased expression of miR-124 is essential for microglial quiescence and that reduced expression of this miR is closely associated with various neuroinflammatory diseases, such as Parkinson's disease (Kanagaraj et al., 2014), dementia (Gascon et al., 2014), and multiple sclerosis (Ponomarev et al., 2011). Mechanisms mediating downregulated expression of miR-124 in inflammatory as well as other disease conditions include epigenetic changes, such as aberrant DNA hypermethylation in the promoter region of primary miR-124 (Koukos et al., 2013; Murray-Stewart et al., 2016; Roy et al., 2017). Mature miR-124 is processed from three primary miR-124; each of these primary miR-124 contains an established CpG island that can be hypermethylated as shown in some disease states (Koukos et al., 2013; Murray-Stewart et al., 2016; Roy et al., 2017). In the current study, we identified a novel epigenetic/molecular mechanism involved in HIV-1 Tat-mediated microglial activation. Our findings suggest HIV-1 Tat-mediated DNA methylation of the miR-124 promoter leads to its downregulation with a concomitant upregulation of the MECP2-STAT3-IL6 that culminates into microglial activation.
Materials and Methods
Reagents.
Endotoxin-free, HIV-1 recombinant Tat (1032–10) was purchased from ImmunoDX. 5-Aza-2′-deoxycytidine (5-Aza; A3656) was purchased from Sigma-Aldrich. TaqMan miR Reverse Transcription Kit (4366596), TaqMan miR assays for miR-124 (001182), TaqMan miR Control Assay-U6 snRNA (4427975), TaqMan Pri-miRNA Assays for miR-124-1, miR-124-2, and miR-124-3 (4427012), and TaqMan Universal PCR Master Mix, no AmpErase UNG (4324018) were purchased from Applied Biosystems. miRIDIAN miR-124-3p mimic (C-310391-05), miRIDIAN miR-124-3p hairpin inhibitor (IH-310391-07), miRIDIAN miR mimic negative control (CN-001000), and miRIDIAN miR hairpin negative control (IN-001005) were purchased from Dharmacon. 5′-DIG and 3′-DIG-labeled, miRCURY locked nucleic acid detection probe for the miR-124 probe was acquired from Exiqon (619867-360). Lipofectamine 2000 Transfection Reagent (11668019) and Opti-MEM I Reduced Serum Media (31985070) were purchased from Invitrogen. ProLong Gold Antifade Mountant with DAPI (P36935) was obtained from Invitrogen. Antibodies were attained from the following sources: CD11b (Novus catalog #NB110–89474, RRID:AB_1216361), Dnmt1 (Santa Cruz Biotechnology catalog #sc-20701, RRID:AB_2293064), Dnmt3a (Santa Cruz Biotechnology catalog #sc-20703, RRID:AB_2093990), Dnmt3b (Santa Cruz Biotechnology catalog #sc-376043, RRID:AB_10988201), p-MeCP2 (Santa Cruz Biotechnology catalog #sc-136171, RRID:AB_2144015), MeCP2 (Cell Signaling Technology catalog #3456S, RRID:AB_2143849), STAT3 (Cell Signaling Technology catalog #4904, RRID:AB_331269), p-STAT3 (Cell Signaling Technology catalog #9131, RRID:AB_331586), Iba-1 (Wako catalog #019-19741, RRID:AB_839504), Peroxidase-AffiniPure Goat Anti-Rabbit IgG (H+L) (Jackson ImmunoResearch Laboratories catalog #111-035-003, RRID:AB_2313567), and Peroxidase-conjugated AffiniPure Goat Anti-Mouse IgG (H+L) (Jackson ImmunoResearch Laboratories catalog #115-035-003, RRID:AB_10015289).
Rhesus macaques and Simian immunodeficiency virus (SIV) infection.
Briefly, both sexes of 2- to 3-year-old Indian rhesus macaques (Macaca mulatta) were purchased from the Caribbean Research Primate Center and individually housed in steel holding cages, in two dedicated rooms within the Association for Assessment and Accreditation of Laboratory Animal Care-approved animal facility at the University of Kansas Medical Center and were randomly divided into saline and SIV groups. The monkeys were daily exposed to 12 h light-dark cycles and given laboratory chow and water ad libitum along with daily snacks. All animal protocols were approved by the local animal care committee (Institutional Animal Care and Use Committee) at the University of Kansas in accordance with the Guide for the Care and Use of Laboratory Animals. The SIV group of rhesus macaques were chronically infected with SIVR71/17E for ∼52 weeks and were antiretroviral naive. At necropsy, tissues were harvested following perfusion using PBS under lethal anesthesia. A comprehensive methodology of SIV infection and information on viral load and other parameters related to disease pathogenesis has been described in our previous publications (Bokhari et al., 2011; Hu et al., 2012; Pendyala et al., 2015; Chivero et al., 2017). Archival frozen basal ganglia brain tissues, as well as formalin-fixed-paraffin-embedded basal ganglia brain tissues from saline and SIV groups, were used in this study.
Mouse primary microglial culture.
Mouse primary microglial cultures were prepared from 1- to 3-d-old newborn pups of either sex bred from C57B1/6 as specified, under standard conditions as described previously (Skaper et al., 2012) with minor modifications. Mixed cell cultures were maintained in DMEM (Corning Cellgro, 10-013-CV) containing 10% heat-inactivated FBS (Atlanta Biologicals, S11050H), and 10 U/ml penicillin-streptomycin (Invitrogen, 15140122) in a 5% CO2-humidified incubator at 37°C. Cell culture medium was changed every 5 d; and after the first medium change, macrophage colony-stimulating factor (0.25 ng/ml; Invitrogen, PHC9504) was added to the flasks to promote microglial proliferation. The confluent mixed glial cultures (∼10 d) were then subjected to shaking at 37°C at 220 × g for 2 h to promote microglial detachment from the flasks. The cell medium, comprising the detached microglia cells, was collected from each flask and centrifuged at 1000 × g for 5 min. The collected cells were plated onto 6-well cell culture plates (3 × 105 cells per well) for all ensuing experiments. Microglia purity was evaluated by immunocytochemistry using the antibody specific for Iba-1 and used if >95% pure.
miR microarray analysis.
Total RNA was isolated from the frozen basal ganglia specimens of both saline and SIV-infected rhesus macaques using TRIzol (Invitrogen, 15596018) and purified using the miRNeasy mini kit (QIAGEN, 217004) as instructed by the manufacturer. Purified total RNA was used for the Affymetrix microarray and was performed by Asuragen. Heatmaps were generated in R using the heatmap.2 function from the gplots package.
TaqMan miR assays for miR-124.
The expression of miR-124 was quantified using TaqMan miR assays as described previously (Guo et al., 2016). Briefly, total RNA was extracted using Quick-RNA MiniPrep Plus (Zymo Research, R1058) as per the manufacturer's protocol and quantified using NanoDrop 2000 spectrophotometer (Thermo Fisher Scientific). Next, the total RNA isolated from each group was reverse-transcribed to synthesize cDNA for individual miR using specific miR primers from the TaqMan miR assays and the TaqMan miR Reverse Transcription kit. Each reverse transcription reaction comprises of 7 μl master mix, 3 μl miR-specific primer (5×), and 5 μl total RNA (10 ng/μl). The tube containing the reaction mixture was briefly centrifuged and positioned onto the thermal cycler for reverse transcription. The reverse transcription product was then diluted 1:10 for the following PCR: TaqMan miR assay (20×) 1 μl, RT reaction product 1.5 μl, TaqMan 2× Universal PCR Master Mix, No AmpErase UNG 10 μl, and nuclease-free water up to 20 μl. Each reaction was performed in triplicate, and six independent experiments were run. TaqMan miR assays were performed using a 7500 Fast Real-Time PCR Systems (Applied Biosystems). The expression level of miR-124 was calculated by normalizing to U6 snRNA.
TaqMan primary miR assay for primary miR-124-1, -2, and -3.
Total RNA was column isolated from the control and treated mouse primary microglial cells using Quick-RNA MiniPrep Plus as per the manufacturer's protocol and quantified using NanoDrop 2000 spectrophotometer (Thermo Fisher Scientific). To determine the expression levels of primary miR-124-1, -2, and -3, reverse transcription and TaqMan qPCR were performed according to the TaqMan Primary miRNA Assay protocols. 18S rRNA was also assessed serving as the endogenous control. PCRs were conducted in six independent triplicates for each sample. Expression of primary miR-124-1, -2, and -3 was normalized to that of 18S rRNA. The relative expression of the primary miR-124 was quantified by the 2−ΔΔCT method.
RNAscope with standard immunohistochemistry.
mRNA expression of IL6 in the basal ganglia of saline and SIV-infected rhesus macaques was determined using a new generation state-of-the-art in situ hybridization (ISH) technology, RNAscope (Advanced Cell Diagnostics). This methodology uses a unique “double Z” probe designed to amplify target-specific signals but not background noise from nonspecific hybridization, thereby enabling the visualization of single transcripts of target RNA within intact cells. RNAscope was performed in the formalin-fixed-paraffin-embedded basal ganglia tissues of saline and SIV-infected rhesus macaques using an RNAscope Fluorescent Multiplex Reagent Kit (Advanced Cell Diagnostics, 320850) and HybEZ Hybridization System (Advanced Cell Diagnostics, 310010) according to the manufacturer's instructions. IL6 mRNA was detected using an RNAscope Made-To-Order Target Probes from Advanced Cell Diagnostics. The RNAscope assay was coupled with standard immunohistochemistry for cell-specific marker CD11b using fluorescent detection.
Transient transfection of miR-124 mimic and inhibitor.
Mouse primary microglial cells were seeded into 6-well plates (3 × 105 cells per well) and were transiently transfected with 30 pmol of miR-124 mimic, miR-124 inhibitor, and miR control using Lipofectamine 2000 as described previously (Guo et al., 2016). Following transfection, cells were exposed to HIV-1 Tat (50 ng/ml) for another 24 h, and total RNA and proteins were extracted for further investigation as indicated.
Small interfering RNA (siRNA) transfection.
Mouse primary microglial cells were transfected with mouse DNMT1 siRNA (Santa Cruz Biotechnology, sc-35203) or mouse MECP2 siRNA (Santa Cruz Biotechnology, sc-35893) using Lipofectamine 2000 Reagent (Invitrogen, 11668019) as per the manufacturer's instructions. Briefly, the mouse primary microglial cells were seeded in a 6-well plate at a density of 3 × 105 cells per well at 37°C in a humidified, 5% CO2 incubator. At 70% confluence, the culture medium was replaced with 1 ml of Opti-MEM I Reduced Serum Medium (Invitrogen, 31985070). Meanwhile, Lipofectamine 2000 Reagent (3 μl/ml) and 120 pmol/ml of individually targeted siRNA were incubated separately with Opti-MEM I Reduced Serum Medium for 5 min at room temperature. Subsequently, the Lipofectamine 2000 mix was added to the individually targeted siRNA mix; this mixture was set aside for 20 min, after which the combined mixture was added to the cells. Scrambled siRNA mixture was also prepared similarly. The culture plate was then shaken gently for 5 s and incubated for 24 h at 37°C in a 5% CO2 incubator (humidified). Knockdown efficiencies were determined by Western blotting.
qPCR.
qPCR experiments were performed according to the protocol described previously (Guo et al., 2016; Periyasamy et al., 2016). Briefly, total RNA was extracted using Quick-RNA MiniPrep Plus (Zymo Research, R1058) as per the manufacturer's protocol and quantified using NanoDrop 2000 spectrophotometer (Thermo Fisher Scientific). Reverse transcription reactions were performed using a Verso cDNA kit (AB-1453/B; Thermo Fisher Scientific), as per the manufacturer's instructions. qPCRs were completed using SYBR Green ROX qPCR Mastermix (QIAGEN, 330510). The 96-well plates were placed into an Applied Biosystems 7500 Fast Real-Time PCR Systems for a program running. Each reaction was performed in triplicate, and six independent experiments were run. GAPDH was used as a housekeeping control for the normalization, and the fold change in expression was obtained by the 2−ΔΔCT method.
Western blotting.
Western blotting was performed using standard procedures as described previously (Guo et al., 2016; Periyasamy et al., 2016). Briefly, the control and treated microglial cells were harvested and lysed using the 200 μl of RIPA buffer (Cell Signaling Technology, 9806). Lysates were centrifuged at 12000 × g for 10 min at 4°C, and the protein content of the supernatant was determined by a BCA assay using Pierce BCA Protein Assay Kit (Thermo Fisher Scientific, 23227) as per the manufacturer's instructions; 10 μg of soluble proteins was resolved in a 10% SDS-PAGE, followed by blotting onto a PVDF membrane (Millipore, IPVH00010). Then, the membranes were blocked with 5% nonfat dry milk (in 1× TTBS buffer) for 1 h at room temperature followed by overnight incubation with the indicated primary antibodies at 4°C. After washing 3 times, the membranes were incubated with a secondary antibody for 1 h at room temperature. Next, the protein signals were visualized using Super Signal West Pico Chemiluminescent Substrate (Thermo Fisher Scientific, 34078). Each band intensity was normalized to the internal control, β-actin (Sigma-Aldrich catalog #A1978, RRID:AB_476692), and the data were presented as a relative fold change by using ImageJ analysis software (Schneider et al., 2012).
miR-124 ISH and CD11b immunostaining.
miR-124 ISH was performed using the methodology described previously (Guo et al., 2016). Briefly, mouse primary microglial cells were seeded into 24-well plate containing sterile glass coverslips (11 mm; 5 × 104 cells per well) at 37°C in a humidified, 5% CO2 incubator for 24 h. Overnight serum-starved mouse primary astrocytes were then exposed with HIV-1 Tat (50 ng/ml) for the next 24 h. Later, the cells were rinsed twice with 1 × PBS at room temperature and fixed with 4% PFA followed by prehybridization with the hybridization buffer (50% formamide, 10 mm Tris-HCl, pH 8.0, 200 μg/mg yeast tRNA, 1× Denhardt's solution, 600 mm NaCl, 0.25% SDS, 1 mm EDTA, 10% dextran sulfate) at 37°C for 1 h. In the meanwhile, locked nucleic acid-modified miR-124, labeled at both the 5′ and 3′ ends with digoxigenin, was diluted to a final concentration of 2 pm in hybridization buffer (heated to 65°C for 5 min) and individually hybridized with the sections at 37°C overnight. The slides were then washed twice in the hybridization buffer (without probe) at 37°C, followed by washing 3 times in 2× SSC and twice in 0.2× SSC at 42°C. The slides were then blocked with 1% BSA, 3% normal goat serum in 1 × PBS for 1 h; after that, slides were incubated with anti-digoxigenin conjugated with HRP and anti-Iba-1 antibodies overnight at 4°C. Following this, the slides were washed twice with 1 × PBS and incubated with AlexaFluor-488-conjugated goat anti-rabbit IgG (H+L) antibody for 1 h at room temperature. This was followed by 2 washes with 1 × PBS and signal amplification using TSA Cy5 kit (PerkinElmer). The coverslips were then mounted on glass slides with ProLong Gold Antifade Reagent with DAPI. Fluorescence images were taken on an Observer using a Z1 inverted microscope (Carl Zeiss), and the acquired images were analyzed using the AxioVs 40 version 4.8.0.0 software (Carl Zeiss).
Bisulfite-converted genomic DNA sequencing.
Bisulfite-converted genomic DNA sequencing was performed using the protocol described previously with slight modifications (Palsamy et al., 2012, 2014a,b,c; Guo et al., 2016). Briefly, genomic DNA extracted from mouse primary microglial cells was exposed to bisulfite conversion by EZ DNA Methylation-Direct Kit (Zymo Research). The bisulfite-modified DNA was amplified by bisulfite sequencing PCR using Platinum PCR SuperMix High Fidelity (Invitrogen) with primers specific to mouse primary miR-124-1, primary miR-124-2, and primary miR-124-3 promoter regions. Subsequently, the amplified PCR products were purified by gel extraction with Zymoclean Gel DNA recovery kit (Zymo Research) followed by cloning into pCR4-TOPO vectors using the TOPO TA Cloning kit (Invitrogen). The recombinant plasmids were transformed into One Shot TOP10 chemically competent Escherichia coli (Invitrogen) using the conventional chemical transformation method. Plasmid DNA was isolated from ∼10 independent clones of each amplicon with PureLink Quick Plasmid Miniprep Kit (Invitrogen) and then sequenced (High-Throughput DNA Sequencing and Genotyping Core Facility, University of Nebraska Medical Center, Omaha, NE) to determine the status of CpG methylation. Only the clones with an insert containing >99.5% bisulfite conversion (i.e., nonmethylated cytosine residues to thymine) were included in this study. The sequence data of each clone were analyzed for methylation in the miR-124 promoter by BISMA software (http://services.ibc.uni-stuttgart.de/BDPC/BISMA/) using default threshold settings.
Dual-luciferase reporter assay.
Mouse primary microglial cells were seeded into 96-well plates and cotransfected with target plasmids, such as pmirGLO-MECP2 3′UTR-miR-124-target or pmirGLO-MECP2 3′UTR-miR-124-target-mutant and miR-124 mimic/miR control in a molar ratio 10:1. The luciferase activity was determined 24 h after transfection, and the reporter assay was performed according to the manufacturer's protocol (Promega). Renilla luciferase activity was normalized to firefly luciferase and was expressed as a percentage of the control.
Ago2 immunoprecipitation.
Mouse primary microglial cells were plated onto 6-well plates and transfected with either miR mimic control or miR-124 mimic for 24 h. After treatment, cells were washed in the cold 1 × PBS, scraped, and then lysed with a buffer containing 0.5% NP40, 150 mm KCL, 25 mm Tris-glycine, pH 7.5, 2 mm EDTA, 0.5 mm DTT, and inhibitors of RNases, proteases, and phosphatases; 10% of total lysate was removed and kept as the input samples, and the remainder used for immunoprecipitation. A total of 10 μg of anti-Ago2 (Sigma-Aldrich catalog #SAB4200085, RRID:AB_10600719) or anti-FLAG (Sigma-Aldrich catalog #F1804, RRID:AB_262044) antibodies were kept overnight with protein A/G agarose beads (Thermo Fisher Scientific, 20423) at 4°C. Precleared lysates were then incubated with the appropriate antibody-bound beads, and the immunoprecipitated proteins were washed and incubated with DNase I (Invitrogen, 18068015) followed by digestion with proteinase K (Zymo Research, D3001-2) for 15 min. RNA extraction was then performed using Quick-RNA MiniPrep Plus (Zymo Research, R1058) and quantified using the NanoDropTM 2000 spectrophotometer (Thermo Fisher Scientific). Total as well as Ago2-immunoprecipitated RNA samples were then used for determining the binding targets of miR-124, such as MECP2 and STAT3 by qPCR.
Experimental design and statistical analyses.
All the data were expressed as mean ± SEM, and appropriate statistical significance was chosen based on the experimental strategy using GraphPad Prism version 6.01. The precise statistical analyses and experimental designs, including tests performed, exact p values, and sample sizes, are provided with the results describing each figure, or within the legend of each figure. Nonparametric Kruskal–Wallis one-way ANOVA followed by Dunn's post hoc test was used to determine the statistical significance between multiple groups, and Wilcoxon matched-pairs signed rank test was used to compare between two groups. For the in vivo experiments, unpaired Student's t test was used to compare between two groups. In all cases, values were statistically significant when p < 0.05.
Results
SIV infection downregulates miR-124 in the basal ganglia of rhesus macaques
Based on the premise that miRs play pivotal roles in brain functioning by regulating gene expression of various CNS cells, we first sought to examine the miR expression profiles in the basal ganglia of saline (n = 4) and SIV-infected rhesus macaques (n = 6) using the Affymetrix microarray platform. Our microarray data showed that SIV infection significantly altered the expression profiles of various miRs, including 84 downregulated miRs, and 21 upregulated miRs in the basal ganglia of rhesus macaques compared with the saline group. Interestingly, most of the miRs altered during SIV infection were found to be associated with neuroinflammation (Fig. 1A). Of these, miR-124 was of interest to us as it was significantly downregulated in the basal ganglia of SIV-infected rhesus macaques compared with the saline group (Fig. 1B; p = 0.0477, unpaired Student's t test). Next, we wanted to validate the downregulation of miR-124 in the basal ganglia of SIV-infected rhesus macaques, by assessing the expression of mature miR-124 using qPCR. As represented in Figure 1C, the expression of mature miR-124 was significantly downregulated in the basal ganglia of SIV-infected rhesus macaques compared with the saline group (p = 0.0001, unpaired Student's t test). Downregulation of miR-124 was not region-specific because other brain regions, such as the cortex, also exhibited a similar downregulation of miR-124 in the SIV-infected rhesus macaques (data not shown). Collectively, these results demonstrated that SIV infection significantly decreased the expression levels of mature miR-124 in the basal ganglia of rhesus macaques.
Figure 1.

SIV infection downregulates miR-124 and increases the DNMTs levels in the basal ganglia of rhesus macaques. A, Unbiased heatmap of the expression profiles of miRNAs in the basal ganglia of saline (n = 4) and SIV-infected rhesus macaques (n = 6) (both sexes). B, Mean fluorescent intensity of miR-124, as measured by miR microarray, in the basal ganglia of saline (n = 4) and SIV-infected rhesus macaques (n = 6). C, qPCR analysis showing the significant downregulation of miR-124 expression in the basal ganglia of SIV-infected rhesus macaques compared with the saline group. D, Quantification of 5-mC using ELISA showing the increased levels of 5-mC in the basal ganglia of SIV-infected rhesus macaques compared with the saline group. E, F, Representative Western blots showing the increased levels of DNMT1, DNMT3a, and DNMT3b in the basal ganglia of SIV-infected rhesus macaques compared with the saline group. β-Actin was probed as a protein loading control for all the experiments. Data are mean ± SEM. *p < 0.05 versus saline (unpaired Student's t test).
SIV infection increases global DNA methylation and DNA methyltransferase (DNMT) expression in the basal ganglia of rhesus macaques
Based on the downregulation of mature miR-124 expression, we next determined the global methylation status of the basal ganglia by measuring the percentage of 5-methylcytosine (5-mC) in the saline and SIV-infected rhesus macaques. Interestingly, the levels of 5-mC were significantly increased in the basal ganglia of SIV-infected rhesus macaques compared with the saline group, thereby confirming increased global DNA methylation in SIV-infected rhesus macaques (Fig. 1D; p = 0.0289, unpaired Student's t test). We next sought to determine the protein expression levels of DNA methylation enzymes, such as DNMT1, DNMT3a, and DNMT3b in the basal ganglia of saline and SIV-infected rhesus macaques. As shown in Figure 1E, F, the protein expression levels of DNMT1 (p = 0.0248, unpaired Student's t test), DNMT3a (p = 0.0220, unpaired Student's t test), and DNMT3b (p = 0.0223, unpaired Student's t test) were significantly upregulated in the basal ganglia of SIV-infected rhesus macaques compared with the saline group.
HIV-1 Tat downregulates miR-124 in mouse primary microglial cells
Based on the findings that the basal ganglia of SIV-infected rhesus macaques exhibited downregulation of miR-124 (higher expression of microRNA-124 is essential for microglial quiescence), the next step was to validate these observations in the purified cultures of mouse primary microglial cells exposed to HIV-1 Tat protein (as a surrogate of HIV-1/SIV infection). Mouse primary microglial cells were exposed to varying doses of HIV-1 Tat (25, 50, 100, and 200 ng/ml; for 24 h) and assessed for the expression of mature miR-124. As shown in Figure 2A, the expression of mature miR-124 was dose-dependently downregulated in mouse primary microglial cells exposed to HIV-1 Tat (p = 0.0001, n = 6, nonparametric Kruskal–Wallis one-way ANOVA followed by Dunn's post hoc test). Based on these findings, the concentration of 50 ng/ml of HIV-1 Tat was chosen for all further experiments and is in keeping with the circulating levels of HIV-1 Tat found in serum and CSF of HIV-1-infected individuals (range, 1–40 ng/ml) (Westendorp et al., 1995; Xiao et al., 2000). Furthermore, it has also been suggested that the local extracellular concentrations of HIV-1 Tat in the CNS could be even higher, especially in the vicinity of HIV-1-infected perivascular cells (Hayashi et al., 2006). As a next step, we performed time course experiments to determine the optimal time when HIV-1 Tat downregulated miR-124 in mouse primary microglial cells. As shown in Figure 2B, exposure of mouse primary microglial cells to HIV-1 Tat significantly downregulated the expression levels of mature miR-124 starting at 3 h onwards (p = 0.0001, n = 6, nonparametric Kruskal–Wallis one-way ANOVA followed by Dunn's post hoc test). As expected, heat-inactivated HIV-1 Tat did not have any effect on mature miR-124 expression (Fig. 2C).
Figure 2.
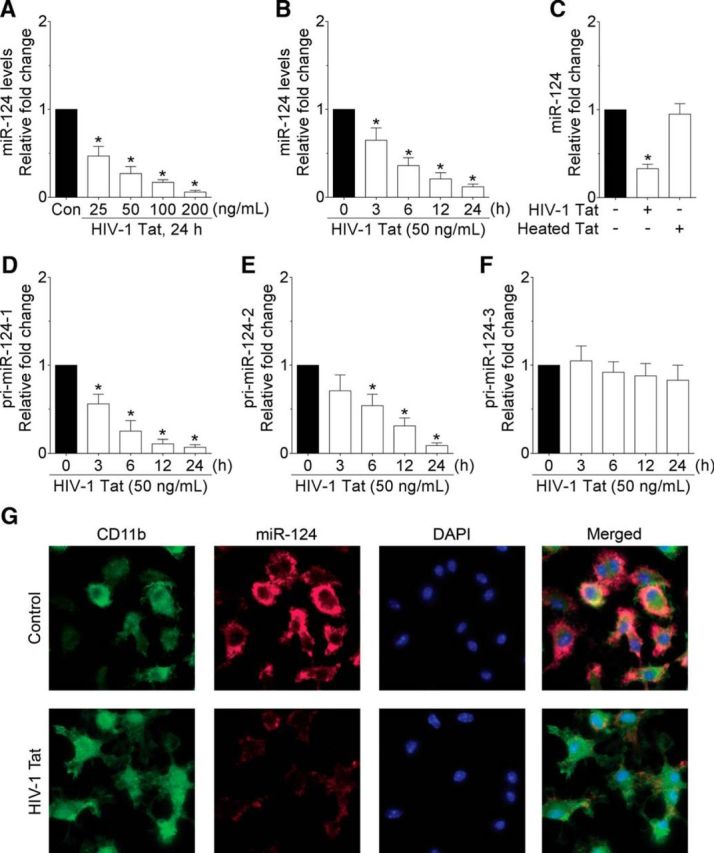
HIV-1 Tat downregulates miR-124 in mouse primary microglial cells. qPCR analysis showing the dose-dependent (A) and time-dependent (B) downregulation of miR-124 expression in HIV-1 Tat exposed to mouse primary microglial cells. C, qPCR analysis showing no change on miR-124 expression in mouse primary microglial cells exposed to heat-inactivated HIV-1 Tat (50 ng/ml) for 24 h. qPCR analysis showing the time-dependent changes on the primary miR-124-1 (D), primary miR-124-2 (E), and primary miR-124-3 (F) in HIV-1 Tat (50 ng/ml) exposed to mouse primary microglial cells. Data are mean ± SEM from six independent experiments. Nonparametric Kruskal–Wallis one-way ANOVA followed by Dunn's post hoc test was used to determine the statistical significance between multiple groups. *p < 0.05 versus control. G, ISH demonstrating decreased expression of miR-124 in HIV-1 Tat (50 ng/ml) exposed mouse primary microglial cells. −, Vehicle treatment (i.e., 1 μl 1 × PBS/ml of medium).
The next step was to determine the expression of primary miR-124s in mouse primary microglial cells exposed to HIV-1 Tat (50 ng/ml) for varying time points. As shown in Figure 2D, E, exposure of mouse primary microglial cells to HIV-1 Tat (50 ng/ml) resulted in a time-dependent decrease in the expression of primary miR-124-1 and -2 starting at 3 h onwards (p < 0.0001, n = 6, nonparametric Kruskal–Wallis one-way ANOVA followed by Dunn's post hoc test). The expression of primary miR-124-3, however, was not altered following HIV-1 Tat exposure to mouse primary microglial cells (Fig. 2F; p = 0.0917, n = 6, nonparametric Kruskal–Wallis one-way ANOVA followed by Dunn's post hoc test).
To further validate that HIV-1 Tat-mediated downregulation of miR-124 leads to microglial activation, we performed ISH for miR-124 followed by immunostaining for CD11b (microglial activation marker) in the mouse primary microglial cells exposed to HIV-1 Tat (50 ng/ml) for 12 h. As shown in Figure 2G, expression of miR-124 was significantly downregulated in mouse primary microglial cells exposed to HIV-1 Tat; this was accompanied by upregulation of microglial activation as evident by CD11b staining.
HIV-1 Tat increases global DNA methylation levels and DNMT expressions in mouse primary microglial cells
To further investigate whether the HIV-1 Tat-mediated downregulation of miR-124 in mouse primary microglial cells resulted from increased DNA methylation, we first assessed the levels of 5-mC in the HIV-1 Tat-exposed mouse microglial cells using ELISA. As expected, the percentage of 5-mC levels was significantly increased in HIV-1 Tat-exposed mouse primary microglial cells compared with the control cells (Fig. 3A; p = 0.0313, n = 6, Wilcoxon matched-pairs signed rank test), thereby underscoring the potential of HIV-1 Tat in increasing global DNA methylation in mouse primary microglial cells.
Figure 3.

HIV-1 Tat increases global DNA methylation levels and DNMT expressions in mouse primary microglial cells. A, Quantification of 5-mC using ELISA showing the increased levels of 5-mC in the HIV-1 Tat-exposed mouse primary microglial cells. Data are mean ± SEM from six independent experiments. Wilcoxon test was used to determine the statistical significance of two groups. *p < 0.05 versus control. qPCR analysis showing time-dependent expressions of DNMT1 (B), DNMT3a (C), and DNMT3b (D) mRNA in HIV-1 Tat (50 ng/ml) exposed mouse primary microglial cells. Representative Western blots showing time-dependent upregulation of DNMT1 (E, F), DNMT3a (E, G), and DNMT3b (E, H) proteins in HIV-1 Tat (50 ng/ml) exposed mouse primary microglial cells. Pretreatment of 5-Aza, an inhibitor of DNMTs, followed by exposure to HIV-1 Tat (50 ng/ml) significantly blocked the DNMT1 (I) levels with concomitant increase in miR-124 (J) levels in mouse primary microglial cells. Gene silencing of DNMT1 by DNMT1 siRNA followed by exposure to HIV-1 Tat (50 ng/ml) significantly blocked the DNMT1 (K) levels with a concomitant increase in miR-124 (L) levels in mouse primary microglial cells. Data are mean ± SEM from six independent experiments. Nonparametric Kruskal–Wallis one-way ANOVA followed by Dunn's post hoc test was used to determine the statistical significance between multiple groups. *p < 0.05 versus control. #p < 0.05 versus HIV-1 Tat. −, Vehicle treatment (i.e., 1 μl 1 × PBS/ml of medium).
Next, we investigated the mRNA expression profile of DNA methylation enzymes, DNMT1, DNMT3a, and DNMT3b in HIV-1 Tat-exposed mouse primary microglial cells using qPCR. Interestingly, as shown in Figure 3B–D, the mRNA expression of DNMT1, DNMT3a, and DNMT3b was significantly increased in a time-dependent manner in mouse primary microglial cells exposed to HIV-1 Tat (50 ng/ml, p < 0.0001, n = 6, nonparametric Kruskal–Wallis one-way ANOVA followed by Dunn's post hoc test). Subsequently, the protein expression levels of DNMT1, DNMT3a, and DNMT3b were also determined. As shown in Figure 3E–H, the protein levels of DNMT1, DNMT3a, and DNMT3b were also time-dependently increased in HIV-1 Tat-exposed mouse primary microglial cells. It is also possible that other viral proteins, such as HIV-1 gp120, could also modulate the expression of miR-124, thereby causing the effect as evident for HIV-1 Tat protein. Our pilot studies on mouse primary microglial cells exposed with HIV-1 YU2 gp120 (50 ng/ml) for 24 h, similar to HIV-1 Tat, also showed significant downregulation of miR-124 with concomitant upregulation of DNMT1, DNMT3a, and DNMT3b mRNA expression (data not shown).
We next sought to determine the expression levels of miR-124 in mouse primary microglial cells pretreated with 5-AZA, a pharmacological inhibitor of DNMTs followed by exposure of cells to HIV-1 Tat (50 ng/ml). Interestingly, pretreatment of mouse primary microglial cells with 5-AZA (5 μm) followed by exposure to HIV-1 Tat (50 ng/ml) significantly blocked HIV-1 Tat-mediated upregulation of DNMT1 levels (Fig. 3I; p = 0.0002, n = 6, nonparametric Kruskal–Wallis one-way ANOVA followed by Dunn's post hoc test). In contrast, mouse primary microglial cells pretreated with 5-AZA followed by exposure to HIV-1 Tat (50 ng/ml) exhibited significant inhibition of HIV-1 Tat-mediated downregulation of miR-124 in mouse primary microglial cells (Fig. 3J; p < 0.0001, n = 6, nonparametric Kruskal–Wallis one-way ANOVA followed by Dunn's post hoc test). Similarly, gene silencing approach using siRNA transfection of cells with DNMT1 siRNA followed by exposure to HIV-1 Tat (50 ng/ml; for 24 h) significantly blocked HIV-1 Tat-mediated increased expression of DNMT1 (Fig. 3K; p = 0.0015, n = 6, nonparametric Kruskal–Wallis one-way ANOVA followed by Dunn's post hoc test) and also exhibited significantly increased levels of miR-124 (Fig. 3L; p = 0.0001, n = 6, nonparametric Kruskal–Wallis one-way ANOVA followed by Dunn's post hoc test). It must be noted that silencing of DNMT1 did not affect the expression levels of DNMT3a and DNMT3b in mouse primary microglial cells (data not shown). Overall, these results suggested that HIV-1 Tat-mediated downregulation of miR-124 in mouse primary microglial cells involved increased DNA methylation via upregulation of DNMTs.
HIV-1 Tat increases DNA methylation of primary miR-124-1 and -2 promoters in mouse primary microglial cells
Based on these findings, we next examined whether HIV-1 Tat-mediated downregulation of miR-124 involved alterations in methylation of primary miR-124 promoter. To determine DNA methylation in primary miR-124s promoter, we performed bisulfite-converted genomic DNA sequencing for the promoters of primary miR-124-1, primary miR-124-2, and primary miR-124-3 in the genomic DNA isolated from the HIV-1 Tat (50 ng/ml) exposed mouse primary microglial cells. Using bioinformatic analyses, we found a dense CpG island in the promoter region of primary miR-124-1 (Fig. 4A), primary miR-124-2 (Fig. 4F), and primary miR-124-3 (Fig. 4K). Next, we designed 2 sets of primers to sequence ∼1 kb of the promoter region (fragments 1 and 2) of each primary miR-124 (primer sequences are shown in Table 1). As shown in Figure 4B–E, exposure of mouse primary microglial cells to HIV-1 Tat significantly increased DNA methylation of fragment 1 (p = 0.0015, n = 10, Wilcoxon matched-pairs signed rank test) and fragment 2 (p = 0.0001, n = 10, Wilcoxon matched-pairs signed rank test) of primary miR-124-1.
Figure 4.
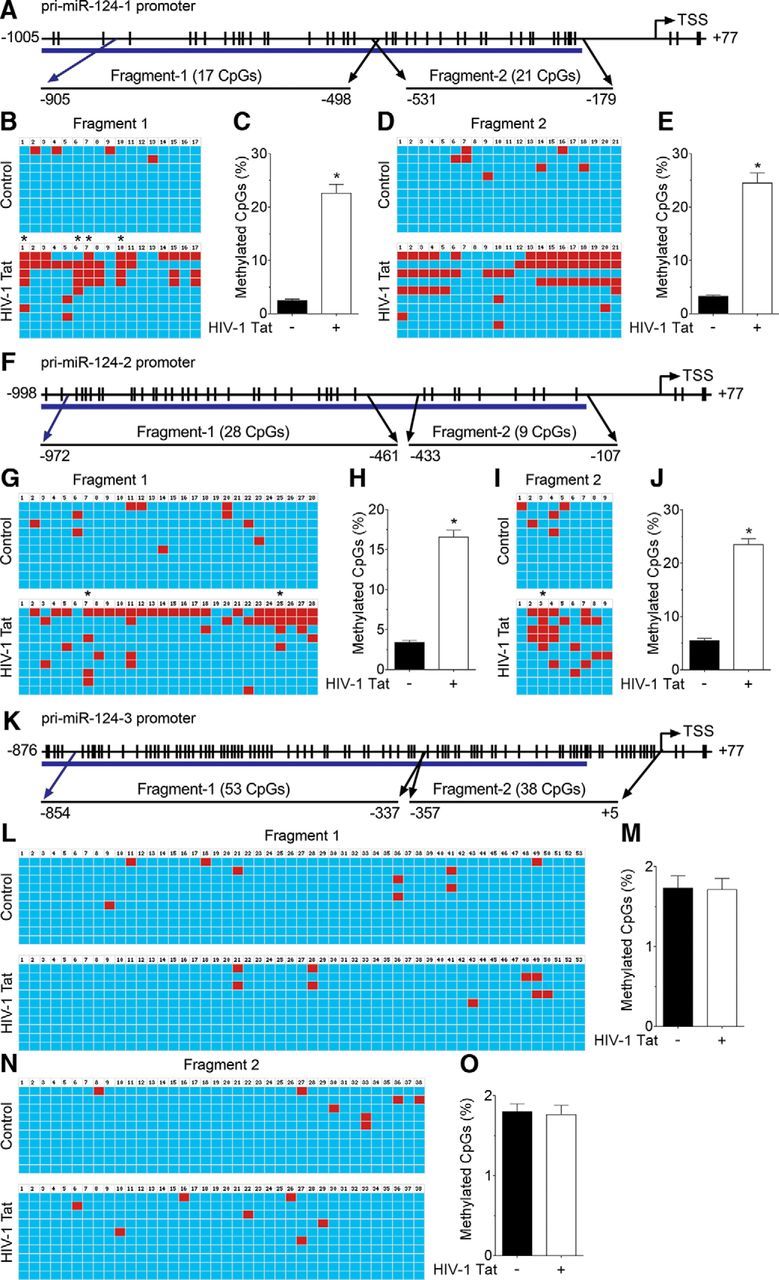
HIV-1 Tat increases DNA methylation of primary miR-124-1 and -2 promoters in mouse primary microglial cells. A, Schematic of CpG islands located in the primary miR-124-1 promoter region. B–E, Bisulfite-converted genomic DNA sequencing showed that HIV-1 Tat (50 ng/ml) significantly increased primary miR-124-1 promoter DNA methylation in mouse primary microglial cells. F, Schematic of CpG islands located in the primary miR-124-2 promoter region. G–J, Bisulfite-converted genomic DNA sequencing showed that HIV-1 Tat (50 ng/ml) significantly increased primary miR-124-2 promoter DNA methylation in mouse primary microglial cells. K, Schematic of CpG islands located in the primary miR-124-3 promoter region. L–O, Bisulfite-converted genomic DNA sequencing showed that HIV-1 Tat (50 ng/ml) did not increase primary miR-124-3 promoter DNA methylation in mouse primary microglial cells. Data are mean ± SEM. Wilcoxon test was used to determine the statistical significance two groups. *p < 0.05 versus control. −, Vehicle treatment (i.e., 1 μl 1 × PBS/ml of medium).
Table 1.
List of primer sequences used for bisulfite genomic DNA sequencing
| Accession no. | Gene description | Forward | Reverse | Product size (bp) |
|---|---|---|---|---|
| NR_029813.1 | Mir124a-1, Fragment 1 | taaggttgatgaggggttaga | cccctacaattccaaacaa | 407 |
| Mir124a-1, Fragment 2 | tgatgtttttttattgtttggaatt | ataaaaaataaaaaaaaaaaaactaac | 352 | |
| NR_029814.1 | Mir124a-2, Fragment 1 | gtaagggatgttgtgaaaagagt | ctcatcccctattaccaaaaatct | 511 |
| Mir124a-2, Fragment 2 | atggtgtggtttttaaagatttg | aacaatactcccacctcaactt | 326 | |
| NR_029538.1 | Mir124a-3, Fragment 1 | tttaggtaaggagagtggga | aatacaaaaccctctccacc | 517 |
| Mir124a-3, Fragment 2 | ggtggagagggttttgtatt | caaaaaaaccctcaaaactac | 362 |
Following this, we next sought to determine the DNA methylation status of primary miR-124-2 in HIV-1 Tat-exposed mouse primary microglial cells. As expected and similar to findings of miR-124-1, HIV-1 Tat exposure also resulted in significant increase in DNA methylation levels of both fragment 1 (Fig. 4G,H; p = 0.0023, n = 1 0, Wilcoxon matched-pairs signed rank test) and fragment 2 (Fig. 4I,J; p < 0.0001, n = 10, Wilcoxon matched-pairs signed rank test) of the primary miR-124-2 promoter. In contrast, HIV-1 Tat (50 ng/ml) exposure failed to alter DNA methylation levels of fragment 1 (Fig. 4L,M; p = 0.8755, n = 10, Wilcoxon matched-pairs signed rank test) and fragment 2 (Fig. 4N,O; p = 0.7500, n = 10, Wilcoxon matched-pairs signed rank test) of the primary miR-124-3. These findings thus confirmed that HIV-1 Tat exposure of mouse primary microglial cells notably increased the methylation levels of primary miR-124-1 and miR-124-2, but not that of the primary miR-124-3 promoter.
HIV-1 Tat-mediated downregulation of miR-124 targets MECP2
Because miRs primarily function through inhibiting their target mRNA by binding to the 3′-UTR region of the mRNA, we next sought to identify a novel miR-124 target that could be critical for microglial activation using online miR target prediction database such as TargetScan (http://www.targetscan.org; Lewis et al., 2005; Betel et al., 2010). From these analyses, we found methyl CpG binding protein 2 (MECP2) as a potential target of miR-124. MECP2 was first identified as a transcriptional repressor protein that selectively bound to the methylated DNA and is known to complex with several other repressor proteins to silence specific gene promoters (Singh et al., 2008). We thus rationalized that HIV-1 Tat-mediated downregulation of miR-124 resulting in upregulation of MECP2 could lead to further repression of miR-124 via a feedback loop.
As shown in Figure 5A, in various species, MECP2 has a conserved miR-124 binding site within its 3′-UTR, indicating that it is a direct target that is regulated by miR-124. To validate this, we cotransfected mouse primary microglial cells with pmirGLO dual luciferase reporter plasmid containing MECP2 wild-type (WT) 3′-UTR and miR-124 mimic followed by an assessment of luciferase activity. Cotransfection of a pmirGLO dual luciferase reporter plasmid containing MECP2 WT 3′-UTR and miR-124 mimics resulted in significant downregulation of luciferase activity (Fig. 5B; p = 0.0034, n = 6, nonparametric Kruskal–Wallis one-way ANOVA followed by Dunn's post hoc test). No such effect was observed in mouse primary microglial cells cotransfected with a pmirGLO dual luciferase reporter plasmid containing MECP2 mutated 3′-UTR and miR-124 mimics (Fig. 5B). Additionally, binding of miR-124 with its target, MECP2, was further confirmed using the Argonaute 2 immunoprecipitation assay (Ago2 IP). Ago2 is an indispensable component of the RNA-induced silencing complex that targets mRNAs by the miR guide strand. This complex was immunoprecipitated, and the mRNA levels were quantified using qPCR (Periyasamy et al., 2018). Briefly, mouse primary microglial cells were transfected with either miR-124 mimic or miR-control for 24 h, followed by pulldown of mRNAs using anti-Ago2 antibody and assessment using qPCR. As shown in Figure 5C, Ago2 IP confirmed MECP2 as a direct target of miR-124 because MECP2 mRNA exhibited significant enrichment in Ago2 protein in cells overexpressed with miR-124 mimic (p = 0.0001, n = 6, nonparametric Kruskal–Wallis one-way ANOVA followed by Dunn's post hoc test). Overall, these findings demonstrated MECP2 as a direct 3′-UTR target regulated by miR-124.
Figure 5.

HIV-1 Tat-mediated downregulation of miR-124 targets MECP2. A, Putative miR-124 binding sites in MECP2 gene. B, Relative luciferase activity of MECP2 3′-UTR as well as MECP2 3′-UTRmut plasmid cotransfected with miR-124 mimic and inhibitor in mouse primary microglial cells. C, Ago2 IP confirmed the enrichment of the miR-124 target mRNA, MECP2 in Ago2 IP compared with total RNA isolated from mouse primary microglial cells transfected with miR-124 mimic and control. D, Representative Western blots showing the protein levels of both p-MECP2 (Ser 80) and MECP2 in mouse primary microglial cells transfected with control and miR-124 mimic following exposure to HIV-1 Tat (50 ng/ml) for 24 h. E, Representative Western blots showing the protein levels of both p-MECP2 (Ser 80) and MECP2 in mouse primary microglial cells transfected with control and miR-124 inhibitor following exposure to HIV-1 Tat (50 ng/ml) for 24 h. F, Representative Western blots showing the protein levels of both p-MECP2 (Ser 80) and MECP2, and (G) qPCR analysis of miR-124 in mouse primary microglial cells transfected with scrambled and MECP2 siRNA following exposure to HIV-1 Tat (50 ng/ml) for 24 h. Representative Western blots showing the protein levels of both p-MECP2 (Ser 80) and MECP2 in mouse primary microglial cells either pretreated with 5-Aza (H) or transfected with scrambled and DNMT1 siRNA (I) following exposure to HIV-1 Tat (50 ng/ml) for 24 h. Data are mean ± SEM from six independent experiments. Nonparametric Kruskal–Wallis one-way ANOVA followed by Dunn's post hoc test was used to determine the statistical significance between multiple groups. *p < 0.05 versus control. #p < 0.05 versus HIV-1 Tat. J, Representative Western blots showing the increased levels of both p-MECP2 (Ser 80) and MECP2 in the basal ganglia of SIV-infected rhesus macaques compared with the saline group. β-Actin was probed as a protein loading control for all the experiments. Data are mean ± SEM. An unpaired Student's t test was used to determine the statistical significance. *p < 0.05 versus saline. −, Vehicle treatment (i.e., 1 μl 1 × PBS/ml of medium).
Next, we wanted to demonstrate the effects of miR-124 on MECP2 expression levels in mouse primary microglial cells transfected either with miR-124 mimic or miR-control followed by Tat exposure for 24 h. Interestingly, as shown in Figure 5D, HIV-1 Tat-mediated increased expression of both p-MECP2 (Ser 80), and MECP2 proteins were significantly inhibited in mouse primary microglial cells transfected with miR-124 mimic (p = 0.0002 for p-MECP2 (Ser 80), p = 0.0001 for MECP2, n = 6, nonparametric Kruskal–Wallis one-way ANOVA followed by Dunn's post hoc test), but not in cells transfected with miR-124 inhibitor (Fig. 5E; p = 0.0008 for p-MECP2 (Ser 80), p = 0.0007 for MECP2, n = 6, nonparametric Kruskal–Wallis one-way ANOVA followed by Dunn's post hoc test), thereby indicating MECP2 as a potential 3′-UTR target of miR-124. Also, miR-124 mimic suppressed, and miR-124 inhibitor increased, both total and phosphorylated MECP2 at similar levels, suggesting that miR-124 regulates MECP2 expression through transcriptionally repressing the total MECP2 expression levels.
Because MECP2 is known to suppress the expression of specific genes, we next wanted to determine whether silencing of MECP2 could lead to increased expression of miR-124 in mouse primary microglial cells. Mouse primary microglial cells were transfected with either scrambled siRNA or MECP2 siRNA followed by exposure to HIV-1 Tat (50 ng/ml) for 24 h. As shown in Figure 5F, cells transfected with MECP2 siRNA showed significant downregulation of both p-MECP2 (Ser 80) and MECP2 proteins (p = 0.0002 for p-MECP2 (Ser 80), p < 0.0001 for MECP2, n = 6, nonparametric Kruskal–Wallis one-way ANOVA followed by Dunn's post hoc test). Next, we assessed the expression of miR-124 in mouse primary microglial cells transfected with either scrambled siRNA or MECP2 siRNA followed by exposure to HIV-1 Tat. As expected, and as shown in Figure 5G, transfection of mouse primary microglial cells with MECP2 siRNA resulted in significantly increased expression of miR-124 both in the presence or absence of HIV-1 Tat (p < 0.0001, n = 6, nonparametric Kruskal–Wallis one-way ANOVA followed by Dunn's post hoc test).
Because DNMT1 and MECP2 regulate the expression levels of miR-124, we next sought to demonstrate the association between DNMT1 and MECP2 in mouse primary microglial cells that were pretreated with either 5-AZA or transfected with DNMT1 siRNA followed by exposure to HIV-1 Tat. Intriguingly, the expression of both p-MECP2 (Ser 80) and MECP2 proteins was significantly downregulated in mouse primary microglial cells pretreated with either 5-Aza (Fig. 5H; p = 0.0002 for p-MECP2 (Ser 80), p < 0.0001 for MECP2, n = 6, nonparametric Kruskal–Wallis one-way ANOVA followed by Dunn's post hoc test) or transfected with DNMT1 siRNA (Fig. 5I; p = 0.0002 for p-MECP2 (Ser 80), p = 0.0002 for MECP2, n = 6, nonparametric Kruskal–Wallis one-way ANOVA followed by Dunn's post hoc test) regardless of HIV-1 Tat exposure. Next, we wanted to validate these in vitro findings in the basal ganglia of saline and SIV-infected rhesus macaques. As shown in Figure 5J and as expected, the protein expression levels of both p-MECP2 (Ser 80) and MECP2 were notably increased in the basal ganglia of SIV-infected rhesus macaques compared with the saline group (p = 0.0166 for p-MECP2 (Ser 80), p = 0.0287 for MECP2, unpaired Student's t test). Overall, these data demonstrated that HIV-1 Tat-mediated downregulation of miR-124 targeted MECP2 both in vitro and in vivo.
HIV-1 Tat-mediated downregulation of miR-124 targets STAT3
In addition to MECP2, we also assessed the expression of the STAT3, a potential direct target of miR-124 (Fig. 6A) in mouse primary microglial cells exposed to HIV-1 Tat. STAT3 is known to play a critical role in microglial activation leading, in turn, to increased secretion of IL6. To validate that STAT3 was a potential target for miR-124, we first performed Ago2 IP in mouse primary microglial cells transfected with miR-124 mimic. As shown in Figure 6B, Ago2 IP confirmed that STAT3 was a direct target of miR-124 because STAT3 mRNA showed significantly increased Ago2 binding in miR-124-transfected mouse primary microglial cells (p < 0.0001, n = 6, nonparametric Kruskal–Wallis one-way ANOVA followed by Dunn's post hoc test). Next, we wanted to demonstrate the effects of miR-124 on STAT3 expression levels in mouse primary microglial cells transfected with either miR-124 mimic or miR-control followed by exposure to HIV-1 Tat (50 ng/ml) for 24 h. Interestingly, as shown in Figure 6C, in cells exposed to HIV-1 Tat, there was a significant increase in the expression of both p-STAT3 (Tyr705) and STAT3 proteins (p = 0.0002 for p-STAT3 (Tyr705), p < 0.0001 for STAT3, n = 6, nonparametric Kruskal–Wallis one-way ANOVA followed by Dunn's post hoc test). Conversely, in mouse primary microglial cells transfected with miR-124 mimic, levels of p-STAT3 (Tyr705) and STAT3 expression were notably decreased in the presence or absence of HIV-1 Tat. As expected, in mouse primary microglial cells transfected with miR-124 inhibitor, expression levels of p-STAT3 (Tyr705) and STAT 3 were significantly upregulated (p = 0.0040 for p-STAT3 (Tyr705), p = 0.0031 for STAT3, n = 6, nonparametric Kruskal–Wallis one-way ANOVA followed by Dunn's post hoc test), and exposure to HIV-1 Tat failed to further upregulate the expression of these proteins (Fig. 6D). Also, miR-124 mimic suppressed, and miR-124 inhibitor increased both total and phosphorylated STAT3 at similar levels, suggesting that miR-124 regulates STAT3 expression through transcriptionally repressing the total STAT3 expression levels.
Figure 6.
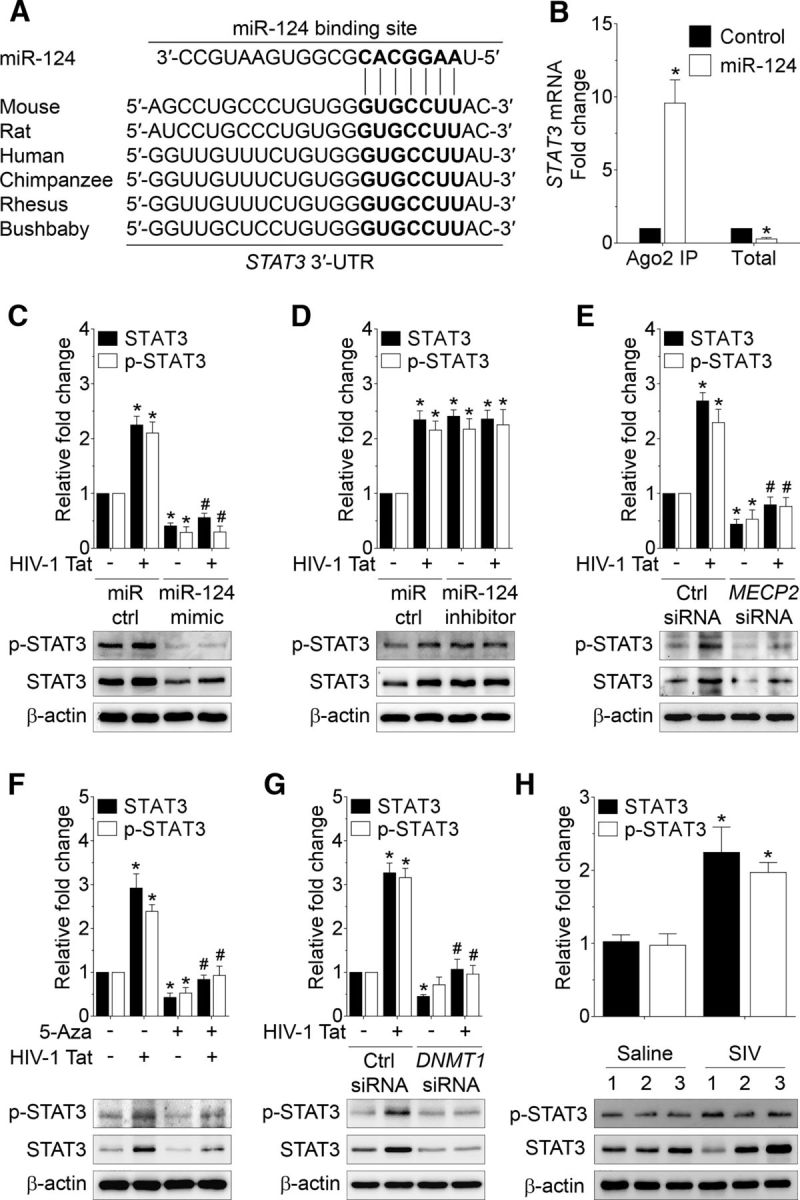
HIV-1 Tat-mediated downregulation of miR-124 targets STAT3. A, Putative miR-124 binding sites in STAT3 gene. B, Ago2 IP confirmed the enrichment of the miR-124 target mRNA, STAT3 in Ago2 IP compared with total RNA isolated from mouse primary microglial cells transfected with miR-124 mimic and control. C, Representative Western blots showing the protein levels of both p-STAT3 (Tyr705) and STAT3 in mouse primary microglial cells transfected with control and miR-124 mimic following exposure to HIV-1 Tat (50 ng/ml) for 24 h. D, Representative Western blots showing the protein levels of both p-STAT3 (Tyr705) and STAT3 in mouse primary microglial cells transfected with control and miR-124 inhibitor following exposure to HIV-1 Tat (50 ng/ml) for 24 h. E, Representative Western blots showing the protein levels of both p-STAT3 (Tyr705) and STAT3 in mouse primary microglial cells transfected with scrambled and MECP2 siRNA following exposure to HIV-1 Tat (50 ng/ml) for 24 h. Representative Western blots showing the protein levels of both p-STAT3 (Tyr705) and STAT3 and in mouse primary microglial cells either pretreated with 5-Aza (F) or transfected with scrambled and DNMT1 siRNA (G) following exposure to HIV-1 Tat (50 ng/ml) for 24 h. Data are mean ± SEM from six independent experiments. Nonparametric Kruskal–Wallis one-way ANOVA followed by Dunn's post hoc test was used to determine the statistical significance between multiple groups. *p < 0.05 versus control. #p < 0.05 versus HIV-1 Tat. H, Representative Western blots showing the increased levels of both p-STAT3 (Tyr705) and STAT3 in the basal ganglia of SIV-infected rhesus macaques compared with the saline group. β-Actin was probed as a protein loading control for all the experiments. Data are mean ± SEM. An unpaired Student's t test was used to determine the statistical significance. *p < 0.05 versus saline. −, Vehicle treatment (i.e., 1 μl 1 × PBS/ml of medium).
Next, we sought to determine the association between MECP2 and STAT3 in HIV-1 Tat-exposed mouse microglial cells. Mouse primary microglial cells were transfected with either MECP2 or scrambled siRNA followed by exposure of cells to HIV-1 Tat for 24 h. As shown earlier, in cells transfected with scrambled siRNA HIV-1 Tat (50 ng/ml), exposure resulted in upregulation of both p-STAT3 (Tyr705) and STAT3 (p = 0.0002 for p-STAT3 (Tyr705), p < 0.0001 for STAT3, n = 6, nonparametric Kruskal–Wallis one-way ANOVA followed by Dunn's post hoc test). However, in mouse primary microglial cells transfected with MECP2 siRNA, HIV-1 Tat exposure failed to upregulate expression levels of both p-STAT3 (Tyr705) and STAT3 (Fig. 6E). Similarly, the expression of p-STAT3 (Tyr705) and STAT3 proteins was significantly downregulated in mouse primary microglial cells pretreated with either 5-Aza (Fig. 6F; p = 0.0001 for p-STAT3 (Tyr705), p = 0.0002 for STAT3, n = 6, nonparametric Kruskal–Wallis one-way ANOVA followed by Dunn's post hoc test) or transfected with DNMT1 siRNA (Fig. 6G; p < 0.0001 for p-STAT3 (Tyr705), p < 0.0001 for STAT3, n = 6, nonparametric Kruskal–Wallis one-way ANOVA followed by Dunn's post hoc test) independent of HIV-1 Tat exposure. Next, we wanted to validate these in vitro findings in the basal ganglia of saline and SIV-infected rhesus macaques. As shown in Figure 6H and as expected, the expression levels of p-STAT3 (Tyr705) and STAT3 proteins were significantly upregulated in the basal ganglia of SIV-infected rhesus macaques compared with the saline group (p = 0.0011 for p-STAT3 (Tyr705), p = 0.0211 for STAT3, unpaired Student's t test).
HIV-1 Tat-mediated activation of microglia involves downregulation of miR-124 with concomitant upregulation of its targets MECP2 and STAT3
To further understand the role of miR-124 in HIV-1 Tat-mediated microglial activation, mouse primary microglial cells were transfected with either miR-124 mimic or miR-124 inhibitor followed by exposure to HIV-1 Tat (50 ng/ml) for 24 h, and assessment of Iba-1 protein, an activation marker for microglia. Interestingly, HIV-1 Tat-mediated increased expression of Iba-1 protein was significantly inhibited in microglial cells transfected with miR-124 mimic (Fig. 7A; p = 0.0002, n = 6, nonparametric Kruskal–Wallis one-way ANOVA followed by Dunn's post hoc test), but not in cells transfected with miR-124 inhibitor (Fig. 7B; p = 0.0034, n = 6, nonparametric Kruskal–Wallis one-way ANOVA followed by Dunn's post hoc test), thereby suggesting the possible involvement of miR-124 in HIV-1 Tat-mediated microglial activation.
Figure 7.

HIV-1 Tat-mediated activation of microglia involves downregulation of miR-124 with concomitant upregulation of its targets, MECP2 and STAT3. A, Representative Western blots showing the protein levels of Iba-1 in mouse primary microglial cells transfected with control and miR-124 mimic following exposure to HIV-1 Tat (50 ng/ml) for 24 h. B, Representative Western blots showing the protein levels of Iba-1 in mouse primary microglial cells transfected with control and miR-124 inhibitor following exposure to HIV-1 Tat (50 ng/ml) for 24 h. C, Representative Western blots showing the protein levels of Iba-1 in mouse primary microglial cells transfected with scrambled and MECP2 siRNA following exposure to HIV-1 Tat (50 ng/ml) for 24 h. Representative Western blots showing the protein levels of Iba-1 in mouse primary microglial cells either pretreated with 5-Aza (D) or transfected with scrambled and DNMT1 siRNA (E) following exposure to HIV-1 Tat (50 ng/ml) for 24 h. Data are mean ± SEM from six independent experiments. Nonparametric Kruskal–Wallis one-way ANOVA followed by Dunn's post hoc test was used to determine the statistical significance between multiple groups. *p < 0.05 versus control. #p < 0.05 versus HIV-1 Tat. F, Representative Western blots showing the increased levels of Iba-1 in the basal ganglia of SIV-infected rhesus macaques compared with the saline group. β-Actin was probed as a protein loading control for all the experiments. Data are mean ± SEM. An unpaired Student's t test was used to determine the statistical significance. *p < 0.05 versus saline. −, Vehicle treatment (i.e., 1 μl 1 × PBS/ml of medium).
Next, we determined whether HIV-1 Tat-mediated activation of microglia involved concomitant upregulation of miR-124 target, MECP2. As shown in Figure 7C, HIV-1 Tat-mediated increased expression of Iba-1 protein was significantly inhibited in mouse primary microglial cells transfected with MECP2 siRNA, thereby suggesting the possible involvement of MECP2 in HIV-1 Tat-mediated microglial activation (p = 0.0029, n = 6, nonparametric Kruskal–Wallis one-way ANOVA followed by Dunn's post hoc test). Similarly, the expression of Iba-1 protein was significantly downregulated in mouse primary microglial cells pretreated with either 5-Aza (Fig. 7D; p < 0.0001, n = 6, nonparametric Kruskal–Wallis one-way ANOVA followed by Dunn's post hoc test) or transfected with DNMT1 siRNA (Fig. 7E; p = 0.0003, n = 6, nonparametric Kruskal–Wallis one-way ANOVA followed by Dunn's post hoc test) independent of HIV-1 Tat exposure. Next, we wanted to validate these in vitro findings in the basal ganglia of saline and SIV-infected rhesus macaques. As shown in Figure 7F, the expression of Iba-1 protein was significantly upregulated in the basal ganglia of SIV-infected rhesus macaques compared with the saline group (p = 0.0368, unpaired Student's t test).
Because miR-124 is involved in maintaining microglial quiescence (Sun et al., 2013; Veremeyko et al., 2013), we next sought to investigate the effect of miR-124 on HIV-1 Tat-mediated expression of proinflammatory cytokine, IL6. Briefly, mouse primary microglial cells were transfected with either miR-124 mimic or miR-124 inhibitor followed by exposure to HIV-1 Tat (50 ng/ml) for 24 h. As shown in Figure 8A, B, expression of IL6 mRNA was significantly upregulated in HIV-1 Tat-exposed mouse primary microglial cells transfected with scrambled siRNA (p = 0.0019, n = 6, nonparametric Kruskal–Wallis one-way ANOVA followed by Dunn's post hoc test). Interestingly, HIV-1 Tat-mediated upregulation of IL6 mRNA expression was significantly inhibited in mouse primary microglial cells transfected with miR-124 mimic (Fig. 8A) but not in cells transfected with miR-124 inhibitor (Fig. 8B). Similarly, gene silencing of MECP2 significantly abolished HIV-1 Tat-mediated upregulation of IL6 mRNA in mouse primary microglial cells (Fig. 8C; p < 0.0001, n = 6, nonparametric Kruskal–Wallis one-way ANOVA followed by Dunn's post hoc test). Additionally, HIV-1 Tat-mediated upregulation of IL6 mRNA expression was significantly inhibited in mouse primary microglial cells pretreated with either 5-Aza (Fig. 8D; p = 0.0002, n = 6, nonparametric Kruskal–Wallis one-way ANOVA followed by Dunn's post hoc test) or transfected with DNMT1 siRNA (Fig. 8E; p = 0.0019, n = 6, nonparametric Kruskal–Wallis one-way ANOVA followed by Dunn's post hoc test). Overall, these results demonstrated that HIV-1 Tat-mediated activation of microglia involves upstream downregulation of miR-124 followed with upregulation of MECP2 and DNMT1.
Figure 8.
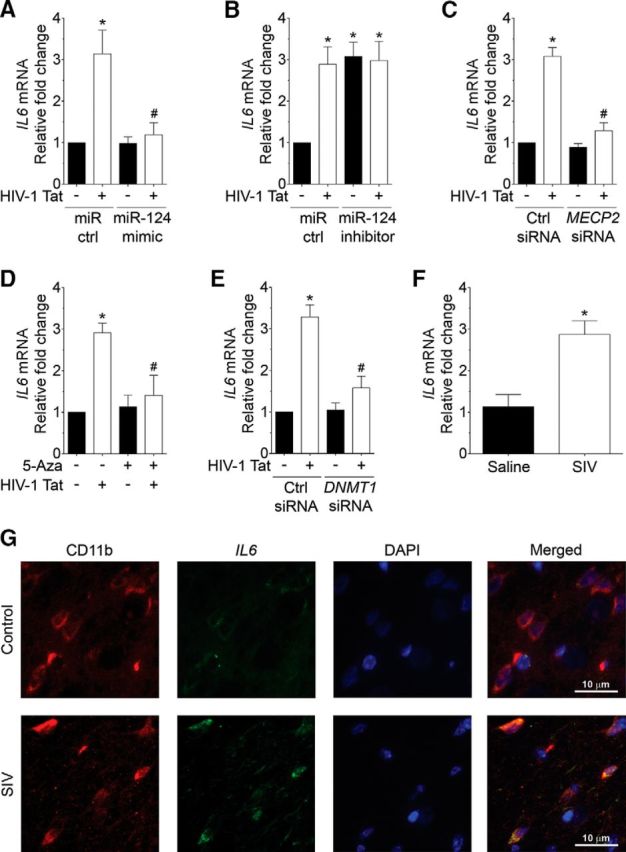
HIV-1 Tat-mediated increased expression of IL6 involves downregulation of miR-124 with concomitant upregulation of its targets, MECP2 and STAT3. A, qPCR analysis showing the IL6 mRNA expression levels in mouse primary microglial cells transfected with control and miR-124 mimic following exposure to HIV-1 Tat (50 ng/ml) for 24 h. B, qPCR analysis showing the IL6 mRNA expression levels in mouse primary microglial cells transfected with control and miR-124 inhibitor following exposure to HIV-1 Tat (50 ng/ml) for 24 h. C, qPCR analysis showing the IL6 mRNA expression levels in mouse primary microglial cells transfected with scrambled and MECP2 siRNA following exposure to HIV-1 Tat (50 ng/ml) for 24 h. qPCR analysis showing the IL6 mRNA expression levels in mouse primary microglial cells either pretreated with 5-Aza (D) or transfected with scrambled and DNMT1 siRNA (E) following exposure to HIV-1 Tat (50 ng/ml) for 24 h. Data are mean ± SEM from six independent experiments. Nonparametric Kruskal–Wallis one-way ANOVA followed by Dunn's post hoc test was used to determine the statistical significance between multiple groups. *p < 0.05 versus control. #p < 0.05 versus HIV-1 Tat. F, qPCR analysis showing the IL6 mRNA expression levels in the basal ganglia of SIV-infected rhesus macaques compared with the saline group. Data are mean ± SEM. An unpaired Student's t test was used to determine the statistical significance. *p < 0.05 versus saline. G, RNAscope analysis demonstrating increased expression of IL6 mRNA in microglia in the basal ganglia of SIV-infected rhesus macaques compared with the saline group. Scale bar, 10 μm. −, Vehicle treatment (i.e., 1 μl 1 × PBS/ml of medium).
These in vitro findings were further validated ex vivo in the basal ganglia tissues of rhesus macaques. As expected, the expression of IL6 mRNA was significantly upregulated in the basal ganglia of SIV-infected rhesus macaques compared with the saline group (Fig. 8F; p = 0.0398, unpaired Student's t test). To assess the cell types critical for expression of IL6 mRNA in the basal ganglia, ISH using RNAscope was performed in archived tissues obtained from both SIV-infected and saline-injected rhesus macaques. As shown in Figure 8G, mRNA expression of IL6 was significantly increased in the microglia of SIV-infected rhesus macaques basal ganglia compared with the saline group. Overall, these findings demonstrated the role of miR-124 in HIV-1 Tat-mediated activation of microglia via MECP2 and STAT3 axis.
Discussion
This study provides new mechanistic insights into the HIV-1 Tat-mediated promoter DNA methylation of miR-124 in regulating microglial activation via targeting the MECP2-STAT3 signaling axis. Herein, we have demonstrated that HIV-1 Tat exposure notably increased DNA methylation of primary miR-124-1 and primary miR-124-2 promoters (no change in primary miR-124-3), thereby downregulating the expression levels of both primary miR-124-1 and primary miR-124-2, and the mature miR-124 in mouse primary microglial cells. HIV-1 Tat-mediated diminished expression of miR-124 resulted in activation of microglia through the MECP2-STAT3 signaling. Thus, modulation of miR-124-MECP2-STAT3 signaling can be envisioned as a therapeutic intervention for diminishing the microglial activation mediated by HIV-1 Tat protein.
In this study, we first performed miR microarray to determine the expression profile of various dysregulated miRs in the basal ganglia of saline and SIV-infected rhesus macaques. It is well known that miR-124 is a key miR responsible for microglia quiescence and that its downregulation results in microglial activation (Ponomarev et al., 2011). Because microglial activation associated with the release of cytotoxic mediators, including proinflammatory cytokines, chemokines, and ROS, is a contributing factor to the pathogenesis of HIV-1-associated neurocognitive disorders (Kaul et al., 2001), in this study, we primarily focused on the involvement of miR-124 in microglial activation during SIV infection as well as following HIV-1 Tat exposure (as a surrogate for HIV infection; see below). Downregulation of miR-124 in the basal ganglia of SIV-infected rhesus macaques was further validated by qPCR as well as ISH, indicating an inverse association between miR-124 and microglial activation. It is well known that dysregulated miR expression is often associated with altered DNA methylation of the respective miR encoding genes, thereby contributing to several disease processes (Gulyaeva and Kushlinskiy, 2016). In this study, we also showed that the homogenates from basal ganglia of SIV-infected rhesus macaques showed increased 5-mC levels (an indicator of global DNA methylation) along with increased expression of DNA methylation enzymes, such as DNMT1, DNMT3a, and DNMT3b. To understand the detailed molecular mechanism(s) involving the role of downregulated miR-124 in microglial activation in the context of epigenetic DNA methylation, we resorted to mouse primary microglial cells exposed to HIV-1 Tat. The rationale for using HIV-1 Tat as a surrogate of infection in cell culture is as follows. It is well accepted that, despite cART, HIV-1 Tat continues to be present in the CNS and lymph nodes of treated patients (Wiley et al., 1996; Hudson et al., 2000). Additionally, there is also evidence of low-level virus replication in the CNS of cART-treated subjects (Wiley et al., 1996; Hudson et al., 2000), thereby providing validity to the use of HIV-1 Tat as a surrogate of HIV-1 infection in cell culture studies.
Findings from our study demonstrate that exposure of mouse primary microglial cells to HIV-1 Tat resulted in downregulated expression of both mature miR-124 as well as the primary miR-124-1 and primary miR-124-2 (but not primary miR-124-3). ISH also validated downregulation of miR-124 in HIV-1 Tat-exposed cells with concomitant activation of microglial cells. Interestingly, exposure of microglial cells to HIV-1 Tat also notably increased global DNA methylation, as evidenced by increased 5-mC levels in addition to increased RNA and protein expression of all DNMTs, thereby indicating a possible involvement of epigenetic DNA modifications in the regulation of miR-124. This was further validated using pharmacological and gene silencing approaches to knockdown of DNMT1. Findings from these studies further confirmed the association between miR-124 expression and DNA methylation of primary miR-124-1 and primary miR-124-2 promoters. One possible explanation for lack of change in DNA methylation of primary miR-124-3 promoter could be its role as a compensatory check that likely functions only in the absence of the other two miRs. Our findings are in agreement with those by others demonstrating that both HIV-1 infection as well as HIV-1 Tat can induce the expression of DNMTs in lymphomas, thereby leading to increased genomic DNA methylation with dysregulated gene/miR expression (Fang et al., 2001; Youngblood and Reich, 2008; Luzzi et al., 2014). Accumulating evidence has also demonstrated downregulation of miR-124 both in the archival brain tissues and in CSF of subjects that are HIV-positive and with HIV-1 encephalitis (Pacifici et al., 2013). Also, DNA methylation in the promoter region of primary miR-124s has also been reported in various disease conditions, including cancers and drug addiction, a comorbidity of HIV-1 infection (Wilting et al., 2010; Koukos et al., 2013; Zhou et al., 2013; Chu et al., 2015; Murray-Stewart et al., 2016; Xu et al., 2016; Periyasamy et al., 2018).
This study, for the first time, reports a novel 3′-UTR target of miR-124, such as MECP2. As mentioned earlier, MECP2 is a transcriptional repressor that selectively binds to the methylated DNA and forms complexes with several other repressor proteins, thereby silencing the expression of specific genes (Singh et al., 2008). It is also documented that MECP2 can suppress the nuclear miR processing by regulating the DGCR8/Drosha complex resulting in decreased expression of several mature miRs, including miR-124 (Cheng et al., 2014). Additionally, numerous reports have also demonstrated that miRNA expression profile was changed in the brains of mecp2 null mice due to the transcriptional repression by MECP2 (Urdinguio et al., 2010; Wu et al., 2010; Cheng et al., 2014). Intriguingly, it has also been demonstrated that phosphorylated MECP2 (Ser80) directly binds to the DGCR8 and releases the Drosha from the DGCR8, thereby blocking miR biogenesis (Cheng et al., 2014). Similar to these reports and in the present study, we found that HIV-1 Tat-mediated downregulation of miR-124 increased the 3′-UTR target protein MECP2 as well as the phosphorylated form of MECP2 (Ser80) in microglial cells. As shown earlier, increased expression of phosphorylated MECP2 (Ser80) can block the miR biogenesis machinery, ultimately leading to further downregulation of miR-124 through a negative regulatory feedback axis.
Another well-established 3′-UTR target of miR-124 is STAT3, which in the current study was shown to be upregulated by HIV-1 Tat. STAT3 is responsible for the production of proinflammatory cytokines, especially, IL6. Our findings are in line with those published by others demonstrating an interrelationship between miR-124-mediated STAT3 activation and IL6 secretion (Koukos et al., 2013; Sun et al., 2013; Cheng et al., 2015). The present study convincingly showed that, in microglial cells, HIV-1 Tat-mediated downregulation of miR-124 upregulated the expression of STAT3, including its activated form, phosphorylated STAT3. Other investigators also have shown that peripheral administration of miR-124 in an experimental murine autoimmune encephalomyelitis caused deactivation of macrophages, reduced activation of myelin-specific T cells, and markedly suppressed disease progression via the downregulation of STAT3 signaling (Koukos et al., 2013). HIV-1 Tat is known to activate astrocytes through activation of STAT3 signaling (Fan et al., 2015). Several reports have indicated the role of activated STAT3 and its phosphorylation form in the generation of proinflammatory cytokines by LPS stimulated macrophages (Sun et al., 2013, 2015). It is well established that proinflammatory cytokines secreted by activated glia or damaged cells play a fundamental role in triggering neuroinflammation. Accordingly, it is likely that HIV-1 Tat-mediated microglial activation leads to overexpression of proinflammatory cytokines, thereby further underpinning the detrimental role of HIV-1 Tat in mediating microglial activation. This study mainly focused on Tat-mediated microglial activation in vitro, and the findings were validated ex vivo using the archival basal ganglia of SIV-infected rhesus macaques. Future studies are, however, warranted to explore the other host and viral components responsible for MECP2 upregulation and microglial activation.
In conclusion, as shown in Figure 9, this study demonstrated that HIV-1 Tat-mediated downregulation of miR-124 involved DNA methylation of primary miR-124-1 and primary miR-124-2 promoters. Furthermore, downregulated miR-124 regulated the MECP2-STAT3 signaling axis, leading, in turn, to increased expression of proinflammatory cytokine IL6, and ensuing microglial activation. These novel results provide functional insights into the interaction of HIV-1 Tat with miR-124 promoter DNA methylation and MECP2-STAT3-IL6 signaling axis, and implicate that inhibition of DNMTs/MECP2, as well as overexpression of miR-124, could be developed as an alternative approach for the treatment of HIV-1 Tat-mediated microglial activation.
Figure 9.

Schematic diagram demonstrating the HIV-1 Tat-mediated downregulation of miR-124 involved DNA methylation of primary miR-124-1 and primary miR-124-2 promoters. Furthermore, downregulated miR-124 regulated the MECP2-STAT3 signaling axis, leading, in turn, to increased expression of proinflammatory cytokine IL6 and ensuing microglial activation.
Footnotes
This work was supported by the National Institutes of Health Grants DA044087 to P.P., DA043138, MH062261, and DA035203 to S.B., and DA033150 to S.B. and G.H.
The authors declare no competing financial interests.
References
- Bagashev A, Sawaya BE (2013) Roles and functions of HIV-1 tat protein in the CNS: an overview. Virol J 10:358. 10.1186/1743-422X-10-358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bansal AK, Mactutus CF, Nath A, Maragos W, Hauser KF, Booze RM (2000) Neurotoxicity of HIV-1 proteins gp120 and tat in the rat striatum. Brain Res 879:42–49. 10.1016/S0006-8993(00)02725-6 [DOI] [PubMed] [Google Scholar]
- Betel D, Koppal A, Agius P, Sander C, Leslie C (2010) Comprehensive modeling of microRNA targets predicts functional non-conserved and non-canonical sites. Genome Biol 11:R90. 10.1186/gb-2010-11-8-r90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bokhari SM, Hegde R, Callen S, Yao H, Adany I, Li Q, Li Z, Pinson D, Yeh HW, Cheney PD, Buch S (2011) Morphine potentiates neuropathogenesis of SIV infection in rhesus macaques. J Neuroimmune Pharmacol 6:626–639. 10.1007/s11481-011-9272-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng TL, Wang Z, Liao Q, Zhu Y, Zhou WH, Xu W, Qiu Z (2014) MeCP2 suppresses nuclear microRNA processing and dendritic growth by regulating the DGCR8/Drosha complex. Dev Cell 28:547–560. 10.1016/j.devcel.2014.01.032 [DOI] [PubMed] [Google Scholar]
- Cheng Y, Li Y, Nian Y, Liu D, Dai F, Zhang J (2015) STAT3 is involved in miR-124-mediated suppressive effects on esophageal cancer cells. BMC Cancer 15:306. 10.1186/s12885-015-1303-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chivero ET, Guo ML, Periyasamy P, Liao K, Callen SE, Buch S (2017) HIV-1 tat primes and activates microglial NLRP3 inflammasome-mediated neuroinflammation. J Neurosci 37:3599–3609. 10.1523/JNEUROSCI.3045-16.2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu M, Chang Y, Guo Y, Wang N, Cui J, Gao WQ (2015) Regulation and methylation of tumor suppressor miR-124 by androgen receptor in prostate cancer cells. PLoS One 10:e0116197. 10.1371/journal.pone.0116197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis GM, Haas MA, Pocock R (2015) MicroRNAs: not “Fine-Tuners” but key regulators of neuronal development and function. Front Neurol 6:245. 10.3389/fneur.2015.00245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falkensammer B, Freissmuth D, Hübner L, Speth C, Dierich MP, Stoiber H (2007) Changes in HIV-specific antibody responses and neutralization titers in patients under ART. Front Biosci 12:2148–2158. 10.2741/2218 [DOI] [PubMed] [Google Scholar]
- Fan Y, Timani KA, He JJ (2015) STAT3 and its phosphorylation are involved in HIV-1 tat-induced transactivation of glial fibrillary acidic protein. Curr HIV Res 13:55–63. 10.2174/1570162X13666150121115804 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang JY, Mikovits JA, Bagni R, Petrow-Sadowski CL, Ruscetti FW (2001) Infection of lymphoid cells by integration-defective human immunodeficiency virus type 1 increases de novo methylation. J Virol 75:9753–9761. 10.1128/JVI.75.20.9753-9761.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gascon E, Lynch K, Ruan H, Almeida S, Verheyden JM, Seeley WW, Dickson DW, Petrucelli L, Sun D, Jiao J, Zhou H, Jakovcevski M, Akbarian S, Yao WD, Gao FB (2014) Alterations in microRNA-124 and AMPA receptors contribute to social behavioral deficits in frontotemporal dementia. Nat Med 20:1444–1451. 10.1038/nm.3717 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grubman A, Kanninen KM, Malm T (2016) Multitasking microglia and Alzheimer's disease: diversity, tools and therapeutic targets. J Mol Neurosci 60:390–404. 10.1007/s12031-016-0825-5 [DOI] [PubMed] [Google Scholar]
- Gulyaeva LF, Kushlinskiy NE (2016) Regulatory mechanisms of microRNA expression. J Transl Med 14:143. 10.1186/s12967-016-0893-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo ML, Periyasamy P, Liao K, Kook YH, Niu F, Callen SE, Buch S (2016) Cocaine-mediated downregulation of microglial miR-124 expression involves promoter DNA methylation. Epigenetics 11:819–830. 10.1080/15592294.2016.1232233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hambardzumyan D, Gutmann DH, Kettenmann H (2016) The role of microglia and macrophages in glioma maintenance and progression. Nat Neurosci 19:20–27. 10.1038/nn.4185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han J, Harris RA, Zhang XM (2017) An updated assessment of microglia depletion: current concepts and future directions. Mol Brain 10:25. 10.1186/s13041-017-0307-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayashi K, Pu H, Andras IE, Eum SY, Yamauchi A, Hennig B, Toborek M (2006) HIV-TAT protein upregulates expression of multidrug resistance protein 1 in the blood–brain barrier. J Cereb Blood Flow Metab 26:1052–1065. 10.1038/sj.jcbfm.9600254 [DOI] [PubMed] [Google Scholar]
- Hu G, Yao H, Chaudhuri AD, Duan M, Yelamanchili SV, Wen H, Cheney PD, Fox HS, Buch S (2012) Exosome-mediated shuttling of microRNA-29 regulates HIV tat and morphine-mediated neuronal dysfunction. Cell Death Dis 3:e381. 10.1038/cddis.2012.114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hudson L, Liu J, Nath A, Jones M, Raghavan R, Narayan O, Male D, Everall I (2000) Detection of the human immunodeficiency virus regulatory protein tat in CNS tissues. J Neurovirol 6:145–155. 10.3109/13550280009013158 [DOI] [PubMed] [Google Scholar]
- Kanagaraj N, Beiping H, Dheen ST, Tay SS (2014) Downregulation of miR-124 in MPTP-treated mouse model of Parkinson's disease and MPP iodide-treated MN9D cells modulates the expression of the calpain/cdk5 pathway proteins. Neuroscience 272:167–179. 10.1016/j.neuroscience.2014.04.039 [DOI] [PubMed] [Google Scholar]
- Kaul M, Garden GA, Lipton SA (2001) Pathways to neuronal injury and apoptosis in HIV-associated dementia. Nature 410:988–994. 10.1038/35073667 [DOI] [PubMed] [Google Scholar]
- Koukos G, Polytarchou C, Kaplan JL, Morley-Fletcher A, Gras-Miralles B, Kokkotou E, Baril-Dore M, Pothoulakis C, Winter HS, Iliopoulos D (2013) MicroRNA-124 regulates STAT3 expression and is down-regulated in colon tissues of pediatric patients with ulcerative colitis. Gastroenterology 145:842–852.e2. 10.1053/j.gastro.2013.07.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le W, Wu J, Tang Y (2016) Protective microglia and their regulation in Parkinson's disease. Front Mol Neurosci 9:89. 10.3389/fnmol.2016.00089 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis BP, Burge CB, Bartel DP (2005) Conserved seed pairing, often flanked by adenosines, indicates that thousands of human genes are microRNA targets. Cell 120:15–20. 10.1016/j.cell.2004.12.035 [DOI] [PubMed] [Google Scholar]
- Luzzi A, Morettini F, Gazaneo S, Mundo L, Onnis A, Mannucci S, Rogena EA, Bellan C, Leoncini L, De Falco G (2014) HIV-1 tat induces DNMT over-expression through microRNA dysregulation in HIV-related non-Hodgkin lymphomas. Infect Agent Cancer 9:41. 10.1186/1750-9378-9-41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maubert ME, Pirrone V, Rivera NT, Wigdahl B, Nonnemacher MR (2015) Interaction between tat and drugs of abuse during HIV-1 infection and central nervous system disease. Front Microbiol 6:1512. 10.3389/fmicb.2015.01512 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mediouni S, Darque A, Baillat G, Ravaux I, Dhiver C, Tissot-Dupont H, Mokhtari M, Moreau H, Tamalet C, Brunet C, Paul P, Dignat-George F, Stein A, Brouqui P, Spector SA, Campbell GR, Loret EP (2012) Antiretroviral therapy does not block the secretion of the human immunodeficiency virus tat protein. Infect Disord Drug Targets 12:81–86. 10.2174/187152612798994939 [DOI] [PubMed] [Google Scholar]
- Michell-Robinson MA, Touil H, Healy LM, Owen DR, Durafourt BA, Bar-Or A, Antel JP, Moore CS (2015) Roles of microglia in brain development, tissue maintenance and repair. Brain 138:1138–1159. 10.1093/brain/awv066 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minghetti L, Visentin S, Patrizio M, Franchini L, Ajmone-Cat MA, Levi G (2004) Multiple actions of the human immunodeficiency virus type-1 tat protein on microglial cell functions. Neurochem Res 29:965–978. 10.1023/B:NERE.0000021241.90133.89 [DOI] [PubMed] [Google Scholar]
- Mosser CA, Baptista S, Arnoux I, Audinat E (2017) Microglia in CNS development: shaping the brain for the future. Prog Neurobiol 149:1–20. 10.1016/j.pneurobio.2017.01.002 [DOI] [PubMed] [Google Scholar]
- Murray-Stewart T, Sierra JC, Piazuelo MB, Mera RM, Chaturvedi R, Bravo LE, Correa P, Schneider BG, Wilson KT, Casero RA (2016) Epigenetic silencing of miR-124 prevents spermine oxidase regulation: implications for Helicobacter pylori-induced gastric cancer. Oncogene 35:5480–5488. 10.1038/onc.2016.91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pacifici M, Delbue S, Ferrante P, Jeansonne D, Kadri F, Nelson S, Velasco-Gonzalez C, Zabaleta J, Peruzzi F (2013) Cerebrospinal fluid miRNA profile in HIV-encephalitis. J Cell Physiol 228:1070–1075. 10.1002/jcp.24254 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palsamy P, Ayaki M, Elanchezhian R, Shinohara T (2012) Promoter demethylation of Keap1 gene in human diabetic cataractous lenses. Biochem Biophys Res Commun 423:542–548. 10.1016/j.bbrc.2012.05.164 [DOI] [PubMed] [Google Scholar]
- Palsamy P, Bidasee KR, Shinohara T (2014a) Valproic acid suppresses Nrf2/Keap1 dependent antioxidant protection through induction of endoplasmic reticulum stress and Keap1 promoter DNA demethylation in human lens epithelial cells. Exp Eye Res 121:26–34. 10.1016/j.exer.2014.01.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palsamy P, Bidasee KR, Shinohara T (2014b) Selenite cataracts: activation of endoplasmic reticulum stress and loss of Nrf2/Keap1-dependent stress protection. Biochim Biophys Acta 1842:1794–1805. 10.1016/j.bbadis.2014.06.028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palsamy P, Bidasee KR, Ayaki M, Augusteyn RC, Chan JY, Shinohara T (2014c) Methylglyoxal induces endoplasmic reticulum stress and DNA demethylation in the Keap1 promoter of human lens epithelial cells and age-related cataracts. Free Radic Biol Med 72:134–148. 10.1016/j.freeradbiomed.2014.04.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pendyala G, Periyasamy P, Callen S, Fox HS, Lisco SJ, Buch SJ (2015) Chronic SIV and morphine treatment increases heat shock protein 5 expression at the synapse. J Neurovirol 21:592–598. 10.1007/s13365-015-0356-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Periyasamy P, Guo ML, Buch S (2016) Cocaine induces astrocytosis through ER stress-mediated activation of autophagy. Autophagy 12:1310–1329. 10.1080/15548627.2016.1183844 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Periyasamy P, Liao K, Kook YH, Niu F, Callen SE, Guo ML, Buch S (2018) Cocaine-mediated downregulation of miR-124 activates microglia by targeting KLF4 and TLR4 signaling. Mol Neurobiol 55:3196–3210. 10.1007/s12035-017-0584-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ponomarev ED, Veremeyko T, Barteneva N, Krichevsky AM, Weiner HL (2011) MicroRNA-124 promotes microglia quiescence and suppresses EAE by deactivating macrophages via the C/EBP-alpha-PU.1 pathway. Nat Med 17:64–70. 10.1038/nm.2266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roy B, Dunbar M, Shelton RC, Dwivedi Y (2017) Identification of microRNA-124–3p as a putative epigenetic signature of major depressive disorder. Neuropsychopharmacology 42:864–875. 10.1038/npp.2016.175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider CA, Rasband WS, Eliceiri KW (2012) NIH image to ImageJ: 25 years of image analysis. Nat Methods 9:671–675. 10.1038/nmeth.2089 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh J, Saxena A, Christodoulou J, Ravine D (2008) MECP2 genomic structure and function: insights from ENCODE. Nucleic Acids Res 36:6035–6047. 10.1093/nar/gkn591 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skaper SD, Argentini C, Barbierato M (2012) Culture of neonatal rodent microglia, astrocytes, and oligodendrocytes from cortex and spinal cord. Methods Mol Biol 846:67–77. 10.1007/978-1-61779-536-7_7 [DOI] [PubMed] [Google Scholar]
- Song GJ, Suk K (2017) Pharmacological modulation of functional phenotypes of microglia in neurodegenerative diseases. Front Aging Neurosci 9:139. 10.3389/fnagi.2017.00139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Y, Li Q, Gui H, Xu DP, Yang YL, Su DF, Liu X (2013) MicroRNA-124 mediates the cholinergic anti-inflammatory action through inhibiting the production of pro-inflammatory cytokines. Cell Res 23:1270–1283. 10.1038/cr.2013.116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Y, Luo ZM, Guo XM, Su DF, Liu X (2015) An updated role of microRNA-124 in central nervous system disorders: a review. Front Cell Neurosci 9:193. 10.3389/fncel.2015.00193 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urdinguio RG, Fernandez AF, Lopez-Nieva P, Rossi S, Huertas D, Kulis M, Liu CG, Croce CM, Calin GA, Esteller M (2010) Disrupted microRNA expression caused by Mecp2 loss in a mouse model of Rett syndrome. Epigenetics 5:656–663. 10.4161/epi.5.7.13055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veremeyko T, Siddiqui S, Sotnikov I, Yung A, Ponomarev ED (2013) IL-4/IL-13-dependent and independent expression of miR-124 and its contribution to M2 phenotype of monocytic cells in normal conditions and during allergic inflammation. PLoS One 8:e81774. 10.1371/journal.pone.0081774 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westendorp MO, Frank R, Ochsenbauer C, Stricker K, Dhein J, Walczak H, Debatin KM, Krammer PH (1995) Sensitization of T cells to CD95-mediated apoptosis by HIV-1 tat and gp120. Nature 375:497–500. 10.1038/375497a0 [DOI] [PubMed] [Google Scholar]
- Wiley CA, Baldwin M, Achim CL (1996) Expression of HIV regulatory and structural mRNA in the central nervous system. AIDS 10:843–847. 10.1097/00002030-199607000-00007 [DOI] [PubMed] [Google Scholar]
- Wilting SM, van Boerdonk RA, Henken FE, Meijer CJ, Diosdado B, Meijer GA, le Sage C, Agami R, Snijders PJ, Steenbergen RD (2010) Methylation-mediated silencing and tumour suppressive function of hsa-miR-124 in cervical cancer. Mol Cancer 9:167. 10.1186/1476-4598-9-167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolf SA, Boddeke HW, Kettenmann H (2017) Microglia in physiology and disease. Annu Rev Physiol 79:619–643. 10.1146/annurev-physiol-022516-034406 [DOI] [PubMed] [Google Scholar]
- Wu H, Tao J, Chen PJ, Shahab A, Ge W, Hart RP, Ruan X, Ruan Y, Sun YE (2010) Genome-wide analysis reveals methyl-CpG-binding protein 2-dependent regulation of microRNAs in a mouse model of Rett syndrome. Proc Natl Acad Sci U S A 107:18161–18166. 10.1073/pnas.1005595107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao H, Neuveut C, Tiffany HL, Benkirane M, Rich EA, Murphy PM, Jeang KT (2000) Selective CXCR4 antagonism by tat: implications for in vivo expansion of coreceptor use by HIV-1. Proc Natl Acad Sci U S A 97:11466–11471. 10.1073/pnas.97.21.11466 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu L, Dai W, Li J, He L, Wang F, Xia Y, Chen K, Li S, Liu T, Lu J, Zhou Y, Wang Y, Guo C (2016) Methylation-regulated miR-124-1 suppresses tumorigenesis in hepatocellular carcinoma by targeting CASC3. Oncotarget 7:26027–26041. 10.18632/oncotarget.8266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Youngblood B, Reich NO (2008) The early expressed HIV-1 genes regulate DNMT1 expression. Epigenetics 3:149–156. 10.4161/epi.3.3.6372 [DOI] [PubMed] [Google Scholar]
- Yu B, Zhou S, Yi S, Gu X (2015) The regulatory roles of non-coding RNAs in nerve injury and regeneration. Prog Neurobiol 134:122–139. 10.1016/j.pneurobio.2015.09.006 [DOI] [PubMed] [Google Scholar]
- Zhou Q, Long L, Shi G, Zhang J, Wu T, Zhou B (2013) Research of the methylation status of miR-124a gene promoter among rheumatoid arthritis patients. Clin Dev Immunol 2013:524204. 10.1155/2013/524204 [DOI] [PMC free article] [PubMed] [Google Scholar]