

Abstract
Severed axon tips reform growth cones following spinal cord injury that fail to regenerate, in part, because they become embedded within an inhibitory extracellular matrix. Chondroitin sulfate proteoglycans (CSPGs) are the major axon inhibitory matrix component that is increased within the lesion scar and in perineuronal nets around deafferented neurons. We have recently developed a novel peptide modulator (intracellular sigma peptide) of the cognate receptor of CSPGs, protein tyrosine phosphatase σ (RPTPσ), which has been shown to markedly improve sensorimotor function, micturition, and coordinated locomotor behavior in spinal cord contused rats. However, the mechanism(s) underlying how modulation of RPTPσ mediates axon outgrowth through inhibitory CSPGs remain unclear. Here, we describe how intracellular sigma peptide modulation of RPTPσ induces enhanced protease Cathepsin B activity. Using DRG neurons from female Sprague Dawley rats cultured on an aggrecan/laminin spot assay and a combination of biochemical techniques, we provide evidence suggesting that modulation of RPTPσ regulates secretion of proteases that, in turn, relieves CSPG inhibition through its digestion to allow axon migration though proteoglycan barriers. Understanding the mechanisms underlying RPTPσ modulation elucidates how axon regeneration is impaired by proteoglycans but can then be facilitated following injury.
SIGNIFICANCE STATEMENT Following spinal cord injury, chondroitin sulfate proteoglycans (CSPGs) upregulate and potently inhibit axon regeneration and functional recovery. Protein tyrosine phosphatase σ (RPTPσ) has been identified as a critical cognate receptor of CSPGs. We have previously characterized a synthetic peptide (intracellular sigma peptide) that targets the regulatory intracellular domain of the receptor to allow axons to regenerate despite the presence of CSPGs. Here, we have found that one important mechanism by which peptide modulation of the receptor enhances axon outgrowth is through secretion of a protease, Cathepsin B, which enables digestion of CSPGs. This work links protease secretion to the CSPG receptor RPTPσ for the first time with implications for understanding the molecular mechanisms underlying neural regeneration and plasticity.
Keywords: axon regeneration, cathepsin B, CSPG, DRG, protease, RPTPσ
Introduction
Chondroitin sulfate proteoglycans (CSPGs) comprise a large family of extracellular matrix glycoproteins with varying numbers and types of glycosaminoglycan (GAG) chains bound to a protein core. During development, CSPGs aid in axon guidance (Snow et al., 1990; Brittis et al., 1992; Carulli et al., 2005) by serving as a molecular guardrail to prevent inappropriate targeting. In adulthood, CSPGs, such as aggrecan, are concentrated in synapse-enwrapping structures called perineuronal nets where they have been implicated in a variety of functions, including limiting axonal sprouting and neuroplasticity (Massey et al., 2006; Kwok et al., 2011; Tran et al., 2018). Following spinal cord injury, CSPGs also become highly upregulated in a gradient radiating outward from the astroglial scar epicenter as a result of (1) various triggering elements that emanate from the leaky blood–brain barrier (Schachtrup et al., 2010), (2) inflammatory cells (Fitch et al., 1999), or (3) fibroblastic-like cells in the lesion core (Okada et al., 2006). The gradient pattern of CSPG expression has been well characterized as a potent impediment to axon regeneration following spinal cord injury (Davies et al., 1997) and modeled in a spot assay in vitro where axons develop stalled, club-like dystrophic end balls once they encounter gradually changing concentrations of aggrecan and laminin (Tom et al., 2004).
The discovery of protein tyrosine phosphatase σ (RPTPσ) as a cognate receptor for the inhibitory actions of CSPGs was a major milestone in the search for potential therapeutic targets to promote regeneration (Shen et al., 2009). Indeed, we have recently reported the recovery of locomotor, sensory, and urinary function in an acute model of rat contusive injury treated for 7 weeks with a daily systemic injection of intracellular sigma peptide (ISP) (Lang et al., 2015). ISP is a synthetic peptide designed to specifically modulate the intracellular wedge domain of RPTPσ. However, the mechanism(s) underlying how ISP modification of RPTPσ signaling can lead to long-term CSPG disinhibition and subsequent axonal outgrowth are still unclear.
While characterizing the regeneration-promoting effects of ISP on adult DRG axons using our in vitro CSPG gradient spot assay, we have observed that ISP treatment markedly reduced the presence of the inhibitory GAG content of the substrate, suggesting possible protease activity. Normal low-level protease release by neurons to digest the extracellular matrix during their migration has been observed since the 1980s (Krystosek and Seeds, 1984). For example, during development, retinal ganglion cell growth cones secrete the protease ADAMTS4, which enables their access to retinotopically appropriate locations within the CSPG-rich lateral geniculate nucleus (Brooks et al., 2013). Furthermore, matrix metalloproteases (MMPs) have been noted for their CSPG-degrading effects (Cua et al., 2013), and MMP-2 knock-out mice showed reduced serotonergic sprouting following spinal cord injury (Hsu et al., 2006). Thus, finely controlled spatiotemporal regulation of protease release by neurons is one strategy by which discrete CSPG degradation may occur to enhance axon regeneration without delivering further damage to the CNS. Here, we present evidence that RPTPσ may be regulating the release of Cathepsin B (CatB), which specifically degrades CSPGs, and that this is one mechanism by which peptide-treated neurons may sustain axon outgrowth in a CSPG-rich environment.
Materials and Methods
Peptide sequences.
Peptides were purchased from GenScript or CS-BIO in lyophilized form (>98% purity) and reconstituted in sterile dH2O at 2.5 mm and aliquoted for storage in −20°C. Peptides were used at 2.5 μm concentration unless stated. ISP, GRKKRRQRRRCDMAEHMERLKANDSLKLSQEYESI; scrambled ISP (S-ISP), GRKKRRQRRRCIREDDSLMLYALAQEKKESNMHES; and TAT, GRKKRRQRRC.
DRG neuron culture.
All culture experiments were performed under sterile conditions. DRGs were cultured from adult female Sprague Dawley rats (Harlan) according to Case Western Reserve University Institutional Animal Care and Use Committee guidelines. Spinal columns were extracted from the animal, and a laminectomy was performed on ice under a dissection microscope (Leica). Forceps (size 3, FST) were used to extract DRGs, which were immediately placed on ice with calcium, magnesium-free media (50 ml 10× Hanks Balanced Salt Solution, 14185-052, Invitrogen, dH2O, 0.174 g sodium bicarbonate, Phenol Red). Extracted DRGs were then axotomized with microscissors (15003-08, FST), delaminated with forceps, and cut in half before they were collected in a 1 ml Eppendorf tube. Media was then replaced with prewarmed dispase (10165859001, Roche, 3.75 U/ml)/collagenase II (4176, Worthington, 200 U/ml) and incubated for 1 h at 37°C, then dissociated with gentle shaking for 30 min at room temperature. Dissociated ganglia were titurated with fire-polished Pasteur pipettes, and excess myelin was filtered at 100 μm (BD Falcon 352360). Following 3 × 5 min 4000 rotation per minute spin (Eppendorf 5415D) washes with calcium, magnesium-free media, DRG pellet was resuspended in prewarmed Neurobasal-A (10888-022, Invitrogen) with B-27 (17504-044, Invitrogen), 0.02% Glutamax (35050061, Invitrogen), 0.01% Pen/Strep (15140-031, Invitrogen) supplements at which point they were ready to be aliquoted for culture with 1 μm Ara-C (C1768, Sigma-Aldrich) to prevent glial outgrowth.
Aggrecan spot assay.
Aggrecan spots were made as previously described by Tom et al. (2004). Briefly, glass coverslips (120545082, Fisher Scientific) were acid-washed (1 m hydrochloric acid) at 55°C overnight and rinsed before use in 24-well plates (35-3047, Falcon). Coverslips were incubated overnight with poly-L-lysine (100 mg/ml, P1274, Sigma-Aldrich), washed 3× with dH2O, and coated with a 40 μl nitrocellulose (1.6 cm2, BA83 Schleicher & Schuell)/methanol (100%, 12 ml, A412-4, Fisher Scientific) mixture on cloning rings; 700 μg/ml aggrecan (A1960, Sigma-Aldrich) and 10 μg/ml laminin (23017-015, Invitrogen) in calcium, magnesium-free media was mixed, and four 2 μl spherical aliquots were carefully plated onto each coverslip and allowed to dry.
Spot assay to assess GAG-CSPG degradation.
DRGs were cultured on poly-L-lysine, 1 μg/ml aggrecan, and 10 μg/ml laminin-coated 6-well plates (35–3046, Falcon) with 1 ml of media and immediately treated with vehicle or peptides for 2 or 4 DIV. Conditioned media (CM) was collected and cell strained through centrifuging or filtration (09-720-3, Fisher Scientific) before plating onto freshly made spots. CM was incubated on spots at 37°C for 2 d after which spots were stained with CS-56 (1:500, C8035, Sigma-Aldrich), a marker for the GAG moieties of CSPGs. Each spot was imaged twice on a standard fluorescent microscope (Leica) using the same gain and exposure settings in a blinded fashion. To assess pixel intensities, an ROI box with a set area was made in ImageJ (National Institutes of Health) and used for all experiments, measured the pixel intensities of the spot rim. Five replicates with ∼20 repeats were performed for each spot experiment. To validate that ISP-treated CM degrades GAG-CSPGs on spots, we incubated 0.1 U Chondroitinase ABC (ChABC, C3667, Sigma-Aldrich) at 37°C for 2 h and quantified CS-56 immunoreactivity as described. To assess how long ISP needed to be incubated with DRGs to degrade spots, we collected media at 2 DIV for controls and 30 min, 1, 4, 24, and 48 h following ISP treatment before staining. To assess whether incubation with media alone would degrade CS-56, we incubated vehicle or peptides in calcium, magnesium-free media for 2 DIV at 37°C before staining. To assess whether boiling CM would rescue CS-56 degradation, we collected CM from DRGs treated with vehicle or peptides for 2 DIV and immediately plated one group on spots and boiled the collected CM from another group at 100°C and added guanidine hydrochloride (5 m) to fully denature the media before plating onto spots for 2 DIV. For spots assaying ISP in conjunction with other drugs, DRGs were treated with vehicle or ISP and the following drugs 24 h after plating for 2 DIV: DMSO (D8418, Sigma-Aldrich), Roche protease inhibitor (0.1%, 05892970001, Roche), GM6001 (25 μm, 364205, Calbiochem), anisomycin (20 μg/ml, A5862, Sigma-Aldrich), cycloheximide (20 μg/ml, C7698, Sigma-Aldrich), α-amanitin (20 μg/ml, A2263, Sigma-Aldrich), and Exo1 (10 μg/ml, ab120292, Abcam).
Spot assays to assess DRG axon outgrowth.
DRGs were collected from one adult female rat and aliquoted among one 24-well plate with fresh spots as described above with Neurobasal-A with supplements. Vehicle or peptides were added immediately after plating for 4 DIV at 37°C, 5% CO2. DRG coverslips were then fixed with 4% PFA, washed with 1× PBS, and incubated with 5% normal goat serum (16210-064, Invitrogen) block, 0.1 g BSA (A3059, Sigma-Aldrich), 0.1% Triton-X (X-100, Sigma-Aldrich) block at room temperature for 1 h. Coverslips were then stained with the following primary antibodies with block overnight at 4°C: CS-56 (1:500, Sigma-Aldrich), β-tubulin III (Tuj1, 1:500, T8660, Sigma-Aldrich). Corresponding secondary antibodies conjugated with fluorophores were then used at overnight, 4°C incubation following 3 × 15 min 1 × PBS washes: anti-mouse IgM AlexaFluor-546 (1:500, A21045, Invitrogen), anti-mouse IgG2b AlexaFluor-488 (1:500, A21121, Invitrogen). Coverslips were then mounted on slides with Citifluor (17971-25, EM Sciences). The number of Tuj1+ axons crossing the CS-56-labeled spot rim was counted and normalized against the total number of Tuj1+ soma present in a blinded fashion on a standard fluorescent microscope (Leica) at 10× objective. For pretreated spots to assess axon outgrowth, the same method was used to culture DRGs, but spots were pretreated with the following for 24 h before DRG plating: 0.1 U/ml ChABC (C3667, Sigma-Aldrich), recombinant CatB (0.5 μg/ml, 965-CY, R&D Systems) activated in 0.1 m sodium acetate, pH 5.0, recombinant Cystatin B (CSTB, 50 μg, 1409-PI, R&D Systems), cell-strained CM collected from another set of 6-well DRGs treated with vehicle or peptides. Three or four replicates with up to 20 repeats were performed.
Western blot assay of GAG degradation.
To confirm GAG degradation, 100 μl CM was collected from DRGs cultured on PLL, laminin/aggrecan as described. Cell-strained CM was incubated with 20 μg/ml aggrecan at 37°C for 2 h; 20 μg protein was mixed with Laemmli/β-mercaptoethanol and denatured at 100°C for 10 min. Samples were loaded onto precast SDS/PAGE gels (456–1094, BD Biosciences) and ran ∼1.5 h at 100 mV, then transferred onto PVDF membranes (Bio-Rad) overnight at 12 mV on ice. Blots were blocked with Super Block (37515, Thermo Fisher Scientific) for 2 h shaking at room temperature before incubation with the following antibodies overnight at 4°C: DIPEN (1:1000, 1042002, MD Biosciences), CS-56 (1:1000, Sigma-Aldrich). Following 3 × 15 min 1 × PBS-0.1% Tween 20 washes, corresponding secondary antibodies were incubated for 2 h at room temperature or 4°C overnight with shaking: anti-mouse IgG-HRP (1:1000, 55563, ICN), anti-mouse IgM-HRP (1:1000, AP128P, Millipore). Blots were developed using chemiluminescent kit (WBKLS0500, Millipore).
DQ Gelatin assay.
DRGs were cultured on PLL-incubated coverslips precoated with 1 μg/ml aggrecan, 10 μg/ml laminin, and 25 μg/ml DQ Gelatin (D12054, Invitrogen), which is a quenched gelatin conjugate that only fluoresces once it has been cleaved by proteases, for 2 DIV. After fixing as previously described, DRGs were then stained with Tuj1 (1:500, Sigma-Aldrich) with a mouse IgG2b AlexaFluor-546 secondary (1:500, Invitrogen). Colocalization was assessed using Just Another Colocalisation Plugin (Bolte and Cordelières, 2006) in ImageJ. Four replicates with 8 repeats each were performed.
Protease activity (EnzChek) assay of DRG CM.
To assay general protease activity in DRG CM, we used EnzChek (E6638, Thermo Fisher Scientific), which uses quenched casein that only fluoresces once it has been cleaved. CM was collected from vehicle, 2.5 μm ISP, or 2.5 μm S-ISP-treated DRGs cultured on poly-L-lysine, 1 μg/ml aggrecan, and 10 μg/ml laminin for 2 or 4 DIV with 1 ml media. CM was cell strained through centrifuging and incubated with 1× EnzChek mixture in 1:4 ratio; 200 μl of CM mixture was aliquoted onto a 96-well plate with 3 replications for each sample overnight at room temperature with gentle shaking. The plate was read at 435/535 nm (excitation/emission), and media-only was used as a blank.
Mass spectrometry of DRG CM.
DRGs were grown on 6-well plates precoated with poly-L-lysine, 10 μg/ml laminin/1 μg/ml aggrecan for 4 DIV with 2.5 μm ISP. This was repeated twice as process replicates for the purpose of screening possible proteases and not for quantification. Cell-strained CM was sent to for mass spectrometry analysis at the Case Western Reserve University Protein Core. Proteome Discoverer version 1.3.0.0339 (Thermo Fisher Scientific) was used to generate peaklist, Mascot version 2.3.00 (MatrixScience) as the search engine, Uniprot Rat Database (December 2012: 7844 sequences) used as the sequence database, and an expectation value of <0.05 was accepted with an estimated false discovery rate of 4.45%.
Western blot assessment of DRG lysate and CM.
DRGs were cultured on poly-L-lysine, laminin/aggrecan-precoated 6-well plates with 1 ml media/well and immediately dosed with vehicle or peptides for 4 DIV. CM was collected, cell-strained, and 100 μg of each sample was processed for Western blot as described above. To process DRG lysates for Western blot analysis, DRGs were cell-scraped from 6-well plates cultured in a similar manner as described above. Lysates were incubated with Protease Inhibitor (Roche Diagnostics) and vortexed at maximum speed for 30 min at 4°C. Lysates were then centrifuged at maximum speed for 20 min at 4°C; 20 μg protein of each sample was processed for Western blot as described above. The following primary antibodies were used for 4°C overnight incubation with gentle shaking: CatB (1:1000, AF953, R&D Systems), CSTB (1:500, AF1408, R&D Systems), GAPDH (1:1000, 2118S, Cell Signaling Technology), histone-3 (1:1000, 4499P, Cell Signaling Technology). The following secondary antibodies were used following washes: anti-mouse IgG-HRP (1:1000, A9044, Sigma-Aldrich), anti-rabbit IgG-HRP (1:1000, P132P, Millipore), anti-goat IgG-HRP (1:1000, 55385, ICN).
DRG immunocytochemistry.
DRGs were grown on poly-L-lysine-incubated coverslips precoated with 1 μg/ml aggrecan and 10 μg/ml laminin in 24-well plate for 2 DIV. Cell fixation and staining were performed similarly as DRG-spot staining with the following primary antibodies: Tuj1 (1:500, Sigma-Aldrich), CatB (1:500, R&D Systems), CSTB (1:500, R&D Systems). Corresponding previously described secondary antibodies were used.
Lentiviral infection of CSTB overexpression on spot assays.
Lentiviral GFP control (sc-108084, Santa Cruz Biotechnology) and CSTB-overexpressing (CSTBo/e) rat gene (LPP-CS-GS498J-Lv165-050, GeneCopoeia) constructs were purchased and aliquoted for storage at −80°C; 0.1 multiplicity of infection (cell number/virus particles in microliters × stock concentration) of each lentiviral particle group was aliquoted to inoculate DRGs on spot assays immediately upon plating for 4 DIV. A subset of DRGs grown on aggrecan/laminin were grown on 6-well plates and inoculated with GFP or CSTBo/e to use for Western blot analyses. Fix and staining were performed as described above. Three replicates with four repeats were performed and analyzed.
Injured spinal cord tissue immunostaining.
All procedures were performed according to Case Western Reserve University Institutional Animal Care and Use Committee guidelines. Female Sprague Dawley rats (225–250 g, Harlan) were injected with ketamine (60 mg/kg)/xylazine (10 mg/kg); thoracic sections T7-T9 were exposed and laminectomized, and the cord was held taut to receive a 250k dyne contusive injury at T8 from an Infinite Horizon Impact Device. Animals were injected with ISP 1 d after surgery (subcutaneous, 22 μg/ml s.c., with 5%) daily for 49 d. Animals were killed 7 weeks after administration of the last ISP injection, perfused, and fixed with 4% PFA followed by 30% sucrose incubation overnight. Animals with the highest Basso, Beattie, and Bresnahan (BBB) score (20, ISP-treated) and lowest BBB score (9, DMSO control) were chosen for tissue processing. Sections (20 μm/section, Hacker cryostat) were blocked with 0.1% BSA, 0.1% Triton-X, and 5% normal goat serum in PBS (81873, Thermo Fisher Scientific). The following primary antibodies were diluted in blocking buffer and incubated overnight at 4°C: serotonin (5-HT, 1:500, 20080, ImmunoStar), Wisteria Floribunda Agglutinin (WFA, 1:50, L1516, Sigma-Aldrich), CatB (1:500, R&D Systems). Following PBS-0.1% Tween 20 washes (3 × 15 min), the following secondary antibodies were diluted in blocking buffer for overnight incubation: anti-rabbit IgG AlexaFluor-546 (1:500, A-11035, Invitrogen) anti-goat IgG AlexaFluor-488 (1:500, A-11055, Invitrogen), and streptavidin AlexaFluor-546 (1:500, S112255, Thermo Fisher Scientific). Imaging took place on a standard fluorescent microscope (Leica). Quantification of images below the site of injury (T8) extending to the lumbar region (∼15 20 μm sections) was performed in ImageJ and normalized by area.
Injured spinal cord Western blotting.
Female Sprague Dawley rats were given T8 contusions as detailed above. One day after injury, subjects were given subcutaneous DMSO vehicle or ISP (22 μg/ml) injections for 14 d before death. Spinal cord was quickly extracted and frozen using liquid nitrogen and stored at −80°C before processing. To process spinal cords for Western blotting, spinal cords from the thoracic to lumbar region were homogenized (D1000, Handheld Tissue Homogenizer, Benchmark Scientific) with 300 μl lysis buffer (89900, Thermo Fisher Scientific) with a tablet of protease inhibitor (11697498001, Sigma-Aldrich) per 10 ml of lysis buffer. Lysates were agitated for 2 h at 4°C and then centrifuged at maximum speed for 20 min before the supernatant was collected; 20 μg protein was loaded for Western blotting as detailed above. The following primary antibodies were used: CatB (1:500, R&D Systems), GAPDH (1:1000, 2118S, Cell Signaling Technology), with the corresponding peroxidase-linked secondary antibodies detailed above.
Transgenic RPTPσ DRG extraction.
RPTPσ-null mutants (Elchebly et al., 1999) and BALB/C WT DRG extractions were performed similarly as rat DRG extractions, with the exception that DRGs provided from 1 animal were used across 8 coverslips with 1 μm FUdR (F0503, Sigma-Aldrich) to prevent glial outgrowth. Experiments for spot assay immunostaining and GAG-CSPG degradation were performed as described above, but with additional concentration of the media for at least 30 min at 4°C (YM-3, Millipore). To assess CatB immunoreactivity, DRGs were plated on poly-L-lysine-incubated coverslips precoated with 1 μg/ml aggrecan and 10 μg/ml laminin in 24-well plate for 2 DIV. Cell fixation and staining were performed similarly as DRG-spot staining with the following primary antibodies: Tuj1 (1:500, Sigma-Aldrich), CatB (1:500, R&D Systems). Corresponding previously described secondary antibodies were used. Axon tips were imaged and quantified using ImageJ normalized by area. Precise sample sizes are listed in the figure legends.
Experimental design and statistics.
Spot degradation assays were typically performed in replicates of up to 5 with ∼20 repeats each (see Materials and Methods subsection). Spot crossing assays were performed with at least 3 replicates and 4 repeats each (see Materials and Methods subsection). Precise n is noted in the figure legends. GraphPad Prism 5 was used to analyze data with the following statistical tests: Pearson's χ test to test for normality, Student's unpaired t test, one-way ANOVA with post hoc Tukey's (to test multiple groups), or Dunnett's multiple comparisons (to test control against other groups) tests.
Results
Serotonergic sprouting correlates with GAG-CSPG digestion in vivo
We had previously observed that, after contusive spinal cord injury, serotonergic (5-HT) sprouting was dramatically enhanced following ISP treatment, which, in turn, allowed for improved urinary and locomotor recovery (Lang et al., 2015). To explore the mechanism(s) responsible for ISP-enhanced fiber sprouting, we immunostained sections of spinal cords derived from animals that had received a thoracic level 8 contusive injury (250k dyne) and were subsequently treated systemically with either vehicle (saline, 5% DMSO) or ISP (22 μg/ml) for 7 weeks beginning 1 d after injury. Serotonergic sprouting was increased distal to the lesion especially in the vicinity of the ventral motor pools and near the central canal of ISP-treated animals compared with vehicle controls (t = 3.320, df = 64, p = 0.0015, unpaired t test; Fig. 1A). Following immunostaining for both serotonin and the GAG moiety of CSPGs (GAG-CSPGs) using the WFA antibody, we found that vehicle-treated animals had profuse GAG-CSPG staining throughout the gray and white matter of the lumbosacral spinal cord compared with ISP-treated animals (t = 4.657, df = 84, p = 0.0001, unpaired t test; Fig. 1B). In comparison, WFA immunoreactivity of spinal cords belonging to animals that received no injury was distributed diffusely across the gray matter with noted immunoreactivity in perineuronal nets surrounding ventrally located motor neurons (Fig. 1A). In vehicle-treated, lesioned animals, serotonergic fibers were present, although in minimal amounts. However, in ISP-treated animals, enhanced serotonin immunostaining occurred in regions where WFA was lacking (Fig. 1B). Indeed, the staining patterns seemed reciprocal with an almost lock-and-key configuration (Fig. 1A). This striking pattern led us to speculate whether ISP treatment enhanced the ability of fibers to sprout through the secretion of proteases, which would diminish the presence of inhibitory GAG-CSPGs. To better explore this hypothesis, we harvested adult DRGs to study in vitro because it was not feasible to extract adult serotonin neurons in significant numbers. Indeed, DRG cultures plated on our aggrecan spot assay and treated with ISP developed “shadows” of CS-56 immunostaining (an antibody specific for the GAG domain of CSPGs) beneath their axons as they extended across a previously inhibitory CSPG gradient (Fig. 1C). This led us to hypothesize that protease activity in DRGs may be induced by ISP treatment.
Figure 1.
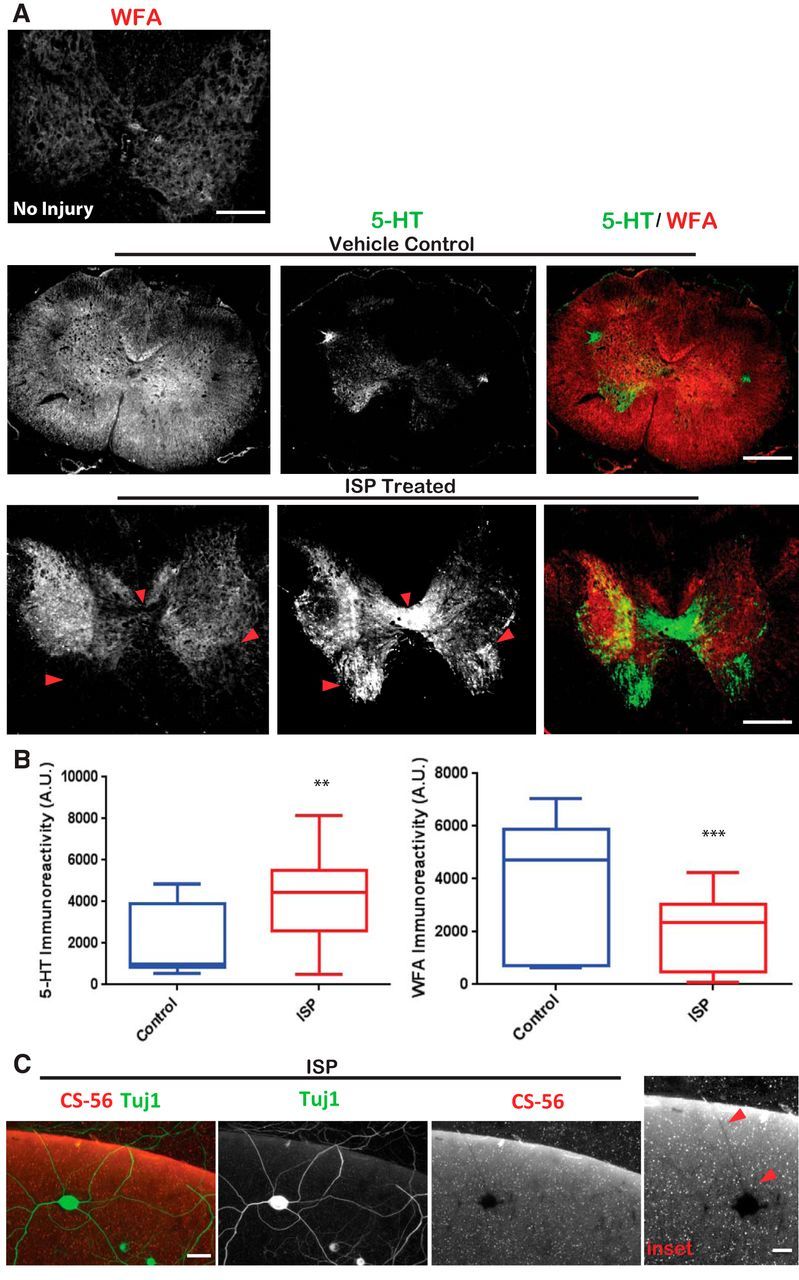
ISP treatment enhances GAG-CSPG degradation by neurons. A, Systemic ISP enhances GAG-CSPG amelioration following spinal cord injury. Rats received 250k dyne thoracic level 8 contusion and treated for 7 weeks with subcutaneous injections of DMSO vehicle or 22 μg/ml ISP beginning 1 d after injury. Tissue was collected 7 weeks after last ISP treatment and stained to visualize serotonergic (5-HT) axons and GAG-CSPGs (WFA). Noninjured spinal cord of an adult rat was immunostained with WFA to visualize normal GAG-CSPG pattern. Scale bar, 500 μm. B, Quantification of 5-HT (n = 25 sections; t = 3.320, df = 64, p = 0.0015, unpaired t test) and WFA (n = 39 sections; t = 4.657, df = 84, p = 0.0001, unpaired t test) immunoreactivity in sections caudal to the injury site. C, DRG axons (Tuj1) leave digested shadows as they cross the high CSPG gradient (CS-56) in our aggrecan spot assays when treated with low concentrations of ISP (1.25 μm). Scale bar, 50 μm. Red arrows indicate regions of absent GAG-CSPGs colocalized with neuronal expression. Lines indicate median. Boxes represent quartiles. Whiskers indicate range. **p < 0.01, ***p < 0.001.
ISP induces DRGs to degrade CSPGs through upregulation of protease secretion
To begin investigating whether protease activity was linked downstream to RPTPσ activity, we cultured DRGs for 4 DIV on a low concentration of aggrecan and laminin and treated the cultures with vehicle control, varying doses of ISP (0.75–5 μm) or S-ISP (2.5 μm). The CM was then collected and incubated with freshly made aggrecan spots for 2 DIV (Fig. 2A–C). These spots were then stained with CS-56, and the pixel intensities of the CSPG gradient were measured. To validate this aggrecan spot assay as a measure for CS-56 immunoreactivity, we additionally incubated spots with 0.1 U/ml ChABC and found that ChABC treatment reduced CS-56 immunoreactivity as expected (Con vs ChABC F(7,173) = 47.21, p = 0.0001, ANOVA Tukey's post hoc; Fig. 2B,C). Interestingly, S-ISP (2.5 μm) also slightly decreased CS-56 immunoreactivity (Con vs S-ISP, p = 0.0006, ANOVA Tukey's post hoc). Importantly however, ISP (2.5 μm) far more dramatically decreased GAG-CSPG over S-ISP (p = 0.0097, ANOVA Tukey's post hoc). The slight amount of apparent protease activity stimulated via the scrambled peptide may be due to the known biological activity that TAT itself (an amino acid sequence included in ISP as well as S-ISP that allows cell penetrance of our peptide) is able to induce, such as enhancing neuroprotection or neuroinflammation through Erk1/2 and Akt signaling (Williams et al., 2010; P. Liu et al., 2014; Soo Youn et al., 2015). We have found no evidence that S-ISP promotes any functional benefits after spinal cord injury (Lang et al., 2015). We did find that CS-GAG digestion was ISP dose-dependent and became significantly enhanced over vehicle control when peptide concentrations reached 2.5 μm (Con vs 2.5 μm ISP, p = 0.0001, ANOVA Tukey's post hoc; Fig. 2C). As a control, we incubated media alone with vehicle, or the same concentrations of ISP, S-ISP, or TAT (Fig. 2E). We found that these peptides alone (no cells present) were unable to significantly digest CSPGs (F(3,107) = 1.684, Con vs ISP, p = 0.1748, ANOVA Tukey's post hoc; Fig. 2E). Instead, ISP degradation of GAG-laden CSPGs increased over time and was dependent on the presence of DRGs (F(6,291) = 30.77, Con vs ISP 48 h, p = 0.0001, ANOVA Tukey's post hoc; Fig. 2D). GAG-CSPG degradation may reflect an enhanced secretion of stored proteases or an increased production of proteases or both.
Figure 2.

ISP promotes GAG chain degradation in coverslip-bound aggrecan/laminin spot assays. A, CM from 4 DIV or 0.1 U/ml ChABC-treated DRGs were incubated with aggrecan spots for 2 DIV, then stained with CS-56 and analyzed. B, Representative aggrecan spot images. Scale bar, 200 μm. C, ISP degrades aggrecan GAG-CSPGs in a dose-dependent manner (n = 120, 73, 69, 92, 77, 59, 50; F(7,173) = 47.21, CON vs 2.5 μm ISP, p = 0.0001; CON vs 5 μm ISP, p = 0.0001; CON vs 2.5 μm S-ISP, p = 0.0006; CON vs ChABC, p = 0.0001; 2.5 μm ISP vs S-ISP, p = 0.0097; ANOVA). D, ISP degradation of GAG chains is time-dependent and significant at 48 h of incubation with DRGs (n = 47, 31, 32, 41, 30, 39, 78; F(6,291) = 30.77, p = 0.001, ANOVA). E, As a control, equal molar concentrations of peptide alone in media do not degrade GAG chains (n = 34, 27, 28, 22; F(3,107) = 1.684, not significant, p = 0.1748, ANOVA). F, Western blots of CM from DRGs treated with varying concentrations of ISP incubated with 20 μg/ml aggrecan confirm CS-56 spot degradation; n = 4 blots. G, DIPEN, a neo-epitope present once aggrecan is cleaved, increases with ISP dose; n = 3. H, Western blots of CM from vehicle control, 2.5 μm ISP, and 2.5 μm S-ISP incubated with 20 μg/ml aggrecan blotted with CS-56 and DIPEN; n = 2. I, Control Western blots of media incubated for varying times show intact CS-56 and no DIPEN signal; n = 3. Lines indicate median. Boxes represent quartiles. Whiskers indicate range. **p < 0.01, ***p < 0.001.
To confirm that aggrecan degradation was occurring independent of our spot assay conditions, we performed Western blot analyses of CM collected from treated DRGs that were then incubated with aggrecan (20 μg/ml) for 2 h (Fig. 2F–H). CS-56 immunoblotting of incubated CM and aggrecan mixtures confirmed a dose-dependent ISP-induced decrease of CS-56 immunoreactivity (Fig. 2F). Interestingly, the immunoreactivity with DIPEN, an antibody specific for a neo-epitope of the aggrecan core protein revealed only once cleaved (Hughes et al., 1995), also increased in an ISP dose-dependent manner (Fig. 2G). As a further control for these assays, we incubated media alone with the same concentration of aggrecan for up to 4 DIV and found that CS-56 was not decreased over time, nor was DIPEN immunoreactivity detected (Fig. 2I). This suggested that a decrease in CS-56 and reciprocal increase in DIPEN expression patterns are ISP-dependent but only in the presence of cells.
To further characterize protease activity, we used a general protease assay (EnzChek) that measures cleaved casein, which also fluoresces once it becomes cleaved by a variety of proteases present in the CM collected from DRG cultures incubated with vehicle control, ISP, or S-ISP for 1, 2, or 4 DIV (Fig. 3C). Just as in our spot assays, we noticed that protease activity increased over time and that ISP (2.5 μm) treatment significantly increased protease activity over vehicle control by 4 DIV (F(8,27) = 1.4, p = 0.0001, ANOVA). In addition, collected CM was thoroughly denatured by boiling at 100°C and incubated with guanidine hydrochloride (5 m). We found that denaturing the CM was able to fully rescue GAG-CSPGs from degradation induced by ISP incubation (F(5,87) = 37.13, ISP vs ISP Boiled, p = 0.0001, ANOVA; Fig. 3D).
Figure 3.
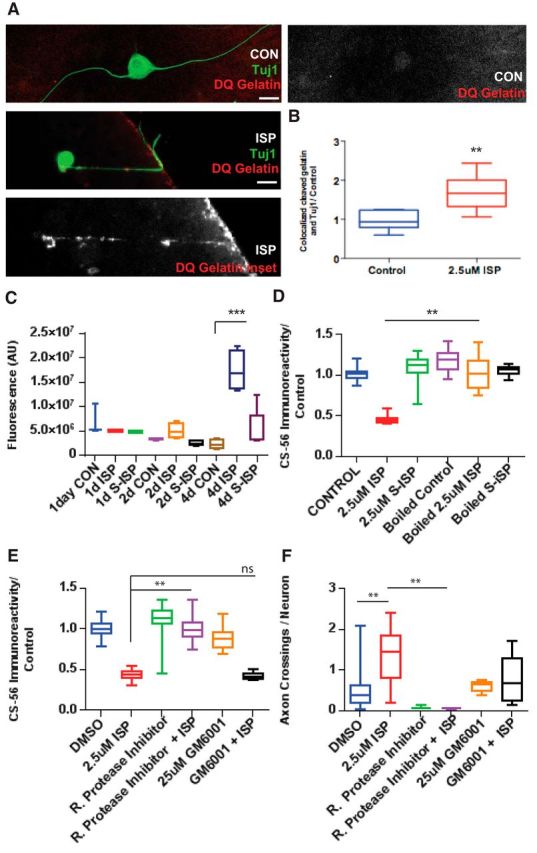
ISP promotes GAG chain degradation through increasing protease activity. A, Representative image of an ISP-treated DRG axon digesting DQ Gelatin as it crosses the CSPG gradient in our spot assay and vehicle control DRG axon. Scale bar, 50 μm. B, Quantification of ratio of cleaved DQ Gelatin overlapping Tuj1-labeled axons (n = 34, 32; t = 4.025, df = 15, p = 0.0011, unpaired t test). C, EnzChek measures fluorescence of cleaved casein of CM from DRGs treated with CON, 2.5 μm ISP or SISP for 1, 2, or 4 DIV (n = 11, 16, 16, 16, 16, 16, 19, 18, 20; F(8,27) = 15.4, p = 0.0001; ANOVA). D, Aggrecan spot GAG chain degradation is rescued following boiling CM, then incubating with guanidine hydrochloride (5 m) before plating (n = 71, 49, 50, 51, 68, 50; F(5,87) = 37.13, p = 0.0001; ANOVA). E, Aggrecan spot degradation was rescued by 0.1% Roche general protease inhibitor mixture, but not broad MMP inhibitor, GM6001 (25 μm) (n = 114, 161, 197, 97, 100, 63; F(5,112) = 82.05, ISP vs RPI+ISP, p = 0.001; ISP vs GM6001+ISP, not significant, p = 0.9964; ANOVA). F, Axon crossings, normalized by the total number of neurons in the spot, were decreased with protease inhibitor treatment (n = 28, 10, 10, 29, 17, 20; F(5,37) = 5.255, DMSO vs ISP, p = 0.0042; ISP vs RPI+ISP, p = 0.0064; ANOVA). Lines indicate median. Boxes represent quartiles. Whiskers indicate range. **p < 0.01, ***p < 0.001.
While we confirmed that GAG-CSPG degradation was enhanced by ISP through our spot assay and Western blots, we sought to corroborate even further that protease(s), in addition to being secreted into the media, were also being focally induced along the regenerating axon. To visualize local GAG degradation by ISP-treated DRG axons, we plated DRGs on modified aggrecan spots that were formulated with 10 μg/ml laminin, 1 μg/ml aggrecan, and 25 μg/ml DQ Gelatin, which is a specially quenched gelatin that fluoresces once cleaved by a protease (Fig. 3A,B). Using this modified aggrecan spot, we found axons that colocalized with a trail of cleaved, fluorescing gelatin as they attempted to extend past the aggrecan gradient (see, e.g., Fig. 3A). Quantification of these modified spots revealed that DRG neurons normally show a baseline of gelatin cleavage; however, treatment with ISP (2.5 μm) significantly increased the amount of cleaved gelatin colocalized with axons labeled with β-tubulin III (Tuj1) (t = 4.025, df = 15, p = 0.0011, unpaired t test; Fig. 3B). We further incubated CM with protease inhibitors, including a low concentration of the globally effective Roche protease inhibitor (0.1%) and a broad MMP inhibitor, GM6001 (25 μm). The nonspecific Roche protease inhibitor was able to rescue ISP-induced GAG degradation (F(5,112) = 82.05, ISP vs RPI+ISP, p = 0.001, ANOVA), but ISP treatment in the presence of GM6001 still allowed for GAG degradation (ISP vs GM6001+ISP, p = 0.9964; Fig. 3E). This suggests that ISP may be upregulating proteases other than the various MMPs that are inhibited by GM6001. The addition of the Roche protease inhibitor also resulted in a decrease in axons crossing the aggrecan gradient in the presence of ISP (F(5,37) = 5.255, ISP vs RPI+ISP, p = 0.0064, ANOVA), suggesting that proteases may, indeed, be playing a role in enhancing axon outgrowth by increasing CSPG digestion (Figs. 2, 3).
ISP upregulates CatB secretion in DRG cultures
We next asked how ISP might be inducing protease activity. To begin exploring whether enhanced transcription or translation of certain proteases was responsible for ISP induction of GAG degradation, we treated DRGs cultured on a low concentration of aggrecan and laminin with vehicle control or ISP (2.5 μm) with translation inhibitors, anisomycin (20 μg/ml) and cycloheximide (20 μg/ml), or a transcription inhibitor, α-amanitin (20 μg/ml), and incubated spots with the collected CM. GAG-CSPG degradation still occurred in all treatment groups once ISP was added, suggesting that neither transcription nor translation was requisite for ISP-dependent GAG-CSPG degradation (F(7,113) = 2.6, ISP vs anisomycin+ISP, p = 0.385; ISP vs cycloheximide+ISP, p = 0.49; ISP vs α-amanatin+ISP, p = 0.6309; ANOVA Tukey's post hoc; Fig. 4A). However, when Exo1 (10 μg/ml), an exocytosis inhibitor (Feng et al., 2003), was added in conjunction with ISP, CS-56 immunoreactivity was restored (F(3109) = 35.18, ISP vs Exo1+ISP, p = 0.0108, ANOVA; Fig. 4B). Thus, ISP seems to be inducing exaggerated protease activity in the CM through enhancing protease secretion.
Figure 4.
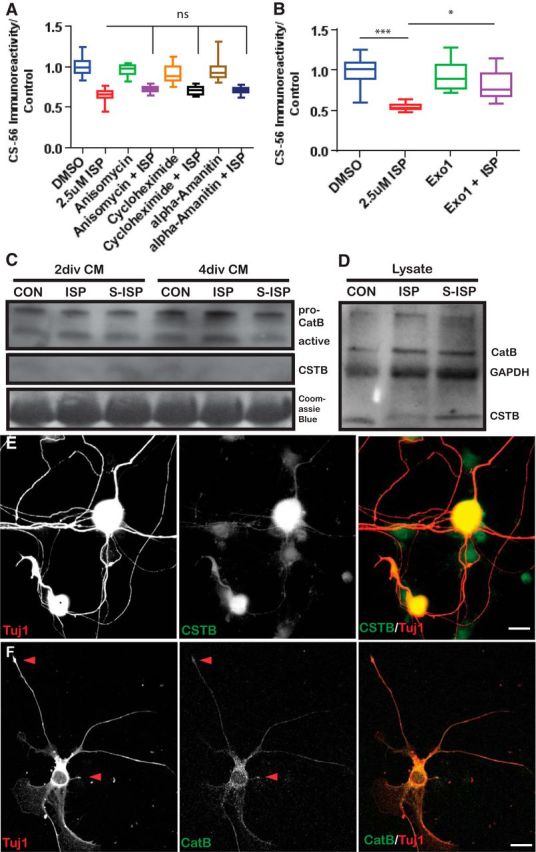
ISP promotes secretion of CatB. A, ISP-dependent aggrecan spot degradation is not dependent on transcription or translation, as shown by anisomycin (20 μg/ml), cycloheximide (20 μg/ml), or α-amanitin (20 μg/ml) treatment of DRGs (n = 103, 91, 54, 50, 73, 57, 72, 60; F(7,113) = 2.6, not significant; ISP vs anisomycin+ISP, p = 0.3855; ISP vs cycloheximide+ISP, p = 0.490; ISP vs α-amanatin, p = 0.6309; ANOVA). B, Inhibiting exocytosis with a high concentration of Exo1 (10 μg/ml) rescues GAG chain degradation by ISP (n = 115, 187, 160, 76; F(3109) = 35.18, DMSO vs ISP, p = 0.0003; ISP vs Exo1+ISP, p = 0.0108; ANOVA). C, Western blot of CatB and CSTB from DRG CM collected after 2 or 4 DIV treated with vehicle control, 2.5 μm ISP, or S-ISP; n = 5. The same blot was stained with total protein dye, Coomassie Blue. D, Western blot of CatB, CSTB, or GAPDH from DRG lysate treated with vehicle control, 2.5 μm ISP, or S-ISP for 4 DIV; n = 3. DRGs stained with Tuj1 and CSTB (E) or CatB (F). Scale bar, 50 μm. Lines indicate median. Boxes represent quartiles. Whiskers indicate range. **p < 0.01, ***p < 0.001.
To discover which protease(s) may be hypersecreted following RPTPσ modulation, we subjected ISP-treated DRG CM to mass spectrophotometry analysis and found that CatB was the most abundant CSPG-degrading protease (Fosang et al., 1992; Mort et al., 1998). Previous work has identified CatB to be enriched primarily in neurons in the gray matter of the spinal cord and brain (Ellis et al., 2005). Western blot analysis of vehicle control, ISP, or S-ISP (2.5 μm) treated CM confirmed that CatB activity increased over time (Fig. 4C) and was, indeed, found in DRG cellular lysates (Fig. 4D). Immunostaining of DRG neurons in vitro (plated on laminin/low aggrecan) additionally revealed that CatB could be found in neuronal somata as well as throughout the axon and axonal tips (Fig. 4F). CatB is a lysosomally derived peptidase that is important in numerous cellular homeostatic processes, including protein degradation and turnover in lysosomes, autophagy, cell signaling, and innumerable other physiological functions (Turk et al., 2012). Interestingly, CatB has been previously reported to also reveal a DIPEN-specific epitope once it cleaves aggrecan much like that which we have observed (Fig. 2G) (Mort et al., 1998). To begin exploring how CatB may be further controlled by RPTPσ modulation, we immunostained for an endogenous inhibitor of CatB, CSTB (Turk and Bode, 1991). While we found trace amounts of CSTB in ISP-treated DRG CM (Fig. 4C), CSTB seemed to be decreased by ISP intracellularly (Fig. 4D). Indeed, CatB (Fig. 4F) as well as CSTB (Fig. 4E) can be detected throughout DRG neurons, including within their axons.
CatB degradation of aggrecan enhances axon outgrowth
We next sought to test whether specific CatB degradation of CSPGs would enhance axon outgrowth past a CSPG gradient. We first tested recombinant CatB and CSTB using a general protease assay (EnzChek) and found that higher concentrations of recombinant CatB were, indeed, able to cleave casein, but that this activity is dampened by the addition of CSTB (F(4,5) = 11.32, recombinant CatB [rCatB] vs rCatB+rCSTB, p = 0.0101, ANOVA; Fig. 5A). Recombinant CatB (0.5 μg/ml) was then used to pretreat aggrecan spots much like ChABC (Fig. 5B). In addition to recombinant CatB, we also pretreated spots with CM from DRGs treated with vehicle, ISP, or S-ISP (2.5 μm) with or without the addition of CSTB (50 μg/ml) that would putatively inhibit any ISP-secreted CatB. As expected, ISP-treated CM yielded GAG-CSPG degradation significantly over media-only positive control (F(12,155) = 30.35, media-only vs ISP, p = 0.0001, ANOVA Tukey's post hoc; Fig. 5B). Additionally, ChABC (0.1 U/ml) was able to decrease CS-56 immunoreactivity as expected (media-only vs ChABC, p = 0.0019, ANOVA Tukey's post hoc). The addition of recombinant CatB was also able to degrade GAG-CSPGs in a similar manner (media-only vs rCatB, p = 0.0001, ANOVA Tukey's post hoc). However, incubating recombinant CSTB with ISP-treated DRG CM was able to rescue CatB-induced GAG-CSPG degradation (ISP vs ISP+rCSTB, p = 0.0123, ANOVA Tukey's post hoc; Fig. 5B). The degradation of GAG-CSPGs by pretreatment of the spot assay also correlated with an increase in axon outgrowth across the outer rim (F(7,32) = 9.719, ISP vs ISP+rCSTB, p = 0.0402, ANOVA; Fig. 5C,D). As expected, ChABC treatment yielded significant axon crossings past the aggrecan gradient compared with media-only pretreatments (p = 0.0018, ANOVA Tukey's post hoc). ISP-treated CM and recombinant CatB pretreatments also similarly enhanced axon crossings (p = 0.0207, ANOVA Tukey's post hoc). However, the addition of recombinant CSTB to recombinant CatB pretreatment attenuated this effect (p = 0.0402, ANOVA Tukey's post hoc; Fig. 5C,D).
Figure 5.

Pretreatment of aggrecan spots with ISP-treated CM or rCatB enhances axon crossings through the aggrecan gradient. A, EnzChek protease assay of recombinant CatB ± recombinant CSTB (rCSTB) (n = 6; F(4,5) = 11.32, p = 0.0101, ANOVA). B, Aggrecan spot degradation with CM from vehicle, 2.5 μm ISP, or S-ISP-treated DRGs, or media-only, 0.1 U/ml ChABC, 50 ng/ml recombinant CatB, 10 ng/ml recombinant CSTB alone or in combination with vehicle, or 2.5 μm ISP-treated DRG CM (n = 106, 157, 141, 136, 21, 45, 11, 63, 47; F(12,155) = 30.35, media-only vs ChABC, p = 0.0019; media-only vs ISP, p = 0.0001; media-only vs rCatB, p = 0.0001; ISP vs ISP+rCSTB, p = 0.0001; ANOVA). C, D, Axon crossings normalized by total neurons present on aggrecan spots pretreated with CM collected from vehicle, 2.5 μm ISP or SISP, or 0.1 U/ml ChABC, recombinant CatB (0.5 μg/ml) with or without recombinant CSTB (50 μg/ml) with representative images. Scale bar, 50 μm. Dotted lines indicate CSPG border. Inset, Axon crossing CSPG gradient (n = 11, 20, 12, 12, 12, 17, 30, 29; F(7,32) = 9.719, media vs ChABC, p = 0.0018; S-ISP vs rCatB, p = 0.0207; ISP CM vs ChABC, not significant, p = 0.5606; ISP vs ISP+rCSTB, p = 0.0402; ANOVA). Lines indicate median. Boxes represent quartiles. Whiskers indicate range. *p < 0.05, **p < 0.01, ***p < 0.001.
To further test whether CSTB inhibition of CatB had any functional effects on axons crossing the CSPG gradient, we overexpressed CSTB (CSTBo/e) using lentiviral particles expressing the CSTB gene under the EFa1 promoter in our DRG culture model. Overexpression of CSTB was confirmed compared with control GFP lentiviral-infected DRG cultures using Western blot analysis (Fig. 6A). Indeed, overexpression of CSTB was able to rescue ISP-induced GAG-CSPG degradation in our spot assays (F(5,115) = 51.03, GFP+ISP vs CSTBo/e+ISP, p = 0.0001, ANOVA; Fig. 6B). Furthermore, overexpression of CSTB attenuated ISP-induced axonal outgrowth past a gradient of aggrecan in our spot assay (F(3,12) = 3.743, GFP+ISP vs CSTBo/e+ISP, p = 0.0415, ANOVA; Fig. 6C,D). Together, this suggests that ISP-induced degradation of GAG-CSPG is, indeed, occurring through CatB expression and that CatB activity is important for axon outgrowth through extracellular matrix laden with CSPGs.
Figure 6.
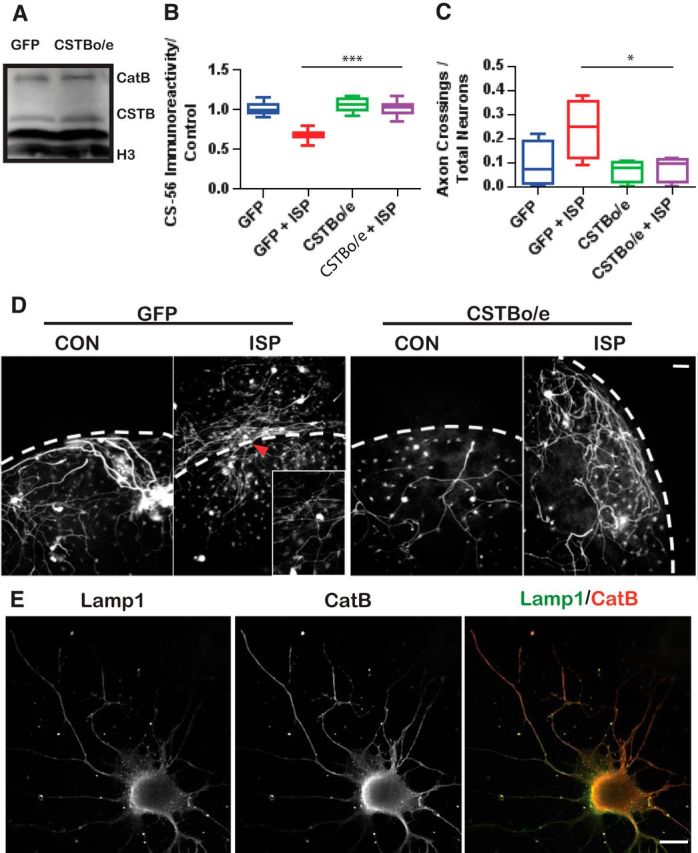
Overexpression of CSTB (CSTBo/e) decreases ISP-treated DRG axon crossings through CSPGs. A, Western blot of DRG lysates inoculated with lentiviral particles expressing GFP or CSTBo/e constructs. B, Aggrecan degradation from CM collected from GFP control or CSTBo/e DRGs treated with vehicle or 2.5 μm ISP (n = 152, 63, 98, 80; F(5,115) = 51.03, p = 0.0001; ANOVA). C, Spot crossings of DRGs treated with lentiviral particles expressing GFP or CSTB overexpressing constructs (n = 12; F(3,12) = 3.743, p = 0.0415; ANOVA). D, Representative images of DRG axons crossing aggrecan spots. Dotted lines indicate CSPG border. Inset, Axon crossing CSPG gradient. Scale bar, 50 μm. E, CatB is associated with lysosomes (Lamp1) in DRGs. Scale bars, 50 μm. Lines indicate median. Boxes represent quartiles. Whiskers indicate range. *p < 0.05, ***p < 0.001.
As CatB is associated with lysosomes (Sloane et al., 1981), we colabeled DRG neurons in vitro (on laminin and low aggrecan) with Lamp1, a lysosomal marker, and CatB to better visualize CatB/lysosomal interactions (Fig. 6E). As expected, the majority of CatB immunoreactivity was colocalized with lysosomes along the axon and soma of DRG neurons. This suggests that lysosomes and CatB are, indeed, in an appropriate region of the neuron to effect GAG-CSPG degradation at the leading edge of the axon as it grows.
To explore whether CatB-induced digestion of the perineuronal net may be occurring in vivo, we colabeled CatB with serotonergic axons in sections from T8-contused spinal cord injury animals that received either daily vehicle or ISP (22 μg/ml) subcutaneous injections for 49 d after injury, with a 24 h delay before treatment was initiated. Interestingly, serotonergic axons seemed to highly express CatB following immunostaining (Fig. 7). It is possible that the pattern of low WFA immunoreactivity and high serotonergic expression (Fig. 1) may be occurring through ISP-enhanced CatB degradation of the proteoglycan-laden perineuronal net and extracellular matrix. To further correlate low WFA immunoreactivity with CatB activity as a result of ISP treatment, we processed (for Western blot analysis) the spinal cords of similarly contused rats that were given 14 d of ISP (22 μg/ml) or vehicle subcutaneous injections. Following Western blotting, we found that ISP treatment induced greater active CatB immunoreactivity over control (Fig. 7B). Through upregulated CatB activity, serotonin fibers may be able to better clear their inhibitory GAG-CSPG-laden environment to allow for enhanced sprouting.
Figure 7.

Serotonergic axons express CatB. A, Spinal cord tissue processed from T8-contused rats that were treated with DMSO vehicle or 22 μg/ml ISP for 7 weeks following injury. At 7 weeks after the last ISP treatment, spinal cord tissue was stained to visualize serotonin (5-HT) and CatB. Scale bar, 500 μm. B, Western blot of injured spinal cord lysates collected from female rats given a thoracic level 8 (T8) contusion and treated with vehicle or 22 μg/ml ISP for 14 d. n = 2.
Linking CatB activity with RPTPσ
Finally, we inquired whether CatB activity was enhanced in RPTPσ-null animals independent of peptide modulation. As the cognate receptor of GAG-CSPGs, the DRGs of RPTPσ-null animals have been extensively reported to show enhanced axonal outgrowth despite the presence of CSPGs (Shen et al., 2009), and we hypothesize that, without RPTPσ, these DRGs may show some elevation of protease activity. Similar to ISP-treated DRGs (Fig. 1C), we found that RPTPσ mutant DRGs plated on aggrecan spots showed digested trails of CS-56 (Fig. 8A). To better quantify this effect, we returned to our CM spot assay and found that CM collected from transgenic RPTPσ animals was able to decrease CS-56 immunoreactivity compared with CM made by their WT littermates (F(7,52) = 71.6, WT vs RPTPσ+/− and RPTPσ−/−, p = 0.0001, ANOVA; Fig. 8B). Interestingly, the reduction of CS-56 immunoreactivity did not synergize with ISP treatment as ISP-treated-null animals yielded a similar level of CS-56 reduction as RPTPσ-null DRGs alone. Finally, to correlate the reduction of CS-56 to CatB, we immunostained and quantified CatB immunoreactivity in the axons of RPTPσ-null DRGs (Fig. 8C,D). We found significantly increased CatB immunoreacivity compared with WT neurons that was found along the axons as well as their growing tips (t = 3.843, df = 30, unpaired t test; Fig. 8C,D). Together, our results link the enhanced activity of CatB to RPTPσ as one of the many underlying mechanisms that may be contributing to enhanced axon outgrowth on CSPG-rich environments.
Figure 8.
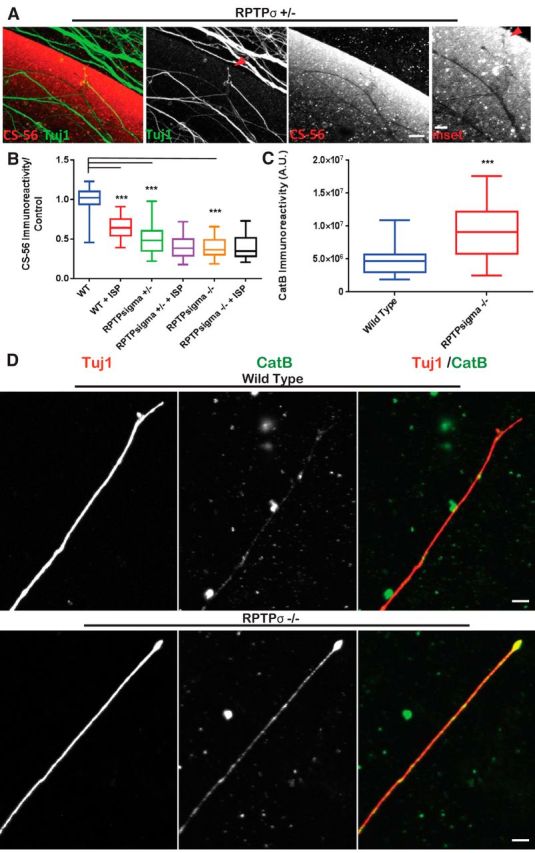
Genetic loss of RPTPσ correlates with CatB activity and CSPG degradation. A, DRGs extracted from RPTPσ+/− animals were cultured on aggrecan spot stained with CS-56 and Tuj1 display digest trails. Arrows point to digested CS-56 left by axon tip. Scale bar, 50 μm. B, CM harvested from BALB/C WT or RPTPσ+/− or −/− animals treated with vehicle or 2.5 μm ISP were incubated onto new aggrecan spots and stained with CS-56 before quantification: n = 100, 56, 51, 32, 63, 64; F(7,52) = 71.6, WT vs ISP, p = 0.0001; WT vs +/−, p = 0.0001; WT vs −/−, p = 0.0001 (ANOVA). C, D, Representative images of CatB in BALB/C WT and RPTPσ−/− DRGs (Tuj1). Scale bar, 20 μm. Quantification of CatB immunoreactivity: n = 45, 30; t = 3.843, df = 30, p = 0.0006 (unpaired t test). Lines indicate median. Boxes represent quartiles. Whiskers indicate range. ***p < 0.001.
Discussion
We have described, for the first time, how ISP modulation of RPTPσ enhances GAG-CSPG degradation, ultimately leading to relief of proteoglycan-mediated axon growth inhibition. Furthermore, we have identified CatB as a major enzyme that is secreted by receptor-modulated DRGs and likely serotonergic axons as well. While initial CGRP staining in our thoracic level 8 contusion model did not show obviously enhanced DRG sprouting following ISP treatment, perhaps more sophisticated labeling techniques that can reveal the detailed anatomy of DRGs will be required to determine whether ISP treatment fosters DRG sprouting in vivo. We propose that exaggerated secretion of CatB may be occurring at the leading edge of ISP-treated axon growth cones as they negotiate their way past an inhibitory CSPG barrier. Peptide modulation of RPTPσ appears to be relatively protease specific and, thus, in stark contrast to the “protease storm” that typically occurs after CNS injury (Noble et al., 2002; Zhang et al., 2010).
The ability of growing neurons to locally secrete extracellular matrix-remodeling proteases is a well-characterized phenomenon (Krystosek and Seeds, 1984; Bai and Pfaff, 2011). For example, peripheral axon regeneration following a sciatic crush lesion is known to be involved with protease remodeling of Schwann cell-derived extracellular matrix (Siconolfi and Seeds, 2001). Also, a preconditioning lesion of the peripheral fibers of DRGs that further improves regeneration after a delayed second lesion is, at least partially, effected by the augmented expression of proteases (Minor et al., 2009). Regenerating olfactory axons have been noted for their ability to release proteases (Cao et al., 2012; Ould-Yahoui et al., 2013), which can continue even after they are transplanted into the spinal cord (Pastrana et al., 2006). Duchossoy et al. (2001) also observed protease-remodeled scar lesions following spinal cord injury that formed pathways for ingrowing neurites. Aside from neurons (Krystosek and Seeds, 1984; Zuo et al., 1998), many different migratory cell types use fine temporal-spatial regulation of proteases to remodel the surrounding extracellular matrix along their prospective pathways. Metastasizing tumor cells, in particular, enhance a program of focalized protease and CatB secretion during migration (Olson and Joyce, 2015).
While we have identified CatB in DRGs as the chief protease secreted following ISP treatment, it is possible that other proteases may be secreted or activated as well. Zuo et al. (1998), for example, have identified DRG neuron activation of MMP-2 that serves to help remodel inhibitory extracellular matrix and enhance axon regeneration by degrading CSPGs in the bands of Bungner while sparing axon-promoting laminin. Given the relatively low densities of our DRG cultures, we were only able to clearly identify robustly secreted CatB but cannot exclude other proteases secreted at lower levels that may act in concert to degrade inhibitory CSPGs surrounding the neuron.
CatB has been described as a “hub” enzyme that can initiate a network of protease activity, including that of MMP-2 and MMP-9 (Gondi and Rao, 2013). For example, Padamsey et al. (2017) have recently described finely regulated exocytosis of CatB at the dendritic spine that, in turn, activates MMP-9 to maintain spine motility. It is possible that enhanced CatB activity seen in our model may also be initiating a network of protease activity (Padamsey et al., 2017). Aside from degrading CSPGs, CatB in neurons has also been described to further digest amyloid-β in mouse models of Alzheimer's disease to functionally improve memory (Yang et al., 2011a).
We have also described that peptide modulation of RPTPσ results in an intracellular decrease in CSTB, an endogenous CatB inhibitor, which may participate to further strengthen CatB activity. Studies by the Nixon group have shown that genetic deletion of CSTB and subsequent enhanced CatB activity works to reverse autophagic dysfunction (Yang et al., 2011b). As inhibition of RPTPσ has been previously linked to increased autophagic flux (Martin et al., 2011), we surmise that a strong link between lysosomal regulation and autophagy may also exist, as autophagosomes must eventually fuse with lysosomes to complete protein degradation and turnover. Furthermore, we have identified lysosomal immunostaining along the DRG axon. It is possible that the bubbling vesicular structures that we first noted in abundance within early developing dystrophic growth cones in our proteoglycan gradient spot assay (Tom et al., 2004) are cycling autophagosomes. This has substantial implications for the possible roles RPTPσ and CSPGs may play in a variety of traumatic but also perhaps neurodegenerative diseases. For example, CSPGs are associated with senile plaques of Alzheimer's disease (DeWitt et al., 1993), and mouse models of Alzheimer's disease develop well-characterized dystrophic axons around senile plaques filled with Cathepsin-laden lysosomes (Lee et al., 2011). Whether these lysosomes are the result of similar CSPG-RPTPσ interactions is an interesting question for future research.
The regulation of protease release by CSPG-RPTPσ interactions may additionally have implications for synaptogenesis during embryonic development or plasticity in adulthood. The family of LAR receptors, including RPTPσ, have been noted for the roles they play in synapse formation during development where they may act as adhesion molecules to structurally stabilize emergent synapses (Mironova and Giger, 2013; Um and Ko, 2013). Given what is known about RPTPσ in inhibiting axon growth and encouraging synaptogenesis (Filous et al., 2014; Lang et al., 2015), it is possible that RPTPσ acts as a switch to regulate these two processes. CSPGs bound to RPTPσ in the context of the growth cone, for example, inhibits axon elongation (receptor on); however, blockade of the receptor (receptor off) enables growth cones to secrete extracellular matrix-remodeling proteases to enhance outgrowth. When a growth cone reaches its target and perineuronal net proteoglycans upregulate, it is conceivable that RPTPσ switches “on” to immediately limit protease secretion to cease rapidly elongating growth and help foster engagement at the synapse. Indeed, there is a rising interest in RPTPσ as a presynaptic receptor acting to inhibit axon branching and initiate synaptogenesis (Horn et al., 2012; Ko et al., 2015). Following injury in adulthood, ISP may be effectively inactivating RPTPσ to allow dystrophic axons to reenter a renewed protease-secreting growth state. Indeed, we found that RPTPσ-null DRGs themselves show enhanced CatB immunoreactivity along their axons. Certainly, there is an important biological need to strategically link and tightly regulate protease secretion at the growth cone depending on whether it is still elongating or engaging with its postsynaptic target.
We further report that, following contusive cord injury and systemic ISP treatment, sprouting serotonergic axons show high expression of CatB within regions denuded of CSPG staining. While the isolation of ample amounts of mature serotonergic neurons for the purpose of in vitro characterization of CSPG degradation was not possible (hence, we turned our attention to mature DRGs), the remarkable reciprocal pattern of dense serotonergic axons and decreased matrix may be the result of enhanced CatB activity as it is in sensory neurons. Recently, Bijata et al. (2017) have linked serotonergic receptor 5-HT7 to MMP-9 release, which resulted in dendritic spine elongation in the presence of the perineuronal net. Following many models of CNS injury, serotonergic axons have additionally been widely reported to be especially plastic and robustly able to sprout (Hawthorne et al., 2011; Jin et al., 2016; Y. Liu et al., 2017). Whether this robust regenerative phenotype is due, at least in part, to their ability to degrade CSPGs through CatB activity will need to be further explored.
It is becoming more evident that proteases play nuanced roles in synaptogenesis, extracellular matrix remodeling, and neuronal plasticity. Our work links RPTPσ modulation with protease activity. Although the precise molecular connections between these processes will need to be investigated, we propose that continued study of RPTPσ modulation would help to elucidate how axon regeneration may be encouraged in many models of CNS trauma or disease.
Footnotes
This work was supported by National Institutes of Health/National Institute of Neurological Disorders and Stroke NS025713, the Kaneko Family Fund, the Brumagin-Nelson Fund, and the Hong Kong Spinal Cord Injury Fund. A patent application (61/621,623) has been filed for ISP compositions and use in neurological diseases.
The authors declare no competing financial interests.
References
- Bai G, Pfaff SL (2011) Protease regulation: the yin and yang of neural development and disease. Neuron 72:9–21. 10.1016/j.neuron.2011.09.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bijata M, Labus J, Guseva D, Stawarski M, Butzlaff M, Dzwonek J, Schneeberg J, Böhm K, Michaluk P, Rusakov DA, Dityatev A, Wilczyński G, Wlodarczyk J, Ponimaskin E (2017) Synaptic remodeling depends on signaling between serotonin receptors and the extracellular matrix. Cell Rep 19:1767–1782. 10.1016/j.celrep.2017.05.023 [DOI] [PubMed] [Google Scholar]
- Bolte S, Cordelières FP (2006) A guided tour into subcellular colocalization analysis in light microscopy. J Microsc 224:213–232. 10.1111/j.1365-2818.2006.01706.x [DOI] [PubMed] [Google Scholar]
- Brittis PA, Canning DR, Silver J (1992) Chondroitin sulfate as a regulator of neuronal patterning in the retina. Science 255:733–736. 10.1126/science.1738848 [DOI] [PubMed] [Google Scholar]
- Brooks JM, Su J, Levy C, Wang JS, Seabrook TA, Guido W, Fox MA (2013) A molecular mechanism regulating the timing of corticogeniculate innervation. Cell Rep 5:573–581. 10.1016/j.celrep.2013.09.041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao L, Rickenbacher GT, Rodriguez S, Moulia TW, Albers MW (2012) The precision of axon targeting of mouse olfactory sensory neurons requires the BACE1 protease. Sci Rep 2:231. 10.1038/srep00231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carulli D, Laabs T, Geller HM, Fawcett JW (2005) Chondroitin sulfate proteoglycans in neural development and regeneration. Curr Opin Neurobiol 15:116–120. 10.1016/j.conb.2005.01.014 [DOI] [PubMed] [Google Scholar]
- Cua RC, Lau LW, Keough MB, Midha R, Apte SS, Yong VW (2013) Overcoming neurite-inhibitory chondroitin sulfate proteoglycans in the astrocyte matrix. Glia 61:972–984. 10.1002/glia.22489 [DOI] [PubMed] [Google Scholar]
- Davies SJ, Fitch MT, Memberg SP, Hall AK, Raisman G, Silver J (1997) Regeneration of adult axons in white matter tracts of the central nervous system. Nature 390:680–683. 10.1038/37776 [DOI] [PubMed] [Google Scholar]
- DeWitt DA, Silver J, Canning DR, Perry G (1993) Chondroitin sulfate proteoglycans are associated with the lesions of Alzheimer's disease. Exp Neurol 121:149–152. 10.1006/exnr.1993.1081 [DOI] [PubMed] [Google Scholar]
- Duchossoy Y, Horvat JC, Stettler O (2001) MMP-related gelatinase activity is strongly induced in scar tissue of injured adult spinal cord and forms pathways for ingrowing neurites. Mol Cell Neurosci 17:945–956. 10.1006/mcne.2001.0986 [DOI] [PubMed] [Google Scholar]
- Elchebly M, Wagner J, Kennedy TE, Lanctôt C, Michaliszyn E, Itié A, Drouin J, Tremblay ML (1999) Neuroendocrine dysplasia in mice lacking protein tyrosine phosphatase sigma. Nat Genet 21:330–333. 10.1038/6859 [DOI] [PubMed] [Google Scholar]
- Ellis RC, O'Steen WA, Hayes RL, Nick HS, Wang KK, Anderson DK (2005) Cellular localization and enzymatic activity of cathepsin B after spinal cord injury in the rat. Exp Neurol 193:19–28. 10.1016/j.expneurol.2004.11.034 [DOI] [PubMed] [Google Scholar]
- Feng Y, Yu S, Lasell TK, Jadhav AP, Macia E, Chardin P, Melancon P, Roth M, Mitchison T, Kirchhausen T (2003) Exo1: a new chemical inhibitor of the exocytic pathway. Proc Natl Acad Sci U S A 100:6469–6474. 10.1073/pnas.0631766100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filous AR, Tran A, Howell CJ, Busch SA, Evans TA, Stallcup WB, Kang SH, Bergles DE, Lee SI, Levine JM, Silver J (2014) Entrapment via synaptic-like connections between NG2 proteoglycan+ cells and dystrophic axons in the lesion plays a role in regeneration failure after spinal cord injury. J Neurosci 34:16369–16384. 10.1523/JNEUROSCI.1309-14.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fitch MT, Doller C, Combs CK, Landreth GE, Silver J (1999) Cellular and molecular mechanisms of glial scarring and progressive cavitation: in vivo and in vitro analysis of inflammation-induced secondary injury after CNS trauma. J Neurosci 19:8182–8198. 10.1523/JNEUROSCI.19-19-08182.1999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fosang AJ, Neame PJ, Last K, Hardingham TE, Murphy G, Hamilton JA (1992) The interglobular domain of cartilage aggrecan is cleaved by PUMP, gelatinases, and cathepsin B. J Biol Chem 267:19470–19474. [PubMed] [Google Scholar]
- Gondi CS, Rao JS (2013) Cathepsin B as a cancer target. Expert Opin Ther Targets 17:281–291. 10.1517/14728222.2013.740461 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hawthorne AL, Hu H, Kundu B, Steinmetz MP, Wylie CJ, Deneris ES, Silver J (2011) The unusual response of serotonergic neurons after CNS injury: lack of axonal dieback and enhanced sprouting within the inhibitory environment of the glial scar. J Neurosci 31:5605–5616. 10.1523/JNEUROSCI.6663-10.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horn KE, Xu B, Gobert D, Hamam BN, Thompson KM, Wu CL, Bouchard JF, Uetani N, Racine RJ, Tremblay ML, Ruthazer ES, Chapman CA, Kennedy TE (2012) Receptor protein tyrosine phosphatase sigma regulates synapse structure, function and plasticity. J Neurochem 122:147–161. 10.1111/j.1471-4159.2012.07762.x [DOI] [PubMed] [Google Scholar]
- Hsu JY, McKeon R, Goussev S, Werb Z, Lee JU, Trivedi A, Noble-Haeusslein LJ (2006) Matrix metalloproteinase-2 facilitates wound healing events that promote functional recovery after spinal cord injury. J Neurosci 26:9841–9850. 10.1523/JNEUROSCI.1993-06.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes CE, Caterson B, Fosang AJ, Roughley PJ, Mort JS (1995) Monoclonal antibodies that specifically recognize neoepitope sequences generated by “aggrecanase” and matrix metalloproteinase cleavage of aggrecan: application to catabolism in situ and in vitro. Biochem J 305:799–804. 10.1042/bj3050799 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin Y, Dougherty SE, Wood K, Sun L, Cudmore RH, Abdalla A, Kannan G, Pletnikov M, Hashemi P, Linden DJ (2016) Regrowth of serotonin axons in the adult mouse brain following injury. Neuron 91:748–762. 10.1016/j.neuron.2016.07.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ko JS, Pramanik G, Um JW, Shim JS, Lee D, Kim KH, Chung GY, Condomitti G, Kim HM, Kim H, de Wit J, Park KS, Tabuchi K, Ko J (2015) PTPσ functions as a presynaptic receptor for the glypican-4/LRRTM4 complex and is essential for excitatory synaptic transmission. Proc Natl Acad Sci U S A 112:1874–1879. 10.1073/pnas.1410138112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krystosek A, Seeds NW (1984) Peripheral neurons and Schwann cells secrete plasminogen activator. J Cell Biol 98:773–776. 10.1083/jcb.98.2.773 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwok JC, Dick G, Wang D, Fawcett JW (2011) Extracellular matrix and perineuronal nets in CNS repair. Dev Neurobiol 71:1073–1089. 10.1002/dneu.20974 [DOI] [PubMed] [Google Scholar]
- Lang BT, Cregg JM, DePaul MA, Tran AP, Xu K, Dyck SM, Madalena KM, Brown BP, Weng YL, Li S, Karimi-Abdolrezaee S, Busch SA, Shen Y, Silver J (2015) Modulation of the proteoglycan receptor PTPσ promotes recovery after spinal cord injury. Nature 518:404–408. 10.1038/nature13974 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S, Sato Y, Nixon RA (2011) Lysosomal proteolysis inhibition selectively disrupts axonal transport of degradative organelles and causes an Alzheimer's-like axonal dystrophy. J Neurosci 31:7817–7830. 10.1523/JNEUROSCI.6412-10.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu P, Liu X, Liou AK, Xing J, Jing Z, Ji X, Liu X, Zhao H, Yan F, Chen J, Cao G, Luo Y (2014) The neuroprotective mechanism of erythropoietin-TAT fusion protein against neurodegeneration from ischemic brain injury. CNS Neurol Disord Drug Targets 13:1465–1474. 10.2174/1871527313666140806155259 [DOI] [PubMed] [Google Scholar]
- Liu Y, Wang X, Li W, Zhang Q, Li Y, Zhang Z, Zhu J, Chen B, Williams PR, Zhang Y, Yu B, Gu X, He Z (2017) A sensitized IGF1 treatment restores corticospinal axon-dependent functions. Neuron 95:817–833.e4. 10.1016/j.neuron.2017.07.037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin KR, Xu Y, Looyenga BD, Davis RJ, Wu CL, Tremblay ML, Xu HE, MacKeigan JP (2011) Identification of PTP as an autophagic phosphatase. J Cell Sci 124:812–819. 10.1242/jcs.080341 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Massey JM, Hubscher CH, Wagoner MR, Decker JA, Amps J, Silver J, Onifer SM (2006) Chondroitinase ABC digestion of the perineuronal net promotes functional collateral sprouting in the cuneate nucleus after cervical spinal cord injury. J Neurosci 26:4406–4414. 10.1523/JNEUROSCI.5467-05.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minor K, Phillips J, Seeds NW (2009) Tissue plasminogen activator promotes axonal outgrowth on CNS myelin after conditioned injury. J Neurochem 109:706–715. 10.1111/j.1471-4159.2009.05977.x [DOI] [PubMed] [Google Scholar]
- Mironova YA, Giger RJ (2013) Where no synapses go: gatekeepers of circuit remodeling and synaptic strength. Trends Neurosci 36:363–373. 10.1016/j.tins.2013.04.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mort JS, Magny MC, Lee ER (1998) Cathepsin B: an alternative protease for the generation of an aggrecan “metalloproteinase” cleavage neoepitope. Biochem J 335:491–494. 10.1042/bj3350491 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noble LJ, Donovan F, Igarashi T, Goussev S, Werb Z (2002) Matrix metalloproteinases limit functional recovery after spinal cord injury by modulation of early vascular events. J Neurosci 22:7526–7535. 10.1523/JNEUROSCI.22-17-07526.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okada S, Nakamura M, Katoh H, Miyao T, Shimazaki T, Ishii K, Yamane J, Yoshimura A, Iwamoto Y, Toyama Y, Okano H (2006) Conditional ablation of Stat3 or Socs3 discloses a dual role for reactive astrocytes after spinal cord injury. Nat Med 12:829–834. 10.1038/nm1425 [DOI] [PubMed] [Google Scholar]
- Olson OC, Joyce JA (2015) Cysteine cathepsin proteases: regulators of cancer progression and therapeutic response. Nat Rev Cancer 15:712–729. 10.1038/nrc4027 [DOI] [PubMed] [Google Scholar]
- Ould-Yahoui A, Sbai O, Baranger K, Bernard A, Gueye Y, Charrat E, Clément B, Gigmes D, Dive V, Girard SD, Féron F, Khrestchatisky M, Rivera S (2013) Role of matrix metalloproteinases in migration and neurotrophic properties of nasal olfactory stem and ensheathing cells. Cell Transplant 22:993–1010. 10.3727/096368912X657468 [DOI] [PubMed] [Google Scholar]
- Padamsey Z, McGuinness L, Bardo SJ, Reinhart M, Tong R, Hedegaard A, Hart ML, Emptage NJ (2017) Activity-dependent exocytosis of lysosomes regulates the structural plasticity of dendritic spines. Neuron 93:132–146. 10.1016/j.neuron.2016.11.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pastrana E, Moreno-Flores MT, Gurzov EN, Avila J, Wandosell F, Diaz-Nido J (2006) Genes associated with adult axon regeneration promoted by olfactory ensheathing cells: a new role for matrix metalloproteinase 2. J Neurosci 26:5347–5359. 10.1523/JNEUROSCI.1111-06.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schachtrup C, Ryu JK, Helmrick MJ, Vagena E, Galanakis DK, Degen JL, Margolis RU, Akassoglou K (2010) Fibrinogen triggers astrocyte scar formation by promoting the availability of active TGF-beta after vascular damage. J Neurosci 30:5843–5854. 10.1523/JNEUROSCI.0137-10.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen Y, Tenney AP, Busch SA, Horn KP, Cuascut FX, Liu K, He Z, Silver J, Flanagan JG (2009) PTPsigma is a receptor for chondroitin sulfate proteoglycan, an inhibitor of neural regeneration. Science 326:592–596. 10.1126/science.1178310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siconolfi LB, Seeds NW (2001) Mice lacking tPA, uPA, or plasminogen genes showed delayed functional recovery after sciatic nerve crush. J Neurosci 21:4348–4355. 10.1523/JNEUROSCI.21-12-04348.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sloane B, Dunn HK, Honn KV (1981) Lysosomal cathepsin B: correlation with metastatic potential. Science 212:1151–1153. 10.1126/science.7233209 [DOI] [PubMed] [Google Scholar]
- Snow DM, Lemmon V, Carrino DA, Caplan AI, Silver J (1990) Sulfated proteoglycans in astroglial barriers inhibit neurite outgrowth in vitro. Exp Neurol 109:111–130. 10.1016/S0014-4886(05)80013-5 [DOI] [PubMed] [Google Scholar]
- Soo Youn G, Ju SM, Choi SY, Park J (2015) HDAC6 mediates HIV-1 tat-induced proinflammatory responses by regulating MAPK-NF-kappaB/AP-1 pathways in astrocytes. Glia 63:1953–1965. 10.1002/glia.22865 [DOI] [PubMed] [Google Scholar]
- Tom VJ, Steinmetz MP, Miller JH, Doller CM, Silver J (2004) Studies on the development and behavior of the dystrophic growth cone, the hallmark of regeneration failure, in an in vitro model of the glial scar and after spinal cord injury. J Neurosci 24:6531–6539. 10.1523/JNEUROSCI.0994-04.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tran AP, Warren PM, Silver J (2018) The biology of regeneration failure and success after spinal cord injury. Physiol Rev 98:881–917. 10.1152/physrev.00017.2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turk V, Bode W (1991) The cystatins: protein inhibitors of cysteine proteinases. FEBS Lett 285:213–219. 10.1016/0014-5793(91)80804-C [DOI] [PubMed] [Google Scholar]
- Turk V, Stoka V, Vasiljeva O, Renko M, Sun T, Turk B, Turk D (2012) Cysteine cathepsins: from structure, function and regulation to new frontiers. Biochim Biophys Acta 1824:68–88. 10.1016/j.bbapap.2011.10.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Um JW, Ko J (2013) LAR-RPTPs: synaptic adhesion molecules that shape synapse development. Trends Cell Biol 23:465–475. 10.1016/j.tcb.2013.07.004 [DOI] [PubMed] [Google Scholar]
- Williams R, Yao H, Peng F, Yang Y, Bethel-Brown C, Buch S (2010) Cooperative induction of CXCL10 involves NADPH oxidase: implications for HIV dementia. Glia 58:611–621. 10.1002/glia.20949 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang DS, Stavrides P, Mohan PS, Kaushik S, Kumar A, Ohno M, Schmidt SD, Wesson D, Bandyopadhyay U, Jiang Y, Pawlik M, Peterhoff CM, Yang AJ, Wilson DA, St George-Hyslop P, Westaway D, Mathews PM, Levy E, Cuervo AM, Nixon RA (2011a) Reversal of autophagy dysfunction in the TgCRND8 mouse model of Alzheimer's disease ameliorates amyloid pathologies and memory deficits. Brain 134:258–277. 10.1093/brain/awq341 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang DS, Stavrides P, Mohan PS, Kaushik S, Kumar A, Ohno M, Schmidt SD, Wesson DW, Bandyopadhyay U, Jiang Y, Pawlik M, Peterhoff CM, Yang AJ, Wilson DA, St George-Hyslop P, Westaway D, Mathews PM, Levy E, Cuervo AM, Nixon RA (2011b) Therapeutic effects of remediating autophagy failure in a mouse model of Alzheimer disease by enhancing lysosomal proteolysis. Autophagy 7:788–789. 10.4161/auto.7.7.15596 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, Adwanikar H, Werb Z, Noble-Haeusslein LJ (2010) Matrix metalloproteinases and neurotrauma: evolving roles in injury and reparative processes. Neuroscientist 16:156–170. 10.1177/1073858409355830 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuo J, Ferguson TA, Hernandez YJ, Stetler-Stevenson WG, Muir D (1998) Neuronal matrix metalloproteinase-2 degrades and inactivates a neurite-inhibiting chondroitin sulfate proteoglycan. J Neurosci 18:5203–5211. 10.1523/JNEUROSCI.18-14-05203.1998 [DOI] [PMC free article] [PubMed] [Google Scholar]