

Abstract
BDNF is a growth factor with important roles in the nervous system in both physiological and pathological conditions, but the mechanisms controlling its secretion are not completely understood. Here, we show that ARMS/Kidins220 negatively regulates BDNF secretion in neurons from the CNS and PNS. Downregulation of the ARMS/Kidins220 protein in the adult mouse brain increases regulated BDNF secretion, leading to its accumulation in the striatum. Interestingly, two mouse models of Huntington's disease (HD) showed increased levels of ARMS/Kidins220 in the hippocampus and regulated BDNF secretion deficits. Importantly, reduction of ARMS/Kidins220 in hippocampal slices from HD mice reversed the impaired regulated BDNF release. Moreover, there are increased levels of ARMS/Kidins220 in the hippocampus and PFC of patients with HD. ARMS/Kidins220 regulates Synaptotagmin-IV levels, which has been previously observed to modulate BDNF secretion. These data indicate that ARMS/Kidins220 controls the regulated secretion of BDNF and might play a crucial role in the pathogenesis of HD.
SIGNIFICANCE STATEMENT BDNF is an important growth factor that plays a fundamental role in the correct functioning of the CNS. The secretion of BDNF must be properly controlled to exert its functions, but the proteins regulating its release are not completely known. Using neuronal cultures and a new conditional mouse to modulate ARMS/Kidins220 protein, we report that ARMS/Kidins220 negatively regulates BDNF secretion. Moreover, ARMS/Kidins220 is overexpressed in two mouse models of Huntington's disease (HD), causing an impaired regulation of BDNF secretion. Furthermore, ARMS/Kidins220 levels are increased in brain samples from HD patients. Future studies should address whether ARMS/Kidins220 has any function on the pathophysiology of HD.
Keywords: ARMS/Kidins220, BDNF, Huntington's disease, secretion, Synaptotagmin IV
Introduction
Brain-derived neurotrophic factor (BDNF), a member of the neurotrophin family, is important in the development and the correct functioning of the nervous system. It contributes to neuronal survival, neurite outgrowth, synaptic activity, learning, and memory (Park and Poo, 2013). Patients with neurodegenerative diseases, such as Huntington's disease (HD) and Parkinson's disease, have altered BDNF levels in the CNS (Lu et al., 2013). BDNF is stored in secretory granules and mainly secreted through a regulated pathway in response to neuronal activity, nerve growth factor (NGF), neurotrophin-3 (NT-3) or neurotrophin-4 (NT-4) (Canossa et al., 1997; Krüttgen et al., 1998; Lessmann, 1998; Dieni et al., 2012). Different studies have measured endogenous BDNF release (Griesbeck et al., 1999; Canossa et al., 2001; Aicardi et al., 2004), and others have identified proteins implicated in the regulation of BDNF secretion, such as Synaptotagmin-IV (Syt-IV) (Dean et al., 2009), Synaptotagmin-VI (Wong et al., 2015), Ca2+-dependent activator protein for secretion 2 (Sadakata et al., 2004), Synaptobrevin, SNAP25, and SNAP47 (Shimojo et al., 2015). However, BDNF secretion is not completely understood. Considering the functional relevance of BDNF, it is essential to elucidate how BDNF is secreted in vivo and whether other proteins are implicated.
HD is a progressive neurodegenerative disorder caused by a polymorphic trinucleotide CAG repeat expansion in exon 1 of the Huntingtin gene. BDNF produced in the cortex and hippocampus is anterogradely transported and released in the striatum, which does not produce BDNF (Altar et al., 1997). In the brain, the huntingtin protein is involved in the synthesis and transport of BDNF from the cortex and hippocampus to the striatum, both of which processes are altered in HD (Zuccato et al., 2001; Gauthier et al., 2004). The huntingtin protein is ubiquitously expressed, but its mutation has detrimental effects particularly in the striatum, where BDNF has neural survival-promoting activity. These facts have led to the premise that reduced endogenous BDNF trophic support may contribute to disease onset and/or progression (Zuccato et al., 2001). Consistent with this, decreased BDNF has been reported in the brains of patients with HD and in various HD mouse models (Ferrer et al., 2000; Zuccato et al., 2001; Ginés et al., 2006). Together, these data support the idea that a BDNF defect is an important component of HD, and BDNF may be a potentially valuable cotherapeutic agent in HD. So far, to our knowledge, no studies have been performed addressing whether BDNF secretion deficits are present in HD.
ARMS (also known as Kidins220) is a scaffold protein with various functions. It was discovered as a protein kinase D substrate (Iglesias et al., 2000) and as an interacting protein between p75 and Trk neurotrophin receptors (Kong et al., 2001). Subsequently, ARMS was identified as important in neurotrophin-dependent and independent signaling and was implicated in the modulation of neuronal activity (for review, see Neubrand et al., 2012). Interestingly, ARMS has also been implicated in neurotensin hormone secretion in BON cells, a cell line derived from a human pancreatic carcinoid tumor (Li et al., 2008). Furthermore, we recently reported that ARMS negatively regulates NGF-mediated secretion in PC12 cells (López-Benito et al., 2016). However, it is unknown whether ARMS modulates BDNF secretion.
Here, we demonstrate that ARMS controls the regulated secretion of BDNF in cultured DRGs and cortical neurons, as well as in ARMS-depleted cortical slices. Therefore, BDNF accumulation is observed in the striatum of mice depleted of ARMS in the cortex and hippocampus. Furthermore, ARMS levels are increased in the hippocampus of two different HD mouse models at symptomatic stages and reducing ARMS expression rescued the impaired regulation of BDNF secretion observed in hippocampal slices of HD mice. Our findings demonstrate that ARMS regulates BDNF secretion through a mechanism that may involve the regulation of Syt-IV protein levels by ARMS.
Materials and Methods
Reagents and antibodies.
NGF was obtained from Alomone Labs and BDNF, NT-3, and NT4 from PreproTech. The following antibodies were used: mouse ARMS monoclonal (MA1-34070, Fisher Scientific), 10 μg/ml for immunofluorescence; rabbit ARMS C terminus polyclonal, used previously (Kong et al., 2001), 0.5 μg/ml for immunoblot and 2 μg/ml for immunohistochemistry; rabbit ARMS C terminus polyclonal GSC16, used previously (Iglesias et al., 2000), 1:500 for immunoblot; mouse GAPDH monoclonal (G8795, Sigma-Aldrich), 1:10,000 for immunoblot; mouse β-actin monoclonal (A5316, Sigma-Aldrich), 1:5000 for immunoblot; rabbit β-tubulin III polyclonal (T8328, Sigma-Aldrich), 1:10,000 for immunoblot; rabbit Cre polyclonal (257003, Synaptic Systems), 1:750 for immunoblot and 1:500 for immunohistochemistry; rabbit Syt-IV polyclonal (sc-30095, Santa Cruz Biotechnology) 1:2000 for immunoblot; BDNF-#1 and #9 were deposited to the DSHB by Y.-A. Barde (DSHB Hybridoma Product BDNF-1 and -9) (Kolbeck et al., 1999), for ELISA, BDNF-1 at 1 μg/well for plate coating, and HRP-labeled BDNF-9 at 12 ng/well.
Postmortem brain tissues.
Hippocampal (4 and 7 male and female controls, respectively, and 5 and 5 male and females HD patients, respectively) and prefrontal cortical (5 and 7 male and female controls, respectively, and 6 and 6 male and female HD patients, respectively) brain tissues from patients with HD Vonsattel Grades 2 and 4 ranging from 43 to 72 years and control cases ranging from 40 to 79 years were supplied by the BT-CIEN and Biobanco Hospital Universitario Fundación Alcorcón. Samples were kept at −80°C and shipped frozen in dry ice. All donors had given informed consent. One male HD sample was excluded because no grade was assigned and one female control sample because it rendered a value with a deviation of >2 times the SD, and her age was 14 years older than the older HD patient. The procedures conducted were approved by the Bioethics Committee of the University of Salamanca, BT-CIEN and Biobanco Hospital Universitario Fundación Alcorcón.
Plasmids.
pLM-CMV-R-Cre was a gift from Michel Sadelain (Addgene plasmid #27546) (Papapetrou et al., 2011); pROSA26–1 was a gift from Philippe Soriano (Addgene plasmid #21714) (Soriano, 1999); pLVTHM, pMD2.G, and psPAX2 were gifts from Didier Trono (Addgene plasmids #12247, #12259, and #12260, respectively) (Wiznerowicz and Trono, 2003); rSyt-IVpIE was a gift from Jane Sullivan (Addgene plasmid #12503) (Ting et al., 2006); shControl (5′-GCGCGCTTTGTAGGATTCG-3′), shARMS-1 (5′-GCCACCAAGATGAGAAATA-3′), and shARMS-2 (5′-GCCGGAACATACGTGAACATAT-3′) sequences, as described previously (Cortés et al., 2007; Higuero et al., 2010; Yu et al., 2011), were cloned in pLVTHM. Flag-BDNF and Syt-IV cDNAs were cloned into pCtetOWP replacing the coding sequence of GFP.
Lentivirus production.
The lentiviruses used in this study were generated by cotransfection using calcium phosphate in 293FT cells as previously described (Yu et al., 2011). The 293FT cells, seeded in a 10 cm plate the day before, were transfected with 20 μg of pCtetOWP-GFP, pCtetOWP-Flag-BDNF, pCtetOWP-Syt-IV, pLM-CMV-R-Cre, or pLVTHM containing the specific shRNA sequence, together with 15 μg of psPAX2 and 6 μg of pMD2.G plasmids. After 8 h, the medium was replaced with a medium without antibiotics. Subsequently, after 48 h, the supernatant containing the lentivirus was collected, centrifuged at 500 × g for 10 min, passed through a 0.45 μm filter, and stored in aliquots at −80°C. DRGs at DIV 4 or cortical neurons at DIV 2 were infected using 50 μl of supernatant containing lentivirus per 1 × 105 cells. Organotypic hippocampal slices were infected at DIV 1 with 100 μl/slice. The expression or reduction in levels of the corresponding protein was assessed using Western blot.
Western blot analysis.
Cells were lysed in a lysis buffer (10 mm Tris, pH 7.4, 150 mm NaCl, 2 mm EDTA, 1% NP-40, 1 mm PMSF, 1 μg/ml aprotinin, 2 μg/ml leupeptin, 1 mm sodium orthovanadate, 10 mm NaF, and 20 mm β-glycerophosphate) for 40 min at 4°C with gentle shaking, and centrifuged at 14,000 × g for 15 min to eliminate the debris. Laemmli buffer was added to lysates and boiled for 7 min to ensure protein denaturation. Proteins were resolved using SDS-PAGE, and Western blots were performed with specific antibodies. To avoid problems with the Ig chains, we used ProtA- or ProtG-conjugated HRP when same species antibodies were used for both immunoprecipitation and Western blot. Images were taken with a MicroChemi 4.2 Chemiluminiscence System using the Gel Capture Software.
qRT-PCR.
Trizol reagent (Invitrogen) was used to isolate total RNA from brain tissues, following the manufacturer's recommendations, and treated with DNase I. Complementary DNA (cDNA) was synthesized with 1 μg of RNA and reverse-transcribed using the Maxima Reverse Transcriptase (Thermo Fisher Scientific) and random primers. The cDNA concentration was determined by measuring absorbance with a Nanodrop 2000c (Thermo Fisher Scientific). Quantitative PCRs were performed in triplicate using SYBR-Green Power Master Mix (Applied Biosystems). The mRNA level of bdnf was normalized using sdha as a reference. The oligonucleotides used to amplify bdnf (bdnf ref NM_007540.4, with an efficiency of 96%) were 5′-gacacattaccttcctgcatct-3′ and 5′-ggatggtcatcactcttctcac-3′ and for sdha (sdha ref NM_023281.1 with an efficiency of 96.33%) were 5′-acacagacctggtggagacc-3′ and 5′-ggatgggcttggagtaatca-3′. A Quant Studio 7 Flex detection system (Applied Biosystems) was used with the following conditions: 10 min at 95°C followed by 40 cycles of 15 s at 95°C and 1 min at 60°C. Following amplification, the melting curves for the products were generated to ensure that a product represented a homogenous species. A comparative cycle of threshold fluorescence (Ct) was used, and the relative transcription level of the bdnf mRNA was normalized to that of sdha mRNA using the 2−ΔΔCt method.
Neuronal and organotypic hippocampal slice cultures.
DRGs were dissected from E15.5 rat or E13.5 mouse embryos. They were incubated and dissociated with 0.25% trypsin in L-15 medium for 45 min at 37°C as previously described (Yu et al., 2011). Briefly, DRG neurons were plated in 12-well plates (1.2 × 105 cells/well) using plating medium (MEM, 10% FBS, 0.4% glucose, 2 mm glutamine, 100 U/ml Pen/Strep), and NGF (100 ng/ml) on Growth Factor Reduced Matrigel (BD Biosciences) coated plates, overnight at 37°C with 5% CO2. The following day, the medium was changed to NB (Neurobasal-A medium, B-27 supplement, 0.4% glucose, 2 mm glutamine), NGF (100 ng/ml), and 5-fluorodeoxyuridine (2.44 μg/ml) and uridine (2.44 μg/ml). Neurons were infected with shControl, shARMS-1, or shARMS-2 lentiviruses at DIV 4 and/or with lentiviruses expressing GFP or Syt-IV at DIV 7. The corresponding experiments were performed at DIV 11.
Cortices were obtained from E18.5 Sprague Dawley rat or E16.5 mouse embryos and incubated with 0.1% 12.5 units/ml of active papain in Earle's Balanced Salt Solution (Sigma) for 20 min at 37°C to obtain individual neurons. These cells were plated in Neurobasal-A medium (Invitrogen) supplemented with NeuroBrew-21 (MACS) and 1 mm l-glutamine on poly-D-lysine-coated plates (Sigma) at 37°C with 5% CO2. Neurons were seeded on 6-well plates (0.8 × 106 cells/well) and 24 h later were infected with shControl, shARMS-1, or shARMS-2. For BDNF secretion experiments, lentiviruses expressing Flag-BDNF were added to the cells at least 2 d before performing the secretion assay.
Organotypic hippocampal slice cultures were prepared from 20- to 30-week-old and 8-month-old R6/1 and KI HD mice (see below), respectively, with the corresponding controls according to the interface method previously described (Stoppini et al., 1991; Vinet et al., 2012), with minor modifications. In brief, slice cultures were prepared under sterile conditions. After decapitation, the hippocampi from both hemispheres were isolated in ice-cold DPBS (Invitrogen). Using a stereo microscope, isolated hippocampi were cut by hand into slices that were subsequently placed in 0.4 μm high-density PET membrane inserts (Falcon) (4 slices per insert). These inserts were transferred to 24-well plates containing 0.4 ml of Neurobasal-A medium (Invitrogen) supplemented with NeuroBrew-21 (MACS) and 1 mm l-glutamine. Slices were infected with 400 μl/well of supernatant containing lentiviruses expressing shControl or shARMS-1. The slice cultures were kept at 35°C in a humidified atmosphere (5% CO2), and the culture medium was refreshed every 72 h. Secretion experiments were performed 7 d after infection.
BDNF immunoassay.
BDNF quantification was performed using an ELISA previously described (Kolbeck et al., 1999), with minor modifications. In brief, 96-well white polystyrene plates (Nunc) were coated with 1 μg of anti BDNF-1 antibody in 100 μl carbonate buffer, pH 9.7, per well overnight at 25°C. Plates were blocked with 4% BSA in PBS and washed three times with TBST. Samples and standards (1–1024 pg of recombinant purified BDNF per well) supplemented with 1% BSA and 1% NP-40 were incubated for 3 h at 30°C together with BDNF-9 antibody coupled to HRP. Plates were washed three times with TBST. SuperSignal ELISA Femto Substrate (Invitrogen), diluted 50% in H2O, was used as the substrate. With these modifications to the assay, we improved its sensitivity to detect 1 pg of BDNF per well (Fig. 1-1A). In addition, using a range of standards with different amounts of recombinant BDNF (1–1024 pg/well), we generated standard curves for each experiment performed, which allowed us to precisely quantify the BDNF present in different cells, tissues, and supernatants (Fig. 1-1B). The amount of secreted BDNF was calculated as the percentage of total BDNF detected in media and cell lysates. There were no differences in basal secretion observed in any of the experiments performed; therefore, basal secretion was set to 100% to express the regulated secretion as a percentage of basal secretion.
To assess BDNF secretion, DRG neurons were infected as described previously; and at DIV 11, the culture media was changed to DMEM-F12 (Invitrogen) and 4 h later secretion experiments were performed. To induce secretion, neurons were first incubated for 30 min with a physiological saline solution (PSS: 15 mm HEPES-NaOH, pH 7.4, 145 mm NaCl, 5.6 mm KCl, 2.2 mm CaCl2, 0.5 mm MgCl2, and 5.6 mm glucose), and the media were collected for the basal condition. Then, neurons were incubated with PSS + NGF (100 ng/ml), and media were collected for the induced condition. Finally, cells were lysed in a buffer containing 10 mm Tris, pH 7.4, 150 mm NaCl, 1% NP-40, and protease inhibitors. BDNF levels in the basal and induced conditions of media and in cell lysates were determined using ELISA.
To assess BDNF secretion, cortical neurons were infected at DIV 2 with shControl, shARMS-1, or shARMS-2. Because we were unable to detect endogenous BDNF in these cultures, neurons were infected with lentiviruses expressing BDNF from DIV 7 to DIV 11, when secretion assays were performed. Before inducing secretion, the culture media was changed to DMEM-F12 for 4 h. Afterward, cortical neurons were incubated for 30 min with PSS, and the solution was collected for the basal condition. These cells were then incubated with PSS + NT-3 (100 ng/ml), PSS + NT-4 (100 ng/ml) or with high potassium depolarizing solution (15 mm HEPES-NaOH, pH 7.4, 95 mm NaCl, 56 mm KCl, 2.2 mm CaCl2, 0.5 mm MgCl2, and 5.6 mm glucose) for 30 min, and solution was collected for the induced condition. Then, cells were lysed as described above for DRG neurons and BDNF levels were determined in the basal and induced media and cell lysates using ELISA.
Cortical slices obtained from 4-month-old male shARMS and shARMS;CamK2a-Cre mice were maintained in PBS for 30 min, and secretion experiments were performed as described above for cortical neurons.
To assess BDNF secretion in organotypic hippocampal slice cultures, shControl or shARMS-1 lentiviruses were added at DIV 1. Media was changed to DMEM-F12 at DIV 7, and 4 h later secretion experiments were performed as described previously for cortical neurons.
Images were taken with a MicroChemi 4.2 Chemiluminiscence System using the Gel Capture Software.
BDNF levels in the brains of mice either exposed or not exposed to physical activity.
A set of male and female mice (1- or 4-month-old) performed 4 sets of 5 min of physical activity in a Rota Rod apparatus (acceleration from 4 to 40 rpm within 5 min) on 4 consecutive days. On the last day, after the final Rota Rod set, mice were placed in activity wheels for 2 h. They were then killed, their brains were obtained, and the cerebral cortex, hippocampus, striatum, and cerebellum were dissected. The tissue was mechanically disaggregated on ice and lysed in cold lysis buffer (as described for the Western blot procedure) supplemented with 0.1% SDS. Cell debris was eliminated using centrifugation. The amount of total protein present in the samples was quantified using the Bradford method before quantifying BDNF levels as described above.
Immunohistochemistry and immunofluorescence.
Mice were anesthetized and perfused with 4% PFA in 0.1 m PB. Before fixing, the circulatory system was washed with saline solution. Once the brains were extracted, they were kept in the fixing solution for 2–4 h and then immersed in 30% sucrose w/v until they sunk to the bottom of the flask. The 40 μm sections were obtained from the freezing microtome, which remained at 0.1 m, pH 7.4, phosphate buffer. Sections to be used for immunohistochemistry were pretreated with hydrogen peroxide and methanol to block peroxidase activity. Those to be used for immunofluorescence were quenched with 50 mm NH4Cl. Then, all sections were treated with 5% normal goat serum to block nonspecific reactions and 0.1% Triton X-100 to permeabilize membranes. Subsequently, the sections were incubated with rabbit anti-ARMS antibody (immunohistochemistry) or mouse anti-ARMS and rabbit anti-Cre antibodies (immunofluorescence), for at least 3 d at 4°C. After washing, sections were incubated with biotin-conjugated anti-rabbit antibody, and proteins were subsequently detected using a diaminobenzidine solution (immunohistochemistry) or with the corresponding fluorescent secondary antibody (AlexaFluor) (immunofluorescence). Images were taken in selected fields using an Olympus ProvisAX70 microscope equipped with a DP Olympus camera (immunohistochemistry) and in Carl Zeiss Time Lapse Model AIXO Observer Z1 equipped with an AIXOCAM MRmCarl Zeiss using ZEN2011 software (immunofluorescence).
shARMS mice.
A cassette was generated to decrease ARMS protein levels in a regulated way using the shARMS-1 (Cortés et al., 2007) subcloned in the pPVIII plasmid. This plasmid contains a cassette that includes the cDNA of enhanced GFP (eGFP) and the H1 promoter both in the 3′ to 5′ orientation and the shARMS-1. The eGFP and H1 promoter are flanked by two pairs of LoxP and Lox2272 sites at opposite orientations (Fig. 2-1A). A neomycin-resistance gene flanked by FRT sites was inserted downstream from shARMS. This cassette, plus the neomycin gene, was subcloned into the pROSA26–1 plasmid (Soriano, 1999) using the XbaI site, generating a targeting vector to allow insertion of the cassette into the endogenous ROSA26 locus by homologous recombination. The pROSA26-1 also contains a diphtheria toxin gene placed at the end of the ROSA26 3′ fragment for negative selection (Fig. 2-1A). The targeting vector was linearized at a unique upstream SacII site and was electroporated into CJ7 embryonic stem (ES) cells, as previously described (Tessarollo, 2001). G418-resistant colonies were screened for homologous recombination using Southern blot with both external and internal probes. The external probe was a 300 bp DNA fragment obtained by PCR from genomic DNA, which hybridizes with the ROSA26 promoter upstream from the homologous recombination region. An internal probe against the neomycin gene was used (Fig. 2-1B). Three targeted ES clones were injected into C57BL/6 blastocysts to generate chimeric mice that were bred with C57BL/6 mice expressing the Flp recombinase driven by the actin promoter to remove the Neo cassette. All three chimeras gave germline transmission. shARMS mouse genotyping was performed routinely using PCR with tail genomic DNA and the following three primers: ROSA26–5′-F (5′-GAGAAGGCCGCACCCTTC-3′), H1 F/R (5′-GGGAATCTTATAAGTTCTG-3′) and ROSA26–3′-R (5′-CACACCAGGTTAGCCTTTAAGCC-3′) (Fig. 2-1B,D,F). A 194 bp product was amplified from the WT allele, and a 271 bp product was amplified from the targeted allele in absence of Cre recombination. Once Cre recombination occurs, this PCR amplifies a 253 bp product (Fig. 2-1D,H).
Mouse lines.
CamK2a-cre transgenic mice in C57BL/6 background, in which Cre recombinase expression begins postnatally (at 3–4 weeks) in excitatory neurons (Tsien et al., 1996) were obtained from The Jackson Laboratory. shARMS (in mixed C57BL6–129c1 background) and CamK2a-cre male and female mice were bred to generate age-matched shARMS;CamK2a-cre as cases and ARMS-shRNA or CamK2a-cre littermates as controls. HdhQ111/Q111 knock-in mutant mice (KI) (in C57BL/6 background) expressing mutant huntingtin with targeted insertion of 109 CAG repeats extending the glutamine segment in murine huntingtin to 111 residues, have been described previously (Wheeler et al., 1999). HdhQ7/Q111 heterozygous male and female mice were intercrossed to generate age-matched HdhQ7/Q111 heterozygous and HdhQ7/Q7 wild-type littermates. Only male mice were used for these experiments. R6/1 heterozygous transgenic mice (Mangiarini et al., 1996) (in C57BL/6 background) expressing exon-1 mutant huntingtin with 115 glutamines under the HD human promoter were obtained from The Jackson Laboratory. Their wild-type littermates were used.
Animals were bred in a specific, pathogen-free facility with freely-available food and water at 20°C-22°C with a 12 h light/dark cycle, and 55%–65% humidity. All animals were housed with a maximum of 5 mice per cage and bred in the SPF Animal Facility of the University of Salamanca. Proper measures were taken to reduce the pain or discomfort of experimental animals, and procedures were conducted in accordance with protocols approved by the Bioethics Committee of the University of Salamanca and following the European Community guidelines.
Statistical analysis.
No statistical methods were used to predetermine sample sizes; however, sample sizes were estimated based on similar experiments reported in previous publications from our laboratory. Data collection and analysis were performed blind to the conditions of the experiments when mice were used. Our data are presented as mean ± SEM and were analyzed using Microsoft Excel and Graph Pad Prism software and quantified with Adobe Photoshop. Comparisons between the means of different groups were performed using paired or unpaired two-tailed Student's t tests because it is the most restrictive method to assess differences.
Data availability.
The data that support the findings of this study are available from the corresponding author upon reasonable request.
Results
ARMS levels regulate BDNF secretion in cultured DRG and cortical neurons
First, we addressed whether ARMS expression regulates BDNF secretion in DRG neurons in response to NGF because we have previously demonstrated that ARMS modulates NGF-mediated secretion (López-Benito et al., 2016). We depleted ARMS using lentiviruses expressing two different shRNAs against ARMS (shARMS-1 or shARMS-2). The reduction of ARMS expression in infected DRG neurons was monitored using Western blot (Fig. 1A) and quantified in several independent experiments (Fig. 1-1A). Then, BDNF secretion was quantified as described (see Material and Methods; Fig. 1-1C,D). Regulated BDNF secretion increased significantly in ARMS-depleted DRG neurons (Fig. 1B), with no differences in basal secretion observed in this experiment. Therefore, ARMS levels affect NGF-regulated BDNF release in DRG neurons. In the CNS, regulated BDNF release is controlled by neuronal activity (Balkowiec and Katz, 2002). To address whether regulated BDNF was regulated in DRG neurons by neural activity, we performed experiments using TTX, a neuronal activity blocker. NGF-mediated BDNF secretion was still present in presence of TTX (Fig. 1-1E). However, basal BDNF secretion was impaired to ∼50% upon TTX treatment (Fig. 1-1E). These data suggest that BDNF secretion from DRG neurons in response to NGF does not require neuronal activity, whereas basal BDNF secretion does need it.
Figure 1.
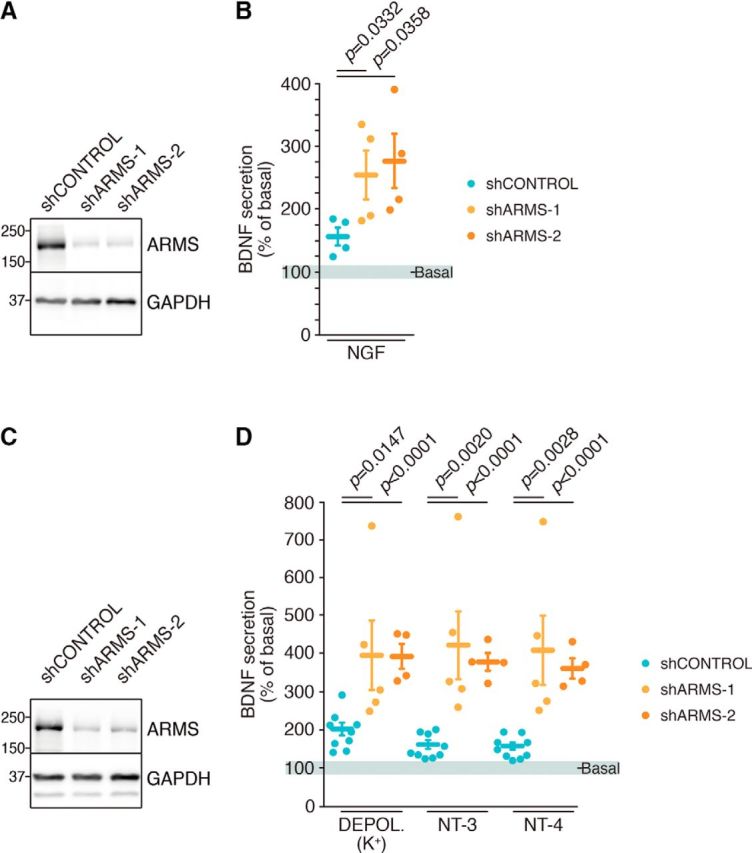
ARMS regulates BDNF release in DRG and cortical neurons. A, ARMS depletion in cultured DRG neurons. Cultured DRG neurons were infected at DIV 4 with control (shControl), ARMS shRNA-1 (shARMS-1), or ARMS shRNA-2 (shARMS-2) lentiviruses and lysates were obtained at DIV 11. Western blot analyses were performed. A representative blot is shown (n = 4). B, BDNF secretion in response to NGF is enhanced in ARMS-depleted DRG neurons. BDNF ELISA was performed using the supernatant of DRG neurons from A that were nonstimulated (basal) and then stimulated with NGF for 30 min at DIV 11. Cell lysates were collected to assess BDNF levels (n = 4). Paired Student's t test, mean ± SEM. shControl versus shARMS-1, t = 3.745, df = 3; shControl versus shARMS-2, t = 3.638, df = 3. C, ARMS knockdown in cultured cortical neurons. Cultured cortical neurons were infected with shControl, shARMS-1, or shARMS-2 lentiviruses at DIV 2 and with lentiviruses expressing BDNF at DIV 7. Cell lysates were obtained at DIV 10. Western blot analyses were performed. A representative blot is shown (n = 4). D, ARMS knockdown potentiates BDNF release in response to different stimuli in cortical neurons. BDNF ELISA was performed using the supernatant of cortical neurons from C that were nonstimulated (basal) and then stimulated with KCl, NT-3, or NT-4 for 30 min at DIV 10 (n = 9, n = 5, and n = 4 for shControl, shARMS-1, and shARMS-2, respectively). Unpaired Student's t test, mean ± SEM. K+ Depol: shControl versus shARMS-1, t = 2.846, df = 12; shControl versus shARMS-2, t = 5.923, df = 11; NT-3: shControl versus shARMS-1, t = 3.917, df = 12; shControl versus shARMS-2, t = 10.03, df = 11; NT-4: shControl versus shARMS-1, t = 3.742, df = 12; shControl versus shARMS-2, t = 9.148, df = 11. See also Figure 1-1.
ARMS downregulation by shRNAs and sensitivity of the BDNF immunoassay. A) Quantification of ARMS depletion in cultured DRG neurons corresponding to the experiments performed in Fig. 1A,B. (n = 4). Paired Student's t-test, mean ± s.e.m. shC vs shARMS-1 t =8.078, df = 3; shC vs shARMS-2 t = 7.03, df = 3. B) Quantification of ARMS depletion in cultured cortical neurons corresponding to the experiments performed in Fig. 1 C,D. (n = 4). Paired Student's t-test, mean ± s.e.m. shC vs shARMS-1 t = 44.19, df = 3; shC vs shARMS-2 t = 111.8, df = 3. C) Different amounts of recombinant BDNF (1-1024 pg/well) were incubated on ELISA plates coated with 1 pg/well of mAb#1. 12.5 ng of mAb#9 HRP were incubated for 3 hours at 30 °C, washed with TBST and developed with SuperSignal ELISA Femto Substrate diluted 50% in water. A representative standard curve is shown. D) Adjusted standard curve of BDNF ELISA using recombinant BON F. E) Neuronal activity is required for BDNF release in response to NGF. BDNF secretion experiments were performed in DRGs in a similar way as described in Fig. 1 B in presence (striped bars) and absence (solid bars) of TTX with or without NGF. (n = 7). Paired Student's t-test, mean± s.e.m. Control vs TTX t = 9.157, df = 6; Controi-NGF vs TTX-NGF t = 4.466, df = 6. Download Figure 1-1, PDF file (304.7KB, pdf)
We then assessed BDNF release in cortical neurons with downregulated ARMS expression upon K+ depolarization or stimulation with NT-3 or NT-4. Reduced ARMS expression in neurons expressing shARMS-1 and shARMS-2 was confirmed using Western blot (Fig. 1C) and quantified (Fig. 1-1B). Depletion of ARMS significantly enhanced BDNF release in response to all three stimuli (Fig. 1D). Together, our results suggest that ARMS levels modulate regulated BDNF secretion in cultured cortical neurons.
Generation of a new conditional ARMS mouse model
To address the role of ARMS in BDNF release in vivo, we generated a new mouse model lacking ARMS using an RNA interference strategy (Dickins et al., 2007) (see Material and Methods). A cassette carrying an expression-inducible shARMS against ARMS was generated (Fig. 2A). To avoid the side effects of random insertion, the cassette was inserted in the ROSA26 locus using homologous recombination in ES cells (Fig. 2-1A). The targeted ES cells were monitored using Southern blot (Fig. 2-1B,C), and mice were generated, which we refer to hereafter as shARMS mice. They were viable, fertile, and had a normal life span. Genotyping was performed using PCR (Fig. 2-1B,D). The functionality of the cassette depleting ARMS levels was tested in cultured cortical neurons (Fig. 2-1E) and DRG neurons (Fig. 2-1F–I) in the presence or absence of Cre recombinase. Therefore, a new mouse model was generated by inserting a cassette carrying an expression-inducible shARMS against ARMS at the ROSA26 locus to allow a conditional, efficient knockdown of ARMS protein upon Cre recombinase expression.
Figure 2.
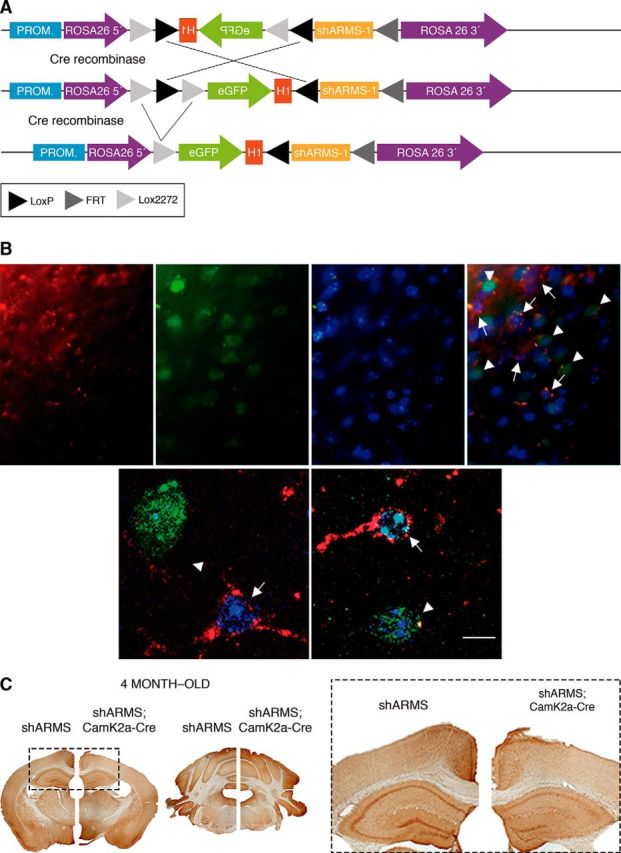
Generation of a mouse model to knock down ARMS expression. A, Schematics of the targeted allele before and after Cre recombination. DTA, Diphtheria toxin; H1, promoter H1; PGK neo, neomycin resistance gene; PROM, promoter ROSA26. B, Immunofluorescence showing the downregulation of ARMS protein (red) in cortical neurons from shARMS;CamK2a-Cre mice. Arrowheads indicate Cre-positive (green) in the cortex. Arrows indicate Cre-negative neurons in the cortex. Nuclei are labeled with Hoechst (blue). Representative pictures are shown (n = 4). Scale bar, 5 μm. C, ARMS expression is downregulated in the cortex and hippocampus, but not in other brain areas, of shARMS;CamK2a-Cre mice at 4 months of age. A representative staining is shown (n = 2). See also Figure 2-1.
Generation, genotyping, and functionality of shARMS mice. A) Schematics of the wild-type ROSA26 allele, the targeting vector and the targeted allele before and after Flp recombination. DTA: diphtheria toxin; eGFP: enhanced GFP; H1: promoter H1; PGK neo: neomycin resistance gene; PROM: promoter ROSA26. B) Schematics of the wild-type ROSA26 allele and the targeted allele before and after Flp recombination with the restriction sites EcoRV and Xbal used to digest genomic DNA and to clone ARMS shRNA cassette, respectively. The size of the fragments generated after EcoRV digestion are shown (blue). For genotyping, primers are denoted with red arrowheads and the fragment sizes amplified after PCR depending on the genotype are: 194 base pairs for WT mice, 271 base pairs and 253 base pairs for shARMS mice before and after Cre recombinase, respectively (red). C) Southern blot analysis of genomic DNA from WT and targeted mice before and after Flip-mediated recombination, upon digestion with EcoRV. As indicated in the schematics in panel B, the 5' external probe detects an 11 kb DNA fragment from WT alleles and a 14.3 kb and 12.5 kb DNA fragment from mutant alleles with or without the Neo cassette, respectively. A representative blot is shown. D) PCR analysis of genomic tail DNA from WT, shARMS, shARMS;shARMS cultured cortical neurons infected with ere-expressing lentiviruses. As indicated in the schematics in B, the fragments amplified were of 194 bp and 271 bp for WT and shARMS mice, respectively, and 253 bp for shARMS mice in presence of Cre recombinase. A representative PCR is shown. E) Efficient ARMS down regulation in cortical neurons from shARMS mouse in the presence of Cre recombinase. Cortical neurons were obtained from E16.5 mouse embryos with the corresponding genotype, and infected or not with lentiviruses expressing Cre recombinase. Western blot analyses were performed to detect ARMS, Cre recombinase and tubulin as a loading control. F) PCR genotyping of E13.5 embryos from shARMS matings. Genomic DNA was obtained from tails and PCR reaction was performed. Samples 2, 5, 7, and 8 are WT; 3 and 4 have one copy, and 1 and 6 two copies of the cassette. A representative PCR is shown. G) ARMS expression is unchanged in shARMS mice. Western blot analyses from lysates of cultured DRG neurons corresponding to E13.5 embryos of panel F. Tubulin was used as loading control. H) PCR analysis of cultured DRGs from E13.5 embryos corresponding to panel F infected with lentivirus expressing Cre recombinase. DRG neurons were cultured and infected with lentiviruses expressing Cre recombinase. Afterwards, genomic DNA was obtained and PCR reaction was carried out. Note the presence of the 253 bp fragment in those samples with the shARMS cassette (samples# 3, 1, 4, and 6). A representative PCR is shown. I) Knockdown of ARMS expression using cultured DRG neurons from shARMS embryos expressing Cre recombinase corresponding to panel H (samples# 3, 1, 4 and 6). Samples were collected and Western blot analyses were performed to assess ARMS expression. Tubulin was used as a loading control. Download Figure 2-1, PDF file (304.7KB, pdf)
Deletion of ARMS expression during nervous system development has detrimental effects on neuronal maturation (Wu et al., 2009; Cesca et al., 2012). Therefore, we depleted ARMS levels in vivo exclusively in neurons from postnatal mice using a CamK2a-Cre mouse. This ensured that ARMS would be depleted specifically in CamK2a-expressing neurons after forebrain development, where BDNF is highly expressed postnatally (Maisonpierre et al., 1990; Phillips et al., 1990). An efficient reduction of ARMS protein was observed in cortical neurons expressing Cre recombinase from the cortex of 4-month-old shARMS;CamK2a-Cre mice (Fig. 2B). Additional immunohistochemistry with ARMS antibodies supported the presence of specific ARMS depletion in the cortex and hippocampus, but not other brain areas, of these mutant mice (Fig. 2C). Therefore, ARMS expression can be regulated in vivo using Cre recombinase in shARMS mice.
ARMS regulates BDNF secretion in the cortex and BDNF levels in the striatum
We next assessed secretion of endogenous BDNF in dissected cortices from 4-month-old control (shARMS) and mutant (shARMS;CamK2a-Cre) mice in response to K+ depolarization, NT-3, and NT-4. Regulated BDNF release increased significantly in mutant compared with control cortices in response to all three stimuli (Fig. 3A, orange vs blue dots). The differences observed in regulated secretion of BDNF were not a consequence of differential BDNF expression in the cortex of control and mutant mice because similar total BDNF levels were observed (Fig. 3B, CORTEX, solid dots). Therefore, ARMS negatively regulates BDNF secretion in vivo in response to different stimuli.
Figure 3.
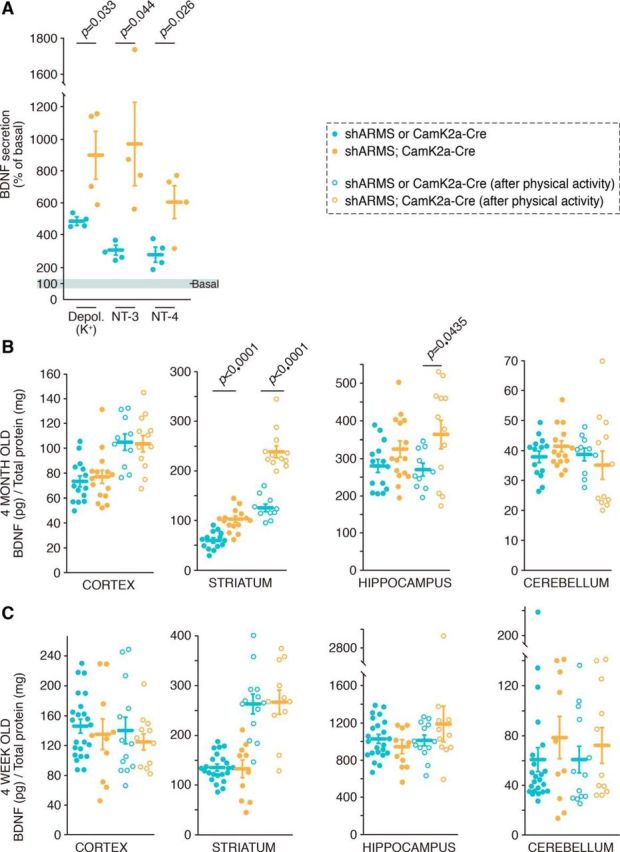
ARMS regulates BDNF secretion in vivo, and its depletion leads to BDNF accumulation in the striatum. A, ARMS levels regulate BDNF secretion in vivo. Cortical slices from control (shARMS) or mutant mice (shARMS;CamK2a-Cre) were obtained and incubated in PBS for 30 min (basal secretion). Then slices were stimulated with KCl, NT-3, or NT-4 for 30 min, the supernatant collected, and the slices were lysed. BDNF ELISA was performed to assess secretion (n = 4). Unpaired Student's t test, mean ± SEM. K+ Depol: shControl versus shARMS-1, t = 2.764, df = 6; NT-3: shControl versus shARMS, t = 2.548, df = 6; NT-4: shControl versus shARMS-1, t = 2.927, df = 6. B, BDNF levels from different brain areas of 4-month-old control (shARMS or CamK2a-Cre) (blue dots) and mutant (shARMS;CamK2a-Cre) mice (orange dots) in resting conditions (solid dots) or after physical activity (empty dots) (n = 14, n = 10, n = 15, and n = 12 for shARMS, shARMS after physical activity, shARMS;CamK2a-Cre, and shARMS;CamK2a-Cre after physical activity, respectively). Unpaired Student's t test, mean ± SEM. shARMS or CamK2a-Cre versus shARMS;CamK2a-Cre: striatum rest conditions, t = 5.748, df = 27; striatum after physical activity, t = 7.850, df = 20; hippocampus after physical activity, t = 2.155, df = 20. C, BDNF levels from different brain areas of 4-week-old controls (shARMS or CamK2a-Cre) (blue dots) and mutant (shARMS;CamK2a-Cre) (orange dots) mice in resting conditions (solid dots) or after physical activity (empty dots) (n = 23, n = 13, n = 10, and n = 11 for shARMS, shARMS after physical activity, shARMS;CamK2a-Cre, and shARMS;CamK2a-Cre after physical activity, respectively). See also Figure 3-1.
A) Quantification of bdnf mRNA levels in mice subjected to physical exercise. Total RNA was obtained, eDNA was synthesized and quantitative PCR was performed as described in Material and Methods. The relative transcription level of bdnf was normalized to that of sdha using the 2-LlLlCt method. Data are presented as means ± SEM. Triplicate determinations from 3-4 control and mutant mice subjected or not to exercise were quantified. P value was calculated using a two-tailed Student's t-test. B) Similar ARMS expression in forebrain of shARMS and shARMS;CamK2a-Cre mice at 4 weeks of age. Immunohistochemistry with ARMS antibodies of control (shARMS) (left hemisphere) and mutant mice (shARMS;CamK2a-Cre) (right hemisphere). A representative staining is shown (n = 2). Download Figure 3-1, PDF file (304.7KB, pdf)
Considering the increased BDNF secretion observed in ARMS-depleted cortices, BDNF protein levels in the striatum and other brain regions were analyzed in control and mutant mice. We observed significantly higher levels of BDNF in the striatum of shARMS;CamK2a-Cre mice compared with control mice (Fig. 3B, orange vs blue, solid dots). This accumulation of BDNF protein in the striatum was specific, as BDNF levels in the cortex, hippocampus, and cerebellum were similar in mutant and control mice (Fig. 3B, orange vs blue, solid dots). Thus, ARMS levels selectively affected BDNF levels in the striatum.
The above experiments were conducted with mice housed in standard conditions (i.e., resting conditions). It is well known that bdnf mRNA and BDNF protein levels increase in response to physical activity (Neeper et al., 1996; Sleiman et al., 2016). To assess whether BDNF levels in the cortex and hippocampus of ARMS-depleted mice were altered in response to physical activity, control and mutant animals were investigated following physical activity (see Material and Methods). BDNF levels were increased in the cortex of 4-month-old mice subjected to physical activity compared with resting conditions (Fig. 3B, empty vs solid dots), but no differences were observed between genotypes (Fig. 3B, blue vs orange, empty dots). Interestingly, BDNF levels in the striatum were augmented in both control and mutant mice undergoing physical activity compared with resting conditions (Fig. 3B, empty vs solid dots), maintaining the significant difference between genotypes (Fig. 3B, blue vs orange dots). In the hippocampus, there were significantly higher BDNF levels in mutant mice than in control mice subjected to exercise (Fig. 3B, orange vs blue, empty dots). No differences in BDNF levels were observed in the cerebellum between genotypes in resting conditions or in response to exercise.
To figure out whether bdnf mRNA levels were affected by physical activity in our mice, we performed additional experiments. Total mRNA was obtained from brain tissues of control and mutant mice in resting conditions or after physical exercise, and bdnf mRNA levels were assessed by qPCR. With the exercise protocol used (see Material and Methods), only the cortex of mutant mice showed a significant increase in bdnf mRNA in response to exercise, although there was also a tendency to increase in the cortex and hippocampus of control mice (Fig. 3-1A). These data suggest that the accumulation observed in the striatum and hippocampus of mutant mice in response to exercise was due to an enhanced secretion. Together, the results regarding BDNF protein and bdnf mRNA amounts indicate that ARMS levels affect regulated secretion of BDNF in vivo in response to exercise, leading to an accumulation of BDNF in the striatum and hippocampus.
To further confirm that the differences in BDNF levels observed were due to ARMS protein levels, we performed similar experiments using control or mutant mice aged 4 weeks. At this age, forebrain ARMS levels have yet not been depleted (Fig. 3-1B). No differences were observed between genotypes in BDNF levels in the striatum, cortex, hippocampus, or cerebellum (Fig. 3C, orange vs blue, solid dots). Moreover, increased BDNF in response to physical activity was similar in both genotypes and, interestingly, was observed exclusively in the striatum (Fig. 3C, empty vs solid dots). Therefore, no differences in BDNF levels in brain tissues were observed between control and mutant mice at 4 weeks of age because ARMS levels were not depleted.
Knockdown of ARMS expression in the hippocampus of HD mice rescues impaired BDNF secretion
Taking into account that reduced BDNF levels and transport deficits have been reported in different mouse models of HD, we investigated whether differences exist in ARMS expression in these mice. We chose two different HD mouse models: the HdhQ7/111 knock-in mice (KI mice) and R6/1. These differ in disease onset and progression, with an earlier onset and faster disease progression in the R6/1 mice than in KI mice (Kim et al., 2011). First, because the cortex and hippocampus are the main regions secreting BDNF, we assessed ARMS expression in these tissues of symptomatic KI and R6/1 (8-month-old and 20- to 30-week-old, respectively) and the corresponding control mice. ARMS levels were increased in the hippocampus of the same KI or R6/1 compared with control mice (Fig. 4A,C), whereas no differences were observed in ARMS expression in cortical lysates from these HD mice (Fig. 4-1A,B). Thus, ARMS levels are elevated in the hippocampus of symptomatic HD mice.
Figure 4.

HD mice have increased ARMS levels and impaired BDNF release, which is partially rescued upon ARMS knockdown. A, Increased ARMS expression in the hippocampus of 8-month-old KI mice (n = 6). A representative blot is shown. Unpaired Student's t test, mean ± SEM. WT versus KI, t = 2.50, df = 10. B, Impaired BDNF secretion in the hippocampus of KI mice and partial rescue by depleting increased ARMS expression (n = 5 and n = 7 for WT and KI mice, respectively). Unpaired Student's t test, mean ± SEM. WT versus KI mice, shC: K+ Depol: t = 9.249, df = 10; NT-3: t = 10.98, df = 10; NT-4: t = 6.643, df = 10; WT versus KI mice, shARMS-1: K+ Depol: t = 3.518, df = 10; NT-3: t = 2.281, df = 10; NT-4: t = 1.925, df = 10. C, Increased ARMS expression in the hippocampus of 20- to 30-week-old R6/1 mice (n = 7 and n = 4; for WT and R6/1, respectively). A representative blot is shown. Unpaired Student's t test, mean ± SEM. WT versus R6/1, t = 5.842, df = 9. D, Impaired BDNF secretion in the hippocampus of R6/1 HD mice and partial rescue by depleting increased ARMS expression (n = 4). Unpaired Student's t test, mean ± SEM. WT versus R6/1 mice, shC: K+ Depol: t = 3.270, df = 6; NT-3: t = 5.852, df = 6; NT-4: t = 3.927, df = 6; WT versus R6/1 mice, shARMS-1: K+ Depol: t = 2.840, df = 6; NT-3: t = 3.390, df = 10; NT-4: t = 13.46, df = 6. See also Figure 4-1.
ARMS levels do not change in the cortex of HD mouse models. A) ARMS expression in the cortex of 8 month-old Kl mice is not altered. Western blot for ARMS in the cortex from WT and Kl mice. Actin was used as a loading control (mean± SEM; n = 7). A representative Western blot is shown. B) ARMS expression in the cortex of 20-30 week-old R6/1 mice is not altered. Western blot for ARMS in the cortex from WT and R6/1 mice. Actin was used as a loading control (mean ± SEM; n = 6 and 6; for WT and R6/1, respectively). A representative Western blot is shown. C) Depletion of ARMS in organotypic hippocampal slices from WT and Kl mice. Quantification of ARMS protein levels upon infection with lentivirus expressing shControl (C) or shARMS-1 (A) (n = 5 and 7 for WT and Kl mice, respectively). Unpaired Student's t-test, mean ± s.e.m. Representative Western blot showing ARMS levels in hippocampal slices of WT and Kl mice. D) Depletion of ARMS in organotypic hippocampal slices from WT and R6/1 mice. Quantification of ARMS protein levels upon infection with lentivirus expressing shControl (C) or shARMS-1 (A) (n = 4). Unpaired Student's t-test, mean ± s.e.m. Representative Western blot showing ARMS levels in hippocampal slices of WT and R6/1 mice. Download Figure 4-1, PDF file (304.7KB, pdf)
To address whether enhanced ARMS expression affects BDNF release, we infected organotypic hippocampal slices obtained from KI or R6/1 and the corresponding control mice with shControl or shARMS-1 lentiviruses. ARMS downregulation from the hippocampal slices infected with shARMS-1 for 7 d was confirmed using Western blot (Fig. 4-1C,D). We performed BDNF secretion assays to assess endogenous BDNF release in the hippocampal slices. We observed a significant regulated BDNF release in the slices from control mice infected with shControl unlike the KI or R6/1 slices, which barely responded above basal secretion (Fig. 4B,D; first vs second dots of each treatment). Depletion of ARMS in the hippocampal slices from control mice further potentiated the release of BDNF (Fig. 4B,D, third dots of each treatment), supporting our previous results (Fig. 3A). Interestingly, ARMS knockdown in the organotypic slices from both HD mice using shARMS-1 partially rescued the deficient BDNF secretion to the levels of wild-type animals in control conditions (Fig. 4B,D; compare fourth and first dots of each treatment). Therefore, increased hippocampal ARMS levels in HD mouse models correlate with deficient regulated BDNF release, whereas reduction of ARMS expression in mutant mice partially rescues hippocampal BDNF secretion deficits.
Increased expression of ARMS in the hippocampus and PFC of patients with HD
To address whether there was any correlation between the altered ARMS levels observed in the HD mice and in patients with HD, we performed Western blot analysis in human postmortem tissues (Fig. 5A). The hippocampus and PFC of patients with HD showed a significant increase in the amount of ARMS compared with controls independently of sex or age (Fig. 5B). Therefore, patients with HD have augmented ARMS levels, which correlate with those observed in HD mouse models.
Figure 5.

HD patients have increased ARMS levels. A, Representative Western blot showing ARMS protein expression. Actin and GAPDH were used as loading controls. C, Control sample; HD, HD sample. B, Quantification of ARMS levels in the hippocampus and PFC of patients with HD (for hippocampus, n = 4, n = 7, n = 5, and n = 5 for male and female controls and male and female patients with HD, respectively; for PFC, n = 5, n = 7, n = 6, and n = 6 for male and female controls and male and female patients with HD, respectively). Unpaired Student's t test, mean ± SEM. Control versus HD patients: hippocampus, t = 2.998, df = 19; PFC, t = 4.510, df = 22. * = nonspecific band.
ARMS regulates BDNF release altering Syt-IV levels
Syt-IV has been described as a regulator of BDNF release (Dean et al., 2009), and it is recruited to mature dense-core vesicles being an NGF-dependent regulator for exocytosis (Fukuda et al., 2003; Mori et al., 2008). Considering these previous findings and that our results indicate that ARMS modulates NGF-mediated BDNF secretion (Fig. 1A), we wondered whether ARMS and Syt-IV function together to regulate BDNF secretion. Coimmunoprecipitation assays indicated that ARMS and Syt-IV are closely associated in cultured DRG and cortical neurons, although the interaction was independent of NGF or BDNF treatment, respectively (Fig. 6A,B). To address whether ARMS and Syt-IV act together to influence BDNF secretion, we performed release assays in response to NGF in DRG neurons. Overexpression of Syt-IV using lentiviruses abolished NGF-mediated secretion of endogenous BDNF, compared with GFP overexpression, without affecting basal secretion (Fig. 6C), which is consistent with previous results in hippocampal neurons (Dean et al., 2009). However, depletion of ARMS levels in neurons infected with lentivirus that overexpressed Syt-IV leads to an increase in NGF-mediated BDNF secretion similar to that obtained by only depleting ARMS (Fig. 6C). To our surprise, we observed that ARMS knockdown hindered overexpression of Syt-IV with lentivirus, and even endogenous Syt-IV levels were significantly reduced when ARMS was knocked down (Fig. 6D). Together, these results suggest that ARMS protein controls regulated BDNF secretion through modulation of Syt-IV levels. Next, we assessed whether Syt-IV depletion modulates NGF-mediated BDNF secretion in DRGs using lentiviruses to deplete Syt-IV protein. We observed that Syt-IV reduction enhanced BDNF release in response to NGF above control conditions (Fig. 6E). Finally, we assessed whether ARMS levels were modified in response to Syt-IV knockdown. ARMS amount was unchanged after Syt-IV depletion (Fig. 6F). Therefore, Syt-IV regulates BDNF secretion in DRG neurons in response to NGF but does not alter ARMS protein levels.
Figure 6.
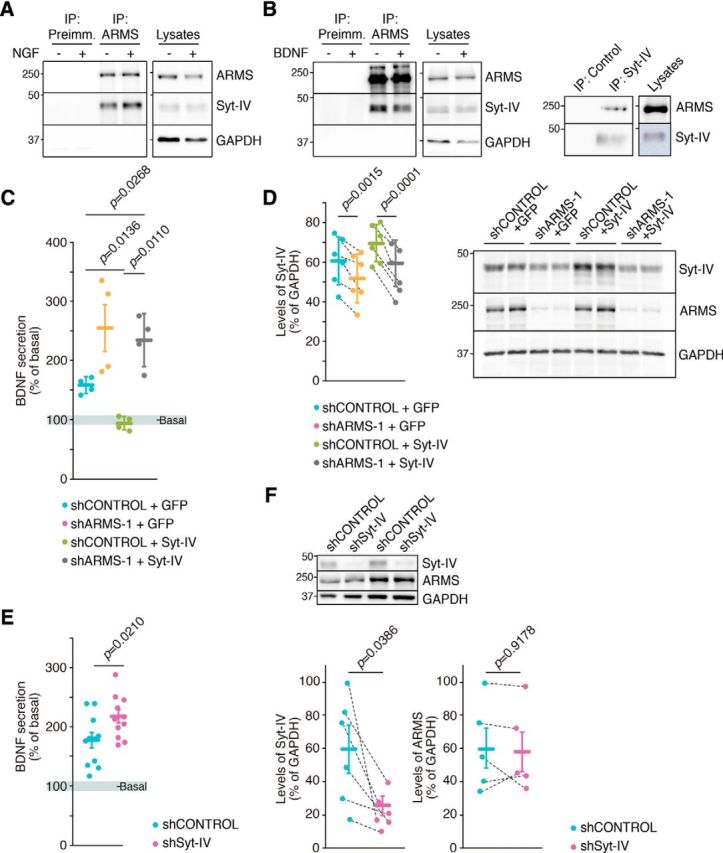
ARMS interacts with and modulates Syt-IV levels controlling regulated BDNF secretion. A, ARMS and Syt-IV interact in DRG neurons. Cell lysates from cells treated or not with NGF for 30 min at DIV 11 were subjected to immunoprecipitation using purified anti-ARMS IgGs (ARMS) or control IgGs (Preimm.). Western blots were performed to detect the presence of Syt-IV and ARMS in the immunoprecipitate. A representative blot is shown (n = 2). B, ARMS and Syt-IV interact in cortical neurons. Immunoprecipitation assays were performed as described in A using lysates from cortical neurons treated or not with BDNF for 30 min at DIV10 (left). Syt-IV was immunoprecipitated using purified anti-Syt-IV IgGs (Syt-IV) or control IgGs (Preimm.), and Western blots were performed to detect ARMS and Syt-IV in the immunoprecipitate (right). A representative blot is shown (n = 3). C, Impaired BDNF secretion in response to NGF in DRG neurons overexpressing Syt-IV is rescued upon ARMS depletion. Cultured DRG neurons were infected at DIV 4 and 7 with lentiviruses expressing shARMS-1 and/or expressing GFP or Syt-IV, and the secretion assays were performed at DIV 11. After NGF starvation for 4 h, the cells were stimulated with NGF for 30 min and BDNF ELISA was performed (n = 4). Paired Student's t test, mean ± SEM. shC + GFP versus shC + SytIV, t = 5.231, df = 3; shC + SytIV versus shA + SytIV t = 5.636, df = 3. The results of shARMS-1 are those presented in Figure 1A and are included here to compare with shControl + GFP and shARMS-1 + Syt-IV. D, ARMS protein regulates Syt-IV levels. Cultured DRG neurons infected with the corresponding lentiviruses as described in C were collected and subjected to Western blot analysis to check ARMS and Syt-IV levels. GAPDH was used as a loading control for quantification. Dotted lines connect results from the same experiment (n = 6). Paired Student's t test, mean ± SEM. shC + GFP versus shA + GFP, t = 6.267, df = 5; shC + SytIV versus shA + SytIV t = 11.09, df = 5. A representative blot is shown. Note the decrease of Syt-IV expression upon ARMS depletion even after infecting with lentiviruses expressing Syt-IV. E, BDNF secretion in response to NGF is increased in Syt-IV-depleted DRG neurons. BDNF ELISA was performed using the supernatant of Syt-IV depleted DRG neurons that were nonstimulated (basal) and then stimulated with NGF for 30 min at DIV 11. Cell lysates were collected to assess BDNF levels (n = 11). Paired Student's t test, mean ± SEM. shControl versus shSyt-IV, t = 21.195, df = 10. F, Syt-IV protein does not regulate ARMS levels. Cultured DRG neurons infected with the corresponding lentiviruses as described in E were collected and subjected to Western blot analysis to check ARMS and Syt-IV levels. GAPDH was used as a loading control for quantification. Dotted lines connect results from the same experiment (n = 5 or 6). A representative blot of Syt-IV depletion and ARMS levels is shown. Paired Student's t test, mean ± SEM. Syt-IV levels in shControl versus shSyt-IV, t = 4.85, df = 5.
Discussion
Low endogenous BDNF levels make the study of its secretion in vivo difficult. As such, researchers have resorted to using exogenously expressed tagged-BDNF and cultured neurons. Therefore, there has been a lack of direct in vivo evidence of endogenous BDNF secretion. Here, we report that endogenous BDNF secretion can be precisely monitored using a finely tuned BDNF immunoassay that has been previously described (Kolbeck et al., 1999). Using several different methods and various mouse models, we have shown that the ARMS protein negatively affects regulated secretion of BDNF under physiological and pathological conditions and that ARMS levels are altered in HD mouse models and patients with HD as follows: (1) using lentiviruses expressing two independent shRNAs against ARMS in cultured DRG or cortical neurons to show that depletion of ARMS expression enhanced BDNF release in response to different stimuli; (2) in ARMS-depleted cortical slices from a newly generated transgenic mouse, we observed increased endogenous BDNF secretion; (3) in ARMS-depleted mice, BDNF accumulates in the striatal region; (4) in the hippocampus of two mouse models of HD, ARMS levels were increased and BDNF secretion was impaired; (5) reduced ARMS levels in the hippocampus of HD mice partially rescued BDNF release in mutant mice; (6) ARMS protein levels are significantly increased in the hippocampus and PFC from human HD postmortem samples; and (7) knocking down ARMS levels leads to a reduction in Syt-IV protein boosting BDNF-regulated secretion.
ARMS is abundant in the developing nervous system. ARMS levels are downregulated during development and by neuronal activity (Cortés et al., 2007), whereas BDNF expression peaks postnatally (Maisonpierre et al., 1990; Phillips et al., 1990) when neuronal differentiation and synaptogenesis are maximal. We have previously described that ARMS expression negatively regulates plasma membrane insertion of the GluA1 AMPAR subunit, suggesting that ARMS levels may be important to keep basal synaptic transmission in check during synaptogenesis (Arévalo et al., 2010). Our current data showing that ARMS expression affects regulated BDNF release, together with the fact that ARMS levels are controlled by neuronal activity, suggest that ARMS might also influence synaptic transmission that directly depends on BDNF. Further experiments will be required to address this.
In this study, we have observed that NGF-mediated secretion of BDNF from DRGs was not blocked by TTX, but basal BDNF secretion was reduced (Fig. 1-1E). Previously, it has been reported that BDNF release in response to different patterns of electrical field stimulations in cultured hippocampal neurons was affected by TTX (Balkowiec and Katz, 2002). A plausible explanation is that on DRGs NGF binding to TrkA activates PLCγ, leading to intracellular release of Ca2+, which triggers BDNF secretion. On the other way, the reduced basal BDNF secretion observed by TTX treatment may respond to the inhibition of TTX-regulated channels and, therefore, network activity on DRGs. Previous studies suggest that TTX chronic treatment reduces the release of a diffusible factor in DRGs (Beaudu-Lange et al., 2000), the secretion of gonadotropin-releasing hormone in cultured neurosecretory neurons (Mellon et al., 1990) and the release of glucagon-like peptide 1 in primary colonic cultures (Rogers et al., 2011) in basal conditions. Therefore, basal and regulated BDNF secretion might be achieved by different mechanisms.
We have previously demonstrated that ARMS negatively regulates NGF-mediated secretion in PC12 cells. The fact that Syt-IV was reported as a negative regulator of BDNF release (Dean et al., 2009), with similar action to ARMS (López-Benito et al., 2016), prompted us to study whether there was a relationship between ARMS and Syt-IV. First, we observed an interaction between ARMS and Syt-IV, suggesting that they may function together. The demonstration of both proteins working together came from the results in which ARMS knockdown rescued the secretion deficits observed upon Syt-IV overexpression. These results suggested that ARMS may control Syt-IV function; and indeed, we found that ARMS modulates Synaptogmin-IV levels. This result was unexpected because, to our knowledge, no effects have been reported of ARMS on the expression of any other protein. Therefore, further experiments will be required to understand how ARMS regulates Syt-IV levels.
Altered BDNF levels in neurons derived from mouse models have been directly linked to HD pathogenesis. The classical view of the HD suggests an important role for an impairment in the corticostriatal pathway (Zuccato and Cattaneo, 2014), but it has also been reported that hippocampal dysfunction is responsible for the cognitive decline observed in mouse models and human patients (Giralt et al., 2012). Interestingly, we observed increased ARMS expression in the hippocampus of symptomatic HD mouse models, which correlated with deficient BDNF release. Remarkably, ARMS levels are increased in the hippocampus and PFC of patients with HD. In addition, knockdown of the ARMS protein in the HD mice reverted the BDNF secretion to the levels of wild-type animals. It has been reported that HD mice show deficits in synaptic activity in the hippocampus, which is believed to underlie cognitive impairment in the disease (Brito et al., 2013; Plotkin et al., 2014). Interestingly, the synaptic deficits in the hippocampus of HD mice can be rescued by BDNF addition (Lynch et al., 2007; Brito et al., 2013). Furthermore, BDNF is required for the induction of LTP (Figurov et al., 1996; Korte et al., 1996; Patterson et al., 1996). Last, a previous report indicated that ARMS heterozygous mice have enhanced hippocampal LTP (Wu et al., 2010). These reports, together with our current results, suggest that LTP deficits reported in HD mice could be a consequence of impaired BDNF secretion and altered ARMS protein levels, supporting a pathological role of ARMS in HD.
Recently, several studies have revealed abnormal levels of ARMS in various diseases, including Alzheimer's disease, Parkinson's disease, and autism spectrum disorder (Scholz-Starke and Cesca, 2016). Different ARMS polymorphisms have also been observed in patients with schizophrenia (Kranz et al., 2015) and psychosis (Kranz et al., 2016). Furthermore, splice variants of ARMS have been reported in patients with spastic paraplegia, intellectual deficit, nystagmus, and obesity (Josifova et al., 2016). Interestingly, all these pathologies have been directly, or indirectly, related to BDNF changes (Adachi et al., 2014). Further research is required to elucidate the potential relationship between BDNF and ARMS in these human diseases.
Footnotes
The authors declare no competing financial interests.
This work was supported by MINECO BFU2011-22898, BFU2014-51846-R, and BFU2017-82667-R to J.C.A. and SAF2013-41177-R to J.P.B., and the EU 7th Framework Program (Marie Curie IRG and PAINCAGE) to J.C.A. S.L.-B. was supported by Consejería de Educación Junta de Castilla y León and the European Social Fund. M.E.P. and L.T. were supported by the National Institutes of Health Intramural Research Program, Center for Cancer Research, National Cancer Institute. We thank Dionisio Martín-Zanca, Pilar Pérez, Moses V. Chao, Rubén Deogracias, and members of the J.C.A. laboratory for helpful discussions; Teresa Iglesias and Giampietro Schiavo for ARMS/Kidins220 antibody (GSC16); Michel Sadelain, Philippe Soriano, Didier Trono, and Jane Sullivan for plasmids; and BT-CIEN and Biobanco Hospital Universitario Fundación Alcorcón for human samples.
The authors declare no competing financial interests.
References
- Adachi N, Numakawa T, Richards M, Nakajima S, Kunugi H (2014) New insight in expression, transport, and secretion of brain-derived neurotrophic factor: implications in brain-related diseases. World J Biol Chem 5:409–428. 10.4331/wjbc.v5.i4.409 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aicardi G, Argilli E, Cappello S, Santi S, Riccio M, Thoenen H, Canossa M (2004) Induction of long-term potentiation and depression is reflected by corresponding changes in secretion of endogenous brain-derived neurotrophic factor. Proc Natl Acad Sci U S A 101:15788–15792. 10.1073/pnas.0406960101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altar CA, Cai N, Bliven T, Juhasz M, Conner JM, Acheson AL, Lindsay RM, Wiegand SJ (1997) Anterograde transport of brain-derived neurotrophic factor and its role in the brain. Nature 389:856–860. 10.1038/39885 [DOI] [PubMed] [Google Scholar]
- Arévalo JC, Wu SH, Takahashi T, Zhang H, Yu T, Yano H, Milner TA, Tessarollo L, Ninan I, Arancio O, Chao MV (2010) The ARMS/Kidins220 scaffold protein modulates synaptic transmission. Mol Cell Neurosci 45:92–100. 10.1016/j.mcn.2010.06.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balkowiec A, Katz DM (2002) Cellular mechanisms regulating activity-dependent release of native brain-derived neurotrophic factor from hippocampal neurons. J Neurosci 22:10399–10407. 10.1523/JNEUROSCI.22-23-10399.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beaudu-Lange C, Colomar A, Israel JM, Coles JA, Amédée T (2000) Spontaneous neuronal activity in organotypic cultures of mouse dorsal root ganglion leads to upregulation of calcium channel expression on remote Schwann cells. Glia 29:281–287. 10.1002/(SICI)1098-1136(20000201)29:3%3C281::AID-GLIA9%3E3.0.CO;2-5 [DOI] [PubMed] [Google Scholar]
- Brito V, Puigdellívol M, Giralt A, del Toro D, Alberch J, Ginés S (2013) Imbalance of p75(NTR)/TrkB protein expression in Huntington's disease: implication for neuroprotective therapies. Cell Death Dis 4:e595. 10.1038/cddis.2013.116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canossa M, Griesbeck O, Berninger B, Campana G, Kolbeck R, Thoenen H (1997) Neurotrophin release by neurotrophins: implications for activity-dependent neuronal plasticity. Proc Natl Acad Sci U S A 94:13279–13286. 10.1073/pnas.94.24.13279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canossa M, Gärtner A, Campana G, Inagaki N, Thoenen H (2001) Regulated secretion of neurotrophins by metabotropic glutamate group I (mGluRI) and trk receptor activation is mediated via phospholipase C signalling pathways. EMBO J 20:1640–1650. 10.1093/emboj/20.7.1640 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cesca F, Yabe A, Spencer-Dene B, Scholz-Starke J, Medrihan L, Maden CH, Gerhardt H, Orriss IR, Baldelli P, Al-Qatari M, Koltzenburg M, Adams RH, Benfenati F, Schiavo G (2012) Kidins220/ARMS mediates the integration of the neurotrophin and VEGF pathways in the vascular and nervous systems. Cell Death Differ 19:194–208. 10.1038/cdd.2011.141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cortés RY, Arévalo JC, Magby JP, Chao MV, Plummer MR (2007) Developmental and activity-dependent regulation of ARMS/Kidins220 in cultured rat hippocampal neurons. Dev Neurobiol 67:1687–1698. 10.1002/dneu.20542 [DOI] [PubMed] [Google Scholar]
- Dean C, Liu H, Dunning FM, Chang PY, Jackson MB, Chapman ER (2009) Synaptotagmin-IV modulates synaptic function and long-term potentiation by regulating BDNF release. Nat Neurosci 12:767–776. 10.1038/nn.2315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dickins RA, McJunkin K, Hernando E, Premsrirut PK, Krizhanovsky V, Burgess DJ, Kim SY, Cordon-Cardo C, Zender L, Hannon GJ, Lowe SW (2007) Tissue-specific and reversible RNA interference in transgenic mice. Nat Genet 39:914–921. 10.1038/ng2045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dieni S, Matsumoto T, Dekkers M, Rauskolb S, Ionescu MS, Deogracias R, Gundelfinger ED, Kojima M, Nestel S, Frotscher M, Barde YA (2012) BDNF and its pro-peptide are stored in presynaptic dense core vesicles in brain neurons. J Cell Biol 196:775–788. 10.1083/jcb.201201038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrer I, Goutan E, Marín C, Rey MJ, Ribalta T (2000) Brain-derived neurotrophic factor in Huntington disease. Brain Res 866:257–261. 10.1016/S0006-8993(00)02237-X [DOI] [PubMed] [Google Scholar]
- Figurov A, Pozzo-Miller LD, Olafsson P, Wang T, Lu B (1996) Regulation of synaptic responses to high-frequency stimulation and LTP by neurotrophins in the hippocampus. Nature 381:706–709. 10.1038/381706a0 [DOI] [PubMed] [Google Scholar]
- Fukuda M, Kanno E, Ogata Y, Saegusa C, Kim T, Loh YP, Yamamoto A (2003) Nerve growth factor-dependent sorting of synaptotagmin IV protein to mature dense-core vesicles that undergo calcium-dependent exocytosis in PC12 cells. J Biol Chem 278:3220–3226. 10.1074/jbc.M208323200 [DOI] [PubMed] [Google Scholar]
- Gauthier LR, Charrin BC, Borrell-Pagès M, Dompierre JP, Rangone H, Cordelières FP, De Mey J, MacDonald ME, Lessmann V, Humbert S, Saudou F (2004) Huntingtin controls neurotrophic support and survival of neurons by enhancing BDNF vesicular transport along microtubules. Cell 118:127–138. 10.1016/j.cell.2004.06.018 [DOI] [PubMed] [Google Scholar]
- Ginés S, Bosch M, Marco S, Gavaldà N, Díaz-Hernández M, Lucas JJ, Canals JM, Alberch J (2006) Reduced expression of the TrkB receptor in Huntington's disease mouse models and in human brain. Eur J Neurosci 23:649–658. 10.1111/j.1460-9568.2006.04590.x [DOI] [PubMed] [Google Scholar]
- Giralt A, Saavedra A, Alberch J, Pérez-Navarro E (2012) Cognitive dysfunction in Huntington's disease: humans, mouse models and molecular mechanisms. J Huntingtons Dis 1:155–173. 10.3233/JHD-120023 [DOI] [PubMed] [Google Scholar]
- Griesbeck O, Canossa M, Campana G, Gärtner A, Hoener MC, Nawa H, Kolbeck R, Thoenen H (1999) Are there differences between the secretion characteristics of NGF and BDNF? Implications for the modulatory role of neurotrophins in activity-dependent neuronal plasticity. Microsc Res Tech 45:262–275. 10.1002/(SICI)1097-0029(19990515/01)45:4/5%3C262::AID-JEMT10%3E3.0.CO;2-K [DOI] [PubMed] [Google Scholar]
- Higuero AM, Sánchez-Ruiloba L, Doglio LE, Portillo F, Abad-Rodríguez J, Dotti CG, Iglesias T (2010) Kidins220/ARMS modulates the activity of microtubule-regulating proteins and controls neuronal polarity and development. J Biol Chem 285:1343–1357. 10.1074/jbc.M109.024703 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iglesias T, Cabrera-Poch N, Mitchell MP, Naven TJ, Rozengurt E, Schiavo G (2000) Identification and cloning of Kidins220, a novel neuronal substrate of protein kinase D. J Biol Chem 275:40048–40056. 10.1074/jbc.M005261200 [DOI] [PubMed] [Google Scholar]
- Josifova DJ, Monroe GR, Tessadori F, de Graaff E, van der Zwaag B, Mehta SG, Study DD, Harakalova M, Duran KJ, Savelberg SM, Nijman IJ, Jungbluth H, Hoogenraad CC, Bakkers J, Knoers NV, Firth HV, Beales PL, van Haaften G, van Haelst MM (2016) Heterozygous KIDINS220/ARMS nonsense variants cause spastic paraplegia, intellectual disability, nystagmus, and obesity. Hum Mol Genet 25:2158–2167. 10.1093/hmg/ddw082 [DOI] [PubMed] [Google Scholar]
- Kim J, Bordiuk OL, Ferrante RJ (2011) Experimental models of HD and reflection on therapeutic strategies. Int Rev Neurobiol 98:419–481. 10.1016/B978-0-12-381328-2.00016-X [DOI] [PubMed] [Google Scholar]
- Kolbeck R, Bartke I, Eberle W, Barde YA (1999) Brain-derived neurotrophic factor levels in the nervous system of wild-type and neurotrophin gene mutant mice. J Neurochem 72:1930–1938. 10.1046/j.1471-4159.1999.0721930.x [DOI] [PubMed] [Google Scholar]
- Kong H, Boulter J, Weber JL, Lai C, Chao MV (2001) An evolutionarily conserved transmembrane protein that is a novel downstream target of neurotrophin and ephrin receptors. J Neurosci 21:176–185. 10.1523/JNEUROSCI.21-01-00176.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korte M, Griesbeck O, Gravel C, Carroll P, Staiger V, Thoenen H, Bonhoeffer T (1996) Virus-mediated gene transfer into hippocampal CA1 region restores long-term potentiation in brain-derived neurotrophic factor mutant mice. Proc Natl Acad Sci U S A 93:12547–12552. 10.1073/pnas.93.22.12547 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kranz TM, Goetz RR, Walsh-Messinger J, Goetz D, Antonius D, Dolgalev I, Heguy A, Seandel M, Malaspina D, Chao MV (2015) Rare variants in the neurotrophin signaling pathway implicated in schizophrenia risk. Schizophr Res 168:421–428. 10.1016/j.schres.2015.07.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kranz TM, Berns A, Shields J, Rothman K, Walsh-Messinger J, Goetz RR, Chao MV, Malaspina D (2016) Phenotypically distinct subtypes of psychosis accompany novel or rare variants in four different signaling genes. EBioMedicine 6:206–214. 10.1016/j.ebiom.2016.03.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krüttgen A, Möller JC, Heymach JV Jr, Shooter EM (1998) Neurotrophins induce release of neurotrophins by the regulated secretory pathway. Proc Natl Acad Sci U S A 95:9614–9619. 10.1073/pnas.95.16.9614 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lessmann V. (1998) Neurotrophin-dependent modulation of glutamatergic synaptic transmission in the mammalian CNS. Gen Pharmacol 31:667–674. 10.1016/S0306-3623(98)00190-6 [DOI] [PubMed] [Google Scholar]
- Li J, Chen LA, Townsend CM Jr, Evers BM (2008) PKD1, PKD2, and their substrate Kidins220 regulate neurotensin secretion in the BON human endocrine cell line. J Biol Chem 283:2614–2621. 10.1074/jbc.M707513200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- López-Benito S, Lillo C, Hernández-Hernández A, Chao MV, Arévalo JC (2016) ARMS/Kidins220 and synembryn-B levels regulate NGF-mediated secretion. J Cell Sci 129:1866–1877. 10.1242/jcs.184168 [DOI] [PubMed] [Google Scholar]
- Lu B, Nagappan G, Guan X, Nathan PJ, Wren P (2013) BDNF-based synaptic repair as a disease-modifying strategy for neurodegenerative diseases. Nat Rev Neurosci 14:401–416. 10.1038/nrn3505 [DOI] [PubMed] [Google Scholar]
- Lynch G, Kramar EA, Rex CS, Jia Y, Chappas D, Gall CM, Simmons DA (2007) Brain-derived neurotrophic factor restores synaptic plasticity in a knock-in mouse model of Huntington's disease. J Neurosci 27:4424–4434. 10.1523/JNEUROSCI.5113-06.2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maisonpierre PC, Belluscio L, Friedman B, Alderson RF, Wiegand SJ, Furth ME, Lindsay RM, Yancopoulos GD (1990) NT-3, BDNF, and NGF in the developing rat nervous system: parallel as well as reciprocal patterns of expression. Neuron 5:501–509. 10.1016/0896-6273(90)90089-X [DOI] [PubMed] [Google Scholar]
- Mangiarini L, Sathasivam K, Seller M, Cozens B, Harper A, Hetherington C, Lawton M, Trottier Y, Lehrach H, Davies SW, Bates GP (1996) Exon 1 of the HD gene with an expanded CAG repeat is sufficient to cause a progressive neurological phenotype in transgenic mice. Cell 87:493–506. 10.1016/S0092-8674(00)81369-0 [DOI] [PubMed] [Google Scholar]
- Mellon PL, Windle JJ, Goldsmith PC, Padula CA, Roberts JL, Weiner RI (1990) Immortalization of hypothalamic GnRH neurons by genetically targeted tumorigenesis. Neuron 5:1–10. 10.1016/0896-6273(90)90028-E [DOI] [PubMed] [Google Scholar]
- Mori Y, Higuchi M, Hirabayashi Y, Fukuda M, Gotoh Y (2008) JNK phosphorylates synaptotagmin-4 and enhances Ca2+-evoked release. EMBO J 27:76–87. 10.1038/sj.emboj.7601935 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neeper SA, Gómez-Pinilla F, Choi J, Cotman CW (1996) Physical activity increases mRNA for brain-derived neurotrophic factor and nerve growth factor in rat brain. Brain Res 726:49–56. 10.1016/0006-8993(96)00273-9 [DOI] [PubMed] [Google Scholar]
- Neubrand VE, Cesca F, Benfenati F, Schiavo G (2012) Kidins220/ARMS as a functional mediator of multiple receptor signalling pathways. J Cell Sci 125:1845–1854. 10.1242/jcs.102764 [DOI] [PubMed] [Google Scholar]
- Papapetrou EP, Lee G, Malani N, Setty M, Riviere I, Tirunagari LM, Kadota K, Roth SL, Giardina P, Viale A, Leslie C, Bushman FD, Studer L, Sadelain M (2011) Genomic safe harbors permit high beta-globin transgene expression in thalassemia induced pluripotent stem cells. Nat Biotechnol 29:73–78. 10.1038/nbt.1717 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park H, Poo MM (2013) Neurotrophin regulation of neural circuit development and function. Nat Rev Neurosci 14:7–23. 10.1038/nrn3379 [DOI] [PubMed] [Google Scholar]
- Patterson SL, Abel T, Deuel TA, Martin KC, Rose JC, Kandel ER (1996) Recombinant BDNF rescues deficits in basal synaptic transmission and hippocampal LTP in BDNF knockout mice. Neuron 16:1137–1145. 10.1016/S0896-6273(00)80140-3 [DOI] [PubMed] [Google Scholar]
- Phillips HS, Hains JM, Laramee GR, Rosenthal A, Winslow JW (1990) Widespread expression of BDNF but not NT3 by target areas of basal forebrain cholinergic neurons. Science 250:290–294. 10.1126/science.1688328 [DOI] [PubMed] [Google Scholar]
- Plotkin JL, Day M, Peterson JD, Xie Z, Kress GJ, Rafalovich I, Kondapalli J, Gertler TS, Flajolet M, Greengard P, Stavarache M, Kaplitt MG, Rosinski J, Chan CS, Surmeier DJ (2014) Impaired TrkB receptor signaling underlies corticostriatal dysfunction in Huntington's disease. Neuron 83:178–188. 10.1016/j.neuron.2014.05.032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogers GJ, Tolhurst G, Ramzan A, Habib AM, Parker HE, Gribble FM, Reimann F (2011) Electrical activity-triggered glucagon-like peptide-1 secretion from primary murine L-cells. J Physiol 589:1081–1093. 10.1113/jphysiol.2010.198069 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sadakata T, Mizoguchi A, Sato Y, Katoh-Semba R, Fukuda M, Mikoshiba K, Furuichi T (2004) The secretory granule-associated protein CAPS2 regulates neurotrophin release and cell survival. J Neurosci 24:43–52. 10.1523/JNEUROSCI.2528-03.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scholz-Starke J, Cesca F (2016) Stepping out of the shade: control of neuronal activity by the scaffold protein Kidins220/ARMS. Front Cell Neurosci 10:68. 10.3389/fncel.2016.00068 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimojo M, Courchet J, Pieraut S, Torabi-Rander N, Sando R 3rd, Polleux F, Maximov A (2015) SNAREs controlling vesicular release of BDNF and development of callosal axons. Cell Rep 11:1054–1066. 10.1016/j.celrep.2015.04.032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sleiman SF, Henry J, Al-Haddad R, El Hayek L, Abou Haidar E, Stringer T, Ulja D, Karuppagounder SS, Holson EB, Ratan RR, Ninan I, Chao MV (2016) Exercise promotes the expression of brain derived neurotrophic factor (BDNF) through the action of the ketone body beta-hydroxybutyrate. eLife 5:e15092. 10.7554/eLife.15092 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soriano P. (1999) Generalized lacZ expression with the ROSA26 cre reporter strain. Nat Genet 21:70–71. 10.1038/5007 [DOI] [PubMed] [Google Scholar]
- Stoppini L, Buchs PA, Muller D (1991) A simple method for organotypic cultures of nervous tissue. J Neurosci Methods 37:173–182. 10.1016/0165-0270(91)90128-M [DOI] [PubMed] [Google Scholar]
- Tessarollo L. (2001) Manipulating mouse embryonic stem cells. Methods Mol Biol 158:47–63. 10.1385/1-59259-220-1:47 [DOI] [PubMed] [Google Scholar]
- Ting JT, Kelley BG, Sullivan JM (2006) Synaptotagmin IV does not alter excitatory fast synaptic transmission or fusion pore kinetics in mammalian CNS neurons. J Neurosci 26:372–380. 10.1523/JNEUROSCI.3997-05.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsien JZ, Chen DF, Gerber D, Tom C, Mercer EH, Anderson DJ, Mayford M, Kandel ER, Tonegawa S (1996) Subregion- and cell type-restricted gene knockout in mouse brain. Cell 87:1317–1326. 10.1016/S0092-8674(00)81826-7 [DOI] [PubMed] [Google Scholar]
- Vinet J, Weering HR, Heinrich A, Kälin RE, Wegner A, Brouwer N, Heppner FL, Rooijen N, Boddeke HW, Biber K (2012) Neuroprotective function for ramified microglia in hippocampal excitotoxicity. J Neuroinflammation 9:27. 10.1186/1742-2094-9-27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wheeler VC, Auerbach W, White JK, Srinidhi J, Auerbach A, Ryan A, Duyao MP, Vrbanac V, Weaver M, Gusella JF, Joyner AL, MacDonald ME (1999) Length-dependent gametic CAG repeat instability in the Huntington's disease knock-in mouse. Hum Mol Genet 8:115–122. 10.1093/hmg/8.1.115 [DOI] [PubMed] [Google Scholar]
- Wiznerowicz M, Trono D (2003) Conditional suppression of cellular genes: lentivirus vector-mediated drug-inducible RNA interference. J Virol 77:8957–8961. 10.1128/JVI.77.16.8957-8951.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong YH, Lee CM, Xie W, Cui B, Poo MM (2015) Activity-dependent BDNF release via endocytic pathways is regulated by synaptotagmin-6 and complexin. Proc Natl Acad Sci U S A 112:E4475–E4484. 10.1073/pnas.1511830112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu SH, Arévalo JC, Sarti F, Tessarollo L, Gan WB, Chao MV (2009) Ankyrin repeat-rich membrane Spanning/Kidins220 protein regulates dendritic branching and spine stability in vivo. Dev Neurobiol 69:547–557. 10.1002/dneu.20723 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu SH, Arévalo JC, Neubrand VE, Zhang H, Arancio O, Chao MV (2010) The ankyrin repeat-rich membrane spanning (ARMS)/Kidins220 scaffold protein is regulated by activity-dependent calpain proteolysis and modulates synaptic plasticity. J Biol Chem 285:40472–40478. 10.1074/jbc.M110.171371 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu T, Calvo L, Anta B, López-Benito S, Southon E, Chao MV, Tessarollo L, Arévalo JC (2011) Regulation of trafficking of activated TrkA is critical for NGF-mediated functions. Traffic 12:521–534. 10.1111/j.1600-0854.2010.01156.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuccato C, Cattaneo E (2014) Huntington's disease. Handb Exp Pharmacol 220:357–409. 10.1007/978-3-642-45106-5_14 [DOI] [PubMed] [Google Scholar]
- Zuccato C, Ciammola A, Rigamonti D, Leavitt BR, Goffredo D, Conti L, MacDonald ME, Friedlander RM, Silani V, Hayden MR, Timmusk T, Sipione S, Cattaneo E (2001) Loss of huntingtin-mediated BDNF gene transcription in Huntington's disease. Science 293:493–498. 10.1126/science.1059581 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
ARMS downregulation by shRNAs and sensitivity of the BDNF immunoassay. A) Quantification of ARMS depletion in cultured DRG neurons corresponding to the experiments performed in Fig. 1A,B. (n = 4). Paired Student's t-test, mean ± s.e.m. shC vs shARMS-1 t =8.078, df = 3; shC vs shARMS-2 t = 7.03, df = 3. B) Quantification of ARMS depletion in cultured cortical neurons corresponding to the experiments performed in Fig. 1 C,D. (n = 4). Paired Student's t-test, mean ± s.e.m. shC vs shARMS-1 t = 44.19, df = 3; shC vs shARMS-2 t = 111.8, df = 3. C) Different amounts of recombinant BDNF (1-1024 pg/well) were incubated on ELISA plates coated with 1 pg/well of mAb#1. 12.5 ng of mAb#9 HRP were incubated for 3 hours at 30 °C, washed with TBST and developed with SuperSignal ELISA Femto Substrate diluted 50% in water. A representative standard curve is shown. D) Adjusted standard curve of BDNF ELISA using recombinant BON F. E) Neuronal activity is required for BDNF release in response to NGF. BDNF secretion experiments were performed in DRGs in a similar way as described in Fig. 1 B in presence (striped bars) and absence (solid bars) of TTX with or without NGF. (n = 7). Paired Student's t-test, mean± s.e.m. Control vs TTX t = 9.157, df = 6; Controi-NGF vs TTX-NGF t = 4.466, df = 6. Download Figure 1-1, PDF file (304.7KB, pdf)
Generation, genotyping, and functionality of shARMS mice. A) Schematics of the wild-type ROSA26 allele, the targeting vector and the targeted allele before and after Flp recombination. DTA: diphtheria toxin; eGFP: enhanced GFP; H1: promoter H1; PGK neo: neomycin resistance gene; PROM: promoter ROSA26. B) Schematics of the wild-type ROSA26 allele and the targeted allele before and after Flp recombination with the restriction sites EcoRV and Xbal used to digest genomic DNA and to clone ARMS shRNA cassette, respectively. The size of the fragments generated after EcoRV digestion are shown (blue). For genotyping, primers are denoted with red arrowheads and the fragment sizes amplified after PCR depending on the genotype are: 194 base pairs for WT mice, 271 base pairs and 253 base pairs for shARMS mice before and after Cre recombinase, respectively (red). C) Southern blot analysis of genomic DNA from WT and targeted mice before and after Flip-mediated recombination, upon digestion with EcoRV. As indicated in the schematics in panel B, the 5' external probe detects an 11 kb DNA fragment from WT alleles and a 14.3 kb and 12.5 kb DNA fragment from mutant alleles with or without the Neo cassette, respectively. A representative blot is shown. D) PCR analysis of genomic tail DNA from WT, shARMS, shARMS;shARMS cultured cortical neurons infected with ere-expressing lentiviruses. As indicated in the schematics in B, the fragments amplified were of 194 bp and 271 bp for WT and shARMS mice, respectively, and 253 bp for shARMS mice in presence of Cre recombinase. A representative PCR is shown. E) Efficient ARMS down regulation in cortical neurons from shARMS mouse in the presence of Cre recombinase. Cortical neurons were obtained from E16.5 mouse embryos with the corresponding genotype, and infected or not with lentiviruses expressing Cre recombinase. Western blot analyses were performed to detect ARMS, Cre recombinase and tubulin as a loading control. F) PCR genotyping of E13.5 embryos from shARMS matings. Genomic DNA was obtained from tails and PCR reaction was performed. Samples 2, 5, 7, and 8 are WT; 3 and 4 have one copy, and 1 and 6 two copies of the cassette. A representative PCR is shown. G) ARMS expression is unchanged in shARMS mice. Western blot analyses from lysates of cultured DRG neurons corresponding to E13.5 embryos of panel F. Tubulin was used as loading control. H) PCR analysis of cultured DRGs from E13.5 embryos corresponding to panel F infected with lentivirus expressing Cre recombinase. DRG neurons were cultured and infected with lentiviruses expressing Cre recombinase. Afterwards, genomic DNA was obtained and PCR reaction was carried out. Note the presence of the 253 bp fragment in those samples with the shARMS cassette (samples# 3, 1, 4, and 6). A representative PCR is shown. I) Knockdown of ARMS expression using cultured DRG neurons from shARMS embryos expressing Cre recombinase corresponding to panel H (samples# 3, 1, 4 and 6). Samples were collected and Western blot analyses were performed to assess ARMS expression. Tubulin was used as a loading control. Download Figure 2-1, PDF file (304.7KB, pdf)
A) Quantification of bdnf mRNA levels in mice subjected to physical exercise. Total RNA was obtained, eDNA was synthesized and quantitative PCR was performed as described in Material and Methods. The relative transcription level of bdnf was normalized to that of sdha using the 2-LlLlCt method. Data are presented as means ± SEM. Triplicate determinations from 3-4 control and mutant mice subjected or not to exercise were quantified. P value was calculated using a two-tailed Student's t-test. B) Similar ARMS expression in forebrain of shARMS and shARMS;CamK2a-Cre mice at 4 weeks of age. Immunohistochemistry with ARMS antibodies of control (shARMS) (left hemisphere) and mutant mice (shARMS;CamK2a-Cre) (right hemisphere). A representative staining is shown (n = 2). Download Figure 3-1, PDF file (304.7KB, pdf)
ARMS levels do not change in the cortex of HD mouse models. A) ARMS expression in the cortex of 8 month-old Kl mice is not altered. Western blot for ARMS in the cortex from WT and Kl mice. Actin was used as a loading control (mean± SEM; n = 7). A representative Western blot is shown. B) ARMS expression in the cortex of 20-30 week-old R6/1 mice is not altered. Western blot for ARMS in the cortex from WT and R6/1 mice. Actin was used as a loading control (mean ± SEM; n = 6 and 6; for WT and R6/1, respectively). A representative Western blot is shown. C) Depletion of ARMS in organotypic hippocampal slices from WT and Kl mice. Quantification of ARMS protein levels upon infection with lentivirus expressing shControl (C) or shARMS-1 (A) (n = 5 and 7 for WT and Kl mice, respectively). Unpaired Student's t-test, mean ± s.e.m. Representative Western blot showing ARMS levels in hippocampal slices of WT and Kl mice. D) Depletion of ARMS in organotypic hippocampal slices from WT and R6/1 mice. Quantification of ARMS protein levels upon infection with lentivirus expressing shControl (C) or shARMS-1 (A) (n = 4). Unpaired Student's t-test, mean ± s.e.m. Representative Western blot showing ARMS levels in hippocampal slices of WT and R6/1 mice. Download Figure 4-1, PDF file (304.7KB, pdf)
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.