

Abstract
β-Amyloid (Aβ) aggregation is causally linked to neuronal pathology in Alzheimer’s disease; therefore, several small molecules, antibodies, and peptides have been tested as anti-Aβ agents. We developed two compounds based on the Aβ-binding domain of transthyretin (TTR): a cyclic peptide cG8 and an engineered protein mTTR, and compared them for therapeutically relevant properties. Both mTTR and cG8 inhibit fibrillogenesis of Aβ, with mTTR inhibiting at a lower concentration than cG8. Both inhibit aggregation of amylin but not of α-synuclein. They both bind more Aβ aggregates than monomer, and neither disaggregates preformed fibrils. cG8 retained more of its activity in the presence of biological materials and was more resistant to proteolysis than mTTR. We examined the effect of mTTR or cG8 on Aβ binding to human neurons. When mTTR was co-incubated with Aβ under oligomer-forming conditions, Aβ morphology was drastically changed and Aβ-cell deposition significantly decreased. In contrast, cG8 did not affect morphology but decreased the amount of Aβ deposited. These results provide guidance for further evolution of TTR-mimetic anti-amyloid agents.
Keywords: aggregation, β-amyloid peptides, cyclic peptides, pluripotent stem cells, transthyretin
Introduction
Alzheimer’s disease (AD) is the most common form of dementia, affecting approximately 5.4 million people in the US alone.[1] With this number expected to triple by 2050,[1] there is a desperate need for AD therapeutics that target the disease pathology; unfortunately, currently available FDA-approved drugs provide only symptomatic relief. β-Amyloid (Aβ) is a key component in AD pathology and therefore has been a main therapeutic target. Aβ is the proteolytic cleavage product of the amyloid precursor protein APP. Variable cleavage releases several different Aβ isoforms, of which the most common are Aβ1–40 and Aβ1–42. Although monomeric Aβ is natively unfolded and nontoxic, the peptide spontaneously self-associates into soluble oligomers and insoluble aggregates with high β-sheet content and fibrillar morphology. The soluble oligomers are believed by many researchers to be the most neurotoxic species, while the insoluble fibrils eventually deposit as the amyloid plaques that are one of the defining features of AD brain pathology. Aβ is one of many amyloidogenic proteins associated with degenerative diseases; other well-known examples including amylin (islet amyloid polypeptide or IAPP) in type II diabetes and α-synuclein in Parkinson’s disease.[2]
Because Aβ aggregation is intrinsically linked to Aβ toxicity, numerous small molecules, such as the polyphenols epigallocatechnin-3-gallate and curcumin, 3-amino-1-propanesulfonic acid and scyllo-inositol, have been developed, with the goal of binding Aβ, inhibiting Aβ aggregation, and decreasing toxicity.[3] However, most of these Aβ aggregation inhibitors have modest affinity and require stoichiometric or greater quantities; furthermore, selectivity and specificity are often unreported, and none have proved to be clinically effective.[3, 4]
Aβ–Aβ interactions leading to aggregate formation are stabilized by a large contact area between adjacent Aβ monomers. Inhibition of protein–protein interactions with small molecules is challenging because the protein–protein surface area is typically large, relatively featureless, and relatively flexible.[5] Thus, small-molecule approaches to inhibiting Aβ aggregation may suffer from poor selectivity and unwanted side effects.[6] Monoclonal antibodies offer higher affinity and excellent specificity ; several anti-Aβ antibodies (such as bapineuzumab and solanezumab) have been developed that showed promise in animal studies but have been largely unsuccessful in clinical trials.[3e, 7] Antibodies that specifically target Aβ oligomers and aggregates, rather than monomers, may be an essential feature for clinical success.[7d] Drawbacks of monoclonal (or other protein therapeutics) include poor oral bioavailability, high cost, and susceptibility to proteolysis.[6] A third approach is to generate peptides and peptidomimetics that bind to Aβ. Peptides and their derivatives occupy a space midway between small-molecule and protein therapeutics. Their larger size affords improved selectivity and affinity over small-molecule drugs, while chemical synthesis provides opportunities to incorporate features that enhance bioavailability and stability while lowering cost of manufacture relative to protein therapeutics.[5, 6]
A common strategy for targeting Aβ is to incorporate a self-recognition domain derived from an Aβ sequence.[3a,d,f–h, 5, 8] Alternatively, Aβ-binding peptides have been identified by screening phage-display libraries.[3f, 9] Our strategy for designing anti-Aβ agents is distinct from these other approaches (Figure 1). We start with evidence, from animal studies, that transthyretin (TTR) overexpression protects against Aβ deposition and toxicity.[10] TTR is a homotetrameric protein that functions as a carrier for thyroxine and forms a complex with retinol binding protein (RBP) for the transport of retinol. TTR has been shown to decrease Aβ toxicity in neuronal cell cultures and in transgenic animal models[11] and is co-localized with amyloid deposits in human AD brain.[11c] This motivated our group and others to investigate TTR-Aβ interactions, leading to the discovery that the main Aβ-binding site of TTR resides in the “inner” β-sheet, including specific residues in strands G and H.[10a,b, 11b,d, 12] Relying on these discoveries, we synthesized peptides that mimicked the Aβ-binding site of TTR. Our first iteration, a linear peptide, was moderately successful at inhibiting Aβ fibrillogenesis and decreasing Aβ toxicity.[13] To increase peptide stability, we then created a cyclic peptide which showed greater inhibitory capability.[14] Further evolution produced cG8, which increased efficacy by increasing the adoption of an antiparallel β-strand structure and decreasing the tendency to self-associate.[15]
Figure 1.
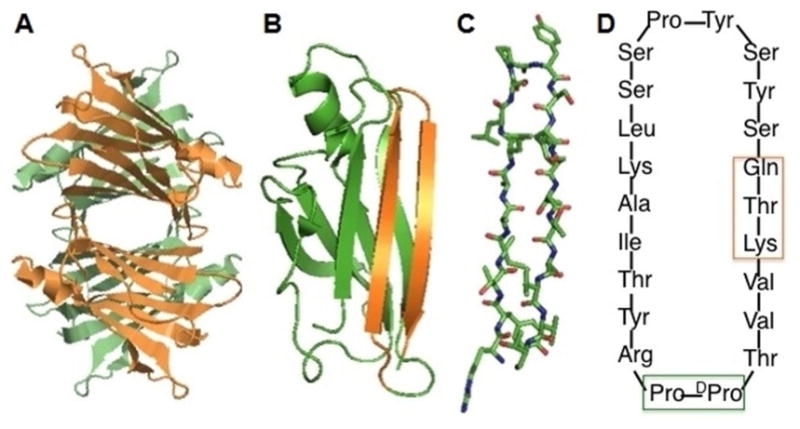
Transthyretin structure and TTR-derived mTTR and cG8. (A) Ribbon structure of TTR tetramer. Created from PDB ID: 1DVG in the RCSB Protein Data Bank. (B) TTR monomer, with strands G and H highlighted in orange. (C) Stick representation of residues in strands G and H. Sequence shown is from wild-type human TTR: RYTIAALLSPYSYSTTAVVT. (D) Sequence of cG8. N-to C-linkage is clockwise. Green box shows template to enforce β-turn to link the N-terminus of strand G to C-terminus of strand H. Orange box shows substitutions to allow cyclization. The linear peptide is synthesized with the trifunctional glutamate attached to resin, which converts into glutamine upon cyclization and release from resin.
As an alternative to synthetic peptides, we also tested an engineered TTR mutant, F87M/L110M (mTTR). Unlike its wild-type tetrameric parent, mTTR is stable as a monomer and does not aggregate.[11d, 16] Our previous studies have shown that, relative to wild-type (tetrameric) TTR, mTTR is more effective at binding to Aβ and decreasing Aβ toxicity, probably because of greater access of Aβ to its putative binding site.[10b, 11d, 12b, 13] mTTR is also unable to bind thyroxine and RBP, so it does not interfere with the normal functions of wild-type TTR. Furthermore binding of RBP to wild-type TTR decreases its ability to inhibit Aβ aggregation (manuscript in preparation).[12a] mTTR does not suffer from that concern.
In the study presented here, cyclic peptide cG8 and engineered protein mTTR were directly compared for their potential as anti-Aβ agents. Specifically, cG8 and mTTR were compared for 1) efficacy at inhibiting Aβ fibrillogenesis at low dose, 2) activity against other amyloidogenic proteins, 3) relative binding of Aβ monomers, oligomers or fibrils, 4) ability to disaggregate preformed Aβ fibrils, 5) interference from other proteins and lipids, and 6) stability against acidic conditions or proteolytic cleavage. Additionally, the association of Aβ to induced pluripotent stem cell (iPSC)-derived neurons as a function of Aβ aggregation state was imaged by confocal fluorescence microscopy, and the impact of cG8 or mTTR on cell association was evaluated. Combined, these findings highlight the relative advantages and disadvantages of using a protein versus peptide as a therapeutic anti-Aβ agent and provide guidance for future development of a therapeutic that combines the advantages of both mTTR and cG8.
Results
cG8 and mTTR are effective inhibitors of Aβ1–40 and Aβ1–42 fibril formation
Aβ fibril formation was measured using a thioflavin T (ThT) assay, a dye that fluoresces in proportion to the mass of amyloid fibril. Freshly prepared Aβ was ThT-negative, as expected. The fluorescence signal increased at 24h and again at 48 h after preparation, indicating aggregation of Aβ into ThT-positive fibrils (Figure 2, white bars). Inhibition of Aβ aggregation by TTR, cG8 or mTTR was compared at varying Aβ/inhibitor molar ratios of 20:1, 50:1, and 75:1. After 24 or 48 h co-incubation, TTR decreased the fibril mass compared with Aβ alone, even at 75-fold excess Aβ (Figure 2A). cG8 was also able to decrease Aβ aggregation at ratios of 20:1 (24 h and 48 h) and 50:1 (48 h, Figure 2B), and was overall similarly effective as TTR. mTTR not only proved effective at decreasing Aβ aggregation at all molar ratios (Figure 2C) but outperformed TTR at 20:1 and 50:1 ratios (p<0.05). The enhanced performance of mTTR vis-/-vis TTR is consistent with previously obtained results, and can be attributed to lowered steric hindrance of Aβ to its binding site.[11d]
Figure 2.

cG8 and mTTR retain TTR,s ability to inhibit Aβ aggregation. Aβ monomer was combined with either TTR (A), cG8 (B), or mTTR (C) and aggregated at 37°C for 48 h. All samples contained 28 μM Aβ with inhibitor/Aβ molar ratio of 1:20 (black), 1:50 (dark grey), or 1:75 (light grey) and are compared to Aβ alone (white). Aggregation was monitored via ThT fluorescence and data was blank (ThT in PBS) subtracted then normalized to the fluorescence of the control at 48 h. Error bars indicate SEM, n=3–4 for samples, n=8 for control. *p<0.05 vs. control.
To ensure aggregation inhibition was not Aβ isoform-dependent, ThT aggregation assays were repeated with Aβ1–42 at Aβ1–42/inhibitor molar ratios of 10:1 and 20:1. Results were similar to that of Aβ1–40: both cG8 and mTTR successfully decreased Aβ1–42 fibril formation (Figure 3A).
Figure 3.

cG8 and mTTR inhibit the aggregation of Aβ1–42 and amylin but not α-synuclein. Aβ1–42 (A), amylin (B), and α-synuclein (C), all at 20 μM final concentration, were combined with cG8 (horizontal lines) or mTTR (vertical lines) at inhibitor/amyloid protein ratios of 1:10 (white bars, black lines), or 1:20 (black bars, white lines) and compared to control (white bars, amyloid protein with no added inhibitor). Aggregation was monitored via ThT fluorescence and data was blank (ThT in PBS) subtracted then normalized to the respective control at 48 h. Error bars indicate SEM, n=2–4. *p<0.05 vs. control.
cG8 and mTTR inhibit some but not all amyloidogenic proteins
A cross-β conformation, a fibrillar morphology, and binding of ThT are features that Aβ shares with other amyloidogenic proteins. Several anti-Aβ agents have been reported to interact with multiple amyloidogenic proteins,[23] likely due to the structural properties shared by amyloid aggregates.[23f, 24] For example, EGCG inhibits fibrillogenesis not only for Aβ, but also for amylin, α-synuclein, huntingtin, and prion.[25] Antibodies such as A11 show broad cross-reactivity with oligomeric amyloid-related intermediates.[23f, 24] While having a therapeutic capable of targeting multiple amyloidogenic proteins has its benefits, it also brings into question the specificity of these compounds. We reasoned that, if cG8 and/or mTTR were recognizing an amyloid-related structural feature on Aβ, they may also recognize other amyloidogenic proteins and inhibit their aggregation. ThT fluorescence intensity was used to determine if cG8 or mTTR could inhibit the aggregation of amylin or α-synuclein. Both cG8 and mTTR significantly decreased amylin aggregation even at 20-fold excess amylin (Figure 3B) but neither had any impact on α-synuclein aggregation (Figure 3C). Thus, cG8 and mTTR are not broad-spectrum anti-amyloid agents. This result suggests that cG8 and mTTR recognize similar epitopes that Aβ and amylin share but that α-synuclein does not possess.
cG8 and mTTR bind more oligomeric and fibrillar than monomeric Aβ but do not disaggregate preformed fibrils
cG8b or mTTRb were immobilized on NeutraAvidin-coated plates, and binding of Aβ monomer (fresh), oligomers (RT, 24 h), and fibrils (37 °C, 24 h) was determined by ELISA, with detection by the anti-Aβ antibody 6E10. A direct quantitative comparison between cG8b and mTTRb should not be made due to potential differences in the amount of peptide versus protein immobilized in each well. However, both cG8b and mTTRb bind substantially more Aβ in oligomer-rich and fibril-rich environments than when there is only Aβ monomer (Figure 4A,B).
Figure 4.

cG8 and mTTR bind to multiple Aβ species but do not disaggregate Aβ fibrils. (A–B) NeutrAvidin plates were coated with 5 μgmL−1 of either cG8b (A) or mTTRb (B) and the relative binding of Aβ monomer (fresh, ○), oligomer (24 h, RT, □) or fibril (24 h, 37°C, △) was measured. Data was blank (PBS, 0 mgmL−1 Aβ) subtracted. Error bars indicate SEM, n=3. *p<0.05 vs. oligomer. #p<0.05 vs. fibril. (C–D) Preformed Aβ fibrils were combined with cG8 (C) or mTTR (D). Final samples contained 28 μM Aβ alone (white bars), or at Aβ/inhibitor molar ratios of 2:1 (grey bars), or 1:1 (black bars). Disaggregation was monitored via ThT fluorescence and data was normalized to the fluorescence of the control at 0 h. Error bars indicate SEM, n=3 for samples, n=6 for control.
Because cG8 and mTTR bound to both Aβ oligomers and fibrils, we asked whether either could disaggregate preformed fibrils. cG8 and mTTR were incubated with preformed Aβ fibrils at 20:1, 10:1, 2:1 and 1:1 Aβ/inhibitor molar ratio for 48 h, after which ThT fluorescence was measured. No significant decrease in ThT fluorescence was observed (Figure 4C,D, data not shown). Thus, despite the fact that coincubation at Aβ/inhibitor ratios as low as 50:1 or 75:1 inhibited formation of Aβ fibrils, even at equimolar concentrations, cG8 and mTTR do not disaggregate fibrils. This feature has also been reported for TTR[26] and is advantageous as it avoids the possibility that fibrils might be inadvertently disaggregated into the more toxic oligomeric form.
cG8 retains Aβ binding in the presence of other biological macromolecules better than mTTR
Anti-amyloid agents will need to target Aβ within a biological environment containing proteins, lipids, carbohydrates and other macromolecules. Therefore, we next checked whether cG8 and mTTR retain Aβ binding in the presence of proteins and lipids. cG8b and mTTRb were immobilized on NeutrAvidin-coated plates. Mouse brain tissue lysate or lysate from human iPSC-derived neurons was spiked with oligomeric Aβ and then added to each well. Final samples contained 5 μgmL−1 Aβ and 0, 1, 5, 15, or 30 mgdL−1 lysate protein. Binding of Aβ decreased in the presence of both tissue and cell lysate. However, Aβ binding to cG8b was significantly less affected than binding to mTTRb (Figure 5A). At 30 mgdL−1 tissue lysate, Aβ binding to cG8b was 52±6% of control (no lysate) while binding to mTTR was only 24±5% of control.
Figure 5.

cG8 and mTTR bind Aβ aggregates in the presence of biological materials. NeutrAvidin plates were coated with 5 μgmL−1 of either cG8b (○) or mTTRb (□) and the relative binding of 5 μgmL−1 of Aβ in the presence of wild-type mouse cortex tissue lysate (A), human neurons (A, closed symbols, n=1), lipids (B), or FBS (C) was measured. Data normalized to binding of Aβ in buffer alone. Error bars indicate SEM, n=3. *p<0.05 vs. mTTRb.
To isolate whether lipids or proteins were responsible for the decrease in Aβ binding, porcine brain total lipid extract or FBS was spiked with Aβ and then added to immobilized cG8b and mTTRb. At typical CSF concentrations (2–4 mgdL−1),[27] lipids had no effect on Aβ binding; however, at high lipid concentrations (600 and 1800 mgdL−1) binding of Aβ to both cG8b and mTTRb was decreased (Figure 5B). As with tissue lysate, lipids affected Aβ binding to cG8b significantly less than binding to mTTRb. Proteins proved to have a greater effect on Aβ binding than lipids, decreasing binding at concentrations typical of CSF (15 and 30 mgdL−1) (Figure 5C). At high levels of FBS (5000 and 15000 mgdL−1), Aβ binding to cG8b reached a plateau corresponding to approximately 60% of control, while Aβ-mTTRb binding was virtually eliminated. Combined, these results demonstrate that most of the loss in Aβ binding is due to interference from protein components in cell homogenates, and that cG8 is less susceptible than mTTR to interference.
cG8 and mTTR differ in stability against protease
cG8 and mTTR were incubated in SGF (pH 1.2) or SIF (pH 6.8) without enzymes for 24h at 37°C. Neither cG8 nor mTTR showed any indication of degradation after exposure to either buffer (mass spectrometry data not shown), nor was there any loss in their ability to inhibit fibril formation (Figure 6A,B). Pepsin was used to evaluate the resistance of cG8 and mTTR to enzyme degradation. mTTR degraded so quickly that even the initial sample contained protein fragments instead of intact protein, and no fragments of 5 kDa or larger were detected after 2 h (Figure 6D). cG8 was intact at t=0 and was only partially degraded after 2 h incubation with pepsin at 37°C (Figure 6C). Besides intact cG8, two fragments were detected of m/z=1244.8 and 1345.8. We used Peptide Cutter (https://web.expasy.org/peptide_cutter/) to predict that pepsin would cleave cG8 between Lys/Leu or between Leu/Ser (between residues 8/9 or 9/10 if DPro is residue 1). Cleavage at Leu/Ser and secondary cleavage at Val/Val or Val/Thr (residues 20/21 or 21/22, SSPYSYSQTKV or SSPYSYSQTKVV) would produce fragments of 1246.3 or 1345.5 Da. The latter cleavage site is not a preferred site for pepsin but could indicate unusual strain induced by the DPro–Pro template. Other possible fragments include VVTdPPRYTIAK (1244.5) and VTdPPRYTIAKLS (1345.6), again suggesting an unusual cleavage site. MS/MS would be required to positively identify fragmentation sites.
Figure 6.

Activity of cG8 and mTTR in biological fluids and stability from enzyme degradation. A–B) cG8 (grey bars) and mTTR (black bars) were incubated in either SGF (A) or SIF (B) for 24 h at 37°C and then combined with Aβ monomer. Final samples contained 28 μM Aβ alone (control, white bars) or at 20:1 Aβ to inhibitor molar ratio. Aggregation was monitored via ThT fluorescence and data was blank (ThT in PBS) subtracted. Error bars indicate SEM, n=3. *p<0.05 vs. control. C–D) cG8 (C) and mTTR (D) were incubated at a 20:1 ratio with pepsin for 0 and 2 h at 37°C. Degradation was assessed via mass spectrometry. Dashed lines indicate correct molecular weight of intact protein or peptide.
mTTR changes the morphology and quantity of cell-associated Aβ
The inhibition of Aβ toxicity by mTTR and TTR-inspired peptides in mouse neuronal cultures has previously been reported, with mTTR showing much greater efficacy.[14] In this work we used human iPSC-derived neurons to detect cellular Aβ binding as a function of aggregation state, and to determine if cG8 and/or mTTR altered the cellular binding and internalization of Aβ. To do this, cells were treated with Aβ monomer, oligomer, or fibrils prepared alone or with cG8 or mTTR. Control treatments tested the effect of cG8 and mTTR added to preformed oligomers and fibrils. The experimental strategy is illustrated in Figure 7. Images were collected after staining for Aβ with both permeabilized and nonpermeabilized cells, to compare total (internalized plus cell surface-associated) versus extracellular (cell surface-associated) Aβ.
Figure 7.
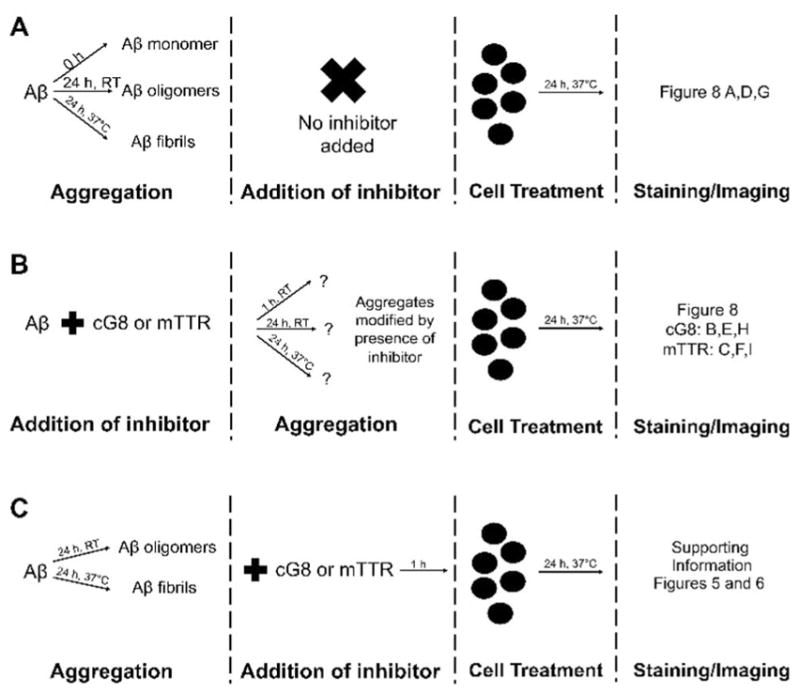
Treatment of human iPSC-derived neurons with Aβ. Aβ monomer, oligomers, and fibrils were formed in the absence (A) or presence (B) of cG8 or mTTR. Additionally, cG8 and mTTR were incubated with preformed oligomers or fibrils prior to cell treatment (C). Cell treatment lasted 24 h before washing, staining, and confocal imaging.
Aβ monomer, oligomers, or fibrils were prepared as illustrated in Figure 7A and added to iPSC-derived human neuronal cultures. Cells were exposed to Aβ (5 μM final concentration) for 24 h before cells were washed, stained and imaged. With all three treatments, some Aβ became cell associated (Figure 8, Supporting Information Figure 3). Treatment with Aβ monomer produced a few small, punctate deposits (Figure 8A). This result suggests that little to no additional aggregation of Aβ monomer occurs during the 24 h cell treatment time, probably because of the decreased Aβ concentration. Noticeably greater amounts of deposited Aβ were observed with oligomer or fibril treatments, and the extensive deposits had a web-like appearance (Figure 8D,G). The fraction of each image containing Aβ, normalized to account for differences in cell density, was estimated as ≈5% for Aβ monomer, ≈63% for Aβ oligomers, and ≈53% for Aβ fibrils (Supporting Information Table 1). The visual appearance of deposited Aβ aggregates for both oligomer and fibril preparations was similar, suggesting that oligomers progressed to fibrils during the 24 h cell exposure (total aggregation time of 48 h). Together with the monomer result (Figure 8A), this is consistent with a distinction between initiation of new aggregates versus growth and maturation of preexisting aggregates. Aβ deposits were clearly cell-associated, as there was no fluorescence in areas of the wells that were cell-free or in control wells that received Aβ treatment but contained no cells (data not shown).
Figure 8.
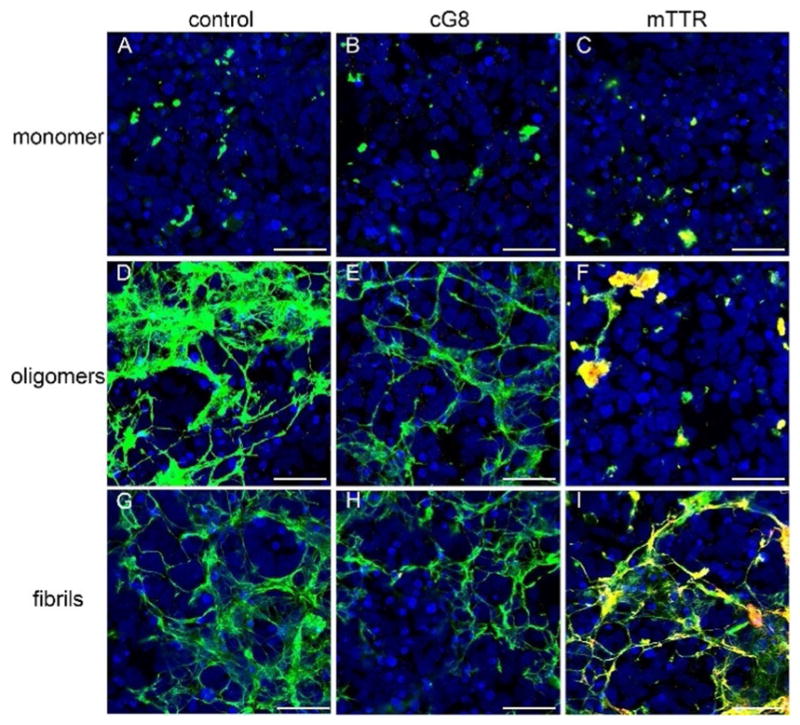
mTTR alters Aβ deposition on human neurons. Aβ monomer (top row), oligomers (middle row), and fibrils (bottom row) were prepared alone (control, left column) or in the presence of cG8 (middle column) or mTTR (right column). After 24 h cell treatment, cells were immunostained to visualize cell nuclei (DAPI, blue), cell associated Aβ (6E10, green), and cell associated cG8b or mTTRb (Neutravidin, red). Co-localization of Aβ and cG8b or mTTRb is shown in yellow. Cells were imaged using a Nikon A1R-Si+ confocal microscope and 20V objective with a zoom factor of 3. Representative images are a max intensity z-projection of 5 slice, z-stack images. Scale bars=50 μm.
Next, cG8 or mTTR was incubated with Aβ monomer (20:1 Aβ/inhibitor) for 1 h and then the neurons were exposed to the Aβ/inhibitor mixture for 24 h. After washing, cells were stained for Aβ (green) and cG8 or mTTR (red). Neither cG8 nor mTTR caused a measurable change in the amount of Aβ deposition or the morphology of the deposited Aβ, relative to Aβ alone (Figures 8B,C). mTTR co-localized with deposited Aβ, as indicated by yellow, whereas there was no observable co-localization of cG8 with Aβ. There was little to no nonspecific association of cG8b or mTTRb to the cells (Supporting Information Figure 4).
Next, cG8 or mTTR was added to Aβ and the mixtures were incubated under conditions that would normally lead to Aβ oligomers (RT, 24 h, Figure 7B). Neurons were exposed to these mixtures for 24 h, after which the cells were washed and stained. cG8 caused a modest decrease in the amount of cell-associated Aβ, from ≈63% in absence of cG8 to ≈47% in presence of cG8 (Figure 8E). There was no evidence that cG8 co-localized with Aβ deposits. In contrast, mTTR significantly decreased the amount of Aβ deposition, to ≈15%. More strikingly, there was a distinct morphological change into smaller, globular aggregates (Figure 8F). Furthermore, co-localization of mTTR with Aβ is clearly evident.
cG8 or mTTR was added to Aβ and the mixtures were incubated under conditions that would normally lead to Aβ fibrils (37 °C, 24 h, Figure 7B), followed by a 24 h exposure of the neurons to the solutions. The physical morphology of the Aβ deposits was not changed by addition of either cG8 or mTTR (Figure 8H,I). The quantity of deposited Aβ was partially decreased by cG8 (from ≈53% to ≈33%) but not by mTTR. mTTR strongly co-localized with cell-associated Aβ (Figure 8I) while there was little to no co-localization of cG8 (Figure 8H). Control experiments in which cG8 or mTTR was added to preformed oligomers or fibrils just prior to cell exposure (Figure 7C) showed no distinct changes in the morphology, and no change in the quantity of cell-associated Aβ, except for a small (≈10%) decrease when cG8 was added to pre-formed fibrils (Supporting Information Figures 5 and 6). Additionally, there was little to no co-localization of either inhibitor to cell-associated Aβ fibrils (Supporting Information Figure 6), suggesting that mTTR incorporates into the growing aggregates rather than binding to fibrils after they are formed. This result is consistent with our finding that mTTR does not break up preformed Aβ fibrillar aggregates (Figure 4).
A comparison of total (internalized plus cell surface-associated) Aβ and extracellular (cell surface-associated) Aβ provides additional insight (Figure 9, Supporting Information Table 1). The comparison is between two separately prepared wells, so the results should be interpreted cautiously. When Aβ monomer or fibrils was added to cells, there was no difference between the morphology or the quantity of extracellular and total cell-associated deposits (Figure 9A,C). With Aβ oligomers, there were also no observable morphological differences between extracellular and total cell-associated deposits (Figure 9B). However, there was a modest decrease in the quantity of extracellular versus total cell-associated deposits (63% versus 48%). Taken together, these results suggest that Aβ oligomers, but not monomers or fibrils, are partially internalized.
Figure 9.

Aβ oligomers are internalized more than Aβ monomer or fibrils. Cells were treated with Aβ monomer (A), oligomers (B), and fibrils (C) for 24 h then stained and images as previously described (Figure 8). Each treatment was done in duplicate to allow for staining of the total amount of Aβ (top row) and just the extracellular Aβ (bottom row). Both a single slice image (left side of panels, scale bars=100 μm) and a z-stack image (right side of panels, scale bars=50 μm) were obtained.
Because our analysis suggested that Aβ oligomers, but not monomers or fibrils, were internalized, we tested whether the inhibitors altered internalization. When Aβ was co-incubated with cG8 for 24 h at room temperature (Figure 7B) prior to cell exposure, the total cell-associated deposited Aβ was greater than extracellular Aβ (≈47% total versus ≈23% extracellular, Figure 10B), suggesting that although cG8 decreases the quantity of both extracellular and internalized Aβ, it does not prevent Aβ oligomer internalization. The notable morphological change induced by mTTR under these conditions, previously described for total cell-associated Aβ (Figure 8F), was also observed for extracellular Aβ (Figure 10C). Additionally, total and extracellular Aβ were similar (≈14% total versus ≈11% extracellular, Figure 10C). Co-incubation of cG8 or mTTR with Aβ as monomer or under fibril-forming conditions prior to cell treatment changed neither the amount nor the morphology of extracellular Aβ relative to total Aβ (Supporting Information Figure 7 and 8).
Figure 10.

cG8 does not prevent internalization of Aβ when present during oligomer formation. Aβ was co-incubated with cG8 (B) or mTTR (C) under oligomer forming conditions. Cells were then treated for 24 h with control oligomers (A) or those formed in the presence of cG8 or mTTR. Cells were stained and imaged as previously described (Figure 8). Each treatment was done in duplicate to allow for staining of the total amount of Aβ (top row) and just the extracellular Aβ (bottom row). Both a single slice image (left side of panels, scale bars=100 μm) and a z-stack image (right side of panels, scale bars=50 μm) were obtained.
Discussion
Previously, we demonstrated that mTTR and TTR-inspired peptides inhibit Aβ fibrillogenesis and decrease Aβ toxicity in mouse neuronal cultures.[13, 15] In this study, we evaluated mTTR and cG8 for their efficacy, stability, selectivity and mode of action. Our aim was to compare critical parameters that influence the choice of protein versus peptide-based compounds for anti-amyloid therapeutic strategies.
mTTR and cG8 were both effective at inhibiting amyloid fibril formation by either Aβ isoform, Aβ1–40 and Aβ1–42 (Figures 2 and 3A). Both were also effective inhibitors of amylin but neither was effective against α-synuclein aggregation (Figure 3B,C). This demonstrates that mTTR and cG8 have a similar degree of selectivity with respect to their interaction with amyloidogenic proteins. Their efficacy against amylin might arise from the fact that amylin is of similar length and shares 24% sequence similarity to Aβ[20] with the greatest degree of similarity in the domains crucial for self-association.[28] α-synuclein monomers are natively disordered, like Aβ, but the protein is much larger (≈14 kDa) and has only 12% sequence similarity.[20] Kellock et al.[23g] recently reported on the design of a peptide that inhibits aggregation of Aβ, amylin and TTR in support of the notion of cross-interaction of these three amyloidogenic proteins.
mTTR and cG8 both bind greater amounts of Aβ oligomers and fibrils than monomers (Figure 4A,B). Aβ oligomers are distinct from monomers in the presence of hydrophobic patches or cavities (as detected by ANS fluorescence) on the former. As oligomers re-arrange and assemble into fibrils, ANS fluorescence partially decreases and ThT fluorescence (indicative of extended cross-β conformation) increases. We hypothesize that selectivity toward oligomers and fibrils occurs because of binding of mTTR or cG8 to hydrophobic patches. As described previously, our data are consistent with the hypothesis that binding of mTTR to oligomers completely arrests both the adoption of extended cross-β conformation and further self-assembly·[11d] The mechanism of action of cyclic peptides is similar, except that cyclic peptides also re-directs the self-association of a fraction of Aβ into large non-fibrillar amorphous clusters.[14] Although both mTTR and cG8 potently inhibit fibrillogenesis of Aβ, neither disaggregates preformed Aβ fibrils (Figure 4C,D). This is a favorable feature of an anti-Aβ compound if intermediate oligomeric aggregates are indeed the most toxic species.
While cG8 and mTTR share many features in common, important differences were also observed. Although cG8 successfully inhibited Aβ fibrillogenesis at substoichiometric ratios, with overall similar dose-dependence as wild-type TTR, mTTR was more effective than cG8, suppressing fibrillogenesis even at a 75-fold excess Aβ (Figure 2). We hypothesize that the greater efficacy of mTTR is attributable to its attaining a stable anti-parallel two-β-strand conformation[15] that fully mimics TTR’s Aβ-binding site, while cG8 displays conformational heterogeneity and its structure is not as constrained. On the other hand, mTTR is more sensitive to the presence of biological materials (cell lysate, lipids or proteins), suffering a greater decrease in Aβ binding in their presence than does cG8 (Figure 5). We hypothesize that the decrease in efficacy in the presence of proteins is attributable to nonspecific binding of the proteins to cG8 or mTTR, and that there is less nonspecific binding to cG8 than to mTTR because of the smaller size of the former. A much larger fraction of the residues in cG8 are part of Aβ-binding domain (the functional part) compared with that in mTTR. An improved therapeutic might therefore be achieved by trimming down the non-essential residues in mTTR while retaining a sufficient scaffold to stabilize the conformation of the Aβ-binding domain.
When exposed to the lower pH of either simulated intestinal or simulated gastric fluids, both inhibitors resisted degradation and retained their ability to inhibit Aβ aggregation (Figure 6). However, mTTR was extremely susceptible to enzymatic degradation while cG8 proved more stable, likely due primarily to its cyclic structure. This reflects a significant stability advantage of synthetic peptides, where protease-resistant features such as D-amino acids, other non-natural amino acids, or cyclization can be readily incorporated into the design.
We next examined association of Aβ with human iPSC-derived neurons as a function of Aβ aggregation state. To our knowledge this is the first such study reported in the literature. With pre-aggregated Aβ (oligomeric or fibrillar), but not with monomeric Aβ, at least half of the cell surface was covered with deposited Aβ. Oligomers, but not monomers or fibrils, appeared to be partially internalized after 24 h cell exposure. This observation, though preliminary, could explain why oligomers are more toxic than fibrils.
When Aβ was co-incubated with mTTR or cG8 prior to contact with neurons, mTTR was tightly integrated into the cell-associated Aβ aggregates whereas there was little to no colocalization of cG8 with Aβ. Under conditions where Aβ alone forms oligomers (presumably the most toxic Aβ species), mTTR substantially decreased the amount of Aβ deposited on cells and drastically altered the physical morphology of the deposits (Figure 8). In contrast, cG8 had no discernable effect on Aβ aggregate morphology, but decreased the quantity of Aβ that deposited on the cells. cG8 did not prevent Aβ internalization, but the amount of Aβ internalized was lower, presumably because the total cell-associated Aβ was reduced.
Under conditions where Aβ alone forms fibrils, neither mTTR nor cG8 affected the morphology of cell-associated Aβ deposits. This was an unexpected result given that both compounds strongly suppressed fibrillogenesis at similar conditions per the ThT assay (Figure 2). There are several possible explanations. First, even if the total Aβ fibril concentration is reduced with cG8 or mTTR, only a small quantity of Aβ fibrils might be needed to coat the cells. Second, contact with cells could catalyze Aβ fibril formation even with inhibitors present. This explanation seems unlikely, because there was no evidence of Aβ fibril formation during the 24 h cell exposure period from the monomer preparation. A third possibility is that cross-β structure (and hence ThT binding) of the aggregates is lost even though there is no observable change in the aggregate morphology. This explanation is unlikely, especially for mTTR, because light scattering and particle tracking experiments have shown that mTTR not only inhibits ThT signal but also prevents aggregates from growing in size.[11d]
Taken together, these data suggest that there are different ways in which mTTR and cG8 interfere with Aβ toxicity. As shown in prior work, mTTR arrests Aβ aggregate growth entirely whereas cG8 redirects aggregation down a non-amyloid pathway.[13, 15] We speculate that mTTR, by retaining Aβ in a smaller and less aggregated state, decreases the quantity of Aβ deposited on cell membranes and inhibits internalization, thereby reducing Aβ-induced cellular toxicity. The striking effect of mTTR on Aβ-cell interactions was observed only when co-incubation occurred under oligomer-forming conditions. mTTR had no influence on morphology or quantity of cell-associated Aβ deposition when co-incubated under fibril-forming conditions even though mTTR was clearly integrated into the deposits. This suggests that mTTR may be the most effective at arresting Aβ growth and preventing Aβ toxicity when the rate of Aβ aggregation is moderate. On the other hand, whether co-incubated under oligomer- or fibril-forming conditions, cG8 decreased Aβ deposition but did not change the morphology nor prevent internalization. Because cG8 binds Aβ (Figure 4) and decreases the quantity of cell-associated Aβ deposits, but is not observed to be integrated into these deposits, we speculate that cG8 acts by preventing binding of a fraction of Aβ aggregates to cells. Experiments are currently underway to explore in greater depth the cellular response to Aβ, in the human iPSC-derived neuron model, and to elucidate the mechanisms by which inhibitors of fibrillogenesis may decrease Aβ toxicity.
Conclusions
This study provides a direct comparison of protein versus peptide, each designed as a mimic of the Aβ-binding domain on wild-type TTR, and each designed to associate with Aβ oligomers and inhibit amyloid fibril formation. By many measures, mTTR and cG8 were similar. Each however has its advantages and disadvantages. Specifically, mTTR is effective to lower concentrations, and has a strong impact on both the morphology and the quantity of Aβ deposited on cells. On the other hand, cG8’s cyclic structure and smaller size results in better stability against proteolysis and less interference from nonspecific biological materials. Studies with human iPSC-derived neurons provide insight into the mechanism by which each inhibitor decreases toxicity. Combined, these results provide guidance for future work to design a molecule that combines the advantages of each.
Experimental Section
Chemicals and reagents
Aβ1–40 and Aβ1–42 were purchased from Bachem (Torrance, CA). Amylin and α-synuclein were purchased from Anaspec (Fremont, CA). Thioflavin T (ThT) and an Amersham ECL Western Blotting Analysis System were purchased from Sigma–Aldrich (St. Louis, MO) and Sulfo-NHS-LC-Biotin was purchased from VWR (Radnor, PA). Anti-Aβ 6E10 antibody was purchased from Covance (Princeton, NJ) and superclonal anti-mouse HRP antibody was purchased from Pierce Chemical (Rockford, IL). All other materials except where noted were purchased from Fisher Scientific (Hampton, NH).
Aβ sample preparation
Aβ1–40 was used in most experiments and will be denoted as Aβ whereas subscripts will be used to indicate when Aβ1–42 was used. Aβ was dissolved to 12 mgmL−1 in urea (8M), glycine (10 mM) buffer (pH 10.0) to monomerize. After 1 h incubation at RT, Aβ stock was aliquoted, snap-frozen, and stored at −80°C until use. Prior to experimentation, NaOH (1M) was added to Aβ at a 1:8 volume ratio and incubated for 20 min at RT to dissolve any Aβ aggregates formed during the freeze-thaw cycle. Aβ was then diluted into phosphate buffered saline with azide (PBSA, 10 mM Na2HPO4/NaH2PO4, 150 mM NaCl, 0.02% NaN3, pH 7.4). For testing of Aβ monomer, Aβ was used immediately after dilution into PBSA. Aβ oligomers were formed by incubating Aβ at RT for 24 h while Aβ fibrils were formed by incubation at 37 °C for 24 h. ANS and ThT fluorescence confirmed monomer (no ANS, no ThT), oligomers (high ANS, low ThT), and fibrils (low ANS, high ThT) (Supporting Information Figure 1).[17] Aβ1–42 was prepared and stored in the same fashion except that a 1:2 NaOH to Aβ1–42 volume ratio was required to remove preformed aggregates.
TTR and mTTR preparation
As previously described in detail,[12b] recombinant human transthyretin (TTR) and transthyretin mutant F87M/L110M (mTTR) was produced and purified. Identity was confirmed by mass spectrometry and purity by gel electrophoresis. Biotinylated mTTR (mTTRb) was produced by combining mTTR with Sulfo-NHS-LC-Biotin (10 mM in H2O) at a 1:5 ratio and incubating for 30 min at RT. Free biotin was removed by centrifugation through 3 kDa MWCO filters (14 000 rpm, 4 °C, 10 min). ThT analysis confirmed that mTTRb retained the ability to inhibit Aβ aggregation (data not shown).
cG8 Preparation cG8 was prepared using Fmoc solid-phase methods and benzotriazol-1-yl-oxytripyrrolidinophosphonium hexafluorophosphate (PyBOP) for head-to-tail cyclization as previously described in detail.[15, 18] Biotinylated cG8 (cG8b) was synthesized by New England Peptide (Gardner, MA) by replacing Val19 with Fmoc-Glu(biotinyl-PEG)-OH. cG8b was received as a linear peptide still on the resin. Peptide was cleaved from resin by diluting in dichloromethane containing hexafluoroisopropanol (HFIP, 20%). The solution was placed on a shaker for 1 h then centrifuged (2000 rpm, 2 min) and the peptide containing supernatant was removed. Additional HFIP/CH2Cl2 was added to the resin and the process was repeated. The combined supernatant was filtered and the HFIP/CH2Cl2 was evaporated under nitrogen. For head-to-tail cyclization, the peptide was dissolved in CH2Cl2 containing PyBoP (25 mg) and diisopropylethylamine (DIPEA, 1.25%) and shaken under nitrogen for 18 h. The solution was evaporated off under nitrogen and cyclization was repeated. After cyclization, the remaining protecting groups were removed by incubating the peptide in Reagent K for 2 h. The peptide was then treated with cold tert-butyl methyl ether (t-BME), pelleted by centrifugation (2000 rpm, 4°C, 10 min), and air dried. cG8b was purified by RP-HPLC on a Vydac C18 column as previously described.[15] ThT analysis confirmed that cG8b retained the ability to inhibit Aβ aggregation (data not shown).
Thioflavin T (ThT) fluorescence assay
ThT is a fluorescent probe commonly used to monitor Aβ aggregation due to its binding to the β-sheet structure that is a hallmark of amyloidogenic proteins.[17b, 19] To compare the inhibitory capability of cG8 and mTTR to that of TTR, Aβ monomer (28 μM) in PBSA was combined with each inhibitor at an Aβ to inhibitor molar ratio of 20:1, 50:1, or 75:1. Aβ aggregated alone was used as a control. To determine the ability of cG8 and mTTR to disaggregate Aβ fibrils, Aβ monomer was incubated in PBSA for 24h at 37°C to allow fibrils to form. Aβ fibrils were then combined with cG8 or mTTR for a final Aβ concentration of 28 μM and at either a 2:1 or 1:1 Aβ to inhibitor molar ratio. Aβ fibrils were used as a control. All samples were incubated for 48 h at 37°C.
Stock ThT solutions were prepared by dissolving ThT into filtered PBS. At times 0, 24, and 48 h, Aβ samples were diluted in ThT stock for final concentrations of 2 μM Aβ and 10 μM ThT. Fluorescence (λexcitation=440 nm, λemission=460–500 nm) was measured using a QuantaMaster spectrometer (PTI, Birmingham, NJ). For each sample, three spectra were collected and the fluorescent intensity at 480 nm was averaged, baseline (PBSA, ThT) subtracted, and normalized to the respective control. TTR, cG8, and mTTR alone were ThT negative. Results are reported as the mean±SEM of 3–4 independent experiments.
Aβ1–42, amylin, and α-synuclein studies
ThT fluorescence was used to determine the ability of cG8 and mTTR to inhibit the aggregation of Aβ1–42, amylin, and α-synuclein. Aβ1–42 was prepared as described above. Amylin was diluted in cold HFIP to 500 μM and incubated for 1 h at RT. Dissolved protein was then aliquoted and HFIP was evaporated off overnight. Dried protein aliquots were stored at −80°C until use. To initiate aggregation, amylin was diluted to 1 mM in NaOH (5 mM) and incubated at RT for 10 min, then combined with cG8 or mTTR in PBS (40 mM Na2HPO4/NaH2PO4, 150 mM NaCl, pH 7.4).
α-Synuclein was prepared per the manufacturer’s instructions. Briefly, the protein was dissolved in 10 mM PBS and, after 10 min incubation at RT, centrifuged (12000 rpm, 4°C, 10 min) to remove any undissolved material. The protein was then aliquoted, snap-frozen, and stored at −80 °C until use. α-synuclein was combined with cG8 or mTTR in 10 mM PBS and agitated (600 rpm) to promote aggregation.
For all three proteins, experiments were conducted at 20 μM protein and at molar ratios of 10:1 and 20:1 protein to inhibitor. All samples were incubated for 48h at 37 °C and ThT fluorescence was measured as described above. Results are reported as the mean±SEM of 2–4 independent experiments.
Sequence similarity comparing Aβ, amylin and α-synuclein was calculated using EMBOSS Needle.[20]
Enzyme-linked immunoassay (ELISA)
Pre-blocked NeutrAvidin protein-coated ELISA plates were used as previously described[15] to determine Aβ binding to cG8b and mTTRb under various conditions. The plates were coated with either cG8b or mTTRb (5 μgmL−1) for 1 h then washed three times with PBST (0.05% Tween20). To assess binding of different Aβ species, Aβ monomer, oligomers, and fibrils were diluted in PBS to 1, 3, and 5 μgmL−1 and added to the plate for 1 h at 37 °C. To assess binding under physiologically relevant conditions, Aβ oligomers were diluted to a final concentration of 5 μgmL−1 Aβ in PBS containing wild-type mouse cortex tissue lysate (0–30 mgdL−1). This experiment was repeated with cell lysate from induced pluripotent stem cell (iPSC)-derived human neurons. To evaluate lipid interference, lipids extracted from porcine brain tissue (Avanti Polar Lipids, Alabaster, AL) were combined with Aβ oligomers. Final samples contained either lipid concentrations similar to the concentration in cerebrospinal fluid (CSF) (2 and 4 mgdL−1) or lipids in excess (600 and 1800 mgdL−1). Similarly, the effect of proteins was tested at biological relevant levels of fetal bovine serum (FBS, 15 and 30 mgdL−1) and at excess FBS concentrations (5000 and 15000 mgdL−1).
After incubation with Aβ, the plate was again washed and anti-Aβ 6E10 antibody (1:3000) was added to the plate for 1 h with gentle shaking. After washing, superclonal anti-mouse HRP antibody (1:3000) was added to the plate for 1 h with gentle shaking. The plate was again washed before addition of Ultra-TMB solution. After full color development, the reaction was stopped by adding equal volume of sulfuric acid (2M). The absorbance (450 nm) was measured using an EL800 Universal Microplate Reader (Biotek Instruments Inc., Winooski, VT). For each sample, four wells were prepared and the absorbance intensity averaged. Control experiments showed that cG8b and mTTRb-coated wells receiving buffer, tissue lysate, FBS, or lipid without Aβ showed negligible signal (<3%) as did non-coated wells that received Aβ (data not shown). For Aβ samples containing biological materials, the absorbance was normalized to the absorbance for Aβ in buffer (PBST or tissue lysis buffer). Results are reported as the mean±SEM of 3 independent experiments.
Tissue lysate preparation
Cortex tissue for use in the ELISA assay was harvested from a 4 month, male, wild-type mouse and was a generous gift from Dr. Jeffery Johnson (School of Pharmacy, UW-Madison, WI). Lysis buffer containing TritonX-100 (1% v/v), dithiothreitol (DTT, 1mM), and protease and phosphatase inhibitors was added at 10 μL of lysis buffer per mg of tissue. Tissue was sonicated until homogenized then centrifuged (16 000 rpm, 4 °C, 30 min) to remove cell debris. Total protein concentration of the supernatant was determined via BCA assay. Lysate aliquots were stored at −80°C until use.
Stability assay
The stability of cG8 and mTTR from pepsin-induced degradation was determined by combining 20-fold excess cG8 or mTTR to pepsin (dissolved in 10 mM HCl) and incubating for 2 h at 37°C. At 0 and 2 h, pepsin was deactivated by adding NaOH (1M) to neutralize the pH.
The stability of cG8 and mTTR was also assessed using simulated gastric fluid (SGF, pH 1.2, without enzyme) and simulated intestinal fluid (SIF, pH 6.8, without enzyme) as previously described.[15] cG8 and mTTR were each diluted into either SGF or SIF to 0.1 mgmL−1 and incubated at 37 °C. At 0 and 24 h, the samples were diluted in equal volume cold H2O and NaOH (1M) was added to SGF samples to neutralize the pH. All samples were prepared for mass spectrometry by being spotted directly onto the plate, dried, and quickly washed with water. Samples were analyzed using MALDI-ToF mass spectrometry.
To test the activity of cG8 and mTTR after incubation in SGF and SIF, inhibitor samples were diluted 35-fold in PBSA to neutralize the pH and combined with Aβ (28 μM) at an Aβ to inhibitor molar ratio of 20:1. To account for any effect the residual biological fluids may have on Aβ aggregation, the same final amount of SGF or SIF present in the Aβ-inhibitor samples (1.4%) was added to Aβ alone. Samples were incubated for 48h at 37°C and ThT fluorescence was measured as described above. Results are reported as the mean±SEM of 3 independent experiments.
Cell culture maintenance and differentiation
CS03iCTRn2 derived EZ spheres[21] were cultured as previously described.[22] EZ spheres were maintained in DMEM/F12 (70:30) media supplemented with epidermal growth factor (100 ngmL−1; Pepro-tech, Rocky Hill, NJ), basic fibroblast growth factor (100 ngmL−1; WiCell Research Institute, Madison, WI), heparin (2 μgmL−1; Sigma–Aldrich), B27 minus Vitamin A (2%), and penicillin/streptomycin (1 %). Cells were grown in an ultra-low attachment flask and approximately two thirds of the media was replaced with fresh media every 2 days. Once a week, cells were passaged using a mechanical dissociation technique as described previously.[22] For differentiation to EZ sphere derived neurons, EZ spheres were dissociated into singular cells using Accutase (Innovative Cell Technologies, San Diego, CA) and seeded onto Matrigel-coated plates (WiCell Research Institute, Madison, WI). Cells were seeded onto a 24-well, black-sided, glass bottom plate at 20000 cells per well for immunostaining. Cells were differentiated into neurons using neuron differentiation media containing DMEM/F12 (70:30) media, heparin (2 μgmL−1), B27 minus Vitamin A (2%) and penicillin/streptomycin (1 %). Neuron differentiation media was replaced every 2 days for 14 days. Successful differentiation was confirmed by staining with β-III tubulin and nestin (Supporting Information Figure 2).
Immunostaining
Immunostaining with confocal microscopy was used to image Aβ bound to iPSC-derived neurons, and to determine whether cG8 and/or mTTR alter the total (internalized plus cell surface associated) Aβ or the ratio of extracellular (cell surface associated) Aβ to total Aβ. Aβ monomer (50 μM) in PBS was incubated alone (control) or at a 20:1 molar ratio with either cG8b or mTTRb under conditions that normally create either fibrils (37 °C, 24 h) or oligomers (RT, 24 h). Biotinylated cG8 and mTTR were used so that they could be visualized using Neutravidin Protein, DyLight 550. Samples were diluted in media for final concentrations of 5 μM Aβ, 0 (controls) or 0.25 μM of either cG8 or mTTR, and <15% PBS. Samples were added to 14 day differentiated neurons for 24 h.
All chemicals and antibodies were diluted in PBS for immunostaining. Following 24 h treatment, cells were washed with PBS, fixed using paraformaldehyde (4%, 15 min, 37°C), and permeabilized using Triton X-100 (0.1 %), glycine (10 mM) buffer (10 min). Cells were blocked (30 min) using goat normal serum (5%) before being probed with DyLight 550 (1:500, 1 h). After additional PBS washes, cells were probed with anti-Aβ 6E10 antibody (1:500, 1 h), washed, and probed with a goat anti-mouse Alexa Fluor 488 antibody (10 μgmL−1, 1 h) to allow visualization of cell associated Aβ. DAPI (1:1000, 10 min) was used for nuclei visualization. For visualization of only extracellular Aβ, cell permeabilization was omitted. After staining, cells were kept in PBS for imaging. Images were obtained with a Nikon A1R-Si+ Confocal Microscope using a three-channel, 1024V1024 scanning format. For each sample. a single mid-slice image was obtained using a 20V objective as well as a 5 slice, z-stack image obtained using a 20V objective with the scan area set to a zoom factor of 3.
ImageJ software (NIH, Bethesda, MD) was used to export the ND2 files into TIFF images and to convert the z-stack images into max intensity z-projections. To provide semi-quantitative analysis, ImageJ was used to determine the fraction of pixels in each image that contained green (AlexaFluor 488, indicating deposited Aβ) or blue (DAPI, indicating cell nuclei). The relative amount of deposited Aβ was normalized by the amount of surface occupied by cells in each image by calculating the ratio (fraction green/fraction blue).
Supplementary Material
Acknowledgments
This work was supported by the US National Institutes of Health (NIH) Grant R01AG033493.
Footnotes
Conflict of interest
The corresponding author holds a US patent on the cyclic peptide described in the manuscript.
Supporting information and the ORCID identification number(s) for the author(s) of this article can be found under : https://doi.org/10.1002/cmdc.201800031.
References
- 1.Alzheimer’s Association. Alzheimer’s Dementia. 2016;12:459–509. doi: 10.1016/j.jalz.2016.03.001. [DOI] [PubMed] [Google Scholar]
- 2.a) Westermark P, Andersson A, Westermark GT. Physiol Rev. 2011;91:795–826. doi: 10.1152/physrev.00042.2009. [DOI] [PubMed] [Google Scholar]; b) El-Agnaf OM, Irvine GB. J Struct Biol. 2000;130:300–309. doi: 10.1006/jsbi.2000.4262. [DOI] [PubMed] [Google Scholar]
- 3.a) Francioso A, Punzi P, Boffi A, Lori C, Martire S, Giordano C, D’Erme M, Mosca L. Bioorg Med Chem. 2015;23:1671–1683. doi: 10.1016/j.bmc.2015.02.041. [DOI] [PubMed] [Google Scholar]; b) Mohamed T, Shakeri A, Rao PP. Eur J Med Chem. 2016;113:258–272. doi: 10.1016/j.ejmech.2016.02.049. [DOI] [PubMed] [Google Scholar]; c) Nie Q, Du XG, Geng MY. Acta Pharmacol Sin. 2011;32:545. doi: 10.1038/aps.2011.14. [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Doig AJ. Curr Opin Drug Discovery Dev. 2007;10:533–539. [PubMed] [Google Scholar]; e) Panza F, Solfrizzi V, Imbimbo BP, Logroscino G. Expert Opin Biol Ther. 2014;14:1465–1476. doi: 10.1517/14712598.2014.935332. [DOI] [PubMed] [Google Scholar]; f) Stains CI, Mondal K, Ghosh I. ChemMedChem. 2007;2:1674–1692. doi: 10.1002/cmdc.200700140. [DOI] [PubMed] [Google Scholar]; g) Rajasekhar K, Chakrabarti M, Govindaraju T. Chem Commun. 2015;51:13434–13450. doi: 10.1039/c5cc05264e. [DOI] [PubMed] [Google Scholar]; h) Rajasekhar K, Madhu C, Govindaraju T. ACS Chem Neurosci. 2016;7:1300–1310. doi: 10.1021/acschemneuro.6b00175. [DOI] [PubMed] [Google Scholar]
- 4.a) Salomone S, Caraci F, Leggio GM, Fedotova J, Drago F. Br J Clin Pharmacol. 2012;73:504–517. doi: 10.1111/j.1365-2125.2011.04134.x. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Mishra P, Ayyannan SR, Panda G. ChemMedChem. 2015;10:1467–1474. doi: 10.1002/cmdc.201500215. [DOI] [PubMed] [Google Scholar]
- 5.Goyal D, Shuaib S, Mann S, Goyal B. ACS Comb Sci. 2017;19:55–80. doi: 10.1021/acscombsci.6b00116. [DOI] [PubMed] [Google Scholar]
- 6.Craik DJ, Fairlie DP, Liras S, Price D. Chem Biol Drug Des. 2013;81:136–147. doi: 10.1111/cbdd.12055. [DOI] [PubMed] [Google Scholar]
- 7.a) Spencer B, Masliah E. Front Aging Neurosci. 2014;6:114. doi: 10.3389/fnagi.2014.00114. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Doody RS, Thomas RG, Farlow M, Iwatsubo T, Vellas B, Joffe S, Kieburtz K, Raman R, Sun X, Aisen PS. N Engl J Med. 2014;370:311–321. doi: 10.1056/NEJMoa1312889. [DOI] [PubMed] [Google Scholar]; c) Vandenberghe R, Rinne JO, Boada M, Katayama S, Scheltens P, Vellas B, Tuchman M, Gass A, Fiebach JB, Hill D. Alzheimer’s Res Ther. 2016;8:18–30. doi: 10.1186/s13195-016-0189-7. [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Sevigny J, Chiao P, BussiHre T, Weinreb PH, Williams L, Maier M, Dunstan R, Salloway S, Chen T, Ling Y, O’Gorman J, Qian F, Arastu M, Li M, Chollate S, Brennan MS, Quintero-Monzon O, Scannevin RH, Arnold HM, Engber T, Rhodes K, Ferrero J, Hang Y, Mikulskis A, Grimm J, Hock C, Nitsch RM, Sandrock A. Nature. 2016;537:50. doi: 10.1038/nature19323. [DOI] [PubMed] [Google Scholar]
- 8.a) Pallitto MM, Ghanta J, Heinzelman P, Kiessling LL, Murphy RM. Biochemistry. 1999;38:3570–3578. doi: 10.1021/bi982119e. [DOI] [PubMed] [Google Scholar]; b) Takahashi T, Mihara H. Acc Chem Res. 2008;41:1309–1318. doi: 10.1021/ar8000475. [DOI] [PubMed] [Google Scholar]
- 9.Orner BP, Liu L, Murphy RM, Kiessling LL. J Am Chem Soc. 2006;128:11882–11889. doi: 10.1021/ja0619861. [DOI] [PubMed] [Google Scholar]
- 10.a) Liu L, Murphy RM. Biochemistry. 2006;45:15702–15709. doi: 10.1021/bi0618520. [DOI] [PubMed] [Google Scholar]; b) Du J, Cho PY, Yang DT, Murphy RM. Prot Eng Des Sel. 2012;25:337–345. doi: 10.1093/protein/gzs026. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Costa DA, Cracchiolo JR, Bachstetter AD, Hughes TF, Bales KR, Paul SM, Mervis RF, Arendash GW, Potter H. Neurobiol Aging. 2007;28:831–844. doi: 10.1016/j.neurobiolaging.2006.04.009. [DOI] [PubMed] [Google Scholar]; d) Stein TD, Anders NJ, DeCarli C, Chan SL, Mattson MP, Johnson JA. J Neurosci. 2004;24:7707–7717. doi: 10.1523/JNEUROSCI.2211-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Stein TD, Johnson JA. J Neurosci. 2002;22:7380–7388. doi: 10.1523/JNEUROSCI.22-17-07380.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]; f) Buxbaum JN, Ye Z, Reixach N, Friske L, Levy C, Das P, Golde T, Masliah E, Roberts AR, Bartfai T. Proc Natl Acad Sci USA. 2008;105:2681–2686. doi: 10.1073/pnas.0712197105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.a) Giunta S, Valli M, Galeazzi R, Fattoretti P, Corder E, Galeazzi L. Clin Biochem. 2005;38:1112–1119. doi: 10.1016/j.clinbiochem.2005.08.007. [DOI] [PubMed] [Google Scholar]; b) Costa R, Goncalves A, Saraiva M, Cardoso I. FEBS Lett. 2008;582:936–942. doi: 10.1016/j.febslet.2008.02.034. [DOI] [PubMed] [Google Scholar]; c) Li X, Masliah E, Reixach N, Buxbaum JN. J Neurosci. 2011;31:12483–12490. doi: 10.1523/JNEUROSCI.2417-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Yang DT, Joshi G, Cho PY, Johnson JA, Murphy RM. Biochemistry. 2013;52:2849–2861. doi: 10.1021/bi4001613. [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Brouillette J, Caillierez R, Zommer N, Alves-Pires C, Benilova I, Blum D, De Strooper B, Bu8e L. J Neurosci. 2012;32:7852–7861. doi: 10.1523/JNEUROSCI.5901-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.a) Li X, Zhang X, Ladiwala ARA, Du D, Yadav JK, Tessier PM, Wright PE, Kelly JW, Buxbaum JN. J Neurosci. 2013;33:19423–19433. doi: 10.1523/JNEUROSCI.2561-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Du J, Murphy RM. Biochemistry. 2010;49:8276–8289. doi: 10.1021/bi101280t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cho PY, Joshi G, Johnson JA, Murphy RM. ACS Chem Neurosci. 2014;5:542. doi: 10.1021/cn500014u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cho PY, Joshi G, Boersma MD, Johnson JA, Murphy RM. ACS Chem Neurosci. 2015;6:778. doi: 10.1021/cn500272a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lu X, Brickson CR, Murphy RM. ACS Chem Neurosci. 2016;7:1264–1274. doi: 10.1021/acschemneuro.6b00150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jiang X, Smith CS, Petrassi HM, Hammarstrçm P, White JT, Sacchettini JC, Kelly JW. Biochemistry. 2001;40:11442–11452. doi: 10.1021/bi011194d. [DOI] [PubMed] [Google Scholar]
- 17.a) Kremer JJ, Pallitto MM, Sklansky DJ, Murphy RM. Biochemistry. 2000;39:10309–10318. doi: 10.1021/bi0001980. [DOI] [PubMed] [Google Scholar]; b) Ladiwala ARA, Litt J, Kane RS, Aucoin DS, Smith SO, Ranjan S, Davis J, Van Nostrand WE, Tessier PM. J Biol Chem. 2012;287:24765–24773. doi: 10.1074/jbc.M111.329763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Murphy RM, Cho PY, Johnson JA, Lu X. University of Wisconsin; Madison: US9809627. US Pat No. 2015
- 19.Biancalana M, Koide S. Biochim Biophys Acta Proteins Proteomics. 2010;1804:1405–1412. doi: 10.1016/j.bbapap.2010.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rice P, Longden I, Bleasby A. Trends Genet. 2000;16:276–277. doi: 10.1016/s0168-9525(00)02024-2. [DOI] [PubMed] [Google Scholar]
- 21.Vatine GD, Al-Ahmad A, Barriga BK, Svendsen S, Salim A, Garcia L, Garcia VJ, Ho R, Yucer N, Qian T, Lim RG, Wu J, Thompson LM, Spivia WR, Chen Z, Van Eyk J, Palecek SP, Refetoff S, Shusta EV, Svendsen CN. Cell Stem Cell. 2017;20:831–843. doi: 10.1016/j.stem.2017.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Canfield SG, Stebbins MJ, Morales BS, Asai SW, Vatine GD, Svendsen CN, Palecek SP, Shusta EV. J Neurochem. 2017;140:874–888. doi: 10.1111/jnc.13923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.a) Rao JN, Dua V, Ulmer TS. Biochemistry. 2008;47:4651–4656. doi: 10.1021/bi8002378. [DOI] [PubMed] [Google Scholar]; b) Ehrnhoefer DE, Bieschke J, Boeddrich A, Herbst M, Masino L, Lurz R, Engemann S, Pastore A, Wanker EE. Nat Struct Mol Biol. 2008;15:558–566. doi: 10.1038/nsmb.1437. [DOI] [PubMed] [Google Scholar]; c) Necula M, Kayed R, Milton S, Glabe CG. J Biol Chem. 2007;282:10311–10324. doi: 10.1074/jbc.M608207200. [DOI] [PubMed] [Google Scholar]; d) Masuda M, Suzuki N, Taniguchi S, Oikawa T, Nonaka T, Iwatsubo T, Hisanaga S-i, Goedert M, Hasegawa M. Biochemistry. 2006;45:6085–6094. doi: 10.1021/bi0600749. [DOI] [PubMed] [Google Scholar]; e) Lendel C, Bertoncini CW, Cremades N, Waudby CA, Vendruscolo M, Dobson CM, Schenk D, Christodoulou J, Toth G. Biochemistry. 2009;48:8322–8334. doi: 10.1021/bi901285x. [DOI] [PubMed] [Google Scholar]; f) Guerrero-MuÇoz MJ, Castillo-Carranza DL, Kayed R. Biochem Pharmacol. 2014;88:468–478. doi: 10.1016/j.bcp.2013.12.023. [DOI] [PubMed] [Google Scholar]; g) Kellock J, Hopping G, Caughey B, Daggett V. J Mol Biol. 2016;428:2317–2328. doi: 10.1016/j.jmb.2016.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kayed R, Head E, Thompson JL, McIntire TM, Milton SC, Cotman CW, Glabe CG. Science. 2003;300:486–489. doi: 10.1126/science.1079469. [DOI] [PubMed] [Google Scholar]
- 25.Attar A, Rahimi F, Bitan G. Trans Neurosci. 2013;4:385–409. [Google Scholar]
- 26.Schwarzman AL, Goldgaber D. The Nature and Origin of Amyloid Fibrils. In: Bock GR, Goode JA, editors. Ciba Foundation Symposium; Chichester: John Wiley & Sons; 1996. pp. 146–164. [Google Scholar]
- 27.a) Fonteh AN, Cipolla M, Chiang J, Arakaki X, Harrington MG. PLoS One. 2014;9:e100519. doi: 10.1371/journal.pone.0100519. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Koch S, Donarski N, Goetze K, Kreckel M, Stuerenburg H-J, Buhmann C, Beisiegel U. J Lipid Res. 2001;42:1143–1151. [PubMed] [Google Scholar]; c) Julk J, van der Boom J, Havekes L. Alzheimer Dis Assoc Disord. 1998;12:198–203. doi: 10.1097/00002093-199809000-00012. [DOI] [PubMed] [Google Scholar]
- 28.Andreetto E, Yan LM, Tatarek-Nossol M, Velkova A, Frank R, Kapurniotu A. Angew Chem Int Ed. 2010;49:3081–3085. doi: 10.1002/anie.200904902. [DOI] [PubMed] [Google Scholar]; Angew Chem. 2010;122:3146–3151. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.