

Abstract
Early coronary artery reperfusion improves outcomes for patients with ST-segment elevation myocardial infarction (STEMI), but morbidity and mortality after STEMI remain unacceptably high. The primary deficits seen in these patients include inadequate pump function, owing to rapid infarction of muscle in the first few hours of treatment, and adverse remodelling of the heart in the months that follow. Given that attempts to further reduce myocardial infarct size beyond early reperfusion in clinical trials have so far been disappointing, effective therapies are still needed to protect the reperfused myocardium. In this Review, we discuss several approaches to preserving the reperfused heart, such as therapies that target the mechanisms involved in mitochondrial bioenergetics, pyroptosis, and autophagy, as well as treatments that harness the cardioprotective properties of inhaled anaesthetic agents. We also discuss potential therapies focused on correcting the no-reflow phenomenon and its effect on healing and adverse left ventricular remodelling.
Early reperfusion with percutaneous transluminal coronary intervention has been shown to be effective in reducing myocardial infarct size and improving cardiac function, but data from clinical trials to evaluate the efficacy of adjunctive therapies in further reducing myocardial infarct size have been disappointing1,2. Consequently, despite the improvements in time-to-reperfusion for ST-segment elevation myocardial infarction (STEMI) and the use of evidence-based therapies during hospitalization in the past decade, in-hospital mortality for STEMI has not declined3. According to the AHA, 30-day mortality in patients presenting with myocardial infarction (MI) is as high as 7.8–11.4%4. Up to 18% of men and 23% of women aged ≥45 years will die within 1 year of their first MI event; these rates increase to 36% and 47%, respectively, up to 5 years after the initial event. In addition, among the survivors in this population, 16% of men and 22% of women will develop heart failure4. Several reasons have been proposed to explain why adjunctive therapies have not been effective in reducing infarct size and improving outcomes after MI in the clinical trial setting, including the lack of reproducibility and rigour in preclinical studies; use of preclinical models that do not adequately reflect clinical comorbidities such as advanced age; and administration of pharmacological agents too late to have an effect on infarct size. Furthermore, antiplatelet agents such as P2Y12-receptor blockers are commonly prescribed to these patients before primary angioplasty, and might mask the protective postconditioning effects of adjunctive therapies5. In the setting of acute MI, these antiplatelet agents help to maintain vessel patency after percutaneous coronary intervention (PCI). In addition to their effects on platelet aggregation, these drugs are powerful postconditioning mimetics, and act on the same protective signalling pathways that are activated during ischaemic preconditioning or postconditioning6,7. As a consequence, therapeutic strategies that target ischaemic preconditioning8 or postconditioning6 as a means for cardioprotection after MI did not reduce infarct size in animal hearts beyond the effects of the antiplatelet agents. The first clinical trial to evaluate the efficacy of ischaemic postconditioning after acute MI reported promising findings, but subsequent trials that were conducted after platelet inhibitors came into widespread use in patients undergoing PCI yielded either neutral results, or demonstrated only marginal benefit with postconditioning5,9,10. Although the use of platelet inhibitors might not be the only confounding factor in these studies, any intervention that cannot provide additional cardioprotective effects beyond that of an antiplatelet agent in the setting of MI (as seen in the animal studies) is unlikely to improve outcomes. Remote ischaemic conditioning — whereby short, serial episodes of ischaemia and reperfusion applied to a (limb) vascular bed confer global protection against subsequent ischaemia–reperfusion injury — has been shown to reduce myocardial infarct size in several contemporary clinical trials and meta-analyses11–14. The mechanisms involved in remote ischaemic conditioning are currently unknown and have not been tested against platelet inhibitors.
In light of these observations, an optimal strategy for preserving the myocardium after MI will be one that mitigates a factor contributing to cell death in the reperfused heart, but is not targeted by platelet inhibitors. For example, mild hypothermia and sodium–hydrogen exchange blockers offer additive protection against infarction when combined with the P2Y12 inhibitor cangrelor in rats; the use of all three strategies in combination is three times as protective against infarction compared with the use of the platelet inhibitor alone8. Unfortunately, mild hypothermia and sodium–hydrogen exchange blockers seem to target ischaemic tissue rather reperfused tissue, and thus these strategies must be applied during ischaemia for optimal efficacy15. Although investigations exploring the effects of rapid cooling (either soon after admission or even in the ambulance to shorten the normothermic ischaemic time) are ongoing16, a better solution would be to develop an intervention that is effective when administered around the time of reperfusion. Future investigations of cardioprotective agents should include experiments in animal models with common clinical comorbidities and in combination with platelet inhibitors to identify therapies most likely to translate to the clinical situation.
Infarct size reduction is not the only therapeutic goal after MI. Long-term therapy with angiotensin-converting-enzyme inhibitors, angiotensin-receptor blockers, and/or β-blockers reduces adverse left ventricular (LV) remodelling, but has only modest effects on mortality17. Whereas stem cell therapy has shown promise in some studies, most of the larger clinical trials yielded either neutral findings or demonstrated very limited improvements in LV ejection fraction after MI18. The promising preclinical results in periprocedural MI stem cell therapy have not yet translated into clinical benefit for patients18. However, cell mimetic therapies such as nanoparticles19 and synthetic stem cells20 engineered to mimic stem cell trophic effects are in preclinical stages of development. This field of research highlights the major unmet need for noncellular therapies that can improve cardiac structure and function after MI, perhaps in association with cell therapy. In this Review, we consider several promising approaches for the preservation of cardiac structure and function after MI that are not currently part of the clinical armamentarium (FIG. 1). This Review takes a broader approach than previously published Reviews on cardioprotection focused on preconditioning, postconditioning, and remote ischaemic conditioning, plus their pharmacological mimetics1,2,21,22. We discuss cardioprotective mechanisms that target mitochondrial bioenergetics, anaesthetic preconditioning, pyroptosis, autophagy, and the no-reflow phenomenon. All these therapies involve mitochondrial function to some extent, such that progress in each of the approaches should inform the others. Indeed, a newly listed clinical trial involving the transplantation of autologous skeletal muscle mitochondria epicardially in children undergoing extracorporeal membrane oxygenation after ischaemic injury points to the emerging importance of bioenergetic approaches for heart disease23.
Figure 1. New and updated approaches to treat the reperfused myocardium.
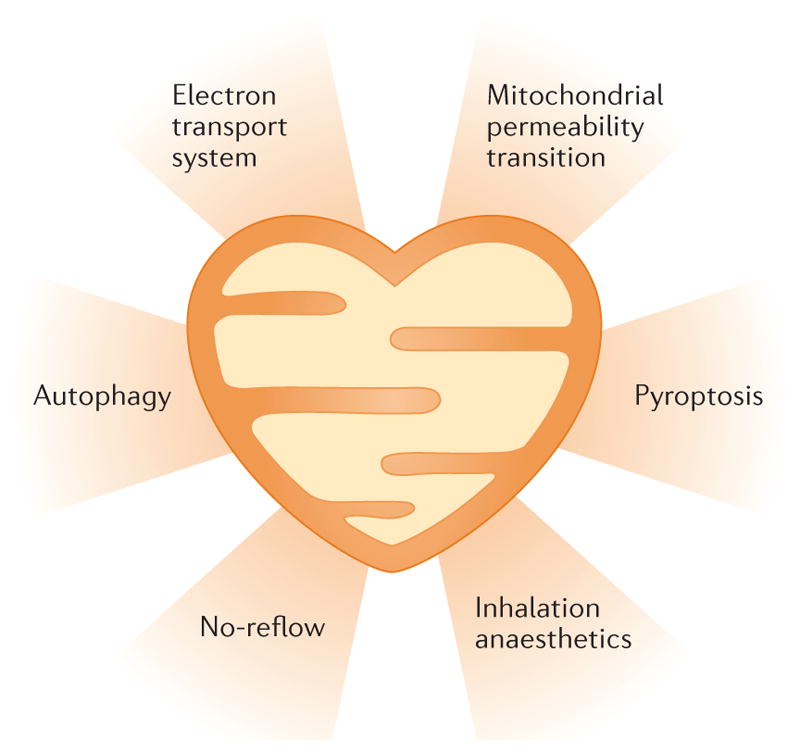
Some of these strategies involve mitochondrial structure and function, whereas others involve anaesthetic preconditioning and reducing the no-reflow phenomenon, which might also involve the mitochondria.
Targeting the mitochondria
Bioenergetics and ROS generation
The pathophysiology of cardiac reperfusion injury is complex, and varies over time after injury. Altered ion homeostasis, inflammation, activation of adaptive signalling pathways, metabolic substrate utilization shifts, and mitochondrial dysfunction are among the many factors that contribute to electromechanical dysfunction and tissue injury. The mitochondria are complex structures that undergo biogenesis, fission, and fusion independent of cell cycling. As central hubs of cellular energetics and redox signalling, the mitochondria influence virtually all cellular functions, particularly cell survival. They have been identified as a drug target in ischaemic heart disease, given the negative effects of increased generation of reactive oxygen species (ROS), abnormal cellular energetics, and impaired mitochondrial ion homeostasis on cardiac structure and function (FIG. 2; BOX 1)24–27. Mitochondrial functional aberrations are often inter-related, such as imbalances in calcium and sodium ion levels, opening of energy-dissipating channels or pores, and increased production of ROS27–30.
Figure 2. Sites of reactive oxygen species (ROS) production within the mitochondria.

Several mitochondrial targets along the outer/inner mitochondrial membranes and within the mitochondrial matrix represent potential approaches to mitigate ischaemia–reperfusion injury. Cyt c, cytochrome c; MAO, monoamine oxidase; NOX-4, NADPH oxidase 4; VDAC, voltage-dependent anion channel.
Box 1. Ischaemia–reperfusion injury: targeting the mitochondria.
TEMPO conjugates: XJB-5-131, mito-TEMPO
Permeability transition pore blockers: cyclosporine A, TRO-40303, platelet inhibitors
Exogenous quinones: idebenone, EPI-743, mitoQ
Transient/reversible electron transport chain inhibition: rotenone, mitoSNO, metformin
Superoxide dismutase mimetics: EUK8, EUK134, MCI-186, M40403, Me2DO2A, Mn-TBA
Attenuate release of mitochondrial DNA to lower pyroptosis
Stimulate mitophagy/autophagy (?)
Sustain mitochondrial membrane potential to support endogenous scavenging defences and ATP production
Formation of excess reactive oxygen intermediates, many of which originate from within the electron transport system and the mitochondrial matrix31,32, NADPH oxidase33, and catecholamine catabolism via monoamine oxidases34,35, cause cellular pathology in many acute and chronic diseases. A small amount of ROS production is essential for normal physiological signalling, but at higher levels, ROS are toxic36–41. During acute reperfusion injury, the rate of ROS production substantially outpaces the capacity of endogenous scavengers to reduce them. A number of strategies to eliminate ROS-mediated damage in the early reperfusion window have been clinically tested, but with limited success42–46. A major source of mitochondrial ROS in early reperfusion seems to be from increased succinate levels during the ischaemic period47, which is postulated to produce ROS by ‘reverse electron transfer’. ROS are released from mitochondria into respiratory complexes called supercomplexes, functional aggregates of the electron transport system that are essential for normal electron flux48–51. Fewer respiratory supercomplexes are formed in the setting of acute52 and chronic53,54 cardiac pathologies. Since ROS production seems to be inversely related to supercomplex formation55, the loss of supercomplexes might be directly linked to augmented ROS production in a ‘vicious cycle’. Interventions that promote sustained generation of supercomplexes might have the potential to reduce myocardial injury during ischaemia and reperfusion25.
Direct scavengers of ROS
Direct scavenging of radicals has long been proposed as a potential treatment for reperfusion injury25,27. Direct scavengers that lessen mitochondrial radical accumulation, such as the scavenger 2,2,6,6-tetramethylpiperidine-1-oxyl (TEMPO; targeted to mitochondria via gramicidin XJB-5-131 (REFS 52,56)) or triphenylphosphonium (Mito-TEMPO)57 are cardioprotective in preclinical models. The hydroxyl scavenger 3--methyl-1-phenyl-2-pyrazolin-5-one (MCI-186)58 and several newer superoxide dismutase mimetics such as EUK8/EUK134 (REFS 59–61), M40403 (REF. 62), Me2DO2A63, and Mn-TBPA64 that convert (via reduction reactions) harmful superoxide molecules into oxygen or hydrogen peroxide are cardioprotective in animal models of ischaemia–reperfusion. Unfortunately, these promising preclinical results have not been replicated in clinical trials65,66. Coenzyme Q10 (also known as ubiquinone) is an endogenous, free-radical-scavenging antioxidant reported to be beneficial in preclinical models of ischaemia–reperfusion67,68. Coenzyme Q10 administration in patients with heart failure significantly reduced cardiac mortality69. Quinones that target mitochondria, such as mitoQ70 and SkQ71, are showing promise in preclinical studies. However, at higher doses, cation conjugates can inhibit mitochondrial function72. Ideally, future exogenous scavengers should be engineered for improved tissue permeability and more specific intracellular or mitochondrial targets.
Interventions that inhibit mitochondrial respiration
Transient, partial inhibition of mitochondrial respiration during the vulnerable early-reperfusion phase might also be cardioprotective by limiting mitochondrial ROS production. Rotenone73 and several different biguanides (such as metformin)74 inhibit complex I of the mitochondrial respiratory chain, and have been shown to be cardioprotective when administered after ischaemia75,76. Notably, however, rotenone is a well-known neurotoxin, and its systemic effects are likely to limit its use in this setting. Likewise, inhibition of complex II (succinate dehydrogenase) with malonate (or the cell-permeable precursor dimethyl malonate) has been shown to reduce ROS and decrease ischaemia–reperfusion injury47,77,78. Furthermore, post-translational modification of electron transport complexes by S-nitrosothiols (mito-SNO) that are engineered to selectively block ROS protects mitochondrial function and lessens reperfusion injury in rodents79–83. The safety of salvaging tissue by transiently blunting mitochondrial bioenergetics must be determined before these approaches can be advanced to clinical trials. Furthermore, methods to target these strategies specifically to the heart (to avoid inhibiting respiration in other organ systems) must also be identified.
Interventions targeting post-translational modifications
Post-translational modifications of electron transport complexes and endogenous ROS-scavengers via sirtuins84 and O-GlcNAc modifications85, or succinylation86 might help in the search for novel pharmacological interventions to treat post-ischaemic injury. Given that the adaptor protein p66Shc catalyses ROS formation via cytochrome c, several studies have investigated the potential role of p66Shc as a regulator of myocardial injury in murine models of cardiac ischaemia and reperfusion. Genetic loss-of-function studies have yielded opposing findings regarding cardiac injury87,88. Isolated hearts from p66Shc knockout mice were protected against acute reperfusion injury88. However, a subsequent investigation assessing ischaemia–reperfusion in vivo found that neither knockout mice nor transient transfection with p66Shc small interfering RNA imparted cardioprotection87, and was actually deleterious. These differences might be related to inherent discrepancies between in vivo versus in vitro studies, as well as differing durations of ischaemic insult across studies. Further research is warranted to fully understand the role of p66Shc signalling and post-translational modifications in reperfusion injury89.
Interventions targeting mitochondrial membrane potential
Interventions to improve mitochondrial function often focus on mitochondrial membrane potential (Δψm). The transmembrane potential of mitochondria is established by electron-dependent proton pumping, and serves as the major energetic driver for the replenishment of ATP, cellular redox buffering, and ion/metabolite transport. Loss of Δψm during cardiac ischaemia–reperfusion is thought to be directly related to cellular injury and electromechanical dysfunction, as seen in in vitro models and intact hearts subjected to ischaemia–reperfusion injury28,43,90–93. Aon and colleagues hypothesized that pathophysiological ROS overflow occurs at both minimal and maximal Δψm94. Highly polarized Δψm is observed in mitochondria that are ‘idling’ (that is, mitochondrial respiration with little to no ATP turnover), suggesting that slight uncoupling might protect mitochondria by reducing ROS formation95. Although this strategy has been used to treat other diseases influenced by mitochondrial dysfunction96, mild uncoupling in the heart did not markedly reduce mitochondrial ROS production97, and actually led to increased ROS production in isolated myocytes98. Numerous studies have shown that cardioprotection is associated with sustained, polarized Δψm; this energized Δψm is essential during metabolic insult to replenish endogenous antioxidants via NADPH-linked reactions, which allows for the downstream detoxification of free radicals99,100. Cardioprotection inferred by sustained Δψm has been observed after exercise preconditioning91, after administering serofendic acid (a cardioprotective substance found in fetal calf serum which inhibits the oxidant-induced mitochondrial death pathway101), or by blocking energy-dissipating ion channels within the mitochondrial inner membrane102,103.
A reduction in Δψm is an early step in programmed cell death104, a process considered irreversible after the apoptotic cascade is initiated. However, even after activation of caspase 3 and DNA nicking, some cells (including primary cardiomyocytes) can recover through a process known as anastasis105. Caspase 3 is used extensively in normal development and in adult organ remodelling, so strategies that can activate the nonapoptotic properties of caspase 3 after cellular injury might provide a potential intervention for cardioprotection after ischaemia–reperfusion106.
The mitochondrial PTP
Potential strategies to preserve mitochondrial bioenergetics by targeting the permeability transition pore (PTP) have received considerable interest, and have been the subject of numerous reviews107–110. Inhibition of PTP at reperfusion is thought to be a primary mechanism by which ischaemic preconditioning and postconditioning protect the heart108,111. As calcium and ROS are postulated to increase the probability of the opening of the PTP, strategies that reduce ROS and lower mitochondrial calcium overload have been extensively explored as cardioprotective interventions25,27. Paradoxically, transient PTP opening seems to reduce mitochondrial calcium overload, analogous to a ‘pressure release valve’, whereby mitochondrial calcium decreases upon transient PTP opening112. This mechanism works to reset mitochondrial calcium concentration, and is postulated to prevent the sustained, large conductance, open conformation of PTP that is associated with pathological injury and cell death. Studies published in the past 6 years suggest that the PTP is also comprised of ATP-synthase (complex V) dimers and associated regulatory proteins113,114.
Use of the PTP blocker cyclosporine A for the treatment of acute coronary syndromes initially showed promise in small trials115,116, but a larger multicentre trial yielded neutral findings117. TRO-40303, a ligand of the putative PTP component translocator protein (also called the peripheral benzodiazepine receptor), did not impart any cardioprotective benefits in patients with MI118. The underlying factors involved in these neutral findings are clearly multifaceted; of note, however, most patients involved in PCI trials since 2005 have received platelet inhibitors as a standard-of-care therapy. As platelet inhibitors can independently protect the heart by inhibiting the PTP6,7,119, patients enrolled in trials that assessed the efficacy of PTP blockers might already be ‘conditioned’ against injury. Importantly, cyclosporine and this ‘conditioning’ effect do not prevent PTP opening, but instead simply raise the threshold for opening. The development of strategies targeting hexokinase (a regulator of PTP opening) to mitochondria120–122, as well as more specific PTP blockers that target mitochondria123 might lead to new therapies for the treatment of ischaemia–reperfusion injury. Several other mitochondrial approaches to protect the heart in this setting are also under development, including methods to deliver autologously derived mitochondria124–126, new mitochondria-targeting peptides43,127–130, strategies that target calcium-activated mitochondrial potassium channels131 or block ATP-sensitive potassium channels132–134, and approaches that block the inflammation caused by mitochondrial DNA after MI135,136.
Challenges and opportunities
Given the role of the mitochondria in reperfusion injury, novel compounds targeted towards altering mitochondrial bioenergetics might reduce myocardial injury. However, delivery of these compounds to mitochondria poses as a daunting problem. Timing is a major concern, because the compound must reach therapeutic levels during the appropriate window, which is often early in reperfusion. Given the importance of minimizing ‘door-to-balloon’ times, achieving therapeutic levels in time with systemic delivery is challenging; direct coronary catheter administration might be more feasible for reducing acute injury. Bioavailability of the compound poses a further challenge, because the compound must resist degradation (or sequestration) in the circulation, traverse the capillary network and cross cell membranes, and then localize to mitochondria. Medicinal chemistry considerations for mitochondria-targeted therapies include whether the targeting moiety is cleaved inside cells, as well as appropriate chemical linkers and positions to which the cargo should be attached on the parent mitochondria-targeting molecule. Despite these obstacles, a number of putative mitochondria-targeting strategies are emerging.
The highly negative Δψm of healthy mitochondria is frequently utilized as an attractant to deliver mitochondrial cargo via positively charged carriers. The most well-characterized conjugate family to date uses triphenylphosphonium, a cationic lipophile, to deliver therapies to mitochondria30,137–139. A similar approach is being developed using plant-derived cell-permeable cations such as berberine or palmatine140,141, although some of these compounds can inhibit respiration at low micromolar concentrations72.
Peptide conjugates are another emerging therapy targeted to mitochondria. Multiple peptide sequences containing alternating cationic and hydrophobic amino acids show cell permeability and mitochondrial localization, and are being developed as delivery vectors142. Examples of these peptides include mitochondria-penetrating peptides143,144 and several of the Szeto-Schiller peptides145. The ROS scavenger XJB-5-131 conjugates the scavenging TEMPO moiety to mitochondria via a gramicidin peptide sequence56; this sequence might be useful to deliver other cargo to mitochondria. Nanoparticles have also been developed to reduce cardiac mitochondrial injury146,147, although the nanoparticle vector might be more important for enhancing cellular uptake in damaged cells than for mitochondrial localization per se. However, cardiac toxicity has been observed with several types of nanoparticles148,149, indicating that more research is needed to develop these agents. Continued investigation into the safety and efficacy of vectors that deliver cargo to mitochondria is an exciting area for cardioprotective therapies.
Anaesthetic preconditioning
Clinical studies of anaesthetic conditioning of the heart have focused almost entirely on preconditioning and on patients undergoing cardiac surgery, for practical reasons — anaesthesiologists are present at the critical time before the coronaries are reperfused. Substantially less work on anaesthetic preconditioning has been conducted in MI models, and on anaesthetic postconditioning after cardiac surgery, despite the technique having therapeutic potential in these settings as well. Similar to ischaemic preconditioning, short exposure of the heart to halogenated volatile anaesthetics offers cardioprotection against subsequent ischaemic or ischaemia–reperfusion injury150. Pharmacological studies suggest that the mechanisms of anaesthetic and ischaemic preconditioning are similar. Volatile anaesthetic preconditioning is complex, and involves agents that increase mitochondrial ROS production; activate protein kinase C (PKC), extracellular signal-related kinase (ERK1/2, also known as mitogen activated protein kinase 3 and 1), and Akt (also known as Rac-α serine/threonine-protein kinase); open KATP channels acutely; and reduce myocardial oxygen consumption151–153. Given these signalling pathways are the same as those targeted by P2Y12 platelet inhibitors, anaesthetic preconditioning is not likely to provide additional protection in patients undergoing PCI who are treated with platelet inhibitors. Patients who are naive to these drugs, such as those undergoing cardiac surgery, are more likely to respond to anaesthetic preconditioning.
Data from preclinical studies suggest that activation of aldehyde dehydrogenase 2 by mitochondrial protein kinase C-ε (PKCε) is necessary for effective anaesthetic preconditioning154, as well as inactivation of cardiac L-type calcium channels155 and increased production of heat shock protein 90 (and its induction of endothelial nitric oxide synthase)156. Longer-term cardiac protection by anaesthetic preconditioning might be mediated through downstream effectors of hypoxia-inducible factor 1 (REF. 157), but its mechanism of action in this setting is unclear. The first double-blind, placebo-controlled trial of anaesthetic preconditioning in patients undergoing cardiac surgery used sevoflurane (5 min inhalation before cardioplegia) on a background of opioid maintenance anaesthesia158. Standard clinical laboratory evidence of MI (cardiac enzymes) were unaffected by this preconditioning regimen, but preconditioning reduced B-type natriuretic peptide levels. Atrial biopsies showed translocation of PKCε to the sarcolemma, mitochondria, and intercalated disks of cardiomyocytes after preconditioning158. Unsurprisingly to anaesthesiologists, treated patients required substantially more pressor support during preconditioning, but still had better glomerular filtration rates after CABG surgery than untreated patients, suggesting a preconditioning effect on the kidneys. Subsequent studies also reported cardioprotection with use of various inhaled anaesthetics after CABG surgery, but these effects were not potent enough to change major outcome variables159,160. In theory, an increase in dosage should lead to an increase in potency, but acute increases in the concentration of inhaled anaesthetics can lead to hypotension and cardiac depression. Similarly, intraoperative remote ischaemic preconditioning (via cuff inflation–deflation on the leg) added to isoflurane preconditioning did not increase the potency of anaesthetic preconditioning, and might even blunt the anaesthetic effect159.
Challenges and potential confounders
With a gradual increase in experience with application of anaesthetic preconditioning during CABG surgery, researchers found that the benefits of anaesthetic preconditioning for cardiac ischaemic events in animal models were not readily translated to humans. The heterogeneity of patients is often unmasked when translating studies from inbred rodent models to the clinic. Furthermore, most animal studies are performed in young animals, whereas most patients undergoing CABG surgery are not young, and senescent hearts are difficult to precondition, even in animal models161. Variables that can affect outcomes after cardiac surgery, such as other drugs, fluids, electrolytes, transfusions, and physiological gases administered, can be very difficult to control for in clinical studies. Furthermore, the inflammatory and oxidative stress-induced damage caused by ischaemia–reperfusion injuries are difficult to avoid in the operating room, as are presurgical comorbidities.
Hyperglycaemia, in particular, blunts anaesthetic preconditioning, in part by disruption of mitochondrial bioenergetics162. A study using human induced pluripotent stem cell (iPSC)-derived cardiomyocytes showed that opening of mitochondrial potassium channels was disrupted in the presence of high glucose levels163. Given the difficulty in designing clinical trials to assess the efficacy of anaesthetic preconditioning, and the slow progress in optimizing this technique owing to the complexity of cardiac surgery, human iPSC-derived cardiomyocytes are an important platform for probing anaesthetic preconditioning. These cells provide an opportunity for rigorous assessment of issues such as timing and amount of preconditioning, changes in molecular mediators of preconditioning as a function of genetic background and the cellular microenvironment, and potential synergistic combinations of anaesthetics and other drugs, all of which are nearly impossible to measure in the clinical setting. Combined with standard preclinical models, human iPSC technology should provide a platform for revisiting clinical anaesthetic preconditioning that allows assessment of the mechanisms underlying the anaesthetic effects.
Pyroptosis and the innate immune system
A study published in 2015 reported that strategies to prevent the escape of mitochondrial DNA (mtDNA) from ruptured mitochondria can be cardioprotective in the setting of infarction when applied just before reperfusion136. Two approaches were tested: the first involved mitochondrial-directed glycosylase, which removes oxidized bases from mtDNA created during reperfusion, thus favouring mtDNA repair over its release; and the second strategy involved use of intravenous DNase I, which degrades any extracellular DNA. The effect of both of these interventions on infarct-related injury were similar to that of conditioning, and most importantly, offers additive protection when combined with a P2Y12 inhibitor in animal models, suggesting that mechanism of mtDNA toxicity cannot be addressed by P2Y12 inhibitor administration alone. Therefore, strategies targeting released mtDNA are likely to be cardioprotective in patients who are already undergoing P2Y12 inhibitor treatment, and warrant further investigation.
The most likely explanation for the toxicity of mtDNA is inflammation, one of the earliest proposed mechanisms of cell death from ischaemia–reperfusion injury. In the 1980s, studies of inflammatory responses concentrated on leukocytes, because they quickly accumulate in large numbers in ischaemic tissue164. Although leukocytes are involved in cell death after MI, most approaches to prevent their accumulation did not limit infarction, and interest in this strategy subsequently waned164. In 2014, a reassessment of inflammation in ischaemia focused specifically on the innate immune system165. FIGURE 3 shows a diagram of the NLRP3 (also known as NACHT, LRR, and PYD domains-containing protein 3) inflammasome, an important component of the innate immune system. Invading pathogens release pathogen-associated molecular patterns (PAMPs) that signal the presence of sepsis. These PAMPs initiate interleukin and interferon production through the Toll-like receptor (TLR) system, whereby TLR4 recognizes lipopolysaccharide and TLR9 recognizes bacterial DNA. This sequence of events is often referred to as signal 1. PAMPs also trigger the assembly of the inflammasome, in a process known as signal 2. NLRP3 combines with the adaptor protein apoptosis-associated speck-like protein containing a CARD (ASC) and pro-caspase 1 to form active caspase 1. Caspase 1, in turn, cleaves pro-interleukins to form active IL-1β and IL-18 (REF. 166). Caspase 1 can also liberate the N-terminal segment of gasdermin D, which creates pores in the cell membrane, in a process known as pyroptosis. These membrane pores allow cytokines to exit the cell and call inflammatory cells to attack the invader, but they also inevitably kill the cell167,168.
Figure 3.

The interplay between Toll-like receptor 9 (TLR9) and the NLRP3 inflammasome. TLR9 ligands such as mitochondrial DNA promote production of IL-1β and IL-18 by activating MyD88 (myeloid differentiation primary response protein 88), which activates the transcription factor nuclear factor-κB (NF-κB) through phosphorylation of the NF-κB inhibitor IκB. This phosphorylation in turn allows p50 (also known as NF-κB1) and p65 (also known as NF-κB p65 subunit) to dimerize, forming active NF-κB. When triggered by oxidized mitochondrial DNA in the cytosol, NLRP3 (NACHT, LRR, and PYD domains-containing protein 3), the adaptor protein ASC (also known as apoptosis-associated speck-like protein containing a CARD), and pro-caspase 1 form an inflammasome complex that results in the release of active caspase 1. The pro-interleukins are cleaved by caspase 1 into their active form. Caspase 1 also cleaves gasdermin D (GSDMD) to form its N-terminal fragment (GSDMD-nt) that creates holes in the membrane allowing the proinflammatory interleukins and caspase 1 to flow into the interstitial space. Membrane failure caused by those holes also kills the cell, a process termed pyroptosis. Finally, caspase 1 can proteolytically degrade many cytosolic enzymes that can cause further injury. Cyto c, cytochrome c; DeUBase, deubiquitinase; PAMPs, pathogen-associated molecular patterns.
Sepsis is not the only trigger for inflammasome formation. Damage-associated molecular patterns (DAMPs) from injured tissue, such as mtDNA, can also trigger the formation of inflammasomes169. Reperfusion causes a burst of oxygen radicals in the mitochondria, which oxidizes bases in the mtDNA and promotes the opening of the PTP through which oxidized mtDNA fragments flow into the cytosol170. With its many unmethylated stretches of DNA in which the frequency of a cytosine immediately followed by a guanine is high, these mtDNA fragments resemble bacterial DNA, are TLR9 ligands, and are potent inducers of the NLRP3 inflammasome171. Extracellular mtDNA is a potent proinflammatory DAMP in traumatic shock172 and in ventilator-induced lung injury173.
Infusing mtDNA into the coronary artery increases infarct size in Krebs buffer-perfused rat hearts136; similarly, mtDNA added to culture medium induces cell death in cultured cardiomyocytes174, indicating that mtDNA triggers cytotoxic pathways in the heart’s parenchymal cells rather than just in the circulating white cells. Investigations into the role of the NLRP3 inflammasome in ischaemic injury have yielded mixed findings, with some studies reporting increased resistance to infarction in hearts in which NLRP3 inflammasome has been genetically deleted175,176, and others reporting neutral findings177,178. Importantly, the NLRP3 inflammasome is not the only activator of caspase 1; at least three other similar inflammasome systems that activate caspase 1 have been described179, which might explain why chronic deletion of the NLRP3 inflammasome might be compensated for by activation of another inflammasome. Several pharmacological NLRP3 inhibitors have been developed, which have all been effective in limiting infarct size in animal models180–182.
Attempts to mitigate caspase 1-mediated damage have yielded more consistently positive findings. In 2001, a study involving human atrial trabeculae subjected to simulated ischaemia–reperfusion showed that inhibition of caspase 1 with a highly selective inhibitor (Ac-YVAD-cmk) preserved contractile function176. IL-18 binding protein and an IL-1 receptor blocker were similarly protective, suggesting that interleukins were involved in mediating the injury. Ac-YVAD-cmk was also found to reduce infarct size in an open-chest rabbit model of surgically induced MI183. Furthermore, caspase 1 overexpression in mice increased infarct size after ischaemia–reperfusion by 50%, whereas complete knock-out of caspase 1 reduced infarct size175,184. The pan-caspase inhibitor z-VAD has also been reported to reduce infarct size185,186; caspase inhibition using z-VAD was assumed to target apoptosis, a process which involves caspase 3 and caspase 9, but the action of caspase 1 would have also been inhibited. Unlike necrosis and pyroptosis, apoptosis does not result in rapid membrane failure and could not have accounted for the washout of dehydrogenase enzymes in the studies of myocardial ischaemia– reperfusion187. However, the apoptotic process seems to be involved in the release of the mtDNA that triggers inflammasome activation188. Caspase 1 not only produces cytotoxic cytokines, but also proteolytically cleaves several important glycolytic enzymes, including glyceraldehyde phosphate dehydrogenase189. Loss of these glycolytic enzymes could contribute to infarction by impairing metabolic recovery during reperfusion. None of the aforementioned caspase 1 studies addressed the problem of possible redundancy with platelet inhibitors, but a study published in 2017 revealed that protection conferred by the highly selective caspase 1 inhibitor VX-765 is additive to the protective effects of the P2Y12 blocker cangrelor190.
Caspase 1 also inhibits autophagy (lysosomal-mediated degradation) of mitochondria (also known as mitophagy)191. Given that mtDNA can promote inflammation in response to ischaemia–reperfusion, mitophagy might be cardioprotective in this setting. Under normal conditions, the body is careful not to release free mtDNA into the cytosol. When senescent mitochondria are eliminated by autophagy, they are enveloped in a vacuole and digested by enzymes such as DNase II. Complete knock-out of DNase II produces a heart failure phenotype, which could be rescued using a TLR9 blocker, indicating that mtDNA that escaped from the vacuole was responsible for the phenotype192. Stimulating mitophagy at a time when mitochondria will be injured by ischaemia–reperfusion might attenuate mtDNA release, and could be yet another explanation as to why autophagy can reduce ischaemic injury.
Autophagy as a new therapeutic target
Role in ischaemic preconditioning
Since the discovery of preconditioning in 1986, investigators have sought to identify pharmacological triggers, elucidate the contributing signal transduction pathways, and define the mechanisms involved193. Autophagy is induced by many of the classic cardioprotective agents, including ischaemic preconditioning itself194, as well as diazoxide194, the adenosine A1 receptor agonist 2-chloro-N(6)-cyclopentyladenosine195, sulfaphenazole194, rapamycin196, valsartan197, resveratrol198, non-sirtuin histone deactylase inhibitors199, and remote ischaemic conditioning200. Many investigators have demonstrated that autophagy is cardioprotective201–203, and several studies have demonstrated an absolute requirement for functional autophagy in cardioprotection194–197,204. Furthermore, selective mitophagy has been shown to be essential for cardioprotection by ischaemic preconditioning205 and statins206. Despite some early contradictory studies207,208, autophagy is now generally regarded as cardioprotective when initiated before ischaemic stress. Although clearance of damaged mitochondria is undoubtedly an important protective consequence of autophagy, autophagy is likely to be cardioprotective through other mechanisms as well, including sequestration and degradation of glycogen, and removal of protein aggregates209,210.
Role in postconditioning
The role of autophagy in postischaemic conditioning is more controversial, in part because of the complex roles and interpretation of autophagy markers. Detection of an increase in autophagosomes by immunostaining or the widely used autophagosome marker LC3-II might indicate upregulation of autophagy, but could also mean impaired autophagosome clearance by lysosomes (impaired flux). Moreover, interventions that restore flux could be misinterpreted as downregulation of autophagy211. Therefore, interpretation of autophagy studies must take into account whether flux was assessed. Most, but not all, reports support a beneficial role for autophagy in postconditioning212–215.
Harmful effects
Autophagy often has a protective role in the heart, but in some settings might also be detrimental. For instance, autophagic cell death, or autosis, has been reported in the setting of cerebral ischaemia, and abrogation of autophagy through gene silencing or chemical inhibitors can reduce cell death216,217. Interestingly, a chemical screen identified cardiac glycosides (digoxin, digitoxigenin, and strophanthidin) as antagonists of autosis218. In the heart, less is known about the settings in which autosis might occur. Beclin 1 has been shown to be important in mediating autosis in the heart219, and haploinsufficiency of Beclin 1 might be cardioprotective220.
Regulatory pathways
Autophagy is primarily regulated by the nutrient-sensing and energy-sensing pathways, namely mechanistic target of rapamycin (mTOR) and AMP-dependent protein kinase (AMPK), respectively. However, multiple other signal transduction pathways are also involved in autophagy, including forkhead box protein O1, p38 mitogen-activated protein kinase, and Akt 2 (also known as RAC-β serine/threonine protein kinase)221. Importantly, autophagy and mitophagy are regulated by sirtuins, a family of NAD-dependent protein deacetylases222–224. Numerous studies have revealed a role for microRNAs (miRNAs) in the regulation of cardiac autophagy; some of these miRNAs are in the circulation or are delivered via exosomes225–240. The term ‘autophagoMIR’ has been used to designate miRNAs that regulate autophagy, and includes both suppressors and activators of autophagy (TABLE 1). Importantly, miR-144 delivered via exosomes has been implicated as a circulating effector of remote preconditioning241. miR-30 targets Beclin 1, which has a dual role in initiation of autophagy, but also can interfere with autophagic flux242. Therefore, downregulation of Beclin 1 can have unpredictable effects on autophagy, and autophagic flux should always be measured to allow for meaningful interpretation of autophagy studies.
Table 1.
MicroRNAs that regulate autophagy in the heart
| MicroRNA | Target | Effect on autophagy | Refs |
|---|---|---|---|
| miR-212/132 | FOXO3a | Suppression | 286 |
| miR-199a | GSK3β | Suppression | 232,236 |
| miR-221 | p27 | Suppression | 236 |
| miR-204 | LC3 | Suppression | 287–289 |
| miR-188-3p | ATG7 | Suppression | 237 |
| miR-497 | LC3b, BCL2 | Suppression | 233 |
| miR-34a | ATG9 | Suppression | 290 |
| miR-451 | TSC1 | Suppression or possibly increased flux | 291 |
| miR-30 | Beclin 1 | Suppression of initiation and/or flux | 292 |
| miR-99a | mTOR | Activation | 293 |
| miR-144 | mTOR | Activation | 241 |
Cardiac remodelling
The role of autophagy in cardiac remodelling is controversial, with numerous studies reporting evidence for a protective role209,243,244, an adverse role220, or both199,245. Autophagy has also been implicated in the pathological role of angiotensin II on hypertrophy246, but its role in this setting is somewhat controversial247, and might be related to a functional dichotomy of autophagy or the nonspecific criteria for defining upregulated autophagy. In the absence of measures of autophagic flux, misinterpretation of LC3 levels or autophagosome abundance is likely211. However, the roles for mitochondrial autophagy in cardiac homeostasis248, adaptation to stress204,249, and handling of misfolded proteins209 are well established.
Lifestyle factors
Autophagy is a homeostatic process that evolved from the need to survive episodes of nutritional deprivation, and is invoked during sleep, when caloric intake is absent; chronic nutritional excess, however, eventually suppresses autophagy. Caloric restriction potently upregulates autophagy, and intermittent fasting — even in the absence of reduced caloric intake — can upregulate autophagy and benefit the heart250,251. Similarly, time-restricted feeding (8 h per day) results in a period of fasting and can protect mice against the ill effects of a high-fat diet252. A high-fat diet has been shown to impair autophagy253,254 and reduce ischaemic tolerance255, underscoring the importance of intact autophagic flux in cardioprotection. Like caloric restriction and intermittent fasting, exercise upregulates autophagy and is associated with cardioprotection256,257. Exercise-induced autophagy is important for clearing protein aggregates and ameliorates cardiac proteinopathy; the beneficial effects of exercise were enhanced in mice with an overexpression of autophagy-related protein 7 (REF. 209).
Cardioprotective effects of autophagy
Autophagy fulfils many roles in the heart. During ischaemia, autophagy provides amino acids and monosaccharides (from glycogenolysis) for ATP production, and is also essential for clearance of protein aggregates that accumulate in both heritable proteinopathies and in acquired heart failure209. During both ischaemia–reperfusion and pressure overload, autophagy serves to sequester and degrade damaged mitochondria that might otherwise trigger apoptosis or necroptosis. Moreover, autophagy is important in preventing the activation of the innate immune system (TLR9 and NLRP3), which can be triggered by mitochondrial cardiolipin, mtDNA, and oxidized mtDNA188,192,258. Finally, mitophagy is important for mitochondrial quality control. Loss of oxidative phosphorylation capacity, whether through protein damage or mtDNA mutations, results in lower membrane potential, and mitophagy selectively eliminates the least functional organelles. Emerging evidence reveals a link between mitophagy and mitochondrial biogenesis259,260, so that after eliminating damaged organelles, a stimulus expands the mitochondrial pool to restore ATP production capacity. One important benefit of mitophagy might be the activation of mitochondrial biogenesis, which would facilitate mitochondrial quality control. Indeed, exercise training, known to upregulate autophagy and protect against MI, has also been shown to improve myocardial energy metabolism and enhance mitochondrial biogenesis260. In addition to intermittent fasting and exercise, drugs that enhance mitophagy and biogenesis are also cardioprotective, and include statins261 and NAD, which activates sirtuins262; this list is likely to expand to include preconditioning and postconditioning agents.
Fragmented mitochondria have been observed in numerous disease states in the heart, and targeting mitochondrial dynamics might be another area for therapeutic development263. Inhibition of mitochondrial fission has been shown to protect cardiac cells from reperfusion injury264. Treatment with a pharmacological inhibitor of Drp1-dependent fission, mitochondrial division inhibitor 1 (Mdivi1), protects the heart when administered either directly264 or as a nanoparticle conjugate146. These strategies are postulated to prevent mitochondrial outer membrane permeabilization, which is triggered by Bax translocation during ischaemia–reperfusion, resulting in cytochrome c release and the onset of cell death cascades265. However, Drp1 (the target of Mdivi1), has been shown to mitigate mitochondrial dysfunction and heart failure in a pressure-overload model243.
No-reflow as a therapeutic target
In the setting of acute MI, no-reflow is defined as a lack of perfusion to portions of the myocardium, despite reopening of the occluded infarct-related epicardial coronary artery266–268. Therapies targeting no-reflow have the potential to further preserve cardiac structure and function following MI. In a canine model of proximal coronary artery occlusion of 90 min, the fluorescent dye thioflavin S, which stains endothelial cells receiving flow, failed to penetrate much of the subendocardium when injected into the vasculature after the proximal coronary artery was reopened269. Perfusion defects were observed as early as 10–12 s after the reopening of the proximal coronary artery and seemed to increase in size as the duration of the reperfusion phase was extended to several hours270, suggesting that no-reflow is a true form of reperfusion injury. Other techniques confirm the phenomenon of no-reflow, including failure of carbon black and radioactive microspheres to penetrate portions of previously ischaemic myocardium269,271.
What causes no-reflow and what is its importance in the pathophysiology of disease- Our group has studied the ultrastructural features of the myocardium within the anatomical no-reflow zone, and found localized swelling or blistering of the endothelium of small blood vessels and capillaries269. Membrane-bound intraluminal blebs, protruding from the endothelial cells, obstructed the lumen of small blood vessels. Mitochondrial abnormalities might contribute to no-reflow. Zhao and colleagues showed that inhibiting the opening of the mitochondrial ATP-sensitive potassium channels prevents simvastatin from exerting a beneficial effect on reducing no-reflow272. Given that the zone of no-reflow is confined within areas of dead cardiomyocytes, and ultrastructural evidence of the microvasculature injury lags behind ultrastructural evidence of cardiomyocyte cell death, no-reflow is unlikely to contribute directly to additional myocyte cell death. However, given that blood-borne trophic factors and cells crucial to the removal of necrotic debris and scar can neither enter nor exit the zone of no-reflow, no-reflow inhibits the healing phase of MI. In our study, we observed that rats subjected to proximal coronary occlusion and reperfusion demonstrated persistent areas of no-reflow at 1 month after acute infarction. Hearts with larger zones of no-reflow and fewer perfused capillaries had greater myocardial infarct expansion (stretching and thinning of the infarcted wall) and dilatation of the left ventricle, signs of poor healing, and adverse LV remodelling273.
The no-reflow phenomenon has been well documented in patients undergoing reperfusion therapy (either thrombolytic or PCI) for acute STEMI268,274–276. Several different noninvasive imaging techniques, such as echocardiography contrast imaging, nuclear imaging with 201 Tl-labelled or 99mTc-labelled albumin microspheres, and MRI with contrast, have been used to document perfusion defects after reperfusion therapy. Preclinical studies verified that MRI-visualized perfusion deficits correlate closely (by location and shape) with perfusion defects observed using thioflavin S staining277,278.
No-reflow has also been associated with adverse LV remodelling in the clinical setting, which is an important observation because the degree of LV remodelling, including dilatation of the LV cavity, is predictive of increased mortality and congestive heart failure after MI279–281. In one study, LV volumes were larger (170 ml) in patients with no-reflow than in patients without no-reflow (115 ml) at 6 months after MI. No-reflow was the most important predictor of LV dilatation, even more so than infarct size, and was the only predictor of cardiac death279. Several clinical studies have also shown that no-reflow in the setting of MI is associated with a poor prognosis275,278–280,282,283. Wu and colleagues demonstrated that patients with no-reflow had more major adverse cardiac events (cardiac death, nonfatal myocardial reinfarction, congestive heart failure, stroke, and unstable angina) over 2 years compared with patients without no-reflow278. This finding was consistent in a multiple regression analysis that accounted for the size of the MI. Ndrepepa and colleagues reported that the presence of no-reflow was predictive of 5-year mortality (OR 1.72–2.01, P = 0.004), again independent of infarct size. They concluded that the presence of no-reflow following intervention for STEMI provides prognostic information independent of and beyond that provided by infarct size282.
Several therapies have been targeted at reducing the size of the no-reflow zone. Experimentally, therapies that reduce MI above and beyond reperfusion often reduce the zone of no-reflow within the infarct. In our experience, such therapies must be given before reperfusion to be of use. However, a major issue limiting application of adjunctive therapies is that stopping to administer a drug or therapy before reperfusion is often seen as a delay in door-to-balloon time, and interventional cardiologists are not willing to risk extending that time. We previously described a therapy that reduces no-reflow independent of any effect on the size of the infarct, which can be administered after the infarct artery is patent284. Delayed therapeutic hypothermia administered locally to the risk zone significantly reduced the zone of no-reflow in both rabbit and rat models of MI. In the rabbit model, local hypothermia applied as late as 30 min after establishing patency of the infarct-related artery markedly reduced the size of the no-reflow zone within the infarct284. Myocardial temperature is lowered to 30–34°C for several hours. We believe that this reduction in temperature is likely to reduce endothelial swelling and inflammation, much like how ice reduces blistering after a burn. We plan to determine the long-term consequences of delayed therapeutic hypothermia and predict that it will reduce adverse LV remodelling and long-term cardiac function by enhancing tissue healing. Targeting the no-reflow phenomenon in a way that will not extend door-to-balloon times and is not dependent on a reduction of infarct size provides a novel therapeutic approach that could help to fill the unmet need of unacceptably high morbidity and mortality following MI, despite successful PCI. However, the mechanisms underlying the protective effects of hypothermia on no-reflow are unknown. Based on our observations of the natural history of no-reflow, as well as studies that we and others have conducted in an attempt to understand how early hypothermia can reduce myocardial infarct size, several potential targets of hypothermia have been postulated, including mitogen-activated protein kinases285, local tissue and endothelial oedema, ROS damage and cellular inflammation, and mitochondrial structure and function.
Conclusions
Despite the success of early reperfusion to reduce myocardial infarct size and improve clinical outcome, the high rates of heart failure and mortality following STEMI remain unacceptable. Given that most clinical trials of adjunctive therapies (adjunctive to therapeutic reperfusion) aimed at further reducing myocardial infarct size have been disappointing, new approaches are needed. In this Review, we presented several new therapeutic targets involved in ischaemia–reperfusion injury that have shown promise in preclinical studies, including therapies to reduce myocardial infarct size based on mitochondrial protective properties; approaches that involve the mechanisms of autophagy, mitophagy, and pyroptosis; strategies that take advantage of the cardioprotective properties of certain anaesthetic agents; treatments that show cardioprotective benefits in addition to the effects of certain antiplatelet agents; and therapies that are focused on correcting the no-reflow phenomenon and its effect on healing and adverse LV remodelling.
Key points.
Mortality after acute ST-segment elevation myocardial infarction (STEMI) has plateaued in the past decade, after a period of continual decline
Beyond early reperfusion, a variety of adjunctive agents to reduce myocardial infarction after reperfusion have been tested in clinical settings, and have yielded largely disappointing results
Before clinical testing, candidate cardioprotective agents should be tested in animal models with comorbidities similar to that in patients, and in combination with P2Y12 inhibitors
Approaches for improving cardiac function following STEMI that have been promising in preclinical studies include those that target mitochondrial bioenergetics, pyroptosis, autophagy, and no-reflow and anaesthetic preconditioning
Acknowledgments
D.A.B. has received funding from the NIH (NHLBI R01-123647). R.A.G. is supported by NIH (P01 HL112730, R01 HL132075).
Footnotes
Author contributions
R.A.K., D.A.B., M.C., W.D., J.M.D., R.G., and J.S. researched data for the article. R.A.K., D.A.B., M.C., J.M.D., and R.G. wrote the article and substantially contributed to the discussion of its content. R.A.K., D.A.B., M.C., J.M.D., R.G., S.L.H., and J.S. reviewed/edited the manuscript before submission.
Competing interests statement
R.A.K. has received grant funding from Faraday Pharmaceuticals and Laboratoires Servier. D.A.B. has received funding from Catabasis Pharmaceuticals and Stealth BioTherapeutics, and has received consulting income from Stealth BioTherapeutics. The other authors declare no competing interests.
References
- 1.Gerczuk PZ, Kloner RA. An update on cardioprotection: a review of the latest adjunctive therapies to limit myocardial infarction size in clinical trials. J Am Coll Cardiol. 2012;59:969–978. doi: 10.1016/j.jacc.2011.07.054. [DOI] [PubMed] [Google Scholar]
- 2.Hausenloy DJ, et al. Targeting reperfusion injury in patients with ST-segment elevation myocardial infarction: trials and tribulations. Eur Heart J. 2016;38:935–941. doi: 10.1093/eurheartj/ehw145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Menees DS, et al. Door-to-balloon time and mortality among patients undergoing primary PCI. N Engl J Med. 2013;369:901–909. doi: 10.1056/NEJMoa1208200. [DOI] [PubMed] [Google Scholar]
- 4.Mozaffarian D, et al. Heart disease and stroke statistics—2015 update: a report from the American Heart Association. Circulation. 2015;131:e29–322. doi: 10.1161/CIR.0000000000000152. [DOI] [PubMed] [Google Scholar]
- 5.Iliodromitis EK, et al. What is wrong with cardiac conditioning? We may shoot moving targets. J Cardiovasc Pharmacol Ther. 2015;20:357–369. doi: 10.1177/1074248414566459. [DOI] [PubMed] [Google Scholar]
- 6.Yang XM, et al. Platelet P2Y12 blockers confer direct postconditioning-like protection in reperfused rabbit hearts. J Cardiovasc Pharmacol Ther. 2013;18:251–262. doi: 10.1177/1074248412467692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yang XM, et al. Two classes of anti-platelet drugs reduce anatomical infarct size in monkey hearts. Cardiovasc Drugs Ther. 2013;27:109–115. doi: 10.1007/s10557-012-6436-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yang XM, Cui L, Alhammouri A, Downey JM, Cohen MV. Triple therapy greatly increases myocardial salvage during ischemia/reperfusion in the in situ rat heart. Cardiovasc Drugs Ther. 2013;27:403–412. doi: 10.1007/s10557-013-6474-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Luz A, et al. Lack of benefit of ischemic postconditioning after routine thrombus aspiration during reperfusion: immediate and midterm results. J Cardiovasc Pharmacol Ther. 2015;20:523–531. doi: 10.1177/1074248415578171. [DOI] [PubMed] [Google Scholar]
- 10.Mentias A, et al. Ischemic postconditioning during primary percutaneous coronary intervention. Catheter Cardiovasc Interv. 2017 doi: 10.1002/ccd.26965. http://dx.doi.org/10.1002/ccd.26965. [DOI] [PubMed]
- 11.Elbadawi A, Ha LD, Abuzaid AS, Crimi G, Azzouz MS. Meta-analysis of randomized trials on remote ischemic conditioning during primary percutaneous coronary intervention in patients with ST-segment elevation myocardial infarction. Am J Cardiol. 2017;119:832–838. doi: 10.1016/j.amjcard.2016.11.036. [DOI] [PubMed] [Google Scholar]
- 12.Gao J, et al. The effects of remote ischemic conditioning in patients with ST-segment elevation myocardial infarction treated with primary percutaneous coronary intervention: a meta-analysis. Minerva Med. 2017 doi: 10.23736/S0026-4806.17.04631-6. http://dx.doi.org/10.23736/s0026-4806.17.04631-6. [DOI] [PubMed]
- 13.Le Page S, Bejan-Angoulvant T, Angoulvant D, Prunier F. Remote ischemic conditioning and cardioprotection: a systematic review and meta-analysis of randomized clinical trials. Bas Res Cardiol. 2015;110:11. doi: 10.1007/s00395-015-0467-8. [DOI] [PubMed] [Google Scholar]
- 14.Man C, Gong D, Zhou Y, Fan Y. Meta-analysis of remote ischemic conditioning in patients with acute myocardial infarction. Sci Rep. 2017;7:43529. doi: 10.1038/srep43529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hale SL, Kloner RA. Myocardial temperature in acute myocardial infarction: protection with mild regional hypothermia. Am J Physiol. 1997;273:H220–227. doi: 10.1152/ajpheart.1997.273.1.H220. [DOI] [PubMed] [Google Scholar]
- 16.Herring MJ, Dai W, Hale SL, Kloner RA. Rapid induction of hypothermia by the thermosuit system profoundly reduces infarct size and anatomic zone of no reflow following ischemia-reperfusion in rabbit and rat hearts. J Cardiovasc Pharmacol Ther. 2015;20:193–202. doi: 10.1177/1074248414535664. [DOI] [PubMed] [Google Scholar]
- 17.Pfeffer MA, et al. Effect of captopril on mortality and morbidity in patients with left ventricular dysfunction after myocardial infarction. Results of the survival and ventricular enlargement trial. The SAVE Investigators. N Engl J Med. 1992;327:669–677. doi: 10.1056/NEJM199209033271001. [DOI] [PubMed] [Google Scholar]
- 18.Fisher SA, Doree C, Taggart DP, Mathur A, Martin-Rendon E. Cell therapy for heart disease: trial sequential analyses of two Cochrane reviews. Clin Pharmacol Ther. 2016;100:88–101. doi: 10.1002/cpt.344. [DOI] [PubMed] [Google Scholar]
- 19.Zhu K, Li J, Wang Y, Lai H, Wang C. Nanoparticles-assisted stem cell therapy for ischemic heart disease. Stem Cells Int. 2016;2016:1384658. doi: 10.1155/2016/1384658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tang J, et al. Therapeutic microparticles functionalized with biomimetic cardiac stem cell membranes and secretome. Nat Commun. 2017;8:13724. doi: 10.1038/ncomms13724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gerczuk PZ, Kloner RA. Protecting the heart from ischemia: an update on ischemic and pharmacologic conditioning. Hosp Pract. 2011;39:35–43. doi: 10.3810/hp.2011.08.577. 1995. [DOI] [PubMed] [Google Scholar]
- 22.Kloner RA, Dai W, Hale SL, Shi J. Approaches to improving cardiac structure and function during and after an acute myocardial infarction: acute and chronic phases. J Cardiovasc Pharmacol Ther. 2015;21:363–367. doi: 10.1177/1074248415616187. [DOI] [PubMed] [Google Scholar]
- 23.US National Library of Medicine. Transplantation of autologous derived mitochondria following ischemia. ClinicalTrialsgov. 2016 https://clinicaltrials.gov/ct2/show/NCT02851758.
- 24.Brown DA, et al. Expert consensus document: mitochondrial function as a therapeutic target in heart failure. Nat Rev Cardiol. 2016;14:238–250. doi: 10.1038/nrcardio.2016.203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Brown DA, Sabbah HN, Shaikh SR. Mitochondrial inner membrane lipids and proteins as targets for decreasing cardiac ischemia/reperfusion injury. Pharmacol Ther. 2013;140:258–266. doi: 10.1016/j.pharmthera.2013.07.005. [DOI] [PubMed] [Google Scholar]
- 26.Murphy E, Steenbergen C. Mechanisms underlying acute protection from cardiac ischemia-reperfusion injury. Physiol Rev. 2008;88:581–609. doi: 10.1152/physrev.00024.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Walters AM, Porter GA, Jr, Brookes PS. Mitochondria as a drug target in ischemic heart disease and cardiomyopathy. Circ Res. 2012;111:1222–1236. doi: 10.1161/CIRCRESAHA.112.265660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Brown DA, O’Rourke B. Cardiac mitochondria and arrhythmias. Cardiovasc Res. 2010;88:241–249. doi: 10.1093/cvr/cvq231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hausenloy DJ, Yellon DM. Targeting myocardial reperfusion injury—the search continues. N Engl J Med. 2015;373:1073–1075. doi: 10.1056/NEJMe1509718. [DOI] [PubMed] [Google Scholar]
- 30.Murphy E, Steenbergen C. Ion transport and energetics during cell death and protection. Physiol (Bethesda, Md ) 2008;23:115–123. doi: 10.1152/physiol.00044.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Murphy MP. How mitochondria produce reactive oxygen species. Biochem J. 2009;417:1–13. doi: 10.1042/BJ20081386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Orr AL, et al. Inhibitors of ROS production by the ubiquinone-binding site of mitochondrial complex I identified by chemical screening. Free Radic Biol Med. 2013;65:1047–1059. doi: 10.1016/j.freeradbiomed.2013.08.170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Siu KL, Lotz C, Ping P, Cai H. Netrin-1 abrogates ischemia/reperfusion-induced cardiac mitochondrial dysfunction via nitric oxide-dependent attenuation of NOX4 activation and recoupling of NOS. J Mol Cell Cardiol. 2015;78:174–185. doi: 10.1016/j.yjmcc.2014.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kaludercic N, Carpi A, Menabo R, Di Lisa F, Paolocci N. Monoamine oxidases (MAO) in the pathogenesis of heart failure and ischemia/reperfusion injury. Biochim Biophys Acta. 2011;1813:1323–1332. doi: 10.1016/j.bbamcr.2010.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kaludercic N, Mialet-Perez J, Paolocci N, Parini A, Di Lisa F. Monoamine oxidases as sources of oxidants in the heart. J Mol Cell Cardiol. 2014;73:34–42. doi: 10.1016/j.yjmcc.2013.12.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Alleman RJ, Katunga LA, Nelson MA, Brown DA, Anderson EJ. The “Goldilocks Zone” from a redox perspective — Adaptive vs. deleterious responses to oxidative stress in striated muscle. Frontiers Physiol. 2014;5:358. doi: 10.3389/fphys.2014.00358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Frasier CR, et al. Redox-dependent increases in glutathione reductase and exercise preconditioning: role of NADPH oxidase and mitochondria. Cardiovasc Res. 2013;98:47–55. doi: 10.1093/cvr/cvt009. [DOI] [PubMed] [Google Scholar]
- 38.Nelson MJ, Harris MB, Boluyt MO, Hwang HS, Starnes JW. Effect of N-2- mercaptopropionyl glycine on exercise-induced cardiac adaptations. Am J Physiol Regul Integr Comp Physiol. 2011;300:R993–R1000. doi: 10.1152/ajpregu.00405.2010. [DOI] [PubMed] [Google Scholar]
- 39.Pain T, et al. Opening of mitochondrial KATP channels triggers the preconditioned state by generating free radicals. Circ Res. 2000;87:460–466. doi: 10.1161/01.res.87.6.460. [DOI] [PubMed] [Google Scholar]
- 40.Rosca MG, Tandler B, Hoppel CL. Mitochondria in cardiac hypertrophy and heart failure. J Mol Cell Cardiol. 2013;55:31–41. doi: 10.1016/j.yjmcc.2012.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Song M, et al. Super-suppression of mitochondrial reactive oxygen species signaling impairs compensatory autophagy in primary mitophagic cardiomyopathy. Circ Res. 2014;115:348–353. doi: 10.1161/CIRCRESAHA.115.304384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bolli R, et al. Direct evidence that oxygen-derived free radicals contribute to postischemic myocardial dysfunction in the intact dog. Proc Natl Acad Sci USA. 1989;86:4695–4699. doi: 10.1073/pnas.86.12.4695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kloner RA, et al. Reduction of ischemia/reperfusion injury with bendavia, a mitochondria-targeting cytoprotective peptide. J Am Heart Assoc. 2012;1:e001644. doi: 10.1161/JAHA.112.001644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zweier JL. Measurement of superoxide-derived free radicals in the reperfused heart. Evidence for a free radical mechanism of reperfusion injury. J Biol Chem. 1988;263:1353–1357. [PubMed] [Google Scholar]
- 45.Zweier JL, Flaherty JT, Weisfeldt ML. Direct measurement of free radical generation following reperfusion of ischemic myocardium. Proc Natl Acad Sci USA. 1987;84:1404–1407. doi: 10.1073/pnas.84.5.1404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zweier JL, et al. Measurement and characterization of postischemic free radical generation in the isolated perfused heart. J Biol Chem. 1989;264:18890–18895. [PubMed] [Google Scholar]
- 47.Chouchani ET, et al. Ischaemic accumulation of succinate controls reperfusion injury through mitochondrial ROS. Nature. 2014;515:431–435. doi: 10.1038/nature13909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Acin-Perez R, Fernandez-Silva P, Peleato ML, Perez-Martos A, Enriquez JA. Respiratory active mitochondrial supercomplexes. Mol Cell. 2008;32:529–539. doi: 10.1016/j.molcel.2008.10.021. [DOI] [PubMed] [Google Scholar]
- 49.Genova ML, Lenaz G. Functional role of mitochondrial respiratory supercomplexes. Biochim Biophys Acta. 2014;1837:427–443. doi: 10.1016/j.bbabio.2013.11.002. [DOI] [PubMed] [Google Scholar]
- 50.Lapuente-Brun E, et al. Supercomplex assembly determines electron flux in the mitochondrial electron transport chain. Science. 2013;340:1567–1570. doi: 10.1126/science.1230381. [DOI] [PubMed] [Google Scholar]
- 51.Schagger H. Respiratory chain supercomplexes of mitochondria and bacteria. Biochim Biophys Acta. 2002;1555:154–159. doi: 10.1016/s0005-2728(02)00271-2. [DOI] [PubMed] [Google Scholar]
- 52.Jang S, et al. Elucidating mitochondrial electron transport chain supercomplexes in the heart during ischemia-reperfusion. Antioxid Redox Signal. 2016;27:57–69. doi: 10.1089/ars.2016.6635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Gomez LA, Monette JS, Chavez JD, Maier CS, Hagen TM. Supercomplexes of the mitochondrial electron transport chain decline in the aging rat heart. Arch Biochem Biophys. 2009;490:30–35. doi: 10.1016/j.abb.2009.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Rosca M, Minkler P, Hoppel CL. Cardiac mitochondria in heart failure: normal cardiolipin profile and increased threonine phosphorylation of complex IV. Biochim Biophys Acta. 2011;1807:1373–1382. doi: 10.1016/j.bbabio.2011.02.003. [DOI] [PubMed] [Google Scholar]
- 55.Maranzana E, Barbero G, Falasca AI, Lenaz G, Genova ML. Mitochondrial respiratory supercomplex association limits production of reactive oxygen species from complex I. Antioxid Redox Signal. 2013;19:1469–1480. doi: 10.1089/ars.2012.4845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Escobales N, et al. Mitochondria-targeted ROS scavenger improves post-ischemic recovery of cardiac function and attenuates mitochondrial abnormalities in aged rats. J Mol Cell Cardiol. 2014;77:136–146. doi: 10.1016/j.yjmcc.2014.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ni R, et al. Therapeutic inhibition of mitochondrial reactive oxygen species with mito-TEMPO reduces diabetic cardiomyopathy. Free Radic Biol Med. 2016;90:12–23. doi: 10.1016/j.freeradbiomed.2015.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Eto M, Kajihara N, Morita S, Tominaga R. A novel electron paramagnetic resonance spin-probe technique demonstrates the relation between the production of hydroxyl radicals and ischemia-reperfusion injury. Eur J Cardiothorac Surg. 2011;39:465–470. doi: 10.1016/j.ejcts.2010.08.011. [DOI] [PubMed] [Google Scholar]
- 59.Liu Y, et al. The alternative crosstalk between RAGE and nitrative thioredoxin inactivation during diabetic myocardial ischemia-reperfusion injury. Am J Physiol Endocrinol Metab. 2012;303:E841–E852. doi: 10.1152/ajpendo.00075.2012. [DOI] [PubMed] [Google Scholar]
- 60.Pucheu S, Boucher F, Malfroy B, De Leiris J. Protective effect of the superoxide scavenger EUK8 against ultrastructural alterations induced by ischemia and reperfusion in isolated rat hearts. Nutrition. 1995;11:582–584. [PubMed] [Google Scholar]
- 61.van Empel VP, et al. EUK-8, a superoxide dismutase and catalase mimetic, reduces cardiac oxidative stress and ameliorates pressure overload-induced heart failure in the harlequin mouse mutant. J Am Coll Cardiol. 2006;48:824–832. doi: 10.1016/j.jacc.2006.02.075. [DOI] [PubMed] [Google Scholar]
- 62.Xu Y, Liu B, Zweier JL, He G. Formation of hydrogen peroxide and reduction of peroxynitrite via dismutation of superoxide at reperfusion enhances myocardial blood flow and oxygen consumption in postischemic mouse heart. J Pharmacol Exp Ther. 2008;327:402–410. doi: 10.1124/jpet.108.142372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Nistri S, et al. A new low molecular weight, MnII-containing scavenger of superoxide anion protects cardiac muscle cells from hypoxia/reoxygenation injury. Free Radic Res. 2015;49:67–77. doi: 10.3109/10715762.2014.979168. [DOI] [PubMed] [Google Scholar]
- 64.Bovo E, Lipsius SL, Zima AV. Reactive oxygen species contribute to the development of arrhythmogenic Ca2+ waves during beta-adrenergic receptor stimulation in rabbit cardiomyocytes. J Physiol. 2012;590:3291–3304. doi: 10.1113/jphysiol.2012.230748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Flaherty JT, et al. Recombinant human superoxide dismutase (h-SOD) fails to improve recovery of ventricular function in patients undergoing coronary angioplasty for acute myocardial infarction. Circulation. 1994;89:1982–1991. doi: 10.1161/01.cir.89.5.1982. [DOI] [PubMed] [Google Scholar]
- 66.Tsujita K, et al. Effects of edaravone on reperfusion injury in patients with acute myocardial infarction. Am J Cardiol. 2004;94:481–484. doi: 10.1016/j.amjcard.2004.05.007. [DOI] [PubMed] [Google Scholar]
- 67.Maulik N, et al. Dietary coenzyme Q(10) supplement renders swine hearts resistant to ischemia-reperfusion injury. Am J Physiol Heart Circ Physiol. 2000;278:H1084–H1090. doi: 10.1152/ajpheart.2000.278.4.H1084. [DOI] [PubMed] [Google Scholar]
- 68.Whitman GJ, et al. The mechanisms of coenzyme Q10 as therapy for myocardial ischemia reperfusion injury. Mol Aspects Med. 1997;18(Suppl):S195–S203. doi: 10.1016/s0098-2997(97)00017-4. [DOI] [PubMed] [Google Scholar]
- 69.Mortensen SA, et al. The effect of coenzyme Q10 on morbidity and mortality in chronic heart failure: results from Q-SYMBIO: a randomized double-blind trial. JACC Heart Fail. 2014;2:641–649. doi: 10.1016/j.jchf.2014.06.008. [DOI] [PubMed] [Google Scholar]
- 70.Adlam VJ, et al. Targeting an antioxidant to mitochondria decreases cardiac ischemia-reperfusion injury. FASEB J. 2005;19:1088–1095. doi: 10.1096/fj.05-3718com. [DOI] [PubMed] [Google Scholar]
- 71.Skulachev VP, et al. An attempt to prevent senescence: a mitochondrial approach. Biochim Biophys Acta. 2009;1787:437–461. doi: 10.1016/j.bbabio.2008.12.008. [DOI] [PubMed] [Google Scholar]
- 72.Lyamzaev KG, et al. Novel mitochondria-targeted antioxidants: plastoquinone conjugated with cationic plant alkaloids berberine and palmatine. Pharm Res. 2011;28:2883–2895. doi: 10.1007/s11095-011-0504-8. [DOI] [PubMed] [Google Scholar]
- 73.Lesnefsky EJ, et al. Blockade of electron transport during ischemia protects cardiac mitochondria. J Biol Chem. 2004;279:47961–47967. doi: 10.1074/jbc.M409720200. [DOI] [PubMed] [Google Scholar]
- 74.Bridges HR, Jones AJ, Pollak MN, Hirst J. Effects of metformin and other biguanides on oxidative phosphorylation in mitochondria. Biochem J. 2014;462:475–487. doi: 10.1042/BJ20140620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Chen Q, Camara AK, Stowe DF, Hoppel CL, Lesnefsky EJ. Modulation of electron transport protects cardiac mitochondria and decreases myocardial injury during ischemia and reperfusion. Am J Physiol Cell Physiol. 2007;292:C137–C147. doi: 10.1152/ajpcell.00270.2006. [DOI] [PubMed] [Google Scholar]
- 76.Yin M, et al. Metformin improves cardiac function in a nondiabetic rat model of post-MI heart failure. Am J Physiol Heart Circ Physiol. 2011;301:H459–H468. doi: 10.1152/ajpheart.00054.2011. [DOI] [PubMed] [Google Scholar]
- 77.Valls-Lacalle L, et al. Succinate dehydrogenase inhibition with malonate during reperfusion reduces infarct size by preventing mitochondrial permeability transition. Cardiovasc Res. 2016;109:374–384. doi: 10.1093/cvr/cvv279. [DOI] [PubMed] [Google Scholar]
- 78.Wojtovich AP, Brookes PS. The endogenous mitochondrial complex II inhibitor malonate regulates mitochondrial ATP-sensitive potassium channels: implications for ischemic preconditioning. Biochim Biophys Acta. 2008;1777:882–889. doi: 10.1016/j.bbabio.2008.03.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Chouchani ET, et al. Identification of S-nitrosated mitochondrial proteins by S-nitrosothiol difference in gel electrophoresis (SNO-DIGE): implications for the regulation of mitochondrial function by reversible S-nitrosation. Biochem J. 2010;430:49–59. doi: 10.1042/BJ20100633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Chouchani ET, et al. Cardioprotection by S-nitrosation of a cysteine switch on mitochondrial complex I. Nat Med. 2013;19:753–759. doi: 10.1038/nm.3212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Methner C, et al. Mitochondria selective S-nitrosation by mitochondria-targeted S-nitrosothiol protects against post-infarct heart failure in mouse hearts. Eur J Heart Fail. 2014;16:712–717. doi: 10.1002/ejhf.100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Nadtochiy SM, Burwell LS, Brookes PS. Cardioprotection and mitochondrial S-nitrosation: effects of S-nitroso-2-mercaptopropionyl glycine (SNO-MPG) in cardiac ischemia-reperfusion injury. J Mol Cell Cardiol. 2007;42:812–825. doi: 10.1016/j.yjmcc.2007.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Prime TA, et al. A mitochondria-targeted S-nitrosothiol modulates respiration, nitrosates thiols, and protects against ischemia-reperfusion injury. Proc Natl Acad Sci USA. 2009;106:10764–10769. doi: 10.1073/pnas.0903250106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Porter GA, Urciuoli WR, Brookes PS, Nadtochiy SM. SIRT3 deficiency exacerbates ischemia-reperfusion injury: implication for aged hearts. Am J Physiol Heart Circ Physiol. 2014;306:H1602–H1609. doi: 10.1152/ajpheart.00027.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Dassanayaka S, Jones SP. O-GlcNAc and the cardiovascular system. Pharmacol Ther. 2014;142:62–71. doi: 10.1016/j.pharmthera.2013.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Boylston JA, et al. Characterization of the cardiac succinylome and its role in ischemia-reperfusion injury. J Mol Cell Cardiol. 2015;88:73–81. doi: 10.1016/j.yjmcc.2015.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Akhmedov A, et al. Genetic deletion of the adaptor protein p66Shc increases susceptibility to short-term ischaemic myocardial injury via intracellular salvage pathways. Eur Heart J. 2015;36:516–526. doi: 10.1093/eurheartj/ehu400. [DOI] [PubMed] [Google Scholar]
- 88.Carpi A, et al. The cardioprotective effects elicited by p66(Shc) ablation demonstrate the crucial role of mitochondrial ROS formation in ischemia/reperfusion injury. Biochim Biophys Acta. 2009;1787:774–780. doi: 10.1016/j.bbabio.2009.04.001. [DOI] [PubMed] [Google Scholar]
- 89.Heusch G. Mitochondria at the heart of cardiovascular protection: p66shc-friend or foe? Eur Heart J. 2015;36:469–471. doi: 10.1093/eurheartj/ehu409. [DOI] [PubMed] [Google Scholar]
- 90.Akar FG, Aon MA, Tomaselli GF, O’Rourke B. The mitochondrial origin of postischemic arrhythmias. J Clin Invest. 2005;115:3527–3535. doi: 10.1172/JCI25371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Alleman RJ, et al. Exercise-induced protection against reperfusion arrhythmia involves stabilization of mitochondrial energetics. Am J Physiol Heart Circ Physiol. 2016;310:H1360–H1370. doi: 10.1152/ajpheart.00858.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Matsumoto-Ida M, Akao M, Takeda T, Kato M, Kita T. Real-time 2-photon imaging of mitochondrial function in perfused rat hearts subjected to ischemia/reperfusion. Circulation. 2006;114:1497–1503. doi: 10.1161/CIRCULATIONAHA.106.628834. [DOI] [PubMed] [Google Scholar]
- 93.Slodzinski MK, Aon MA, O’Rourke B. Glutathione oxidation as a trigger of mitochondrial depolarization and oscillation in intact hearts. J Mol Cell Cardiol. 2008;45:650–660. doi: 10.1016/j.yjmcc.2008.07.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Aon MA, Cortassa S, O’Rourke B. Redox-optimized ROS balance: a unifying hypothesis. Biochim Biophys Acta. 2010;1797:865–877. doi: 10.1016/j.bbabio.2010.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Brand MD. Uncoupling to survive? The role of mitochondrial inefficiency in ageing. Exp Gerontol. 2000;35:811–820. doi: 10.1016/s0531-5565(00)00135-2. [DOI] [PubMed] [Google Scholar]
- 96.Smith BK, et al. Salsalate (salicylate) uncouples mitochondria, improves glucose homeostasis, and reduces liver lipids independent of AMPK-β1. Diabetes. 2016;65:3352–3361. doi: 10.2337/db16-0564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Quarrie R, et al. Mitochondrial uncoupling does not decrease reactive oxygen species production after ischemia-reperfusion. Am J Physiol Heart Circ Physiol. 2014;307:H996–H1004. doi: 10.1152/ajpheart.00189.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Brennan JP, et al. Mitochondrial uncoupling, with low concentration FCCP, induces ROS-dependent cardioprotection independent of KATP channel activation. Cardiovasc Res. 2006;72:313–321. doi: 10.1016/j.cardiores.2006.07.019. [DOI] [PubMed] [Google Scholar]
- 99.Nickel AG, et al. Reversal of mitochondrial transhydrogenase causes oxidative stress in heart failure. Cell Metab. 2015;22:472–484. doi: 10.1016/j.cmet.2015.07.008. [DOI] [PubMed] [Google Scholar]
- 100.O’Rourke B. Metabolism: beyond the power of mitochondria. Nat Rev Cardiol. 2016;13:386–388. doi: 10.1038/nrcardio.2016.95. [DOI] [PubMed] [Google Scholar]
- 101.Ioroi T, et al. Serofendic acid protects against myocardial ischemia-reperfusion injury in rats. J Pharmacol Sci. 2014;126:274–280. doi: 10.1254/jphs.14139fp. [DOI] [PubMed] [Google Scholar]
- 102.Brown DA, et al. Cardiac arrhythmias induced by glutathione oxidation can be inhibited by preventing mitochondrial depolarization. J Mol Cell Cardiol. 2010;48:673–679. doi: 10.1016/j.yjmcc.2009.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Solhjoo S, O’Rourke B. Mitochondrial instability during regional ischemia-reperfusion underlies arrhythmias in monolayers of cardiomyocytes. J Mol Cell Cardiol. 2015;78:90–99. doi: 10.1016/j.yjmcc.2014.09.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Zamzami N, et al. Reduction in mitochondrial potential constitutes an early irreversible step of programmed lymphocyte death in vivo. J Exp Med. 1995;181:1661–1672. doi: 10.1084/jem.181.5.1661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Tang HL, et al. Cell survival, DNA damage, and oncogenic transformation after a transient and reversible apoptotic response. Mol Biol Cell. 2012;23:2240–2252. doi: 10.1091/mbc.E11-11-0926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Ding AX, et al. CasExpress reveals widespread and diverse patterns of cell survival of caspase-3 activation during development in vivo. eLife. 2016;5:e10936. doi: 10.7554/eLife.10936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Alam MR, Baetz D, Ovize M. Cyclophilin D and myocardial ischemia-reperfusion injury: a fresh perspective. J Mol Cell Cardiol. 2015;78:80–89. doi: 10.1016/j.yjmcc.2014.09.026. [DOI] [PubMed] [Google Scholar]
- 108.Baines CP. The mitochondrial permeability transition pore and ischemia-reperfusion injury. Bas Res Cardiol. 2009;104:181–188. doi: 10.1007/s00395-009-0004-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Bernardi P, Di Lisa F. The mitochondrial permeability transition pore: molecular nature and role as a target in cardioprotection. J Mol Cell Cardiol. 2015;78:100–106. doi: 10.1016/j.yjmcc.2014.09.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Ong SB, Samangouei P, Kalkhoran SB, Hausenloy DJ. The mitochondrial permeability transition pore and its role in myocardial ischemia reperfusion injury. J Mol Cell Cardiol. 2015;78:23–34. doi: 10.1016/j.yjmcc.2014.11.005. [DOI] [PubMed] [Google Scholar]
- 111.Hausenloy DJ, Yellon DM, Mani-Babu S, Duchen MR. Preconditioning protects by inhibiting the mitochondrial permeability transition. Am J Physiol Heart Circ Physiol. 2004;287:H841–H849. doi: 10.1152/ajpheart.00678.2003. [DOI] [PubMed] [Google Scholar]
- 112.Lu X, Kwong JQ, Molkentin JD, Bers DM. Individual cardiac mitochondria undergo rare transient permeability transition pore openings. Circ Res. 2016;118:834–841. doi: 10.1161/CIRCRESAHA.115.308093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Carraro M, et al. Channel formation by yeast F-ATP synthase and the role of dimerization in the mitochondrial permeability transition. J Biol Chem. 2014;289:15980–15985. doi: 10.1074/jbc.C114.559633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Giorgio V, et al. Dimers of mitochondrial ATP synthase form the permeability transition pore. Proc Natl Acad Sci USA. 2013;110:5887–5892. doi: 10.1073/pnas.1217823110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Mewton N, et al. Effect of cyclosporine on left ventricular remodeling after reperfused myocardial infarction. J Am Coll Cardiol. 2010;55:1200–1205. doi: 10.1016/j.jacc.2009.10.052. [DOI] [PubMed] [Google Scholar]
- 116.Piot C, et al. Effect of cyclosporine on reperfusion injury in acute myocardial infarction. N Engl J Med. 2008;359:473–481. doi: 10.1056/NEJMoa071142. [DOI] [PubMed] [Google Scholar]
- 117.Cung TT, et al. Cyclosporine before PCI in Patients with Acute Myocardial Infarction. N Engl J Med. 2015;373:1021–1031. doi: 10.1056/NEJMoa1505489. [DOI] [PubMed] [Google Scholar]
- 118.Atar D, et al. Effect of intravenous TRO40303 as an adjunct to primary percutaneous coronary intervention for acute ST-elevation myocardial infarction: MITOCARE study results. Eur Heart J. 2015;36:112–119. doi: 10.1093/eurheartj/ehu331. [DOI] [PubMed] [Google Scholar]
- 119.Bell RM, et al. 9th Hatter Biannual Meeting: position document on ischaemia/reperfusion injury, conditioning and the ten commandments of cardioprotection. Bas Res Cardiol. 2016;111:41. doi: 10.1007/s00395-016-0558-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Calmettes G, et al. Hexokinases and cardioprotection. J Mol Cell Cardiol. 2015;78:107–115. doi: 10.1016/j.yjmcc.2014.09.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Halestrap AP, Pereira GC, Pasdois P. The role of hexokinase in cardioprotection - mechanism and potential for translation. Br J Pharmacol. 2015;172:2085–2100. doi: 10.1111/bph.12899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Nederlof R, Eerbeek O, Hollmann MW, Southworth R, Zuurbier CJ. Targeting hexokinase II to mitochondria to modulate energy metabolism and reduce ischaemia-reperfusion injury in heart. Br J Pharmacol. 2014;171:2067–2079. doi: 10.1111/bph.12363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Dube H, et al. A mitochondrial-targeted cyclosporin A with high binding affinity for cyclophilin D yields improved cytoprotection of cardiomyocytes. Biochem J. 2012;441:901–907. doi: 10.1042/BJ20111301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Cowan DB, et al. Intracoronary delivery of mitochondria to the ischemic heart for cardioprotection. PLoS ONE. 2016;11:e0160889. doi: 10.1371/journal.pone.0160889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Kaza AK, et al. Myocardial rescue with autologous mitochondrial transplantation in a porcine model of ischemia/reperfusion. J Thorac Cardiovasc Surg. 2017;153:934–943. doi: 10.1016/j.jtcvs.2016.10.077. [DOI] [PubMed] [Google Scholar]
- 126.Masuzawa A, et al. Transplantation of autologously derived mitochondria protects the heart from ischemia-reperfusion injury. Am J Physiol Heart Circ Physiol. 2013;304:H966–H982. doi: 10.1152/ajpheart.00883.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Brown DA, et al. Reduction of early reperfusion injury with the mitochondria-targeting peptide Bendavia. J Cardiovasc Pharmacol Ther. 2013;153:934–943. doi: 10.1177/1074248413508003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Dai W, et al. Cardioprotective effects of mitochondria-targeted peptide SBT-20 in two different models of rat ischemia/reperfusion. Cardiovasc Drugs Ther. 2016;30:559–566. doi: 10.1007/s10557-016-6695-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Shi J, et al. Bendavia restores mitochondrial energy metabolism gene expression and suppresses cardiac fibrosis in the border zone of the infarcted heart. Life Sci. 2015;141:170–178. doi: 10.1016/j.lfs.2015.09.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Zhao K, et al. Cell-permeable peptide antioxidants targeted to inner mitochondrial membrane inhibit mitochondrial swelling, oxidative cell death, and reperfusion injury. J Biol Chem. 2004;279:34682–34690. doi: 10.1074/jbc.M402999200. [DOI] [PubMed] [Google Scholar]
- 131.Tano JY, Gollasch M. Calcium-activated potassium channels in ischemia reperfusion: a brief update. Front Physiol. 2014;5:381. doi: 10.3389/fphys.2014.00381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Foster DB, et al. Mitochondrial ROMK channel is a molecular component of mitoKATP. Circ Res. 2012;111:446–454. doi: 10.1161/CIRCRESAHA.112.266445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Foster MN, Coetzee WA. KATP channels in the cardiovascular system. Physiol Rev. 2016;96:177–252. doi: 10.1152/physrev.00003.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Lefer DJ, Nichols CG, Coetzee WA. Sulfonylurea receptor 1 subunits of ATP-sensitive potassium channels and myocardial ischemia/reperfusion injury. Trends Cardiovasc Med. 2009;19:61–67. doi: 10.1016/j.tcm.2009.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.West AP, et al. Mitochondrial DNA stress primes the antiviral innate immune response. Nature. 2015;520:553–557. doi: 10.1038/nature14156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Yang XM, et al. Mitochondrially targeted Endonuclease III has a powerful anti-infarct effect in an in vivo rat model of myocardial ischemia/reperfusion. Bas Res Cardiol. 2015;110:3. doi: 10.1007/s00395-014-0459-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Skulachev MV, et al. Mitochondrial-targeted plastoquinone derivatives. Effect on senescence and acute age-related pathologies. Curr Drug Targets. 2011;12:800–826. doi: 10.2174/138945011795528859. [DOI] [PubMed] [Google Scholar]
- 138.Smith RA, Hartley RC, Cocheme HM, Murphy MP. Mitochondrial pharmacology. Trends Pharmacol Sci. 2012;33:341–352. doi: 10.1016/j.tips.2012.03.010. [DOI] [PubMed] [Google Scholar]
- 139.Smith RA, Hartley RC, Murphy MP. Mitochondria-targeted small molecule therapeutics and probes. Antioxid Redox Signal. 2011;15:3021–3038. doi: 10.1089/ars.2011.3969. [DOI] [PubMed] [Google Scholar]
- 140.Chernyak BV, et al. Novel mitochondria-targeted compounds composed of natural constituents: conjugates of plant alkaloids berberine and palmatine with plastoquinone. Biochem Biokhimiia. 2012;77:983–995. doi: 10.1134/S0006297912090040. [DOI] [PubMed] [Google Scholar]
- 141.Pustovidko AV, et al. Derivatives of the cationic plant alkaloids berberine and palmatine amplify protonophorous activity of fatty acids in model membranes and mitochondria. Mitochondrion. 2013;13:520–525. doi: 10.1016/j.mito.2012.09.006. [DOI] [PubMed] [Google Scholar]
- 142.Yousif LF, Stewart KM, Horton KL, Kelley SO. Mitochondria-penetrating peptides: sequence effects and model cargo transport. Chembiochem. 2009;10:2081–2088. doi: 10.1002/cbic.200900017. [DOI] [PubMed] [Google Scholar]
- 143.Horton KL, Pereira MP, Stewart KM, Fonseca SB, Kelley SO. Tuning the activity of mitochondria-penetrating peptides for delivery or disruption. Chembiochem. 2012;13:476–485. doi: 10.1002/cbic.201100415. [DOI] [PubMed] [Google Scholar]
- 144.Horton KL, Stewart KM, Fonseca SB, Guo Q, Kelley SO. Mitochondria-penetrating peptides. Chem Biol. 2008;15:375–382. doi: 10.1016/j.chembiol.2008.03.015. [DOI] [PubMed] [Google Scholar]
- 145.Szeto HH, Schiller PW. Novel therapies targeting inner mitochondrial membrane—from discovery to clinical development. Pharm Res. 2011;28:2669–2679. doi: 10.1007/s11095-011-0476-8. [DOI] [PubMed] [Google Scholar]
- 146.Ikeda G, et al. Nanoparticle-mediated targeting of cyclosporine A enhances cardioprotection against ischemia-reperfusion injury through inhibition of mitochondrial permeability transition pore opening. Sci Rep. 2016;6:20467. doi: 10.1038/srep20467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Ishikita A, et al. Nanoparticle-mediated delivery of mitochondrial division inhibitor 1 to the myocardium protects the heart from ischemia-reperfusion injury through inhibition of mitochondria outer membrane permeabilization: a new therapeutic modality for acute myocardial infarction. J Am Heart Assoc. 2016;5:e003872. doi: 10.1161/JAHA.116.003872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Guerrero-Beltran CE, et al. Silica nanoparticles induce cardiotoxicity interfering with energetic status and Ca2+ handling in adult rat cardiomyocytes. Am J Physiol Heart Circ Physiol. 2017;312:H645–H661. doi: 10.1152/ajpheart.00564.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Thompson LC, et al. Pulmonary instillation of multi-walled carbon nanotubes promotes coronary vasoconstriction and exacerbates injury in isolated hearts. Nanotoxicology. 2014;8:38–49. doi: 10.3109/17435390.2012.744858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Cope DK, Impastato WK, Cohen MV, Downey JM. Volatile anesthetics protect the ischemic rabbit myocardium from infarction. Anesthesiology. 1997;86:699–709. doi: 10.1097/00000542-199703000-00023. [DOI] [PubMed] [Google Scholar]
- 151.De Hert SG, et al. Sevoflurane but not propofol preserves myocardial function in coronary surgery patients. Anesthesiology. 2002;97:42–49. doi: 10.1097/00000542-200207000-00007. [DOI] [PubMed] [Google Scholar]
- 152.Mellidis K, et al. Activation of prosurvival signaling pathways during the memory phase of volatile anesthetic preconditioning in human myocardium: a pilot study. Mol Cell Biochem. 2014;388:195–201. doi: 10.1007/s11010-013-1910-5. [DOI] [PubMed] [Google Scholar]
- 153.Riess ML, Stowe DF, Warltier DC. Cardiac pharmacological preconditioning with volatile anesthetics: from bench to bedside? Am J Physiol Heart Circ Physiol. 2004;286:H1603–H1607. doi: 10.1152/ajpheart.00963.2003. [DOI] [PubMed] [Google Scholar]
- 154.Lang XE, et al. Isoflurane preconditioning confers cardioprotection by activation of ALDH2. PLoS ONE. 2013;8:e52469. doi: 10.1371/journal.pone.0052469. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 155.Tampo A, Hogan CS, Sedlic F, Bosnjak ZJ, Kwok WM. Accelerated inactivation of cardiac L-type calcium channels triggered by anaesthetic-induced preconditioning. Br J Pharmacol. 2009;156:432–443. doi: 10.1111/j.1476-5381.2008.00026.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Amour J, et al. Role of heat shock protein 90 and endothelial nitric oxide synthase during early anesthetic and ischemic preconditioning. Anesthesiology. 2009;110:317–325. doi: 10.1097/ALN.0b013e3181942cb4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Hieber S, Huhn R, Hollmann MW, Weber NC, Preckel B. Hypoxia-inducible factor 1 and related gene products in anaesthetic-induced preconditioning. Eur J Anaesthesiol. 2009;26:201–206. doi: 10.1097/eja.0b013e3283212cbb. [DOI] [PubMed] [Google Scholar]
- 158.Julier K, et al. Preconditioning by sevoflurane decreases biochemical markers for myocardial and renal dysfunction in coronary artery bypass graft surgery: a double-blinded, placebo-controlled, multicenter study. Anesthesiology. 2003;98:1315–1327. doi: 10.1097/00000542-200306000-00004. [DOI] [PubMed] [Google Scholar]
- 159.Lucchinetti E, et al. Remote ischemic preconditioning applied during isoflurane inhalation provides no benefit to the myocardium of patients undergoing on-pump coronary artery bypass graft surgery: lack of synergy or evidence of antagonism in cardioprotection? Anesthesiology. 2012;116:296–310. doi: 10.1097/ALN.0b013e318242349a. [DOI] [PubMed] [Google Scholar]
- 160.Suleiman MS, Zacharowski K, Angelini GD. Inflammatory response and cardioprotection during open-heart surgery: the importance of anaesthetics. Br J Pharmacol. 2008;153:21–33. doi: 10.1038/sj.bjp.0707526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Nguyen LT, et al. Attenuation of isoflurane-induced preconditioning and reactive oxygen species production in the senescent rat heart. Anesthesia Analgesia. 2008;107:776–782. doi: 10.1213/ane.0b013e318180419d. [DOI] [PubMed] [Google Scholar]
- 162.Kehl F, et al. Hyperglycemia prevents isoflurane-induced preconditioning against myocardial infarction. Anesthesiology. 2002;96:183–188. doi: 10.1097/00000542-200201000-00032. [DOI] [PubMed] [Google Scholar]
- 163.Canfield SG, et al. Marked hyperglycemia attenuates anesthetic preconditioning in human-induced pluripotent stem cell-derived cardiomyocytes. Anesthesiology. 2012;117:735–744. doi: 10.1097/ALN.0b013e3182655e96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Saxena A, Russo I, Frangogiannis NG. Inflammation as a therapeutic target in myocardial infarction: learning from past failures to meet future challenges. Transl Res. 2016;167:152–166. doi: 10.1016/j.trsl.2015.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Takahashi M. NLRP3 inflammasome as a novel player in myocardial infarction. Int Heart J. 2014;55:101–105. doi: 10.1536/ihj.13-388. [DOI] [PubMed] [Google Scholar]
- 166.Franchi L, Munoz-Planillo R, Nunez G. Sensing and reacting to microbes through the inflammasomes. Nat Immunol. 2012;13:325–332. doi: 10.1038/ni.2231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Aglietti RA, et al. GsdmD p30 elicited by caspase-11 during pyroptosis forms pores in membranes. Proc Natl Acad Sci USA. 2016;113:7858–7863. doi: 10.1073/pnas.1607769113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Liu X, et al. Inflammasome-activated gasdermin D causes pyroptosis by forming membrane pores. Nature. 2016;535:153–158. doi: 10.1038/nature18629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Arnoult D, Soares F, Tattoli I, Girardin SE. Mitochondria in innate immunity. EMBO Rep. 2011;12:901–910. doi: 10.1038/embor.2011.157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Garcia N, Chavez E. Mitochondrial DNA fragments released through the permeability transition pore correspond to specific gene size. Life Sci. 2007;81:1160–1166. doi: 10.1016/j.lfs.2007.08.019. [DOI] [PubMed] [Google Scholar]
- 171.Man SM, Karki R, Kanneganti TD. DNA-sensing inflammasomes: regulation of bacterial host defense and the gut microbiota. Pathog Dis. 2016;74:ftw028. doi: 10.1093/femspd/ftw028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Zhang Q, et al. Circulating mitochondrial DAMPs cause inflammatory responses to injury. Nature. 2010;464:104–107. doi: 10.1038/nature08780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Simmons JD, et al. Mitochondrial DNA damage associated molecular patterns in ventilator-associated pneumonia: prevention and reversal by intratracheal DNase I. J Trauma Acute Care Surg. 2017;82:120–125. doi: 10.1097/TA.0000000000001269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Bliksoen M, et al. Extracellular mtDNA activates NF-κB via toll-like receptor 9 and induces cell death in cardiomyocytes. Bas Res Cardiol. 2016;111:42. doi: 10.1007/s00395-016-0553-6. [DOI] [PubMed] [Google Scholar]
- 175.Kawaguchi M, et al. Inflammasome activation of cardiac fibroblasts is essential for myocardial ischemia/reperfusion injury. Circulation. 2011;123:594–604. doi: 10.1161/CIRCULATIONAHA.110.982777. [DOI] [PubMed] [Google Scholar]
- 176.Pomerantz BJ, Reznikov LL, Harken AH, Dinarello CA. Inhibition of caspase 1 reduces human myocardial ischemic dysfunction via inhibition of IL-18 and IL-1β. Proc Natl Acad Sci USA. 2001;98:2871–2876. doi: 10.1073/pnas.041611398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Jong WM, et al. Nlrp3 plays no role in acute cardiac infarction due to low cardiac expression. Int J Cardiol. 2014;177:41–43. doi: 10.1016/j.ijcard.2014.09.148. [DOI] [PubMed] [Google Scholar]
- 178.Sandanger O, et al. NLRP3 inflammasome activation during myocardial ischemia reperfusion is cardioprotective. Biochem Biophys Res Commun. 2016;469:1012–1020. doi: 10.1016/j.bbrc.2015.12.051. [DOI] [PubMed] [Google Scholar]
- 179.Schroder K, Tschopp J. The inflammasomes. Cell. 2010;140:821–832. doi: 10.1016/j.cell.2010.01.040. [DOI] [PubMed] [Google Scholar]
- 180.Marchetti C, et al. A novel pharmacologic inhibitor of the NLRP3 inflammasome limits myocardial injury after ischemia-reperfusion in the mouse. J Cardiovasc Pharmacol. 2014;63:316–322. doi: 10.1097/FJC.0000000000000053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Mastrocola R, et al. Pharmacological inhibition of NLRP3 inflammasome attenuates myocardial ischemia/reperfusion injury by activation of RISK and mitochondrial pathways. Oxid Med Cell Longev. 2016;2016:5271251. doi: 10.1155/2016/5271251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.van Hout GP, et al. The selective NLRP3- inflammasome inhibitor MCC950 reduces infarct size and preserves cardiac function in a pig model of myocardial infarction. Eur Heart J. 2016;38:828–836. doi: 10.1093/eurheartj/ehw247. [DOI] [PubMed] [Google Scholar]
- 183.Holly TA, et al. Caspase inhibition reduces myocyte cell death induced by myocardial ischemia and reperfusion in vivo. J Mol Cell Cardiol. 1999;31:1709–1715. doi: 10.1006/jmcc.1999.1006. [DOI] [PubMed] [Google Scholar]
- 184.Syed FM, et al. Proapoptotic effects of caspase-1/interleukin-converting enzyme dominate in myocardial ischemia. Circ Res. 2005;96:1103–1109. doi: 10.1161/01.RES.0000166925.45995.ed. [DOI] [PubMed] [Google Scholar]
- 185.Koshinuma S, Miyamae M, Kaneda K, Kotani J, Figueredo VM. Combination of necroptosis and apoptosis inhibition enhances cardioprotection against myocardial ischemia-reperfusion injury. J Anesthesia. 2014;28:235–241. doi: 10.1007/s00540-013-1716-3. [DOI] [PubMed] [Google Scholar]
- 186.Mocanu MM, Baxter GF, Yellon DM. Caspase inhibition and limitation of myocardial infarct size: protection against lethal reperfusion injury. Br J Pharmacol. 2000;130:197–200. doi: 10.1038/sj.bjp.0703336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Duprez L, Wirawan E, Vanden Berghe T, Vandenabeele P. Major cell death pathways at a glance. Microbes Infection. 2009;11:1050–1062. doi: 10.1016/j.micinf.2009.08.013. [DOI] [PubMed] [Google Scholar]
- 188.Shimada K, et al. Oxidized mitochondrial DNA activates the NLRP3 inflammasome during apoptosis. Immunity. 2012;36:401–414. doi: 10.1016/j.immuni.2012.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Shao W, Yeretssian G, Doiron K, Hussain SN, Saleh M. The caspase-1 digestome identifies the glycolysis pathway as a target during infection and septic shock. J Biol Chem. 2007;282:36321–36329. doi: 10.1074/jbc.M708182200. [DOI] [PubMed] [Google Scholar]
- 190.Yang X-M, et al. The highly selective caspase-1 inhibitor VX-765 provides additive protection against myocardial infarction in rat hearts when combined with a platelet inhibitor. J Cardiovasc Pharmacol Ther. 2017 doi: 10.1177/1074248417702890. http://dx.doi.org/10.1177/1074248417702890. [DOI] [PMC free article] [PubMed]
- 191.Yu J, et al. Inflammasome activation leads to Caspase-1-dependent mitochondrial damage and block of mitophagy. Proc Natl Acad Sci USA. 2014;111:15514–15519. doi: 10.1073/pnas.1414859111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Oka T, et al. Mitochondrial DNA that escapes from autophagy causes inflammation and heart failure. Nature. 2012;485:251–255. doi: 10.1038/nature10992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Murry CE, Jennings RB, Reimer KA. Preconditioning with ischemia: a delay of lethal cell injury in ischemic myocardium. Circulation. 1986;74:1124–1136. doi: 10.1161/01.cir.74.5.1124. [DOI] [PubMed] [Google Scholar]
- 194.Huang C, et al. Autophagy and protein kinase C are required for cardioprotection by sulfaphenazole. Am J Physiol Heart Circ Physiol. 2010;298:H570–H579. doi: 10.1152/ajpheart.00716.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Yitzhaki S, et al. Autophagy is required for preconditioning by the adenosine A1 receptor-selective agonist CCPA. Bas Res Cardiol. 2009;104:157–167. doi: 10.1007/s00395-009-0006-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Wang LQ, Cheng XS, Huang CH, Huang B, Liang Q. Rapamycin protects cardiomyocytes against anoxia/reoxygenation injury by inducing autophagy through the PI3k/Akt pathway. J Huazhong Univ Sci Technolog Med Sci. 2015;35:10–15. doi: 10.1007/s11596-015-1381-x. [DOI] [PubMed] [Google Scholar]
- 197.Wu X, et al. Induction of autophagy contributes to the myocardial protection of valsartan against ischemiareperfusion injury. Mol Med Rep. 2013;8:1824–1830. doi: 10.3892/mmr.2013.1708. [DOI] [PubMed] [Google Scholar]
- 198.Gurusamy N, et al. Cardioprotection by resveratrol: a novel mechanism via autophagy involving the mTORC2 pathway. Cardiovasc Res. 2010;86:103–112. doi: 10.1093/cvr/cvp384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Xie M, et al. Histone deacetylase inhibition blunts ischemia/reperfusion injury by inducing cardiomyocyte autophagy. Circulation. 2014;129:1139–1151. doi: 10.1161/CIRCULATIONAHA.113.002416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Rohailla S, et al. Acute, delayed and chronic remote ischemic conditioning is associated with downregulation of mTOR and enhanced autophagy signaling. PLoS ONE. 2014;9:e111291. doi: 10.1371/journal.pone.0111291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Gurusamy N, Lekli I, Gherghiceanu M, Popescu LM, Das DK. BAG-1 induces autophagy for cardiac cell survival. Autophagy. 2009;5:120–121. doi: 10.4161/auto.5.1.7303. [DOI] [PubMed] [Google Scholar]
- 202.Park HK, et al. Autophagy is involved in the ischemic preconditioning. Neurosci Lett. 2009;451:16–19. doi: 10.1016/j.neulet.2008.12.019. [DOI] [PubMed] [Google Scholar]
- 203.Yan L, Sadoshima J, Vatner DE, Vatner SF. Autophagy in ischemic preconditioning and hibernating myocardium. Autophagy. 2009;5:709–712. doi: 10.4161/auto.5.5.8510. [DOI] [PubMed] [Google Scholar]
- 204.Huang C, et al. Autophagy induced by ischemic preconditioning is essential for cardioprotection. J Cardiovasc Transl Res. 2010;3:365–373. doi: 10.1007/s12265-010-9189-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Huang C, et al. Preconditioning involves selective mitophagy mediated by Parkin and p62/SQSTM1. PLoS ONE. 2011;6:e20975. doi: 10.1371/journal.pone.0020975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Andres AM, et al. Mitophagy is required for acute cardioprotection by simvastatin. Antioxid Redox Signal. 2014;21:1960–1973. doi: 10.1089/ars.2013.5416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Matsui Y, et al. Distinct roles of autophagy in the heart during ischemia and reperfusion: roles of AMP-activated protein kinase and Beclin 1 in mediating autophagy. Circ Res. 2007;100:914–922. doi: 10.1161/01.RES.0000261924.76669.36. [DOI] [PubMed] [Google Scholar]
- 208.Schiattarella GG, Hill JA. Therapeutic targeting of autophagy in cardiovascular disease. J Mol Cell Cardiol. 2016;95:86–93. doi: 10.1016/j.yjmcc.2015.11.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Bhuiyan MS, et al. Enhanced autophagy ameliorates cardiac proteinopathy. J Clin Invest. 2013;123:5284–5297. doi: 10.1172/JCI70877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Delbridge LM, Mellor KM, Taylor DJ, Gottlieb RA. Myocardial autophagic energy stress responses—macroautophagy, mitophagy, and glycophagy. Am J Physiol Heart Circ Physiol. 2015;308:H1194–H1204. doi: 10.1152/ajpheart.00002.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Gottlieb RA, Andres AM, Sin J, Taylor DPJ. Untangling autophagy measurements: all fluxed up. Circ Res. 2015;116:504–514. doi: 10.1161/CIRCRESAHA.116.303787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Han Z, et al. Autophagy is involved in the cardioprotection effect of remote limb ischemic postconditioning on myocardial ischemia/reperfusion injury in normal mice, but not diabetic mice. PLoS ONE. 2014;9:e86838. doi: 10.1371/journal.pone.0086838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Ke J, Yao B, Li T, Cui S, Ding H. A2 adenosine receptor-mediated cardioprotection against reperfusion injury in rat hearts is associated with autophagy downregulation. J Cardiovasc Pharmacol. 2015;66:25–34. doi: 10.1097/FJC.0000000000000239. [DOI] [PubMed] [Google Scholar]
- 214.Su J, Zhang T, Wang K, Zhu T, Li X. Autophagy activation contributes to the neuroprotection of remote ischemic perconditioning against focal cerebral ischemia in rats. Neurochem Res. 2014;39:2068–2077. doi: 10.1007/s11064-014-1396-x. [DOI] [PubMed] [Google Scholar]
- 215.Zhang YL, et al. Restoration of autophagic flux in myocardial tissues is required for cardioprotection of sevoflurane postconditioning in rats. Acta Pharmacol Sin. 2014;35:758–769. doi: 10.1038/aps.2014.20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Koike M, et al. Inhibition of autophagy prevents hippocampal pyramidal neuron death after hypoxic-ischemic injury. Am J Pathol. 2008;172:454–469. doi: 10.2353/ajpath.2008.070876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Xing S, et al. Beclin 1 knockdown inhibits autophagic activation and prevents the secondary neurodegenerative damage in the ipsilateral thalamus following focal cerebral infarction. Autophagy. 2012;8:63–76. doi: 10.4161/auto.8.1.18217. [DOI] [PubMed] [Google Scholar]
- 218.Liu Y, et al. Autosis is a Na+, K+-ATPase-regulated form of cell death triggered by autophagy-inducing peptides, starvation, and hypoxia-ischemia. Proc Natl Acad Sci USA. 2013;110:20364–20371. doi: 10.1073/pnas.1319661110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Elgendy M, Sheridan C, Brumatti G, Martin SJ. Oncogenic Ras-induced expression of Noxa and Beclin-1 promotes autophagic cell death and limits clonogenic survival. Mol Cell. 2011;42:23–35. doi: 10.1016/j.molcel.2011.02.009. [DOI] [PubMed] [Google Scholar]
- 220.Rothermel BA, Hill JA. Autophagy in load-induced heart disease. Circ Res. 2008;103:1363–1369. doi: 10.1161/CIRCRESAHA.108.186551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Wang Q, Ren J. mTOR-Independent autophagy inducer trehalose rescues against insulin resistance-induced myocardial contractile anomalies: Role of p38 MAPK and Foxo1. Pharmacol Res. 2016;111:357–373. doi: 10.1016/j.phrs.2016.06.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Jang SY, Kang HT, Hwang ES. Nicotinamide-induced mitophagy: event mediated by high NAD+/NADH ratio and SIRT1 protein activation. J Biol Chem. 2012;287:19304–19314. doi: 10.1074/jbc.M112.363747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.Lee IH, et al. A role for the NAD-dependent deacetylase Sirt1 in the regulation of autophagy. Proc Natl Acad Sci USA. 2008;105:3374–3379. doi: 10.1073/pnas.0712145105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.Liu G, et al. Loss of NAD-dependent protein deacetylase sirtuin-2 alters mitochondrial protein acetylation and dysregulates mitophagy. Antioxid Redox Signal. 2016;26:849–863. doi: 10.1089/ars.2016.6662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225.Bo L, et al. Autophagic program is regulated by miR-325. Cell Death Differ. 2014;21:967–977. doi: 10.1038/cdd.2014.18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.Chen Q, Zhou Y, Richards AM, Wang P. Up-regulation of miRNA-221 inhibits hypoxia/reoxygenation-induced autophagy through the DDIT4/mTORC1 and Tp53inp1/p62 pathways. Biochem Biophys Res Commun. 2016;474:168–174. doi: 10.1016/j.bbrc.2016.04.090. [DOI] [PubMed] [Google Scholar]
- 227.Gottlieb RA, Pourpirali S. Lost in translation: miRNAs and mRNAs in ischemic preconditioning and ischemia/reperfusion injury. J Mol Cell Cardiol. 2016;95:70–77. doi: 10.1016/j.yjmcc.2015.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228.Gupta SK, Thum T. Non-coding RNAs as orchestrators of autophagic processes. J Mol Cell Cardiol. 2016;95:26–30. doi: 10.1016/j.yjmcc.2015.11.012. [DOI] [PubMed] [Google Scholar]
- 229.Higashi K, et al. MicroRNA-145 repairs infarcted myocardium by accelerating cardiomyocyte autophagy. Am J Physiol Heart Circ Physiol. 2015;309:H1813–1826. doi: 10.1152/ajpheart.00709.2014. [DOI] [PubMed] [Google Scholar]
- 230.Huang J, Huang C, Luo Y, Liu S, Chen X. Role of MiR.-30a in cardiomyocyte autophagy induced by Angiotensin, II. J Renin Angiotensin Aldosterone Syst. 2015;16:1–5. doi: 10.1177/1470320314562060. [DOI] [PubMed] [Google Scholar]
- 231.Li D, et al. Salvianolic acid B induced upregulation of miR-30a protects cardiac myocytes from ischemia/reperfusion injury. BMC Complement Alternative Med. 2016;16:336. doi: 10.1186/s12906-016-1275-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232.Li G, et al. miR-22 regulates starvation-induced autophagy and apoptosis in cardiomyocytes by targeting p38α. Biochem Biophys Res Commun. 2016;478:1165–1172. doi: 10.1016/j.bbrc.2016.08.086. [DOI] [PubMed] [Google Scholar]
- 233.Li X, et al. Inhibition of microRNA-497 ameliorates anoxia/reoxygenation injury in cardiomyocytes by suppressing cell apoptosis and enhancing autophagy. Oncotarget. 2015;6:18829–18844. doi: 10.18632/oncotarget.4774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234.Nandi SS, et al. Induction of autophagy markers is associated with attenuation of miR-133a in diabetic heart failure patients undergoing mechanical unloading. Am J Transl Res. 2015;7:683–696. [PMC free article] [PubMed] [Google Scholar]
- 235.Su M, et al. Cardiac-specific overexpression of miR-222 induces heart failure and inhibits autophagy in mice. Cell Physiol Biochem. 2016;39:1503–1511. doi: 10.1159/000447853. [DOI] [PubMed] [Google Scholar]
- 236.Su M, et al. MicroRNA-221 inhibits autophagy and promotes heart failure by modulating the p27/CDK2/mTOR axis. Cell Death Differ. 2015;22:986–999. doi: 10.1038/cdd.2014.187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237.Wang K, et al. APF lncRNA regulates autophagy and myocardial infarction by targeting miR-188-3p. Nat Commun. 2015;6:6779. doi: 10.1038/ncomms7779. [DOI] [PubMed] [Google Scholar]
- 238.Wu D, Jiang H, Chen S, Zhang H. Inhibition of microRNA-101 attenuates hypoxia/reoxygenationinduced apoptosis through induction of autophagy in H9c2 cardiomyocytes. Mol Med Rep. 2015;11:3988–3994. doi: 10.3892/mmr.2015.3215. [DOI] [PubMed] [Google Scholar]
- 239.Yang Y, et al. Exosomal transfer of miR-30a between cardiomyocytes regulates autophagy after hypoxia. J Mol Med (Berlin, Germany) 2016;94:711–724. doi: 10.1007/s00109-016-1387-2. [DOI] [PubMed] [Google Scholar]
- 240.Zou Y, Liu W, Zhang J, Xiang D. miR-153 regulates apoptosis and autophagy of cardiomyocytes by targeting Mcl-1. Mol Med Rep. 2016;14:1033–1039. doi: 10.3892/mmr.2016.5309. [DOI] [PubMed] [Google Scholar]
- 241.Li J, et al. MicroRNA-144 is a circulating effector of remote ischemic preconditioning. Bas Res Cardiol. 2014;109:423. doi: 10.1007/s00395-014-0423-z. [DOI] [PubMed] [Google Scholar]
- 242.Ma X, et al. Impaired autophagosome clearance contributes to cardiomyocyte death in ischemia-reperfusion injury. Circulation. 2012;125:3170–3181. doi: 10.1161/CIRCULATIONAHA.111.041814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 243.Shirakabe A, et al. Drp1-dependent mitochondrial autophagy plays a protective role against pressure overload-induced mitochondrial dysfunction and heart failure. Circulation. 2016;133:1249–1263. doi: 10.1161/CIRCULATIONAHA.115.020502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 244.Foyil SA, et al. BNIP3-induced aurophagy plays a protective role against pressure overload-induced mitochondrial dysfunction and heart failure. J Cardiac Fail. 2010;16:S35. [Google Scholar]
- 245.Cao DJ, et al. Histone deacetylase (HDAC) inhibitors attenuate cardiac hypertrophy by suppressing autophagy. Proc Natl Acad Sci USA. 2011;108:4123–4128. doi: 10.1073/pnas.1015081108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 246.Huang J, et al. LC3B, a protein that serves as an autophagic marker, modulates angiotensin ii-induced myocardial hypertrophy. J Cardiovasc Pharmacol. 2015;66:576–583. doi: 10.1097/FJC.0000000000000306. [DOI] [PubMed] [Google Scholar]
- 247.Zhou L, Ma B, Han X. The role of autophagy in angiotensin II-induced pathological cardiac hypertrophy. J Mol Endocrinol. 2016;57:R143–R152. doi: 10.1530/JME-16-0086. [DOI] [PubMed] [Google Scholar]
- 248.Ikeda Y, et al. Endogenous Drp1 mediates mitochondrial autophagy and protects the heart against energy stress. Circ Res. 2015;116:264–278. doi: 10.1161/CIRCRESAHA.116.303356. [DOI] [PubMed] [Google Scholar]
- 249.Shen C, et al. Acetaldehyde dehydrogenase 2 (ALDH2) deficiency exacerbates pressure overload-induced cardiac dysfunction by inhibiting Beclin-1 dependent autophagy pathway. Biochim Biophys Acta. 2015;1852:310–318. doi: 10.1016/j.bbadis.2014.07.014. [DOI] [PubMed] [Google Scholar]
- 250.Ahmet I, Wan R, Mattson MP, Lakatta EG, Talan M. Cardioprotection by intermittent fasting in rats. Circulation. 2005;112:3115–3121. doi: 10.1161/CIRCULATIONAHA.105.563817. [DOI] [PubMed] [Google Scholar]
- 251.Godar RJ, et al. Repetitive stimulation of autophagy-lysosome machinery by intermittent fasting preconditions the myocardium to ischemia-reperfusion injury. Autophagy. 2015;11:1537–1560. doi: 10.1080/15548627.2015.1063768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 252.Hatori M, et al. Time-restricted feeding without reducing caloric intake prevents metabolic diseases in mice fed a high-fat diet. Cell Metab. 2012;15:848–860. doi: 10.1016/j.cmet.2012.04.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 253.Hsu HC, Chen CY, Lee BC, Chen MF. High-fat diet induces cardiomyocyte apoptosis via the inhibition of autophagy. Eur J Nutr. 2016;55:2245–2254. doi: 10.1007/s00394-015-1034-7. [DOI] [PubMed] [Google Scholar]
- 254.Hsu HC, et al. Time-dependent cellular response in the liver and heart in a dietary-induced obese mouse model: the potential role of ER stress and autophagy. Eur J Nutr. 2016;55:2031–2043. doi: 10.1007/s00394-015-1017-8. [DOI] [PubMed] [Google Scholar]
- 255.Andres AM, et al. Discordant signaling and autophagy response to fasting in hearts of obese mice: Implications for ischemia tolerance. Am J Physiol Heart Circ Physiol. 2016;311:H219–H228. doi: 10.1152/ajpheart.00041.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 256.Powers SK, Quindry JC, Kavazis AN. Exercise-induced cardioprotection against myocardial ischemia-reperfusion injury. Free Radic Biol Med. 2008;44:193–201. doi: 10.1016/j.freeradbiomed.2007.02.006. [DOI] [PubMed] [Google Scholar]
- 257.Tam BT, et al. Autophagic adaptations to long-term habitual exercise in cardiac muscle. Int J Sports Med. 2015;36:526–534. doi: 10.1055/s-0034-1398494. [DOI] [PubMed] [Google Scholar]
- 258.Iyer SS, et al. Mitochondrial cardiolipin is required for Nlrp3 inflammasome activation. Immunity. 2013;39:311–323. doi: 10.1016/j.immuni.2013.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 259.Stevens DA, et al. Parkin loss leads to PARIS-dependent declines in mitochondrial mass and respiration. Proc Natl Acad Sci USA. 2015;112:11696–11701. doi: 10.1073/pnas.1500624112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 260.Tao L, et al. Exercise training protects against acute myocardial infarction via improving myocardial energy metabolism and mitochondrial biogenesis. Cell Physiol Biochem. 2015;37:162–175. doi: 10.1159/000430342. [DOI] [PubMed] [Google Scholar]
- 261.Bouitbir J, et al. Opposite effects of statins on mitochondria of cardiac and skeletal muscles: a ‘mitohormesis’ mechanism involving reactive oxygen species and PGC-1. Eur Heart J. 2012;33:1397–1407. doi: 10.1093/eurheartj/ehr224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 262.Yamamoto T, et al. Nicotinamide mononucleotide, an intermediate of NAD+ synthesis, protects the heart from ischemia and reperfusion. PLoS ONE. 2014;9:e98972. doi: 10.1371/journal.pone.0098972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 263.Ong SB, Hausenloy DJ. Mitochondrial dynamics as a therapeutic target for treating cardiac diseases. Handb Exp Pharmacol. 2016 doi: 10.1007/164_2016_7. http://dx.doi.org/10.1007/164_2016_7. [DOI] [PubMed]
- 264.Ong SB, et al. Inhibiting mitochondrial fission protects the heart against ischemia/reperfusion injury. Circulation. 2010;121:2012–2022. doi: 10.1161/CIRCULATIONAHA.109.906610. [DOI] [PubMed] [Google Scholar]
- 265.Gustafsson AB, Gottlieb RA. Bcl-2 family members and apoptosis, taken to heart. Am J Physiol Cell Physiol. 2007;292:C45–C51. doi: 10.1152/ajpcell.00229.2006. [DOI] [PubMed] [Google Scholar]
- 266.Kloner RA. No-reflow phenomenon: maintaining vascular integrity. J Cardiovasc Pharmacol Ther. 2011;16:244–250. doi: 10.1177/1074248411405990. [DOI] [PubMed] [Google Scholar]
- 267.Reffelmann T, Kloner RA. The “no-reflow” phenomenon: basic science and clinical correlates. Heart. 2002;87:162–168. doi: 10.1136/heart.87.2.162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 268.Rezkalla SH, Kloner RA. No-reflow phenomenon. Circulation. 2002;105:656–662. doi: 10.1161/hc0502.102867. [DOI] [PubMed] [Google Scholar]
- 269.Kloner RA, Ganote CE, Jennings RB. The “no-reflow” phenomenon after temporary coronary occlusion in the dog. J Clin Invest. 1974;54:1496–1508. doi: 10.1172/JCI107898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 270.Reffelmann T, Kloner RA. Microvascular reperfusion injury: rapid expansion of anatomic no reflow during reperfusion in the rabbit. Am J Physiol Heart Circ Physiol. 2002;283:H1099–H1107. doi: 10.1152/ajpheart.00270.2002. [DOI] [PubMed] [Google Scholar]
- 271.Kloner RA, Alker KJ. The effect of streptokinase on intramyocardial hemorrhage, infarct size, and the no-reflow phenomenon during coronary reperfusion. Circulation. 1984;70:513–521. doi: 10.1161/01.cir.70.3.513. [DOI] [PubMed] [Google Scholar]
- 272.Zhao JL, Yang YJ, Cui CJ, You SJ, Gao RL. Pretreatment with simvastatin reduces myocardial no-reflow by opening mitochondrial KATP channel. Br J Pharmacol. 2006;149:243–249. doi: 10.1038/sj.bjp.0706862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 273.Reffelmann T, Hale SL, Dow JS, Kloner RA. No-reflow phenomenon persists long-term after ischemia/reperfusion in the rat and predicts infarct expansion. Circulation. 2003;108:2911–2917. doi: 10.1161/01.CIR.0000101917.80668.E1. [DOI] [PubMed] [Google Scholar]
- 274.Ito H. Etiology and clinical implications of microvascular dysfunction in patients with acute myocardial infarction. Int Heart J. 2014;55:185–189. doi: 10.1536/ihj.14-057. [DOI] [PubMed] [Google Scholar]
- 275.Rezkalla SH, Dharmashankar KC, Abdalrahman IB, Kloner RA. No-reflow phenomenon following percutaneous coronary intervention for acute myocardial infarction: incidence, outcome, and effect of pharmacologic therapy. J Interv Cardiol. 2010;23:429–436. doi: 10.1111/j.1540-8183.2010.00561.x. [DOI] [PubMed] [Google Scholar]
- 276.Schwartz BG, Kloner RA. Coronary no reflow. J Mol Cell Cardiol. 2012;52:873–882. doi: 10.1016/j.yjmcc.2011.06.009. [DOI] [PubMed] [Google Scholar]
- 277.Judd RM, et al. Physiological basis of myocardial contrast enhancement in fast magnetic resonance images of 2-day-old reperfused canine infarcts. Circulation. 1995;92:1902–1910. doi: 10.1161/01.cir.92.7.1902. [DOI] [PubMed] [Google Scholar]
- 278.Wu KC, et al. Prognostic significance of microvascular obstruction by magnetic resonance imaging in patients with acute myocardial infarction. Circulation. 1998;97:765–772. doi: 10.1161/01.cir.97.8.765. [DOI] [PubMed] [Google Scholar]
- 279.Bolognese L, et al. Impact of microvascular dysfunction on left ventricular remodeling and long-term clinical outcome after primary coronary angioplasty for acute myocardial infarction. Circulation. 2004;109:1121–1126. doi: 10.1161/01.CIR.0000118496.44135.A7. [DOI] [PubMed] [Google Scholar]
- 280.Galiuto L, et al. The extent of microvascular damage during myocardial contrast echocardiography is superior to other known indexes of post-infarct reperfusion in predicting left ventricular remodeling: results of the multicenter AMICI study. J Am Coll Cardiol. 2008;51:552–559. doi: 10.1016/j.jacc.2007.09.051. [DOI] [PubMed] [Google Scholar]
- 281.White HD, et al. Left ventricular end-systolic volume as the major determinant of survival after recovery from myocardial infarction. Circulation. 1987;76:44–51. doi: 10.1161/01.cir.76.1.44. [DOI] [PubMed] [Google Scholar]
- 282.Ndrepepa G, et al. 5-year prognostic value of no-reflow phenomenon after percutaneous coronary intervention in patients with acute myocardial infarction. J Am Coll Cardiol. 2010;55:2383–2389. doi: 10.1016/j.jacc.2009.12.054. [DOI] [PubMed] [Google Scholar]
- 283.Niccoli G, Burzotta F, Galiuto L, Crea F. Myocardial no-reflow in humans. J Am Coll Cardiol. 2009;54:281–292. doi: 10.1016/j.jacc.2009.03.054. [DOI] [PubMed] [Google Scholar]
- 284.Hale SL, Herring MJ, Kloner RA. Delayed treatment with hypothermia protects against the no-reflow phenomenon despite failure to reduce infarct size. J Am Heart Assoc. 2013;2:e004234. doi: 10.1161/JAHA.112.004234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 285.Yang X, et al. Cardioprotection by mild hypothermia during ischemia involves preservation of ERK activity. Bas Res Cardiol. 2011;106:421–430. doi: 10.1007/s00395-011-0165-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 286.Ucar A, et al. The miRNA-212/132 family regulates both cardiac hypertrophy and cardiomyocyte autophagy. Nat Commun. 2012;3:1078. doi: 10.1038/ncomms2090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 287.Jian X, et al. MiR-204 regulate cardiomyocyte autophagy induced by hypoxia-reoxygenation through LC3-II. Int J Cardiol. 2011;148:110–112. doi: 10.1016/j.ijcard.2011.01.029. [DOI] [PubMed] [Google Scholar]
- 288.Mikhaylova O, et al. VHL-regulated MiR-204 suppresses tumor growth through inhibition of LC3B-mediated autophagy in renal clear cell carcinoma. Cancer Cell. 2012;21:532–546. doi: 10.1016/j.ccr.2012.02.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 289.Xiao J, et al. MiR-204 regulates cardiomyocyte autophagy induced by ischemia-reperfusion through LC3-II. J Biomed Sci. 2011;18:35. doi: 10.1186/1423-0127-18-35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 290.Huang J, et al. miR-34a modulates angiotensin II-induced myocardial hypertrophy by direct inhibition of ATG9A expression and autophagic activity. PLoS ONE. 2014;9:e94382. doi: 10.1371/journal.pone.0094382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 291.Song L, et al. MiR-451 is decreased in hypertrophic cardiomyopathy and regulates autophagy by targeting TSC1. J Cell Mol Med. 2014;18:2266–2274. doi: 10.1111/jcmm.12380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 292.Pan W, et al. MiR-30-regulated autophagy mediates angiotensin II-induced myocardial hypertrophy. PLoS ONE. 2013;8:e53950. doi: 10.1371/journal.pone.0053950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 293.Li Q, et al. Overexpression of microRNA-99a attenuates heart remodelling and improves cardiac performance after myocardial infarction. J Cell Mol Med. 2014;18:919–928. doi: 10.1111/jcmm.12242. [DOI] [PMC free article] [PubMed] [Google Scholar]