

Abstract
Aims
To identify gestational diabetes mellitus exposure-associated DNA methylation changes and assess whether such changes are also associated with adiposity-related outcomes.
Methods
We performed an epigenome-wide association analysis, using Illumina 450k methylation arrays, on whole blood collected, on average, at 10.5 years of age from 81 gestational diabetes-exposed and 81 unexposed offspring enrolled in the EPOCH (Exploring Perinatal Outcomes in Children) study, and on the cord blood of 31 gestational diabetes-exposed and 64 unexposed offspring enrolled in our own ‘Healthy Start’ cohort. Validation was performed by pyrosequencing.
Results
We identified 98 differentially methylated positions associated with gestational diabetes exposure at a false discovery rate of <10% in peripheral blood, with 51 loci remaining significant (plus additional 40 loci) after adjustment for cell proportions. We also identified 2195 differentially methylation regions at a false discovery rate of <5% after adjustment for cell proportions. We prioritized loci for pyrosequencing validation and association analysis with adiposity-related outcomes based on strengths of association and effect size, network and pathway analysis, analysis of cord blood, and previous publications. Methylation in six out of nine (67%) gestational diabetes-associated genes was validated and we also showed that methylation of SH3PXD2A was significantly (P<0.05) associated with multiple adiposity-related outcomes.
Conclusions
Our findings suggest that epigenetic marks may provide an important link between in utero exposure to gestational diabetes and obesity in childhood, and add to the growing body of evidence that DNA methylation is affected by gestational diabetes exposure.
Introduction
With obesity now affecting one in five children in the USA and gestational diabetes mellitus (GDM) increasing among all racial and ethnic groups, it is becoming increasingly important to understand how events that happen early in life, even before birth, can alter the obesity trajectory of individuals [1]. The fetal over-nutrition hypothesis posits that the intrauterine environment of diabetic and obese pregnancies may permanently alter the offspring’s long-term risk of obesity and metabolic diseases, through developmental programming [2]. This risk is hypothesized to operate through permanent changes in cell and tissue structure and function induced by changes in expression of target genes. Epigenetic modifications are mediators of the effect of the environment on gene expression and are likely to be a crucial mechanism involved in these processes
The epigenome is particularly susceptible to alterations during gestation because the DNA synthesis rate is high and the DNA methylation patterning required for normal tissue development is established during this period. In fact, previous studies have demonstrated an association of DNA methylation changes with in utero GDM exposure in maternal peripheral blood [3], cord blood [4–7], placenta [3,5,8–10] and offspring peripheral blood DNA [11]. Importantly, changes in the epigenome are also hypothesized to play a role in later-life disease susceptibility [12]. Accordingly, DNA methylation changes have been associated with Type 2 diabetes [13–15] and obesity [16–18].
We aimed to identify GDM-exposure-associated DNA methylation changes and assess whether such changes are also associated with adiposity-related outcomes, using a sample of participants from the EPOCH (Exploring Perinatal Outcomes in Children) study in Colorado, and sensitive measures of childhood adiposity and fat distribution using state-of-the-art techniques. While a number of previous studies have identified DNA methylation changes associated with GDM, only one other study to date has tested the association of these epigenetic marks with childhood adiposity-related outcomes [11].
Methods
In the following sections, we provide an overview of the methods and details are given in the Supporting Information (File S1).
Study populations
EPOCH
The EPOCH study is a historical prospective cohort that enrolled children aged 10.5 years, on average, who were exposed (n=100) or not exposed (n=504) to maternal GDM during their intrauterine life [19]. Previously, we found that exposure to maternal GDM was associated with multiple adiposity markers and a more centralized fat distribution pattern [19]. The present analysis included a sample of 285 mother–child pairs: 81 offspring who had been exposed to maternal GDM during intrauterine life and 204 unexposed offspring, frequency-matched on age and sex.
Healthy Start
Because of a lack of cord blood sample availability in the EPOCH cohort, our analysis also included 31 GDM-exposed and 64 unexposed babies (frequency-matched on age and sex) enrolled in ‘Healthy Start’, a prospective pre-birth cohort that used the same methods for assessment of GDM exposure [20] and that had cord blood samples available.
Exposure definition
Physician-diagnosed maternal diabetes status was ascertained from the Kaiser Permanente perinatal database, as previously described [19]. At 24–28 weeks, all pregnant women were offered screening for gestational diabetes with a 1-h, 50-g oral glucose tolerance test (OGTT). Women with a value ≥7.7 mmol/l were asked to undergo a 3-h, 100-g diagnostic OGTT. Gestational diabetes was diagnosed when two or more glucose values during the diagnostic OGTT met or exceeded the criteria for a positive test [21]. Exposure to diabetes in utero was defined as maternal diabetes diagnosed during the index pregnancy.
Measures of childhood adiposity and fat distribution
Childhood height and weight were measured in light clothing and without shoes, and BMI was calculated as weight (kg)/height (m2). Waist circumference was measured according to the National Health and Nutrition Examination Survey (NHANES) protocol. Skinfold thickness was measured in triplicate using Holtain calipers and averaged. The ratio of subscapular to triceps skinfold thickness was calculated to assess regional differences in subcutaneous fat distribution. MRI of the abdominal region was used to quantify visceral adipose tissue and subcutaneous adipose tissue with a 3-T HDx Imager (General Electric, Waukashau, WI, USA).
Laboratory analyses of adiposity-related markers
Blood samples were drawn after an overnight fast for measurement of glucose (measured using the Olympus AU400 advanced chemistry analyser system), insulin (measured by a radioimmunoassay method), adiponectin and leptin (measured with enzyme-linked immunosorbent assay kits). Homeostatic model assessment of insulin resistance (HOMA-IR) was calculated as fasting glucose (mmol/l)×insulin (pmol/l)]/156.3).
Genome-wide methylation assessment
We measured genome-wide DNA methylation using Illumina’s Infinium Human Methylation 450k BeadChip on bisulfite-treated samples using published methods [22].
Data quality controls
We performed several quality control steps that are standard in the analysis of Illumina 450k array data, as previously published [22]. We removed probes with high detection P values (443), probes with fewer than three beads in at least 10% of the samples (510), probes with known single-nucleotide polymorphisms (SNPs) within the CpG motif (17415), and cross-reactive probes (29 233) [23]. We removed one sample with low median intensity and two samples with inconsistency between reported and methylation-predicted sex. Two samples that were >3 standard deviation points away from the mean of the top principal component were also removed.
Overview of DNA methylation analyses
The overall analytical strategy for analysis of DNA methylation data is presented in Fig. 1. In all analyses of epigenetic data, we adjusted for child age, sex and race/ethnicity. We estimated and adjusted for cell counts in whole blood to characterize methylation changes as those that are attributable to the presence of different proportions of cell types in peripheral blood vs those that are inherently differentially methylated in cells. Differentially methylated positions (DMPs) were prioritized using a combination of our own data and published DNA methylation changes associated with GDM exposure at birth. Differentially methylated regions (DMRs) were prioritized using pathway enrichment analysis; this strategy was used in the case of DMRs because of the large number of significant associations.
FIGURE 1.
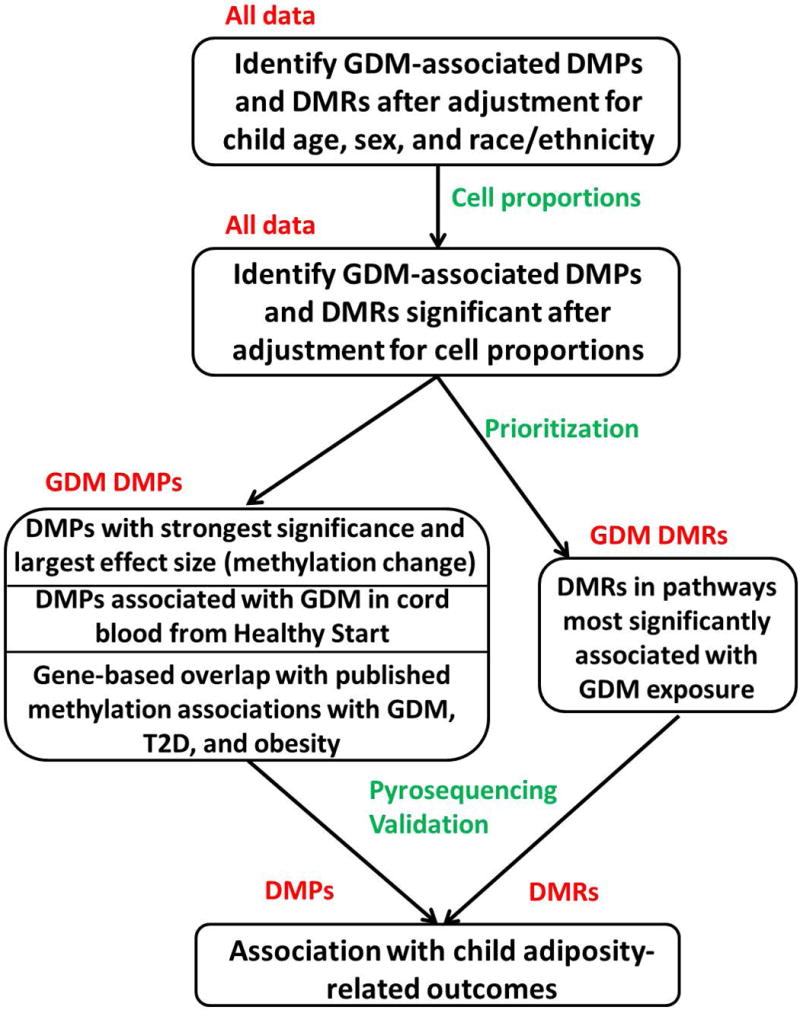
Overall strategy used to identify gestational diabetes (GDM)-associated DNA methylation changes, prioritize and validate them, and test for association with adiposity outcomes. DMP, differentially methylated position; DMR, differentially methylated region.
Prioritization was followed by pyrosequencing analysis for technical validation of GDM-associated epigenetic marks and testing for association with adiposity-related outcomes. To maximize power for detecting associations with adiposity outcomes, pyrosequencing was performed on the same 81 GDM-exposed offspring used for the genome-wide methylation assessment and 204 unexposed offspring, consisting of 81 used in the genome-wide assessment and an additional 123 unexposed offspring; this provided a ~2.5 ratio of unexposed to exposed participants included in further analyses.
Statistical analysis of DNA methylation data
Data from the methylation array were normalized using the stratified quantile method and batch-corrected. Linear regression models were used to test for association between CpG sites and GDM exposure, adjusting for child sex, child age (in the EPOCH cohort) and child race/ethnicity. We ran two sets of models, adjusting or not adjusting for cell proportions in peripheral or cord blood. DMPs were identified by calculating false discovery rate (FDR)-adjusted P values, from linear model P values using the method of Benjamini and Hochberg. DMRs were identified with DMRCate software using the same model and covariates as the DMP analysis. Linear regression analyses were also conducted to test for associations between childhood adiposity outcomes and pyrosequencing methylation data, with childhood outcomes as the dependent variables, while adjusting for child sex, child age, child race/ethnicity and Tanner stage.
Network and enrichment analyses
We used the Network Analyst and GeneMANIA servers to perform network analysis, and the GREAT webserver for enrichment of functional gene categories.
Pyrosequencing
Bisulfite-converted DNA was PCR-amplified using primers designed in the Pyromark Assay Design Software (Qiagen, Germantown, MD, USA), with details of the assay and quality control previously published [22]. PCR and sequencing primers are listed in Table S1.
Results
The characteristics of the EPOCH participants included in the present study are shown in Table 1. These characteristics are representative of the entire EPOCH cohort (Table S2) [19]. After adjusting for child age, sex, race/ethnicity and batch effects, and removing probes with known SNPs at the CpG motif, we identified 105 CpG sites associated with GDM exposure [false discovery rate (FDR)-adjusted P <0.1; Fig. 2A and Table S3]. Of the 105 loci, 55 remained significant at this same FDR value after adjustment for cell proportions. Furthermore, analysis adjusted for cell proportions identified 43 additional significant DMPs (FDR-adjusted P <0.1) that were not significant in the unadjusted analysis (Fig. 2B and Table S3). The median (range) absolute percent methylation change between exposed and unexposed children was 1.1 (0.44–5.0)%, with an almost equal number of hypo- and hypermethylayed loci (56% hypomethylated). The distribution of CpG sites in relation to genes is roughly equal in gene promoters and gene bodies (Table S3). Protein–protein interaction analysis, using Network Analyst, of the genes nearest to the 91 GDM exposure-associated DMPs after adjustment for cell proportions identified several of the differentially methylated genes as hubs in a significant protein–protein interaction; this includes transcription factors CEBPG, CALCOCO2 (NDP52) and E2F6, protein kinases MAP3K11, NTRK1, PRKCE and RYK, and methyltransferases EHMT1 and SHMT1 (Fig. 3). We performed an additional network analysis using GeneMANIA, which showed that all genes except for three are connected based on gene co-expression (58%), physical interactions (32%), genetic interactions (7%), co-localization (1%), shared protein domains (1%) and prediction (1%; Fig. S1). These results suggest that our analysis identified a module of epigenetic marks, rather than multiple unrelated methylation changes, that are likely to act in concert in affecting gene expression as a result of in utero exposure to GDM.
Table 1.
Characteristics of EPOCH participants included in the present study
| GDM unexposed (N=204) |
GDM exposed (N=81) |
P | |
|---|---|---|---|
| Mean (SD) age at visit, years | 10.3 (1.3) | 9.6 (1.7) | 0.0017* |
| Child sex: female, n (%) | 102 (50) | 40 (49.4) | 1† |
| Non-Hispanic white race/ethnicity, n (%) | 128 (62.8) | 53 (65.4) | 0.685† |
| Mean (SD) child birth weight, g | 3369 (460) | 3358(538) | 0.869* |
| Mean (SD) maternal age at delivery, years | 30.9 (5.2) | 33.3 (5.6) | 0.0011* |
| Tanner stage, n (%) | 0.0011† | ||
| 1 | 105 (51.7) | 57 (70.4) | |
| 2 | 78 (38.4) | 13 (16.1) | |
| 3 | 14 (6.9) | 6 (7.4) | |
| 4 | 6 (3.0) | 4 (4.9) | |
| 5 | 0 (0.0) | 1 (1.2) | |
GDM, gestational diabetes.
Two sample t-test.
Fisher’s exact test.
FIGURE 2.
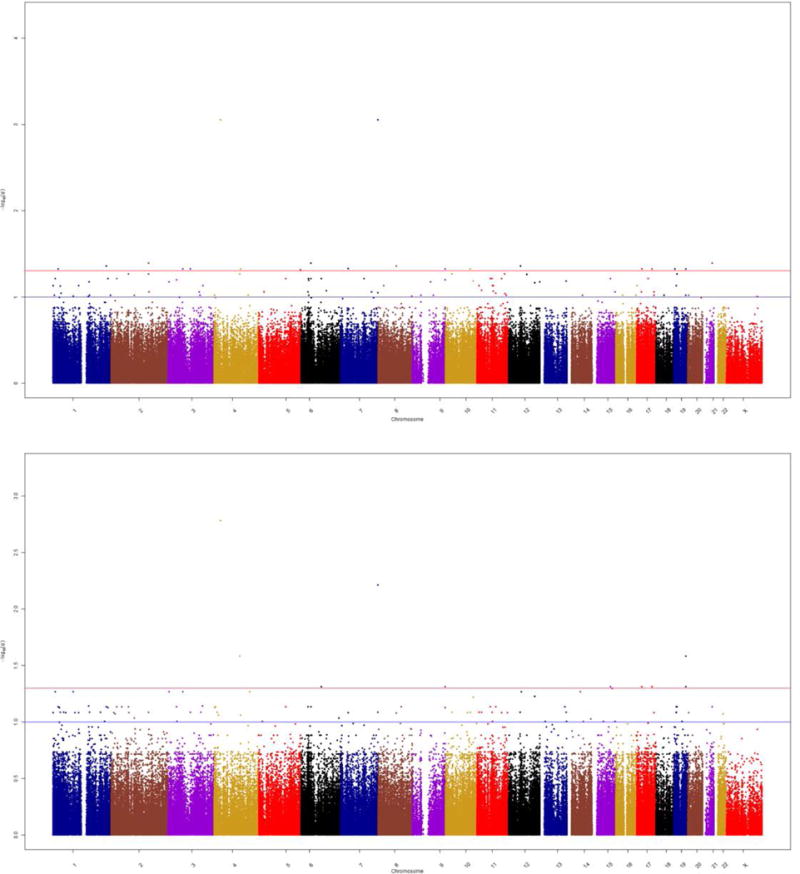
Manhattan plots of differentially methylated positions (a) before and (b) after adjustment for cell proportions. q values are plotted, calculated from P values from the linear model that tested association of CpG sites on the Illumina 450k array with gestational diabetes exposure, adjusting for child sex, child age and child race/ethnicity. Blue line represents q<0.05 (5% false discovery rate [FDR]) and red q<0.1 (10% FDR).
FIGURE 3.
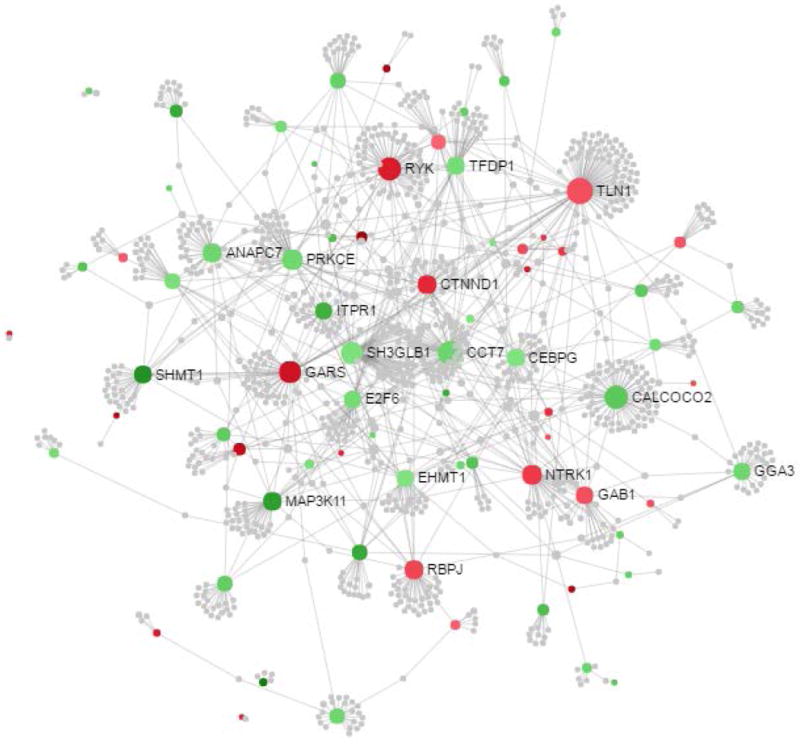
Protein–protein interactome analysis of genes nearest 98 gestational diabetes exposure-associated differentially methylated positions after adjustment for cell proportions. The interactome was created using NetworkAnalyst and the InnateDB protein–protein interaction dataset. The nodes are coloured based on their methylation (red, hypermethylated; green, hypomethylated). Sizes of nodes are proportional to their betweenness centrality values.
We next prioritized DMPs for pyrosequencing using several criteria, namely, strengths of association and effect size, network analysis, analysis of cord blood and previous publications. We first selected three loci based on strength of statistical association and magnitude of methylation change: PTPRN2 as the most significantly associated with GDM exposure, NPHP4 as the most hypomethylated locus and DAPL1 as the most hypermethylated locus. Secondly, we examined whether the GDM exposure-associated DMPs were also differentially methylated in cord blood from our Healthy Start pre-birth cohort. Two EPOCH GDM-associated DMPs with FDR-adjusted P<0.2 were identified: E2F6 and PTPRN2 (Table S4). Based on the presence in both network analysis (Fig. 3) and this analysis of Healthy Start cord blood, the E2F6 transcription factor locus was prioritized. We next examined DNA methylation loci/genes that have been previously identified as associated with GDM exposure in maternal peripheral blood [3], cord blood [4–7] and placenta [3,5,8–10], as well as those associated with Type 2 diabetes [13–15] and obesity [16,17]. This analysis showed that 27 of the genes associated with GDM exposure in EPOCH have been previously identified as associated with GDM exposure (16 genes), Type 2 diabetes (11 genes) and obesity (three genes; Table S5). Among these, differential methylation of PTPRN2 and SH3PXD2A has been associated with GDM in cord blood [5] and placenta [5,8], as well as Type 2 diabetes in pancreatic islets [14]. SH3PXD2A was therefore prioritized for pyrosequencing validation in addition to PTPRN2, which was already included based on strength of association criterion. We prioritized an additional gene based on network analysis (SHMT1) and another two based on overlap with previous studies (RNF39 and ST5) because of their potential functional relevance. This resulted in a total of eight genes prioritized for pyrosequencing validation based on DMP analysis.
Because differential methylation is often regional, meaning that multiple CpG sites in a genomic region are differentially methylated, we also identified DMRs. Using the DMR finding algorithm DMRCate, there were 2195 DMRs (FDR-adjusted P <0.05) after adjustment for child age, sex, race/ethnicity, cell proportions, and batch effects (Table S6). GDM exposure-associated DMRs consist of a median (range) of 5 (2–80) probes and 44% of the DMRs have consistent directionality of methylation changes across the majority of probes (>80%; Table S6). Of the 2195 DMRs, 1264 (58%) were hypomethylated in GDM-exposed compared with GDM-unexposed offspring, with a median (range) absolute change in methylation of 1.3 (0.2–15.5)%.
Prioritization of the large number of DMRs using the same strategy as we used for DMPs was impractical; instead we used enrichment analysis of nearby genes, which is often used in genome-wide studies. The set of DMRs compared with a background region set representing the array contained 3638 and 17 675 nearest genes, respectively. At an FDR-adjusted P value < 0.05, the top Gene Ontology Biological Processes were related to development and transcription regulation (Table S7). We then examined individual genes within these categories and identified those with search terms ‘insulin’, ‘diabetes’ or ‘growth’ in PubMed abstracts, which narrowed the list down to eight genes with 15 associated DMRs, many of which were transcription factors. We selected two transcription factors for pyrosequencing, ISL1 because it binds to the enhancer region of the insulin genes and regulates insulin gene expression [24] and POU5F1 because it has been identified as a Type 2 diabetes genetic susceptibility locus by a transethnic genome-wide association study meta-analysis [25]; this brings the total number of genes selected for pyrosequencing to 10.
We next employed pyrosequencing to validate selected GDM exposure-associated CpG sites and to test association with adiposity-related outcomes. Primers for pyrosequencing were successfully designed for nine of the 10 genes. Association with GDM exposure was validated for five of the nine (56%) genes: DAPL1 (three CpGs in two separate DMPs), POU5F1, PTPRN2, RNF39 and SHMT1 at the FDR-adjusted P value <0.05 significance threshold. Additionally, SH3PXD2A approached significance with P<0.1 (Table 2).
Table 2.
Pyrosequencing validation of selected CpG sites identified as differentially methylated in offspring exposed to gestational diabetes (GDM) vs offspring not exposed to GDM, stratifed by differentially methylated position and differentially methylated region analysis
| Gene | Prioritization | Mean (SD) methylation in GDM-exposed offspring, % | Mean (SD) methylation in GDM-unexposed offspring, % | Methylation change*, % | P* | FDR-adjusted P value |
|---|---|---|---|---|---|---|
| DAPL1 DMP1 cg09357934 |
Effect size | 40.3 (4.4) | 37.3 (4.9) | 3 | 4.21 ×10−6 | 2.74×10−5 |
| DAPL1 DMP2 CpG 1 cg21992932 |
Effect size | 23.1 (4.7) | 20.3 (4.8) | 3 | 7.06×10−6 | 3.06×10−5 |
| DAPL1 DMP2 CpG 2 cg21992932 |
29.5 (5.1) | 26.1 (5.0) | 3 | 2.80×10−6 | 2.74×10−5 | |
| E2F6 CpG 1 cg09914581 |
HS cord blood; network analysis | 0.9 (0.8) | 1.0 (1.2) | -0.1 | 0.603 | 0.713 |
| E2F6 CpG 2 cg09914581 |
1.0 (1.1) | 1.1 (1.5) | −0.1 | 0.826 | 0.868 | |
| NPHP4 cg09275986 |
Effect size; previous studies | 40.0 (5.9) | 39.9 (6.2) | –0.1 | 0.868 | 0.868 |
| POU5F1 cg00357273 |
DMR transcription factor | 87.6 (4.6) | 86.0 (4.3) | 1.6 | 4.82×10−3 | 0.0125 |
| PTPRN2 cg05874166 |
Significance; HS cord blood; previous studies | 93.7 (1.03) | 93.3 (1.54) | 0.5 | 0.010 | 0.0217 |
| RNF39 cg14417687 |
Previous studies | 36.1 (3.3) | 35.4 (3.2) | 0.9 | 0.024 | 0.0446 |
| SH3PXD2A cg10521450 |
Previous studies | 74.3 (2.6) | 74.7 (2.5) | –0.5 | 0.087 | 0.1414 |
| SHMT1 cg26763362 |
Network analysis | 18.8 (5.7) | 20.8 (5.2) | –2.3 | 8.60×10−4 | 0.00280 |
| ST5 CpG1 cg20303245 |
Previous studies | 91.8 (1.6) | 92.1 (1.5) | –0.3 | 0.249 | 0.360 |
| ST5 CpG2 cg20303245 |
85.6 (1.2) | 85.8 (1.3) | –0.1 | 0.397 | 0.516 |
DMP, differentially methylated position; DMR, differentially methylated region; FDR, false discovery rate; GDM, gestational diabetes; HS, Healthy Start cohort.
Methylation change was calculated as the coefficient from the linear model adjusted for child age, sex, race/ethnicity, and pyrosequencing plate.
From linear model adjusted for child age, sex, race/ethnicity and pyrosequencing plate.
We next tested for an association between DNA methylation in these six genes and 12 adiposity-related outcomes collected in the EPOCH cohort at the same visit as the blood draw (Table 3) while adjusting for child age, sex, race/ethnicity, Tanner stage and pyrosequencing plate. This analysis showed that methylation at the DMP in SH3PXD2A is significantly (FDR-adjusted P<0.05) associated with multiple adiposity-related outcomes: BMI, waist circumference, triceps, suprailiac and subscapular skinfold thickness, subcutaneous adipose tissue and leptin levels (Table 3). Additionally, association of SH3PXD2A methylation with visceral adipose tissue approached statistical significance (FDR-adjusted P<0.1). DNA methylation in E2F6 was also associated (FDR-adjusted P<0.05) with fasting insulin and HOMA-IR (Table S8).
Table 3.
DNA methylation of the SH3PXD2A gene is associated with multiple adiposity-related outcomes at the same visit
| Methylation change | P | FDR-adjusted P value | |
|---|---|---|---|
| BMI | –0.1171 | 0.0108 | 0.0312 |
| Waist circumference | –0.0875 | 0.0144 | 0.0312 |
| Tricep skinfold | –9.6835 | 0.01 | 0.0312 |
| Subscapular skinfold | –0.3132 | 0.0194 | 0.0360 |
| Suprailiac skinfold | –14.6405 | 0.0099 | 0.0312 |
| Subcutaneous adipose tissue | –0.5433 | 0.0049 | 0.0312 |
| Visceral adipose tissue | –0.2816 | 0.0589 | 0.0957 |
| Subscapular to triceps ratio | –0.1083 | 0.3819 | 0.4513 |
| Fasting glucose | –0.9342 | 0.8209 | 0.8209 |
| Fasting insulin | –0.1814 | 0.2129 | 0.2768 |
| HOMA-IR | –0.1994 | 0.1996 | 0.2768 |
| Adiponectin | 1.0438 | 0.7333 | 0.7944 |
| Leptin | –0.5061 | 0.0131 | 0.0312 |
FDR, false discovery rate; HOMA-IR, homeostatic model assessment of insulin resistance.
From linear model adjusted for child age, sex, race/ethnicity, Tanner stage and pyrosequencing plate. Methylation change is calculated as the coefficient from the linear model adjusted for child age, sex, race/ethnicity, Tanner stage and pyrosequenicng plate.
Discussion
In the present study we identified DNA methylation changes in childhood that are influenced by in utero exposure to GDM, with some of the GDM-associated DNA methylation changes also having an effect on childhood adiposity-related outcomes. These findings suggest that epigenetic marks may provide an important link between in utero exposure to GDM and childhood obesity, and adds to the growing body of evidence that DNA methylation is affected by GDM exposure.
Previous studies have identified DNA methylation changes associated with GDM during pregnancy [3] and at birth [3–10]. More recently, a study in PIMA Indians identified DNA methylation marks influenced by in utero exposure to GDM in offspring peripheral blood, and showed that some of these methylation marks also influence insulin secretory function and BMI [11]. The present study was the first to identify GDM-exposure-associated DNA methylation changes that are also associated with measures of childhood adiposity and fat distribution collected using state-of-the-art techniques. Epigenetic marks we identified differed from those identified by the PIMA Indian study [11], probably because of differences in population (i.e. racial/ethnic background), methodological approach and focus on different outcomes.
DNA methylation changes associated with GDM exposure were in genes that are related to each other by multiple network and pathway analyses, suggesting that our study identified modules of differential methylation that are more likely to be biologically relevant than unrelated genes. By using the Healthy Start cord blood data and published GDM-associated DNA methylation changes at birth (in cord blood and placenta [3–10]) for prioritization of DNA methylation changes identified in this report, we attempted to reduce confounding by postnatal exposures. Our results are in concordance with previous reports that have shown both hypo- and hypermethylation of specific genes in GDM exposure [3–10]. Similarly, GDM-associated DNA methylation marks were equally distributed in the gene promoters and gene body [26]. Methylation changes we identified in peripheral blood are small in magnitude but may be reflective of larger changes in target tissues. Alternatively, they may be small because of the presence of multiple cell types in whole blood; however, further studies will be needed to demonstrate this. Regardless, small methylation changes have been shown to have an effect on transcription and are viewed as important in children’s health [27].
Of the prioritized genes, DNA methylation in SH3PXD2A was associated with several measures of childhood adiposity and fat distribution in the EPOCH cohort. SH3PXD2A, also known as TKS5, is an adapter protein involved in invadopodia/podosome formation. SH3PXD2A hypomethylation (same direction as in our study) has been associated with GDM exposure in cord blood at birth [5], and with Type 2 diabetes in pancreatic islets [14]. Moreover, SH3PXD2A is hypomethylated and overexpressed in the placenta of pre-eclamptic pregnancies [28]. Finally, a CpG site within its homologue SH3PXD2B has also been shown to be hypomethylated in severe childhood obesity [29]. While the biological function of SH3PXD2A involvement in diabetes and obesity is unclear at this time, these multiple studies strongly suggest that it may be a crucial player in molecular mechanism(s) of metabolic disorders.
The main strength of the present study was the availability of sensitive measures of childhood adiposity and fat distribution using state-of-the-art techniques. An additional strength of our approach was the complementary use of single CpG and regional analyses, as previously reported in other diseases [22]. The main limitations were the use of peripheral blood for assessment of DNA methylation and unavailability of cord blood sample in the EPOCH cohort. We mitigated these limitations by estimating cell proportions for methylation data and adjusting for them in statistical models, profiling cord blood from Healthy Start, and using available published data in placenta and adipose tissue to prioritize GDM-associated methylation marks for further investigation. However, prospectively obtained data at birth and later in life, with replication across other cohorts, will be needed to conclusively validate our findings. A large epigenome-wide association study of BMI showed that the associations of methylation with BMI were independent of variation in cell subset composition and replicated in both isolated white blood cells and isolated adipocytes [18]. Furthermore, Type 2 diabetes-associated methylation changes in peripheral have been shown to be on the causal pathway to disease later in life by using genotype as an anchor for causality [30]. Another limitation that restricts biological interpretation of GDM-associated methylation changes is unavailability of RNA for gene expression profiling. Our ongoing analysis using the second EPOCH visit (on average, at 16 years of age) will allow us to test stability of GDM-associated methylation marks and their impact on gene expression. Since this is an observational analysis, causality cannot be inferred with certainty. We are currently exploring the use of mediation and two-step Mendelian randomization analyses to further address this issue. Similarly, the present study did not address the influence of maternal pre-pregnancy BMI on GDM-associated methylation marks. We did not adjust for pre-pregnancy BMI in our models because such adjustment may obscure the effects we are trying to assess, i.e. effects of in utero exposure to hyperglycaemia on cord blood DNA methylation patterns, because obese pregnancies have higher glucose levels than non-obese ones, even if some are below the threshold used to diagnose GDM [31]. Future mechanistic studies in cell and animal models will be needed to delineate the effects of these related variables.
Despite these limitations, identification of GDM-associated DNA methylation changes that are also associated with adiposity-related outcomes is a significant advancement in the field. Epigenetic marks may provide an important mechanistic link for the association between in utero exposure to diabetes and development of obesity and Type 2 diabetes later in life. Moreover, given that epigenetic marks are reversible and that locus-specific epigenetic therapies are being developed, methylation changes identified in the present study may become important therapeutic targets.
Supplementary Material
Table S1. Primers used for pyrosequencing.
Table S2. Characteristics of the selected children compared to the entire EPOCH cohort (after exclusion of IUGR and T1D exposed children).
Table S3. 138 GDM‐associated DMPs.
Table S4. 459 GDM‐associated DMPs after adjustment for child sex, race, and cell proportions in Healthy Start cord blood.
Table S5. Genes woth GDM associated DMPs in EPOCH analysis that have been previously identified as differnetially methylated in GDM exposed offspring.
Table S6. GDM‐associated DMRs after adjustment for child age, sex, race, and cell proportions.
Table S7. Gene Ontology Biological Processes enriched at q<0.05 among GDM‐associated DMRs.
Table S8. Association of DNA methylation changes with child adiposity‐related outcomes at the same visit.
File S1. Supplemental methods.
What’s new?
We identified DNA methylation changes in childhood that are influenced by in utero exposure to gestational diabetes mellitus (GDM).
Some of the GDM-associated DNA methylation changes also have an effect on childhood adiposity-related outcomes.
This study adds to the growing body of evidence that DNA methylation is affected by GDM exposure.
These findings also suggest that epigenetic marks may provide an important link between in utero exposure to GDM and childhood obesity.
Acknowledgments
We would like to thank Curtis Harrod and Brandy Ringham for database assistance, Mercedes Martinez for assistance with project management, and Okyong Cho in the University of Colorado Genomics core for Illumina array data collection.
Funding sources
This study was funded by the National Institutes of Health (NIH) National Institute for Diabetes and Digestive and Kidney Diseases (NIDDK) R01DK100340 (Dabelea/Kechris/Yang) and R01DK068001 (Dabelea).
Footnotes
DR IVANA V YANG (Orcid ID: 0000-0002-1018-489X)
Competing interests
None declared.
Supporting information
Additional Supporting Information may be found in the online version of this article:
References
- 1.Monteiro LJ, Norman JE, Rice GE, Illanes SE. Fetal programming and gestational diabetes mellitus. Placenta. 2016;48(Suppl 1):S54–S60. doi: 10.1016/j.placenta.2015.11.015. [DOI] [PubMed] [Google Scholar]
- 2.Hanson MA, Gluckman PD. Developmental origins of health and disease: new insights. Basic Clin PharmacolToxicol. 2008;102:90–93. doi: 10.1111/j.1742-7843.2007.00186.x. [DOI] [PubMed] [Google Scholar]
- 3.Houde AA, St-Pierre J, Hivert MF, Baillargeon JP, Perron P, Gaudet D, et al. Placental lipoprotein lipase DNA methylation levels are associated with gestational diabetes mellitus and maternal and cord blood lipid profiles. J Dev Orig Health Dis. 2014;5:132–141. doi: 10.1017/S2040174414000038. [DOI] [PubMed] [Google Scholar]
- 4.Quilter CR, Cooper WN, Cliffe KM, Skinner BM, Prentice PM, Nelson L, et al. Impact on offspring methylation patterns of maternal gestational diabetes mellitus and intrauterine growth restraint suggest common genes and pathways linked to subsequent type 2 diabetes risk. FASEB J. 2014;28:4868–4879. doi: 10.1096/fj.14-255240. [DOI] [PubMed] [Google Scholar]
- 5.Finer S, Mathews C, Lowe R, Smart M, Hillman S, Foo L, et al. Maternal gestational diabetes is associated with genome-wide DNA methylation variation in placenta and cord blood of exposed offspring. Hum Mol Genet. 2015;24:3021–3029. doi: 10.1093/hmg/ddv013. [DOI] [PubMed] [Google Scholar]
- 6.Chen D, Zhang A, Fang M, Fang R, Ge J, Jiang Y, et al. Increased methylation at differentially methylated region of GNAS in infants born to gestational diabetes. BMC Med Genet. 2014;15:108. doi: 10.1186/s12881-014-0108-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Blue EK, Sheehan BM, Nuss ZV, Boyle FA, Hocutt CM, Gohn CR, et al. Epigenetic Regulation of Placenta-Specific 8 Contributes to Altered Function of Endothelial Colony-Forming Cells Exposed to Intrauterine Gestational Diabetes Mellitus. Diabetes. 2015;64:2664–2675. doi: 10.2337/db14-1709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Petropoulos S, Guillemin C, Ergaz Z, Dimov S, Suderman M, Weinstein-Fudim L, et al. Gestational Diabetes Alters Offspring DNA Methylation Profiles in Human and Rat: Identification of Key Pathways Involved in Endocrine System Disorders, Insulin Signaling, Diabetes Signaling, and ILK Signaling. Endocrinology. 2015;156:2222–2238. doi: 10.1210/en.2014-1643. [DOI] [PubMed] [Google Scholar]
- 9.Liu L, Zhang X, Rong C, Rui C, Ji H, Qian YJ, et al. Distinct DNA methylomes of human placentas between pre-eclampsia and gestational diabetes mellitus. Cell Physiol Biochem. 2014;34:1877–1889. doi: 10.1159/000366386. [DOI] [PubMed] [Google Scholar]
- 10.Cote S, Gagne-Ouellet V, Guay SP, Allard C, Houde AA, Perron P, et al. PPARGC1alpha gene DNA methylation variations in human placenta mediate the link between maternal hyperglycemia and leptin levels in newborns. Clin Epigenetics. 2016;8:72. doi: 10.1186/s13148-016-0239-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chen P, Piaggi P, Traurig M, Bogardus C, Knowler WC, Baier LJ, et al. Differential methylation of genes in individuals exposed to maternal diabetes in utero. Diabetologia. 2017;60:645–655. doi: 10.1007/s00125-016-4203-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bateson P, Barker D, Clutton-Brock T, Deb D, D’Udine B, Foley RA, et al. Developmental plasticity and human health. Nature. 2004;430:419–421. doi: 10.1038/nature02725. [DOI] [PubMed] [Google Scholar]
- 13.Prokopenko I, Poon W, Magi R, Prasad BR, Salehi SA, Almgren P, et al. A central role for GRB10 in regulation of islet function in man. PLoS Genetics. 2014;10:e1004235. doi: 10.1371/journal.pgen.1004235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dayeh T, Volkov P, Salo S, Hall E, Nilsson E, Olsson AH, et al. Genome-wide DNA methylation analysis of human pancreatic islets from type 2 diabetic and non-diabetic donors identifies candidate genes that influence insulin secretion. PLoS Genetics. 2014;10:e1004160. doi: 10.1371/journal.pgen.1004160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yuan W, Xia Y, Bell CG, Yet I, Ferreira T, Ward KJ, et al. An integrated epigenomic analysis for type 2 diabetes susceptibility loci in monozygotic twins. Nat Communications. 2014;5:5719. doi: 10.1038/ncomms6719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dick KJ, Nelson CP, Tsaprouni L, Sandling JK, Aissi D, Wahl S, et al. DNA methylation and body-mass index: a genome-wide analysis. Lancet. 2014;383:1990–1998. doi: 10.1016/S0140-6736(13)62674-4. [DOI] [PubMed] [Google Scholar]
- 17.Demerath EW, Guan W, Grove ML, Aslibekyan S, Mendelson M, Zhou YH, et al. Epigenome-wide association study (EWAS) of BMI, BMI change and waist circumference in African American adults identifies multiple replicated loci. Hum Mol Genet. 2015;24:4464–4479. doi: 10.1093/hmg/ddv161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wahl S, Drong A, Lehne B, Loh M, Scott WR, Kunze S, et al. Epigenome-wide association study of body mass index, and the adverse outcomes of adiposity. Nature. 2017;541:81–86. doi: 10.1038/nature20784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Crume TL, Ogden L, West NA, Vehik KS, Scherzinger A, Daniels S, et al. Association of exposure to diabetes in utero with adiposity and fat distribution in a multiethnic population of youth: the Exploring Perinatal Outcomes among Children (EPOCH) Study. Diabetologia. 2011;54:87–92. doi: 10.1007/s00125-010-1925-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Shapiro AL, Schmiege SJ, Brinton JT, Glueck D, Crume TL, Friedman JE, et al. Testing the fuel-mediated hypothesis: maternal insulin resistance and glucose mediate the association between maternal and neonatal adiposity, the Healthy Start study. Diabetologia. 2015;58:937–941. doi: 10.1007/s00125-015-3505-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Classification and diagnosis of diabetes mellitus and other categories of glucose intolerance. National Diabetes Data Group. Diabetes. 1979;28:1039–1057. doi: 10.2337/diab.28.12.1039. [DOI] [PubMed] [Google Scholar]
- 22.Yang IV, Pedersen BS, Liu AH, O’Connor GT, Pillai D, Kattan M, et al. The nasal methylome and childhood atopic asthma. J Allergy Clin Immunol. 2017;139:1478–1488. doi: 10.1016/j.jaci.2016.07.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chen YA, Lemire M, Choufani S, Butcher DT, Grafodatskaya D, Zanke BW, et al. Discovery of cross-reactive probes and polymorphic CpGs in the Illumina Infinium HumanMethylation450 microarray. Epigenetics. 2013;8:203–209. doi: 10.4161/epi.23470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wang W, Shi Q, Guo T, Yang Z, Jia Z, Chen P, et al. PDX1 and ISL1 differentially coordinate with epigenetic modifications to regulate insulin gene expression in varied glucose concentrations. Mol Cell Endocrinol. 2016;428:38–48. doi: 10.1016/j.mce.2016.03.019. [DOI] [PubMed] [Google Scholar]
- 25.Mahajan A, Go MJ, Zhang W, Below JE, Gaulton KJ, Ferreira T, et al. Genome-wide trans-ancestry meta-analysis provides insight into the genetic architecture of type 2 diabetes susceptibility. Nat Genet. 2014;46:234–244. doi: 10.1038/ng.2897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jones PA. Functions of DNA methylation: islands, start sites, gene bodies and beyond. Nat Rev Genet. 2012;13:484–492. doi: 10.1038/nrg3230. [DOI] [PubMed] [Google Scholar]
- 27.Breton CV, Marsit CJ, Faustman E, Nadeau K, Goodrich JM, Dolinoy DC, et al. Small-Magnitude Effect Sizes in Epigenetic End Points are Important in Children’s Environmental Health Studies: The Children’s Environmental Health and Disease Prevention Research Center’s Epigenetics Working Group. Environ Health Perspect. 2017;125:511–526. doi: 10.1289/EHP595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Xiang Y, Cheng Y, Li X, Li Q, Xu J, Zhang J, et al. Up-regulated expression and aberrant DNA methylation of LEP and SH3PXD2A in pre-eclampsia. PloS One. 2013;8:e59753. doi: 10.1371/journal.pone.0059753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Huang RC, Garratt ES, Pan H, Wu Y, Davis EA, Barton SJ, et al. Genome-wide methylation analysis identifies differentially methylated CpG loci associated with severe obesity in childhood. Epigenetics. 2015;10:995–1005. doi: 10.1080/15592294.2015.1080411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Elliott HR, Shihab HA, Lockett GA, Holloway JW, McRae AF, Smith GD, et al. Role of DNA Methylation in Type 2 Diabetes Etiology: Using Genotype as a Causal Anchor. Diabetes. 2017;66:1713–1722. doi: 10.2337/db16-0874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lawlor DA. The Society for Social Medicine John Pemberton Lecture 2011. Developmental overnutrition–an old hypothesis with new importance? Int J Epidemiol. 2013;42:7–29. doi: 10.1093/ije/dys209. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Primers used for pyrosequencing.
Table S2. Characteristics of the selected children compared to the entire EPOCH cohort (after exclusion of IUGR and T1D exposed children).
Table S3. 138 GDM‐associated DMPs.
Table S4. 459 GDM‐associated DMPs after adjustment for child sex, race, and cell proportions in Healthy Start cord blood.
Table S5. Genes woth GDM associated DMPs in EPOCH analysis that have been previously identified as differnetially methylated in GDM exposed offspring.
Table S6. GDM‐associated DMRs after adjustment for child age, sex, race, and cell proportions.
Table S7. Gene Ontology Biological Processes enriched at q<0.05 among GDM‐associated DMRs.
Table S8. Association of DNA methylation changes with child adiposity‐related outcomes at the same visit.
File S1. Supplemental methods.