

Abstract
Background & objectives:
Bardet–Biedl syndrome (BBS) is a genetically heterogeneous autosomal recessive disorder characterized by multiple organ defects involving retina, kidney, liver and brain. Disease-causing mutations in BBS genes narrowed down by homozygosity mapping in small consanguineous and non-consanguineous pedigrees were reported in 80 per cent of the study population. This study was aimed to screen these genes (BBS3, BBS10) and specific exons of BBS genes (BBS1, BBS5, MKKS, BBS9, BBS11 and BBS12) for recurrent mutations in a selected sample of BBS patients.
Methods:
The recurrent mutations in BBS genes were screened in the BBS affected individuals by PCR based direct sequencing. The pathogenicity of the observed mutations were confirmed by co-segregation analysis, screening of healthy unrelated controls and in silico analysis.
Results:
In the 64 BBS patients (44 males, 20 females) were studied, mutations were predominant in BBS10 and ARL6 genes; the c.272T>C; p.(I91T) mutation in ARL6 gene was a recurrent mutation. One novel non-sense mutation c.425T>G; p(L142*) was obtained in BBS5 gene (family BSI-31).
Interpretation & conclusions:
BBS10 gene mutations clustered in exon 2 of the gene suggesting the exon as a probable hotspot for mutations in Indian population. A cost- and time-effective strategy for the molecular diagnosis of BBS was designed based on these results.
Keywords: ARL6, Bardet–Biedl syndrome, BBS10, India
Bardet– Biedl syndrome (BBS) is a genetically heterogeneous autosomal recessive disorder manifesting with major clinical features of retinal degeneration, obesity, hypogonadism, polydactyly, renal dysfunction and learning disability, and minor features such as cardiac abnormalities, diabetes, hypertension, hearing defects and anosmia. The diagnosis for BBS is well established by the clinical manifestations; the presence of three primary with two secondary or four primary features confirms the diagnosis of BBS1. The prevalence of BBS is estimated as 1 in 150000 individuals worldwide, but its incidence increases in isolated/inbred/consanguineous populations2. This syndrome is mapped to 20 genes (BBS1-21)3,4,5; these genes code for proteins involved in ciliary biogenesis and functions. Mutation in these genes affect the structure and function of cilia, the characteristics of any ciliopathy disorders and BBS is grouped one among them.
Mutations in BBS genes (BBS1-18) account for about 70-80 per cent of the disease with frequent recurrent BBS1 and BBS10 gene mutations as reported in certain populations of European and Caucasian descents6. The role of founder effect has also been indicated for mutations such as the M390R in BBS1 and C91fs*95 in BBS10 genes in specific populations7. Mutations in BBS4, BBS5 and TTC8/BBS8 are most commonly seen in the Middle East and North African populations8. BBS4 gene is reported as the most prevalent gene among Turks and Pakistanis3. Among Saudi BBS patients, there is an unusual predominance of ARL6/ARL6 gene mutations, all of which are novel, in striking contrast to the BBS reports from Caucasians in which ARL6 gene mutations are extremely rare9.
The divergent clinical/syndromic features of BBS and variable age of onset often complicates the diagnosis of BBS. Retinal degeneration involving the macula by the second to third decade, with decreased visual acuity 20/200 or worse is the major primary feature of the disease10. In patients with progressive renal impairment, end-stage renal disease renal transplantation is required in 10 per cent of affected individuals11. A higher frequency of sterility was observed in male BBS patients when compared to the female BBS patients2. Cardiovascular and renal diseases are the major causes of death and morbidity12. Hence, diagnosing the disease as early as possible will have an impact on disease management for the patients as there are no treatment options available for BBS.
Screening of Indian BBS families, we have earlier observed disease-causing mutations in 80 per cent of the study population13. In Indian population, ARL6 gene accounted for 18 per cent of the mutations, as against worldwide reports (0.4%), followed by BBS10 (11%), BBS9/PTHB1 and BBS6/MKKS gene (8% each)13. The frequently reported mutations C91fsX95 (BBS10) and M390R (BBS1) genes observed in Caucasians were not more common in Indians13. To validate our earlier observation that >10 per cent of mutations were in ARL6 (inclusive of a recurrent mutation c.272T>C; p.(I91T)) and BBS10 genes, the current study was undertaken in a larger sample.
Material & Methods
Clinical examination was performed on all the patients and family members included in the study. All the patients manifesting at least four primary features or three primary features with at least two minor symptoms14 were included. The study protocol was approved by the Institutional Research and Ethics Committee of Vision Research Foundation, Chennai, India. The patients were selected by consecutive sampling method during December 2012 to May 2015 after a detailed clinical evaluation at the vitreoretina clinic of Sankara Nethralaya, Chennai, or department of Paediatrics, Amrita Institute of Medical Sciences, Kochi. The ophthalmological and other morphological features were documented in a questionnaire based on the details provided by the probands/parents medical history. Power of 80 and 96.1 per cent was achieved for the sample size of 50 cases and 100 controls for screening the BBS3 and BBS10 genes when calculated (http://osse.bii.a-star.edu.sg) based on the allele frequency observed in our initial study13.
Blood collection and DNA extraction: The pedigree was constructed followed by blood collection (8 ml of whole blood collected in Acid Citrate Dextrose Vacutainer) from the proband and the family members after their written consent. The genomic DNA of the patients was extracted using NucleoSpin XL DNA extraction kit as per the manufacturer's instruction (Machery-Nagel, Duren, Germany).
Mutational analysis: Polymerase chain reaction (PCR)-based direct sequencing was performed inABI Prism3100 AVANTGenetic analyzer (Applied Biosystems, USA) for the entire coding regions of ARL6, BBS10 and the specific exons harbouring the reported mutations from Indian patients (BBS1, BBS5, MKKS, BBS9, BBS11 and BBS12). The sequences were compared with the reference sequences NM_001278293.1 and ENST00000463745 (ARL6), NM_024685.3 and ENST00000393262 (BBS10), NM_018848.3 and ENST00000347364 (MKKS), NM_152384.2 and ENST00000295240 (BBS5) and ENST00000450136 and NM_001099679 (BBS11). The primer sequences and PCR conditions were as mentioned earlier13.
Bioinformatic analysis: The mutation nomenclature was in accordance with the guidelines of Human Genome Variation Society (HGVS v.2.0, http://www.hgvs.org/) and checked with the Mutalyzer. The functional effect of the novel missense mutations was evaluated using PolyPhen (http://genetics.bwh.harvard.edu/pph2/), SIFT (http://sift.bii.a-star.edu.sg/www/SIFT_seq_submit2.html) and Consurf server (http://consurf.tau.ac.il/).
Results
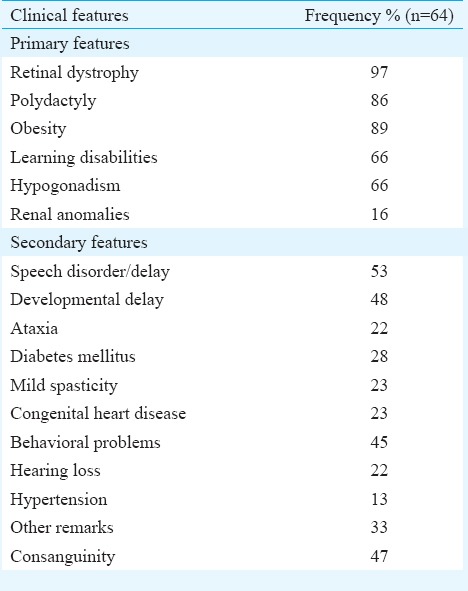
In total, 64 BBS patients (males 44 and females 20) with a mean age of 13.4±6.42 yr (range 6-30) were studied. Table I describes the clinical features of these patients. Thirty one of the 64 families showed a history of consanguinity. Retinal dystrophy was seen in 97 per cent. The frequencies of obesity, polydactyly and brachydactyly were 89, 86 and 29 per cent, respectively. Twenty two per cent of the cases had hearing loss, and 48 per cent showed delayed milestones.
Table I.
Frequency of primary and secondary clinical features in the study population
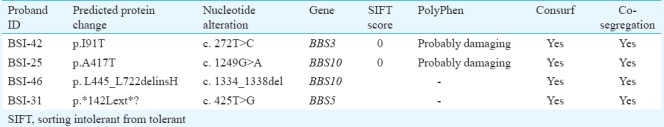
Mutational analysis of ARL6 and BBS10 genes: Three mutations (3/64) were observed in the study population [c.272T>C; p.(I91T) ARL6 gene (family BSI-42); c.1249G>A; p.(A417T) (family BSI- 25), and c.1334_1338del; p. (L445_L722delinsH) (family BSI-46) in BBS10 gene]. The novel mutations co-segregated with the disease phenotype (Fig. 1A–C) and predicted to be pathogenic and deleterious by the bioinformatic tools. (Table II). The c.1334_1338del; p. (L445_L722delinsH) resulted in 446 amino acid (aa) product as against the 721 aa in wild-type protein. Mutation screening of specific exons in BBS1, BBS5, MKKS, BBS9, TRIM32/BBS11 and BBS12 genes revealed one novel non-sense mutation c.425T>G; p.(L142*) in BBS5 gene (family BSI-31) (Fig. 1D). These mutations segregated with the disease and the parents were heterozygous for the same; none of the novel mutations were seen in 100 healthy individuals.
Fig. 1.

Co-segregation analysis for Bardet–Biedl syndrome gene mutations: (A) c.272T>C in ARL6 gene (BSI-42 family) (B) c.1249G>A in BBS10 gene (family BSI-25) (C) c.1334_1338del in BBS10 gene (family BSI-46) (D) c.425T>G in BBS5 gene (family BSI-31).
Table II.
In silico analysis of BBS gene mutations identified in this study
Discussion
The clinical diagnosis of BBS is often made when the patient reports for their visual distress. Some of the features prompting a clinical diagnosis of BBS include (i) childhood-onset retinal dystrophy that occurs in more than 90 per cent of BBS patients, (ii) prenatal appearance of enlarged hyperechoic kidneys without corticomedullary differentiation (CMD) along with polydactyly15, and (iii) positive family history of BBS. Even in families without a history of BBS, in utero findings of CMD and polydactyly has always prompted a differential diagnosis for BBS. However, these primary or secondary features, such as polydactyly (congenital) and obesity (that develops by 2-3 yr), are often ignored. Due to the high incidence of renal developmental anomalies and renal cell carcinoma in relatives of BBS patients, a combination of renal ultrasound study and intravenous pyelography is advised for early identification of renal anomalies in these families16. A detailed clinical history of other systemic features has also been recommended17. There are reports on diagnosis of BBS through prenatal ultrasonography which was subsequently confirmed postnatally through clinical evaluation and molecular confirmation18.
Genetic diagnosis often helps in disease confirmation, effective clinical management, severity prediction, carrier risk identification with potential counselling benefits. A practical approach by prioritizing the frequently mutated genes has been earlier reported for an efficient genetic screening8. High throughput sequencing by panel testing has proven to be very effective in identifying the disease-causing mutations in a single platform19; however, a strategic and cost-effective approach is necessary for countries like India.
On screening 30 Indian families using homozygosity-based gene mapping, we reported a different mutation profile earlier13. ARL6 and BBS10 gene mutations were predominant with a common variant, p.I91T, in ARL6 gene. The present study was performed to validate these observations in an increased sample number that might result in strategical mutation screening system at an affordable cost. To achieve this, a direct sequencing was adopted for entire coding regions of ARL6 and BBS10 genes contributing to >10 per cent of mutations and specific exons in genes (BBS1, BBS5, MKKS, BBS9, BBS11 and BBS12) that were mutated in <10 per cent of the study population.
The study revealed that BBS10 gene mutations were more common when compared to the other genes as has been reported earlier18. However, we were unable to replicate the results of other genes, probably due to genetic heterogeneity of the disease. The c.272T>C; p.(I91T) mutation in ARL6 gene recurred in the present study similar to our earlier results13 thus suggesting the probability of I91T being a recurrent mutation in Indian population; p.(M390R) in BBS1 gene was not seen in any of the BBS patients.
It was observed that patients with BBS10 gene mutations manifested retinitis punctata albescens, polydactyly, obesity and learning difficulties, and those with ARL6/BBS5 gene mutations correlated with retinal dystrophy, polydactyly, obesity and hypogonadism. With these data, we propose here a mutation screening strategy for the genetic diagnosis of BBS in Indian population that has potential counselling benefits (Fig. 2) in Indian population. The feasibility of the strategy has, however, to be tested after screening all the BBS genes in larger sample size. On successful validation of this strategy, an affordable molecular test for patients and their afflicted family would be available having potential for a timely assistance in prenatal diagnosis and genetic counselling benefits.
Fig. 2.

Strategy designed for Bardet–Biedl syndrome diagnosis in Indian Bardet–Biedl syndrome patients.
The limitation of the study was sample size. By increasing the sample size the mutation data observed in our previous study13 could have been validated. On screening the exons of other BBS genes, the complete mutation spectrum for 100 families could have been achieved.
To summarize, the c.272T>C; p.(I91T) mutation in ARL6 gene was a recurrent mutation, and the intermediate domain of exon-2 of BBS10 gene as a probable hotspot region for Indian population. Varying frequency of clinical features and the novel mutations might be attributed to the ethnicity/a different mutation spectrum. Similar studies in larger samples representing various parts of the country are required to validate the observed results and also to increase the knowledge on varying expression of clinical features in Indian BBS patients.
Footnotes
Financial support & sponsorship: The first author (SPC) acknowledges the Indian Council of Medical Research (ICMR), New Delhi, for providing financial support as Senior Research Fellowship (Project number: 45/08/2012-HUM-BMS)
Conflicts of Interest: None.
References
- 1.Beales PL, Elcioglu N, Woolf AS, Parker D, Flinter FA. New criteria for improved diagnosis of Bardet-Biedl syndrome: Results of a population survey. J Med Genet. 1999;36:437–46. [PMC free article] [PubMed] [Google Scholar]
- 2.Forsythe E, Beales PL. Bardet-Biedl syndrome. Eur J Hum Genet. 2013;21:8–13. doi: 10.1038/ejhg.2012.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Fattahi Z, Rostami P, Najmabadi A, Mohseni M, Kahrizi K, Akbari MR, et al. Mutation profile of BBS genes in Iranian patients with Bardet-Biedl syndrome: Genetic characterization and report of nine novel mutations in five BBS genes. J Hum Genet. 2014;59:368–75. doi: 10.1038/jhg.2014.28. [DOI] [PubMed] [Google Scholar]
- 4.Schaefer E, Stoetzel C, Scheidecker S, Geoffroy V, Prasad MK, Redin C, et al. Identification of a novel mutation confirms the implication of IFT172 (BBS20) in Bardet-Biedl syndrome. J Hum Genet. 2016;61:447–50. doi: 10.1038/jhg.2015.162. [DOI] [PubMed] [Google Scholar]
- 5.Heon E, Kim G, Qin S, Garrison JE, Tavares E, Vincent A, et al. Mutations in C8ORF37 cause Bardet Biedl syndrome (BBS21) Hum Mol Genet. 2016;25:2283–94. doi: 10.1093/hmg/ddw096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.M’hamdi O, Ouertani I, Chaabouni-Bouhamed H. Update on the genetics of Bardet-Biedl syndrome. Mol Syndromol. 2014;5:51–6. doi: 10.1159/000357054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zaghloul NA, Katsanis N. Mechanistic insights into Bardet-Biedl syndrome, a model ciliopathy. J Clin Invest. 2009;119:428–37. doi: 10.1172/JCI37041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Billingsley G, Deveault C, Héon E. BBS mutational analysis: A strategic approach. Ophthalmic Genet. 2011;32:181–7. doi: 10.3109/13816810.2011.567319. [DOI] [PubMed] [Google Scholar]
- 9.Abu Safieh L, Aldahmesh MA, Shamseldin H, Hashem M, Shaheen R, Alkuraya H, et al. Clinical and molecular characterisation of Bardet-Biedl syndrome in consanguineous populations: The power of homozygosity mapping. J Med Genet. 2010;47:236–41. doi: 10.1136/jmg.2009.070755. [DOI] [PubMed] [Google Scholar]
- 10.Klein D, Ammann F. The syndrome of laurence-moon-Bardet-Biedl and allied diseases in Switzerland. Clinical, genetic and epidemiological studies. J Neurol Sci. 1969;9:479–513. doi: 10.1016/0022-510x(69)90091-4. [DOI] [PubMed] [Google Scholar]
- 11.Mihai CM, Marshall JD, Stoicescu RM. Bardet-Biedl syndrome with end-stage kidney disease in a four-year-old Romanian boy: A case report. J Med Case Rep. 2011;5:378. doi: 10.1186/1752-1947-5-378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Imhoff O, Marion V, Stoetzel C, Durand M, Holder M, Sigaudy S, et al. Bardet-Biedl syndrome: A study of the renal and cardiovascular phenotypes in a French cohort. Clin J Am Soc Nephrol. 2011;6:22–9. doi: 10.2215/CJN.03320410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sathya Priya C, Sen P, Umashankar V, Gupta N, Kabra M, Kumaramanickavel G, et al. Mutation spectrum in BBS genes guided by homozygosity mapping in an Indian cohort. Clin Genet. 2015;87:161–6. doi: 10.1111/cge.12342. [DOI] [PubMed] [Google Scholar]
- 14.Katsanis N, Lupski JR, Beales PL. Exploring the molecular basis of Bardet-Biedl syndrome. Hum Mol Genet. 2001;10:2293–9. doi: 10.1093/hmg/10.20.2293. [DOI] [PubMed] [Google Scholar]
- 15.Cassart M, Eurin D, Didier F, Guibaud L, Avni EF. Antenatal renal sonographic anomalies and postnatal follow-up of renal involvement in Bardet-Biedl syndrome. Ultrasound Obstet Gynecol. 2004;24:51–4. doi: 10.1002/uog.1086. [DOI] [PubMed] [Google Scholar]
- 16.Ashkinadze E, Rosen T, Brooks SS, Katsanis N, Davis EE. Combining fetal sonography with genetic and allele pathogenicity studies to secure a neonatal diagnosis of Bardet-Biedl syndrome. Clin Genet. 2013;83:553–9. doi: 10.1111/cge.12022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Beales PL, Reid HA, Griffiths MH, Maher ER, Flinter FA, Woolf AS, et al. Renal cancer and malformations in relatives of patients with Bardet-Biedl syndrome. Nephrol Dial Transplant. 2000;15:1977–85. doi: 10.1093/ndt/15.12.1977. [DOI] [PubMed] [Google Scholar]
- 18.Pasinska M, Dudarewicz L, Jakubowski L, Haus O. Prenatal and postnatal diagnostics of a child with Bardet-Biedl syndrome: Case study. J Mol Genet Med. 2015;9:189. [Google Scholar]
- 19.Glöckle N, Kohl S, Mohr J, Scheurenbrand T, Sprecher A, Weisschuh N, et al. Panel-based next generation sequencing as a reliable and efficient technique to detect mutations in unselected patients with retinal dystrophies. Eur J Hum Genet. 2014;22:99–104. doi: 10.1038/ejhg.2013.72. [DOI] [PMC free article] [PubMed] [Google Scholar]