

Abstract
The osteopontin (OPN) protein is found expressed at high level in several processes including fibrotic evolution of organ injuries, tumorigenesis, and immune response. The molecular mechanisms that underly overexpression, especially at the transcriptional level, have been only partially clarified. Therefore, this study was undertaken in search for additional DNA elements in the regulatory regions of the OPN gene and cognate transcription factors. Our results on the region upstream of the transcription start site confirmed that essential elements are located within the first 100 bp. Analysis of the sequence that includes the first untranslated exon and first intron revealed that it could enhance the promoter activity. Experiments of transfection of constructs containing different fragments of this sequence showed that most of the enhancer activity was confined in the terminal 30-bp tract of the first intron, although it was not functioning in a myofibroblast cell line. DNA/protein binding assays and cotransfection experiments showed that the C/EBP-beta transcription factor was able to bind a recognition sequence in this 30-bp segment. We found a bi-allelic sequence polymorphism at +245 in the first intron, which did not show a significant functional effect, but is a useful tool for future association studies.
Keywords: Osteopontin, Transcription, Enhancer, C/EBP-beta
OSTEOPONTIN (OPN) is a protein secreted in the extracellular matrix by different types of cells in different tissues, such as osteoblasts and osteoclasts, tubular kidney cells, mammary gland epithelial cells, macrophages, and T lymphocytes (4). This protein is a sialic acid-rich glycophosphoprotein that contains an arginine-glycine-aspartic acid (RGD) amino acid motif found in other extracellular matrix proteins and responsible for cell binding by interaction with integrin receptors and the CD44 receptor (5). OPN pattern of expression and adhesive properties explain how it has been found involved in several disease mechanisms, among which are tumor invasiveness and metastasis, angiogenesis, injury responses leading to organ fibrosis in kidney, heart, and lung, bone destruction in arthritis, and cell-mediated immune response (5). Overexpression is the common finding in such different conditions, raising the question how this overexpression is achieved and which factors activate OPN gene transcription.
Moreover, studies in animals have shown an important role of OPN in different processes, such as early embryonic development (3), development of the mammary gland (12), bone remodeling in ovariectomized mice (21), wound healing (11), and postinfarct myocardial remodeling (18).
The human OPN encoding gene is located on the human chromosome 4 and spans approximately 8 kb. Similar to the mouse gene, it is composed of 7 exons, with the ATG start codon located in the second exon and a 5′-untranslated sequence that includes the first exon and first intron (20). The OPN gene expression is subjected to various types of posttranscriptional and posttranslational modifications that lead to the presence of multiple molecular forms in vivo (10). Previous work has provided some information on the transcriptional regulation of this gene (19,20), although a comprehensive analysis of basal and inducible OPN gene transcription is still to be completed. The gene structure and previous evidence coming from studies on the human and mouse genes (3,20) suggest that elements important for transcriptional activity may be present in the untranslated region that includes first exon and first intron. We present here the results of an analysis of promoter and intronic elements in different cell lines, which show the presence of a short region at the 3′ end of the first intron of the human OPN gene, able to enhance promoter activity. Our results also suggest that this enhancer ability is not ubiquitous in the tested cell lines, suggesting that it might contribute to restrict the pattern of transcriptional control.
MATERIALS AND METHODS
Plasmids and DNA Constructions
Plasmid 2235-LUC contains the human OPN gene fragment from −2235 to +95 relative to the transcription start site, obtained by PCR from genomic DNA and cloned upstream of the luciferase reporter gene in the SmaI site of the pGL3 basic vector plasmid (Promega).
Plasmid 1206-LUC contains a promoter fragment from −1206 to +40 obtained by restriction digestion with SacI and AvaII of the pCat-1206 plasmid (generous gift of Professor S. Yamamoto) (20) and cloning in the pGL3 Basic Vector (Promega) plasmid between the SacI and SmaI sites in the polylinker.
Plasmids 124-LUC and 62-LUC, containing shorter promoter fragments from −124 and −62 to +32, were prepared by PCR amplification from the 1206-LUC plasmid with sense primers specific for the respective 5′ ends (primer 124: 5′-GGGGAAGTGTGGGAGCAGGT-3′; primer 62: 5′-CCTCCCTGTGTTGGTGGAGGAT-3′) and an antisense primer designed on the vector sequence (primer GL2: 5′-CTTTATGTTTTTGGCGTCTTCCA-3′). The amplified fragments were first made blunt, then digested with HindIII to obtain a sticky 3′ end, and finally cloned in the pGL3 basic vector plasmid between the SmaI and HindIII sites of the polylinker.
The fragment containing the first exon and first intron was prepared by PCR amplification from genomic DNA using the sense primer 62 and an antisense complementary to the terminal end of the first intron and carrying a HindIII site at its 5′ end (primer intro: 5′-GATCAAGCTTCTGCAAAATATTTCA-3′). The 1245-bp amplified fragment was digested with PstI (at −33 in the promoter) and HindIII, which allowed directional cloning in 2235-LUC, 124-LUC, and 62-LUC, resulting in the respective 2235i-LUC, 124i-LUC, and 62i-LUC that contain promoter sequence, first untranslated exon, and first intron upstream of luciferase.
Plasmids 124i/delXba-LUC (intron suquence up to nucleotide +260) and 124i/delAsp-LUC (intron sequence up to nucleotide +1059) were prepared by cloning intronic fragments, obtained by restriction digestion of 124i-LUC with PstI/XbaI and PstI/Asp700, respectively, into the 124i-LUC plasmid after removal of the PstI/HindIII fragment and blunting of the fragments’ 3′ ends and vector’s HindIII end.
124i/del32-LUC (entire intron sequence but the last 32 bp) was prepared by PCR amplification using the 124i-LUC plasmid as template, the sense primer 62, upstream of the PstI intronic site, and an anti-sense primer carrying a HindIII site containing tail (primer intro 32: 5′-AAGCTTCATAGGTTACAACAGTGATACC-3′). This 1206-bp fragment was inserted into a TA cloning vector (Invitrogen), digested with PstI/HindIII, and cloned into the 124i-LUC plasmid.
Site-specific mutagenesis was performed according to the PCR-based procedure of Higuchi et al. (8) to obtain 124i-CEBPbetaMut-LUC and 124i-1TG-LUC. In all cases a mutagenized PstI/HindIII fragment was inserted into the 124i-LUC plasmid, after restriction digestion.
The pCEP4/cebp plasmid (16), containing the rat c/EBP-beta cDNA under the control of the CMV promoter, was a kind gift of Dr. Valeria Poli (Dipartimento di Genetica, Biologia e Biochimica, University of Torino, Italy).
All the plasmids were carefully verified by restriction analysis and DNA sequencing.
Cell Culture and Transfections
Cell lines were maintained in Dulbecco’s modified Eagle’s medium supplemented with 10% fetal calf serum, 2 mM L-glutamine, 100 U/ml penicillin, 100 μg/ml streptomycin in a humidified atmosphere with 5% CO2. All cell lines were available in the laboratory and originally obtained from the American Type Culture Collection.
Transfections were performed using the polyethy-lenimine (PEI) cationic polymer, as previously described (15).
Luciferase activity was measured using the Dual-Luciferase Reporter Assay System (Promega), according to the manufacturer’s instructions, on a Turner Luminometer.
The data obtained represent the average ± SD of at least three independent experiments, each performed either in triplicate or in duplicate.
Preparation of Nuclear Extracts and Gel Retardation Assay
Nuclear extracts were prepared as previously described (6) from the U2OS and COS7 cell lines. All steps were carried out at +4°C, in the presence of a protease inhibitor cocktail (Roche Molecular Bio-chemicals).
Gel retardation experiments were performed essentially as described (2). Briefly, 5–10 μg of nuclear extract protein was incubated in a binding mixture containing 10 mM HEPES buffer, pH 7.9, 0.1 mM EDTA, 50 mM KCl, 0.5 mM dithiothreitol, 10% glycerol, 1 μg of poly(dI-dC)/poly(dI-dC), and 1× protease inhibitor cocktail. The incubation lasted for 20 min at room temperature and, when required, competitor oligonucleotides were added as indicated in text and figures. Supershift experiments were performed by including specific antibodies in the incubation mixture. The sequence of the oligonucleotide used as labeled probe or unlabeled competitor and its mutated version are reported in Figure 4B.
Figure 4.
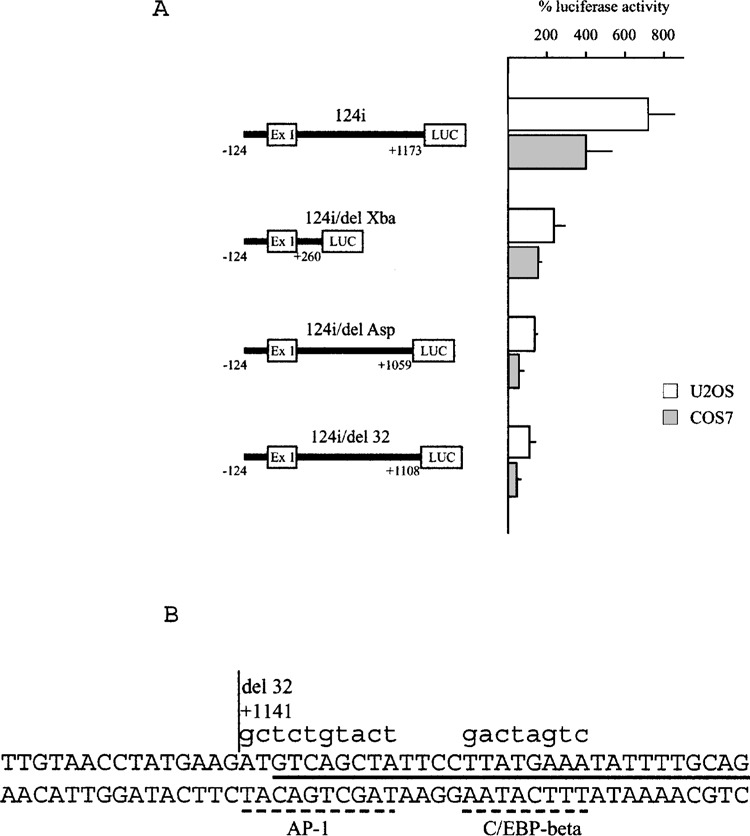
Effect of deletions in the first exon/first intron region on enhancer activity. (A) Results of luciferase activity of cell lines transfected with constructs containing fragments of different length of the first exon/first intron region are reported as percentage of the 124-LUC plasmid. The results are mean ± SD of at least three experiments performed in duplicate. (B) Sequence of the first intron 3′ terminal sequence: the site of the deletion present in the 124i-del32 plasmid is marked by a vertical line; the putative recognition sites of the C/EBP-beta and AP-1 transcription factors are marked by hatched lines; the sequence of the oligonucleotide used for gel retardation assays is marked by a black full line; lowercase letters indicate substitutions in mutated plasmids and in the oligonucleotide mutant probe.
Analysis of an Intronic Polymorphism
PCR was performed on genomic DNA using a specific pair of primers to amplify an intronic fragment including a polymorphic variant with two alleles differing for a 2 nucleotide repetition (TG/TGTG). We used a 5′ fluorescence-labeled sense primer (5′-6FAMTGGGTTGTGCATTCAGCTG-3′) in order to analyze the repetition variant on an automated ABI PRISM 377 DNA sequencer. Four different antisense primers (primer R1: 5′-TTAGCATCGGTGGTTTCCG-3′; R2: 5′-TTTTGAGGACCCAGTGGAAG-3′; R3: 5′-CCTGCACAGTCACCCACTG-3′; R4: 5′-CAGTGGCATATTCAGAAAGGG-3′) were combined with the same sense primer to obtain PCR products of different length, therefore increasing the number of analyzable samples in the same electrophoretic lane.
RESULTS
The OPN Promoter Activity Is Enhanced by Regulatory Sequences Downstream of the Transcription Start Site
Transfection of plasmids containing fragments of different length derived from the OPN 5′ upstream genomic region, from −2235 to −62 relative to the transcription start site, fused to the luciferase reporter gene, were performed in different cell lines: HeLa (epithelial, derived from human uterine cervical carcinoma), MCF-7 (epithelial, derived from human breast adenocarcinoma), H9c2(2-1) (myofibroblast, derived from embryonic rat heart), U2OS (osteoblast-like, derived from human osteogenic sarcoma), NRK-52E (epithelioid, derived from rat kidney), COS-7 (fibroblast, derived from monkey kidney). These cell lines were chosen because of the known OPN expression pattern in different organs and tissues and in tumors. Transcriptional activity of the 2.3-kb 5′-flanking sequence was well appreciable in all tested cell lines, although at low level if compared with activity of the pGL3-Control Vector (Promega), containing SV40 promoter and enhancer sequences, and at different rates in different cell lines, as shown in Figure 1. Figure 2 shows that, after deletion of the 5′ sequence, transcriptional activity was maintained up to nucleotide −124 in all transfected cell lines, although with quantitative differences, and was strongly reduced after deleting sequence between −124 and −62, confirming in part previously described results (19,20). Moreover, DNA elements with negative effect on transcriptional activity were detectable between −2235 and −1206, as shown by an increase of activity of 1206-LUC plasmid, particularly evident in COS7, HeLa, MCF7, and H9c2(2-1) cell lines.
Figure 1.
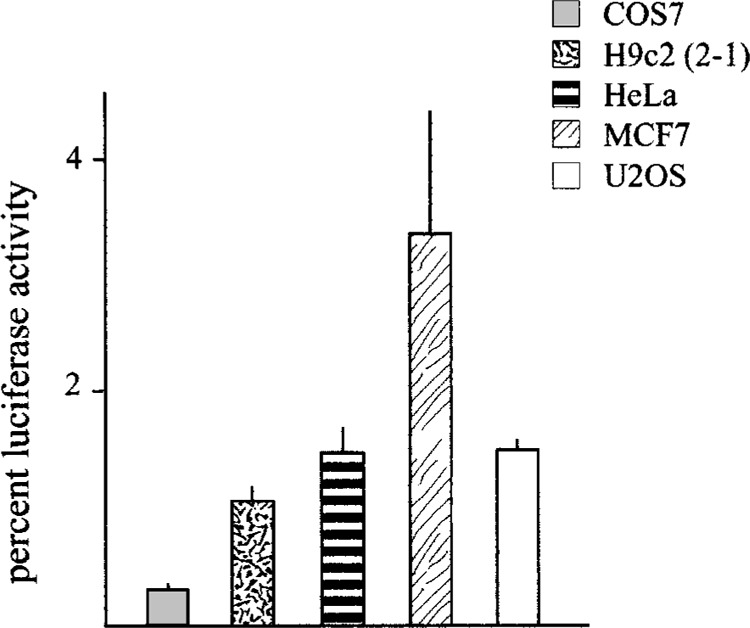
OPN promoter activity in different cell lines compared to a viral promoter/enhancer construct. Results of luciferase activity after transfection of the 2235-LUC plasmid in the indicated cell lines are expressed as percentage of the pGL3-Control Vector containg the SV40 promoter and enhancer. The results are mean ± SD of at least three experiments performed in duplicate.
Figure 2.
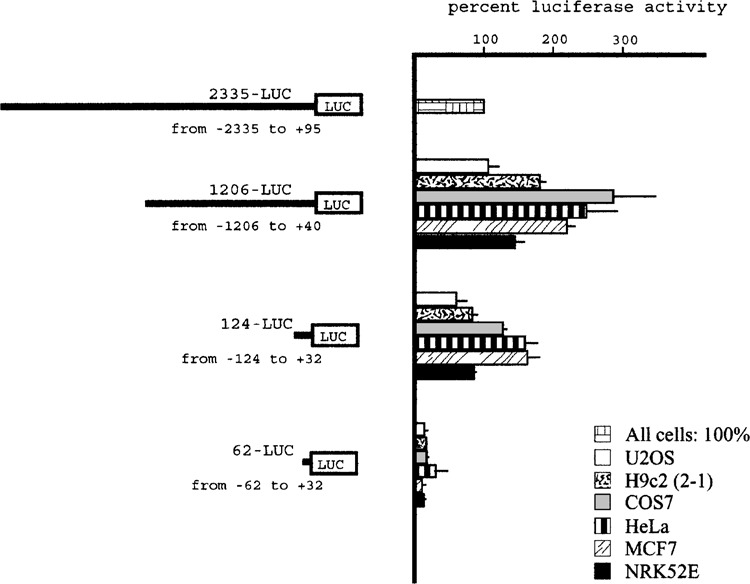
Activity of OPN promoter fragments. Results of luciferase activity after transfection of the 1206-LUC, 124-LUC, and 62-LUC plasmids in the indicated cell lines are reported as percentage of the 2235-LUC plasmid taken as 100% for any cell line. The results are mean ± SD of at least three experiments performed in duplicate.
Given the observation that the OPN gene has a 1.1-kb untranslated transcribed region that includes the first exon and first intron, structurally conserved in human and mouse, and given previously reported evidence indicating that regulatory elements were acting in this region (3,20), we studied the entire untranslated sequence looking for DNA elements able to exert a regulatory function on transcription. Transfection of plasmids 2335i-LUC, 124i-LUC, and 62i-LUC, in which the entire region was inserted downstream of the respective 5′-flanking sequence, indicated that the exon/intron sequence was able to enhance transcriptional activity (Fig. 3). This enhancer effect was particularly evident with the 124-bp promoter fragment but, contrary to the ubiquitous type of activity of the promoter region, was not detectable in the H9c2(2-1) cell line, suggesting that it could contribute to restrict the pattern of gene expression in tissues.
Figure 3.
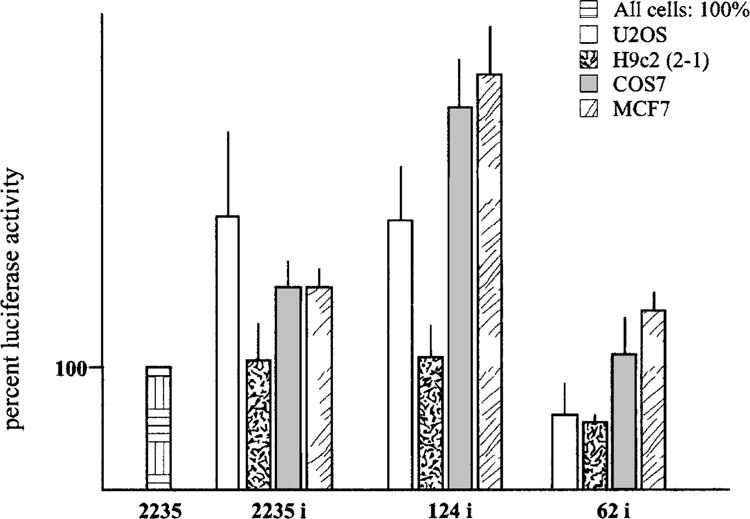
Enhancing effect of the first exon/first intron sequence on OPN promoter activity. Luciferase activity is expressed as percentage of the 2235-LUC plasmid activity, taken as 100% for any cell line. The results are mean ± SD of at least three experiments performed in triplicate.
In all tested cell lines, the addition of the 1.1-kb region was able to confer transcriptional activity to the 62-LUC plasmid, which was virtually inactive in the absence of this region (see Fig. 1). Similarly, when this DNA fragment was inserted upstream of the luciferase gene in the promoterless pGL3-Basic Vector, the resulting pGL3-i plasmid showed transcriptional activity in COS7 and U2OS cells at a 50% and 75% rate, respectively, compared with the 2235-LUC plasmid. These results raised the hypothesis that an alternative promoter could be present inside the 1.1-kb region. However, our attempts to detect OPN transcripts with alternative start sites by 5′ RACE experiments using cDNA from human kidney (Marathon Ready cDNA, Clontech) failed to provide data in favor of such hypothesis (not shown).
Identification and Characterization of the Region Responsible for Enhancer Activity
To identify in more detail the DNA elements responsible for enhancer activity, we made deletions of segments of the 1.1-kb untranslated region that are present in the following plasmids: 124i/delXba-LUC, containing sequence up to +260; 124i/delAsp-LUC, containing sequence up to +1059 and lacking the last 114 bp intronic segment. Both these plasmids lost their enhancer ability, showing transcription activity close to the 124-LUC plasmid, which contains only promoter sequence, in transfection experiments in U2OS and COS7 cells (Fig. 4A). Therefore, our work was focused on the search for a DNA element inside the terminal region of the first intron. Computer analysis of this sequence to find putative transcription factor binding sites indicated the presence of a C/EBP-beta site, 5′-TTATGAAA-3′ (Fig. 4B), inside the terminal 30 bp of the intron. To verify whether this C/EBP-beta binding site could be involved in the enhancer effect, we prepared the 124i/del32-LUC plasmid, containing the entire intron sequence but the last 32 bp. This plasmid showed comparable loss of enhancer activity, indicating that the 32-bp terminal sequence might contain the functional element (Fig. 4A).
We performed gel retardation assay (GRA) using a 30-bp double-stranded oligonucleotide, which included the putative C/EBP-beta binding site, as labeled probe (Fig. 4B). Figure 5A shows that this probe bound a nuclear factor(s) present in U2OS cell nuclear extract in a sequence-specific way, because specific retarded complexes could be competed by the same unlabeled oligonucleotide and not by a mutated oligonucleotide in which 8 nucleotides were substituted in the putative C/EBP-beta binding site (see Fig. 4B for sequence details). Comparable results were obtained using nuclear extracts from COS7 cells. When the mutated oligonucleotide was used as a labeled probe, it was unable to bind the specific factor(s) (not shown). Addition of a specific anti-C/EBP-beta antibody in the GRA caused disappearance of the specific complexes and an evident supershifted complex (Fig. 5B, lane 4), whereas another control antibody and preimmune serum (not shown) did not affect the GRA pattern. The supershifted band could be efficiently competed by the specific unlabeled oligonucleotide, thus confirming the specificity of protein/DNA interaction (Fig. 5B, lane 5).
Figure 5.
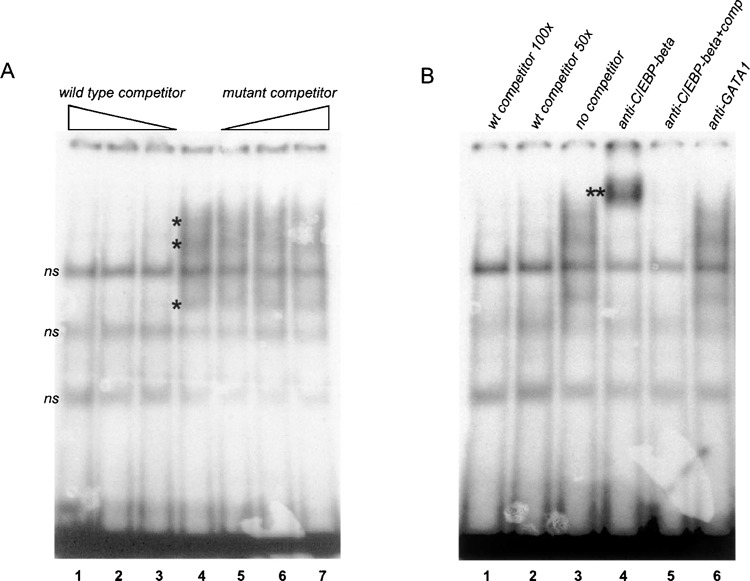
The 32-bp terminal sequence of the OPN first intron contains a C/EBP-beta binding site. (A) Gel retardation assay with a double-stranded oligonucleotide that reproduces the last 30-bp sequence of the first intron and nuclear extract from U2OS cells, in the absence (lane 4) or in the presence of wild-type competitor oligonucleotide at 50-, 100-, or 250-fold molar excess (lanes 1–3) or mutant competitor (lanes 5–7). The asterisk indicates sequence-specific retarded complexes; ns indicates nonspecific bands. (B) Gel retardation assay with same oligonucleotide and nuclear extract as in (A), in the absence of competitor and antibody (lane 3), in the presence of competitor oligonucleo-tide (lanes 1, 2, and 5), with anti-C/EBP-beta antibody (lane 4), with anti-GATA1 antibody (lane 6). The double asterisk indicates a supershifted complex.
Because the nucleotide substitution abolished the ability to bind the C/EBP-beta factor, we introduced the same substitution mutation in the 124i-CEBP/ betaMUT-LUC plasmid and verified its functional properties by transfection in U2OS and COS7 cells, in comparison with the wild-type 124i-LUC plasmid. As shown in Figure 6A, in both cell lines the mutation caused a significant reduction of enhancer activity.
Figure 6.
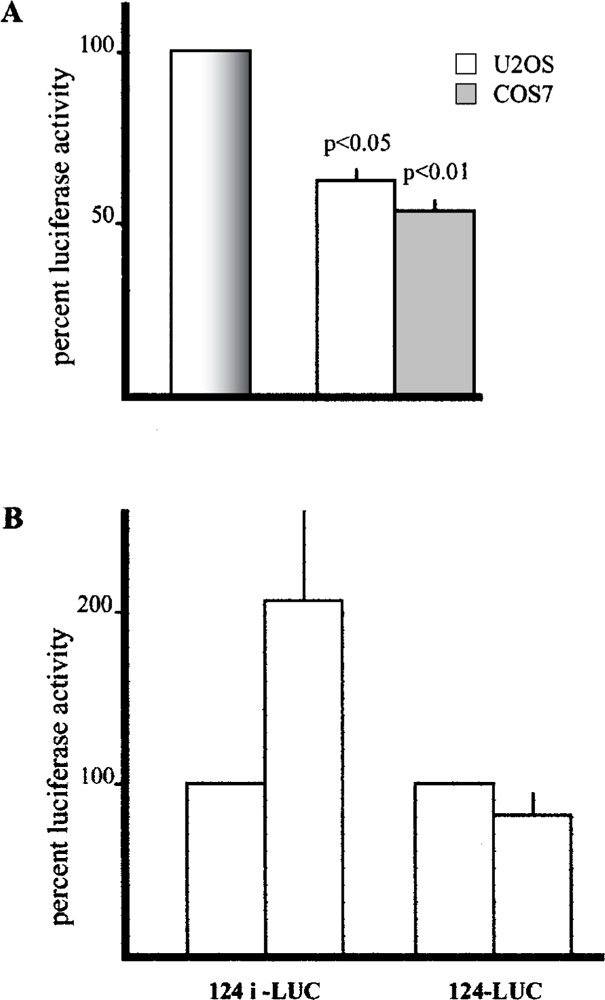
Effect of site- specific mutagenesis in the C/EBP-beta binding site and cotransfection with a C/EBP-beta expression vector. (A) Luciferase activity of plasmid 124i-CEBPbetaMut-LUC containing a substitution mutation in the C/EBP-beta binding site expressed as percentage of the wild-type 124i-LUC plasmid (bar on the left indicated as 100% in both cell lines). The results are mean ± SD of at least three experiments performed in duplicate. (B) Luciferase activity of 124i-Luc and 124-Luc plasmids in U2OS cells cotransfected with the pCEP4/cebp plasmid expressed as percentage of cells cotransfected with the empty pCEP4 vector (left bar for each cotransfection result). The results are mean ± SD of at least three experiments performed in triplicate.
Our computer analysis of the terminal intronic sequence indicated the presence of a putative AP1 binding site flanking the C/EBP-beta site. To verify the hypothesis that this sequence could cooperate with the C/EBP-beta site for the enhancer activity, we mutagenized by nucleotide substitution another 8 nucleotides in the putative AP1 site, producing the 124i-CEBP/AP1MUT-LUC plasmid in which both sites were mutated. However, this additional mutation did not cause further loss of enhancer activity compared with the mutation in the C/EBP-beta site alone, failing to demonstrate that this flanking sequence could be involved in the C/EBP-beta enhancing activity (not shown).
Finally, cotransfection of the 124i-LUC plasmid with an expression vector containing the C/EBP-beta cDNA showed a twofold activation of reporter expression that was not observed with the 124-LUC plasmid, as shown in Figure 6B.
Identification and Analysis of an Intronic Polymorphism
While studying the 1.1-kb region containing first untranslated exon and first intron of the human OPN gene we identified a bi-allelic variant in position +245 consisting of the TG dinucleotide in one or two repetition (TG/TGTG). We genotyped 207 individuals of Caucasian origin for this variant and found that it is a polymorphism with a frequency of 0.67 for the TG allele and 0.33 for the TGTG allele. Because our method (see Materials and Methods for details) to genotype was based on size detection, it also could detect other dinucleoltide repetitions if present; however, no additional alleles other than TG or TGTG were present in our sample population. Considering the properties of the OPN first intron in transcriptional regulation, we wanted to verify whether this variant could influence the intronic enhancer function. This was investigated by both transfection and gel retardation assay experiments. Comparison of the 124i-LUC plasmid, which carries the TGTG allele, with the 124i/1TG-LUC plasmid in transfection experiments in U2OS and COS7 cells did not show any significant difference in enhancer ability (not shown).
GRA in which two labeled double-stranded oligonucleotide probes, with the two different allelic sequences, were compared for binding of nuclear factors from COS7 nuclear extracts, showed quantitative and not qualitative differences in retarded complexes (not shown).
Taken together, these data indicate that this polymorphic variant, although very useful for genetic studies, is unlikely to influence regulatory function.
DISCUSSION
The importance of understanding the mechanisms that regulate OPN gene transcription in the context of cellular systems involved in different functions of its protein product prompted us to investigate in more detail this largely incomplete aspect of OPN regulation. The analysis of sequences at the 5′-flanking region, upstream of the transcription start site, substantially confirmed results obtained by others who found that essential elements for reporter gene transcription were located in a sequence interval within the first 100 bp upstream of the transcription start point (19,20). In particular, it seems that a 30-bp promoter fragment from −94 to −62 is critical for transcriptional activity. However, when we compared the regulatory activity of this region with that of a construct IP: 103.62.30.226 On: Wedcontaining SV40 promoter and enhancer, we detected low level of OPN promoter activity in different cell lines, even those that might be envisaged as good models of OPN-expressing tissues and cell types. This suggests that this weak basal promoter activity can be increased in all conditions in which OPN reaches high levels of expression and therefore DNA elements able to respond to different stimuli should be taken in great consideration. Moreover, other elements, in addition to those located in the 5′-flanking region, might contribute to basal and induced gene transcription. Previous reports, although lacking a systematic analysis and identification of cis- and trans-acting factors, had indicated that sequences in the first intron could play some role in transcriptional regulation (3,20). Here we show that sequences inside a region that includes first untranslated exon and first intron enhance the promoter transcriptional activity and that the 3′ terminal end of the first intron is critical in such regulation. A C/EBP-beta binding site appears to play an important role in this enhancer region, as shown by the significant reduction of enhancer activity after mutation of this binding site and the twofold activation observed after cotransfection with a C/EBP-beta expression plasmid, only detected when the OPN/luciferase plasmid contained the enhancer sequence.
The enhancer activity was negligible in a rat cardiac myofibroblast cardiac cell line, H9c2(2-1), which suggests that this enhancer might also contribute to regulate the basal level of gene transcription in a cell type-restricted manner.
When cloned into a promoterless vector, the 1.1kb sequence was able to direct transcription to the same activity as occurred in plasmids containing the 5′-flanking region. Although we might hypothesize that this potential promoter function is relevant in vivo under particular physiological circumstances (e.g., during embryonic development), leading to alternative transcripts with much shorter untranslated 5′-flanking sequence, we did not collect any evidence supporting such hypothesis.
C/EBP-beta belongs to a family of transcription factors characterized by a basic DNA binding domain and a basic leucine zipper dimerization motif. Some of the members of the C/EBP family, including C/ EBP-beta, act as transcriptional activators; others, such as C/EBP-gamma, liver-enriched transcriptional inhibitory protein (LIP), and C/EBP-homologous protein 10 (CHOP-10), act as negative regulators (17). Because all the members of the family can form het-erodimers with each other, a balance between positive and negative regulation in different physiological and/or pathological conditions would provide a fine means to modulate the expression of target genes. In the case of OPN, considering the high level of C/ EBP-beta expression after stimulation by molecules such as bacterial LPS, IL-6, and IL-1 cytokines (1), regulation by this factor and eventually its homologs fits very well with the role that this protein has in inflammatory processes. Indeed, enhanced expression of C/EBP-beta and OPN has been described in rheumatoid arthritis at the same sites of the rheumatoid synovium (13,14), suggesting that the production of OPN may be regulated in part by C/EBP-beta.
Recent results highlight the role of C/EBP transcription factors, in particular C/EBP-beta in cooperation with the Runx2 transcription factor, in supporting osteoblast-specific gene expression and playing an important regulatory role during osteoblast differentiation (7).
During the extensive work of cloning, we had the chance to identify a polymorphic variant in the first intron, which, due to the regulatory role of intronic sequences, might have some effect on transcriptional regulation. Our experiments, although showing some difference in functional activity of constructs carrying the two alleles of this polymorphism and some quantitative difference in the ability to bind nuclear factors, did not result in statistically significant data. Therefore, we conclude that this polymorphism does not have a major effect, if any, in functional regulation of gene expression. However, the identification of an intragenic polymorphic variant in a gene that is involved in several different biological processes is useful to genetic studies in search for association between alleles and pathological conditions. In particular, we might expect that OPN plays some role in the pathogenesis of bone diseases, of fibrogenic progression of organ injuries, of abnormal immune response, and tumor invasiveness and metastasis. It is curious that, at the same nucleotide position in the first intron, another polymorphic variant has been reported in the Japanese population (9), which we did not observe in our population sample of Caucasian origin.
ACKNOWLEDGMENTS
We thank Prof. Shunsuke Yamamoto, Department of Pathology, Oita Medical University, for the generous gift of the plasmid −1206/+90CAT. The gift of the pCEP4/cebp plasmid by Dr. Valeria Poli (University of Torino, Italy) is greatly acknowledged. This work was supported by grants from CNR-Target Project on Biotechnology to R.R., by the Italian Ministry of University and Research PRIN and FIRB to R.R., and by the CEBR-Center of Excellence at the University of Genova, Italy. The secretarial assistance of Mrs. Loredana Velo is greatly acknowledged.
REFERENCES
- 1. Akira S.; Isshiki H.; Sugita T.; Tanabe O.; Kinoshita S.; Nishio Y.; Nakajima T.; Hirano T.; Kishimoto T. A nuclear factor for IL-6 expression (NF-IL6) is a member of a C/EBP family. EMBO J. 9:1897–1906; 1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Bobola N.; Hirsch E.; Albini A.; Altruda F.; Noonan D.; Ravazzolo R. A single cis-acting element in a short promoter segment of the gene encoding the interphotoreceptor retinoid-binding protein confers tissue-specific expression. J. Biol. Chem. 270:1289–1294; 1995. [DOI] [PubMed] [Google Scholar]
- 3. Botquin V.; Hess H.; Fuhrmann G.; Anastassiadis C.; Gross M. K.; Vriend G.; Scholer H. R. New POU dimer configuration mediates antagonistic control of an osteopontin preimplantation enhancer by Oct-4 and Sox-2. Genes Dev. 12:2073–2090; 1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Denhardt D. T.; Guo X. Osteopontin: A protein with diverse function. FASEB J. 7:475–482; 1993. [PubMed] [Google Scholar]
- 5. Denhardt D. T.; Noda M.; O’Regan A. W.; Pavlin D.; Berman J. S. Osteopontin as a means to cope with environmental insults: Regulation of inflammation, tissue remodeling, and cell survival. J. Clin. Invest. 107:1055–1061; 2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Dignam J. D.; Lebovitz R. M.; Roeder R. G. Accurate transcription initiation by RNA polymerase II in a soluble extract from isolated nuclei. Nucleic Acids Res. 11:1475–1489; 1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Gutierrez S.; Javed A.; Tennant D. K.; van Rees M.; Montecino M.; Stein G. S.; Stein J. L.; Lian J. B. CCAAT/enhancer-binding proteins (C/EBP) beta and delta activate osteocalcin gene transcription and synergize with Runx2 at the C/EBP element to regulate bone-specific expression. J. Biol. Chem. 277:1316–1323; 2002. [DOI] [PubMed] [Google Scholar]
- 8. Higuchi R.; Krummel B.; Saiki R. K. A general method of in vitro preparation and specific mutagenesis of DNA fragments: Study of protein and DNA interactions. Nucleic Acids Res. 16:7351–7367; 1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Iwasaki H.; Shinohara Y.; Ezura Y.; Ishida R.; Kodaira M.; Kajita M.; Nakajima T.; Shiba T.; Emi M. Thirteen single-nucleotide polymorphisms in the human osteopontin gene identified by sequencing of the entire gene in Japanese individuals. J. Hum. Genet. 46:544–546; 2001. [DOI] [PubMed] [Google Scholar]
- 10. Kon S.; Maeda M.; Segawa T.; Hagiwara Y.; Horikoshi Y.; Chikuma S.; Tanaka K.; Rashid M. M.; Inobe M.; Chambers A. F.; Uede T. Antibodies to different peptides in osteopontin reveal complexities in the various secreted forms. J. Cell. Biochem. 77:487–498; 2000. [PubMed] [Google Scholar]
- 11. Liaw L.; Birk D. E.; Ballas C. B.; Whitsitt J. S.; Davidson J. M.; Hogan B. L. M. Altered wound healing in mice lacking a functional osteopontin gene (spp1). J. Clin. Invest. 101:1468–1478; 1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Nemir M.; Bhattacharyya D.; Li X.; Singh K.; Mukherjee A. B.; Mukherjee B. B. Targeted Inhibition of osteopontin expression in the mammary gland causes abnormal morphogenesis and lactation deficiency. J. Biol. Chem. 275:969–976; 2000. [DOI] [PubMed] [Google Scholar]
- 13. Nishioka K.; Ohshima S.; Umeshita-Sasai M.; Yamaguchi N.; Mima T.; Nomura S.; Murata N.; Shimizu M.; Miyake T.; Yoshizaki K.; Suemura M.; Kishimoto T.; Saeki Y. Enhanced expression and DNA binding activity of two CCAAT/enhancer-binding protein isoforms, C/EBPbeta and C/EBPdelta, in rheumatoid synovium. Arthritis Rheum. 43:1591–1596; 2000. [DOI] [PubMed] [Google Scholar]
- 14. Ohshima S.; Kobayashi H.; Yamaguchi N.; Nishioka K.; Umeshita-Sasai M.; Mima T.; Nomura S.; Kon S.; Inobe M.; Uede T.; Saeki Y. Expression of osteopontin at sites of bone erosion in a murine experimental arthritis model of collagen-induced arthritis: Possible involvement of osteopontin in bone destruction in arthritis. Arthritis Rheum. 46:1094–2101; 2002. [DOI] [PubMed] [Google Scholar]
- 15. Patrone G.; Puliti A.; Bocciardi R.; Ravazzolo R.; Romeo G. Sequence and characterisation of the RET proto-oncogene 5′ flanking region: Analysis of retinoic acid responsiveness at the transcriptional level. FEBS Lett. 419:76–82; 1997. [DOI] [PubMed] [Google Scholar]
- 16. Poli V.; Mancini F. P.; Cortese R. IL-6DBP, a nuclear protein involved in interleukin-6 signal transduction, defines a new family of leucine zipper proteins related to C/EBP. Cell 63:643–653; 1990. [DOI] [PubMed] [Google Scholar]
- 17. Rosati M.; Valentin A.; Patenaude D. J.; Pavlakis G. N. CCAAT-enhancer-binding protein beta (C/EBP beta) activates CCR5 promoter: Increased C/EBP beta and CCR5 in T lymphocytes from HIV-1-infected individuals. J. Immunol. 167:1654–1662; 2001. [DOI] [PubMed] [Google Scholar]
- 18. Trueblood N. A.; Xie Z.; Communal C.; Sam F.; Ngoy S.; Liaw L.; Jenkins A. W.; Wang J.; Sawyer D. B.; Bing O. H.; Apstein C. S.; Colucci W. S.; Singh K. Exaggerated left ventricular dilation and reduced collagen deposition after myocardial infarction in mice lacking osteopontin. Circ. Res. 88:1080–1087; 2001. [DOI] [PubMed] [Google Scholar]
- 19. Wang D.; Yamamoto S.; Hijiya N.; Benveniste E.; Gladson C. Transcriptional regulation of the human osteopontin promoter: Functional analysis and DNA-protein interactions. Oncogene 19:5801–5809; 2000. [DOI] [PubMed] [Google Scholar]
- 20. Yamamoto S.; Hijiya N.; Setoguchi M.; Matsuura K.; Ishida T.; Higuchi Y.; Akizuki S. Structure of the osteopontin gene and its promoter. Ann. NY Acad. Sci. 760:44–58; 1995. [DOI] [PubMed] [Google Scholar]
- 21. Yoshitake H.; Rittling S. R.; Denhardt D. T.; Noda M. Osteopontin-deficient mice are resistant to ovariectomy-induced bone resorption. Proc. Natl. Acad. Sci. USA 96:8156–8160; 1999. [DOI] [PMC free article] [PubMed] [Google Scholar]