

Abstract
To date, the phylogeny of Triatoma dimidiata sensu lato (s. l.) (Hemiptera: Reduviidae: Triatominae), the epidemiologically most important Chagas disease vector in Central America and a secondary vector in Mexico and northern South America, has only been investigated by one multi-copy nuclear gene (Internal Transcribed Spacer – 2) and a few mitochondrial genes. We examined 450 specimens sampled across most of its native range from Mexico to Ecuador using reduced representation next-generation sequencing encompassing over 16,000 single nucleotide polymorphisms (SNPs). Using a combined phylogenetic and species delimitation approach we uncovered two distinct species, as well as a well-defined third group that may contain multiple species. The findings are discussed with respect to possible drivers of diversification and the epidemiological importance of the distinct species and groups.
Keywords: Triatoma dimidiata, species delimitation, biogeography, phylogeny, Chagas disease, insect vector
1. Introduction
Triatoma dimidiata sensu lato (s. l.) (Hemiptera: Reduviidae: Triatominae) is currently the epidemiologically most important Chagas disease vector in Central America and a secondary vector in Mexico and northern South America. It is one of the most widespread Triatominae taxa with a native range extending from Mexico southwards into Peru (Galvão et al., 2003). Across this extensive geographic range it exhibits considerable morphological diversity, which has led to multiple splitting and merging of the species (reviewed in Dorn et al., 2007). In addition, its behavior varies across its range, most notably in ways that affect vector capacity, for example, degree of anthropophily. The tendency to enter houses varies between Yucatan (Dumonteil et al., 2007) and Jutiapa, Guatemala (Stevens et al., 2015). Differences in food preferences have been demonstrated (Zeledón and Rabinovich 1981) as well as in blood source profiles (Lima-Cordón, et al. in review).
These differences in both the tendency to enter houses and blood meal sources influence the frequency of human-vector contact and thus Chagas disease transmission rates. Thus, delimiting the genetic boundaries of populations and species through a well-resolved phylogeny would benefit control efforts by indicating how much migration and interbreeding occur among locations, the potential for the spread of insecticide-resistance, and barriers to gene flow, especially when genetically different subpopulations also differ in vector capacity (Stevens and Dorn 2017). The epidemiological importance and variability of T. dimidiata s. l. prompted the need for a well-resolved phylogeny to evaluate the epidemiological relevance of different lineages and to efficiently direct vector control efforts.
Many investigators have sought to understand the phylogeny of this taxon. Early on, morphotypes from the extremes of the range were considered different species by some authors (e.g. T. maculipennis in Mexico [originally Conorhinus maculipennis, Stahl, 1859] (Pinto, 1931) and T. capitata in Colombia (Usinger, 1941)), reviewed in Dorn, et al. 2007). However, all were synonymized as the same species following a comprehensive review of specimens across the geographic range (Lent and Wygodzinsky, 1979); the authors concluded that the differences reflect clinal variation, with the exception of morphologically unique specimens from caves in Central America. Later studies of antenna sensilla, head morphometry, cytogenetics, and cuticular hydrocarbons have divided T. dimidiata into two to four taxa, including at least one cryptic taxon (reviewed in Dorn et al., 2016).
To resolve these conflicting systematic hypotheses, some of which may reflect environmental influence rather than evolutionary history, several investigators have turned to DNA sequence-based phylogenetic inference. These phylogenies, based on individual gene sequences, have shown that Triatoma hegneri falls within T. dimidiata s. l. (Groups 1 and 2; Monteiro et al., 2013), and have also been valuable in separating morphologically similar species (Fig. 1). The first distinct species was identified from Yucatan, Mexico and later found in Petén, Guatemala (Bargues et al., 2008; Dorn et al., 2009, 2007; Marcilla et al., 2001) and referred to as T. sp. aff. dimidiata (Group 3; Bargues et al., 2008) (Table 1). Importantly, Petén specimens were confirmed as biological species by experimental crosses (García et al., 2013). With expanded sampling, another species was identified in the Rio Frio cave, Belize (Dorn et al., 2016; Monteiro et al., 2013) and referred to as T. sp. aff. dimidiata (Group 4 - cave) (Dorn et al., 2016). And although all gene trees tested so far are in agreement dividing T. dimidiata s. l. into three evolutionarily independent clades (T. dimidiata s. s. and the two newly identified species) their relative placement differed between phylogenies inferred from mitochondrial compared to the nuclear gene Internal Transcribed Spacer-2 (ITS-2), reflecting limitations of a gene-based approach (Table 1; Dorn et al., 2016; Takahata and Nei, 1985). Because the nuclear phylogeny is based on a single marker, ITS-2, additional data should help clarify relationships amongst the clades.
Fig. 1.
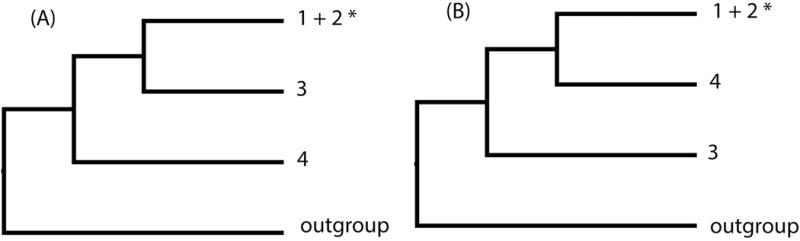
Summary of the relationships observed in published phylogenies of T. dimidiata s. l. Groups 1 and 2 are T. dimidiata s. s. (sensu stricto), Group 3 is T. sp. aff. dimidiata (groups based on Bargues et al., 2008), Group 4 is T. sp. aff. dimidiata (cave) (Dorn et al., 2016). (A) Relationships based on mitochondrial cyt b and ND4 genes (Dorn et al., 2016; Monteiro et al., 2013), (B) Relationships based on the multi-copy nuclear ITS-2 gene (Dorn et al., 2016). Note the conflict between the nuclear gene and mitochondrial gene topologies. * indicates T. hegneri was recovered within that clade.
Table 1.
Systematics of Triatoma dimidiata based on various DNA markers
| Summary | Markers | Conclusions | Reference |
|---|---|---|---|
| T. dimidiata + 1 cryptic species | ITS-2 | First suggestion of cryptic species in Yucatan, Mexico, suggest Ecuadorean T. dimidiata introduced from Central America. | (Marcilla et al., 2001) |
| T. dimidiata + 1 cryptic species | ITS-2 | T. dimidiata from Peten, Guatemala identical to cryptic species from Yucatan, Mexico | (Dorn et al., 2007) |
| Revived 3 subspecies, added T. hegneri as a subspecies + 1 cryptic species | ITS-2 | Named cryptic species from Yucatan, T. sp. aff. dimidiata; Subspecies: Group 1A (T. dimidiata dimidiata-Central Am.), 1B (T. dimidiata capitata - Colombia), 2 T. dimidiata maculipennis - Mexico), 3 (T. sp. aff. dimidiata); T. dimidiata hegneri – Cozumel, Mexico | (Bargues et al., 2008) |
| T. dimidiata s.s. + 1 cryptic species | ITS-2, Cytb (some bugs typed with both) | Subspecies not supported by mitochondrial marker, T. sp. aff. dimidiata confirmed by two markers, nuclear and mitochondrial | (Dorn et al., 2009) |
| 5 species including T. hegneri + 2 cryptic species | Cytb, ND-4 with Cytb | Rejected ITS-2 results, concludes 5 species: 4 T. dimidiata, plus T. hegneri; first report of cryptic species in Rio Frio, Belize | (Monteiro et al., 2013) |
| T. dimidiata s.l., includes T. hegneri | ITS-2 | North and Central Am. T. dimidiata in a monophyletic clade including divergent T. sp. aff. dimidiata within this clade; T. hegneri within T. dimidiata s.s. clade. | (de la Rua et al., 2014) |
| T. dimidiata s.s. + 2 cryptic species | ITS-2, Cytb, COI, ND4 | All markers support T. dimidiata s.s., T. sp. aff. dimidiata, T. sp. aff. dimidiata (cave); basal clade differs in mitochondrial and nuclear genes | (Dorn et al., 2016) |
Approaches using genome-wide markers can improve the robustness of phylogenies. Although whole genome sequencing of a large number of specimens is often cost-prohibitive, reduced representation sequencing (Davey et al., 2011) is an affordable method for including information from across the entire genome. And clearly, the broader the taxon sampling, the more complete the picture resolving issues of clinal variation vs. discrete taxa and uncovering rare cryptic species. It is also important to test different methods of aligning/mapping given that method-specific assumptions and algorithms may produce different results.
To test the hypothesis that T. dimidiata s. l. is a species complex, in this study we combined the most extensive specimen sampling for T. dimidiata s. l. across Mexico, Central and South America (n > 600) with the first genome-wide DNA sequence data for the reconstruction of a Triatominae phylogeny and species delimitation. Reduced-representation genotyping-by-sequencing (GBS) allowed us to sample single nucleotide polymorphisms (SNPs) across the genome and recover robust results. To test the robustness of the SNP calling we compared the three most popular aligners (BWA. Bowtie and Bowtie2, Toland et al., 2013). Furthermore, we combined the phylogenetic reconstruction with species delimitation. This is the first study to use genomic and species delimitation approaches to understand the phylogenetic relationships among Chagas disease vector taxa.
2. Methods
2.1. Taxon Sampling
Specimens belonging to T. dimidiata s. l, T. hegneri and Triatoma nitida (outgroup) were collected from domestic, peridomestic and sylvatic ecotopes encompassing as much as possible of the species distribution from southern Mexico to northern South America (Fig. 2; Table S1) by personnel trained in the safe handling of infectious agents, preserved in 95% ethanol, and stored at room temperature. For most specimens, the geographic location was recorded using a handheld Garmin 76X device (Olathe, KS, USA). When not recorded, GPS coordinates were estimated with Google maps (https://maps.google.com). T. nitida, a closely related species that lives in sympatry with T. dimidiata, was chosen as the outgroup.
Fig. 2.
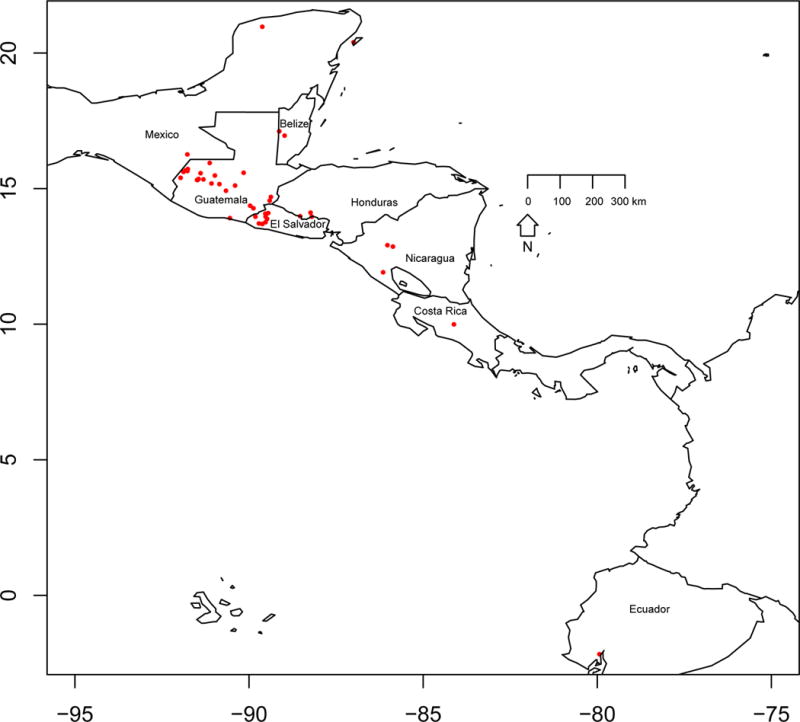
Map of southern Mexico to northern South America indicating the collection locations for specimens used in this study.
2.2. DNA extraction and Illumina sequencing
High molecular weight DNA was extracted from surface sterilized legs, thorax, or abdominal tissue (Table S1) using the DNeasy extraction kit (Qiagen, USA). DNA quantity and quality were assessed using a Nanodrop spectrophotometer (Thermo Scientific ®, Germany), Qubit ® Flurometric Quantitation (Life Technologies, USA) and gel electrophoresis. DNA from 622 specimens (30 μl of 10–30 ng/μl high molecular weight DNA) was sent to the Genome Core Facility at Cornell University, Ithaca, NY, USA for 48-plex, 100bp single end HiSeq reduced representation sequencing (Elshire et al., 2011). Libraries were generated using the restriction enzyme PstI (5′-C TGCA↓G-3′-3′-G↑ACGT C - 5′) based on the facility recommendation. Individually barcoded specimens were demultiplexed using the program sabre (https://github.com/najoshi/sabre), allowing for up to a single base pair mismatch in the barcoding sequence. After trimming the barcodes and recognition sites with FastX-trimmer in the FASTX Toolkit v. 0.0.14 (http://hannonlab.cshl.edu/fastx_toolkit/index.html, Gordon and Hannon et al. 2010), the 85 bp sequences were filtered with FastQ-quality-filter removing sequences with a quality score below 10 at any point along the sequence.
2.3. Construction of the reference catalog
Since there are no available genomes for the genus Triatoma, and analysis from a previous study found no T. dimidiata tags matched the genome of Rhodnius prolixus (Orantes et al., in review) which is estimated to have diverged over 30 million years ago (Justi et al. 2016), sequences generated from 12 T. dimidiata s. l. (supplementary Table 1) surface-sterilized legs were used to construct a reference catalog of putative T. dimidiata loci. Leg sequences were used because the thorax and abdomen were likely to contain gut microbiome, and possibly T. cruzi and blood meal DNA, in addition to the insects’ DNA.
The reference catalog sequences were assembled into homologous tags using the STACKS denovo_map.pl pipeline (Catchen et al., 2013, 2011). Because of the wide genetic diversity of the specimens, the parameters for de novo assembly were chosen to identify loci with enough sequencing representation across the tree for downstream analysis. A minimum of a single read was required to create a stack (−m = 1), a maximum of three mismatches among tags within an individual (−M = 3), and three mismatches among individuals when building the catalog (−n = 3). To filter out false positives, only tags which met four criteria were retained. (1) Zero to six SNPs were present across the reference specimens. The maximum of six SNPs on an 85bp read reduces the chance of including distant homologs (i.e. same gene in a different species) since the maximum variability allowed is ~7%. (2) At least six of the 11 reference specimens contained one or more reads of that tag. Accepting tags present in half the specimens allowed for variability in the sequencing depth among these 12 specimens. A higher value at the catalog stage would be too stringent and not representative of the diversity sampled. (3) All SNPs were biallelic. This allowed the catalog to comprised SNPs variable enough to cover the diversity investigated. (4) All the specimens contained no more than two alternate haplotypes. This ensured that all retained tags were, at most, representative of heterozygous individuals, lowering the chance of false SNP discovery. Because the sequencing depth was higher and the maximal number of mis-matches is lower for the mapping step (see below), and all specimens including the reference specimens were mapped to the reference catalog to identify SNPs, any low-frequency assembly errors in the catalog-building stage would not be carried through to the final dataset.
Contamination of the retained tags was assessed using the available Bowtie2 indicies for Archaea, Bacteria, Fungi and Virus genomes and the human genome. The indicies were downloaded as a step of the implementation for Taxoner (Langmead and Salzberg, 2012, see https://code.google.com/archive/p/taxoner/).
2.4. Mapping to reference catalog and SNP calling
As a control, the specimens used to construct the reference catalog were mapped against the reference catalog using Bowtie (Langmead et al., 2009), Bowtie2 and BWA (Li and Durbin, 2009). We used Bowtie, with default settings, to map the raw reads obtained for all 622 initial specimens to the reference catalog. Resulting mapped tags (sam files) were used as input to the ref_map.pl pipeline in STACKS, and SNPs were called using the genotypes pipeline, also in STACKS, both using default parameters (Catchen et al., 2013, 2011).
2.5. Phylogenetic reconstruction
Prior to phylogenetic reconstruction, R package phrynomics (https://github.com/bbanbury/phrynomics.git) was used to remove data interpreted by RaxML as invariant (e.g. fixed SNPs or sites with missing data).
Phylogenetic reconstruction was performed under the maximum likelihood (ML) algorithm in RaxML-HPC v.8 (Stamatakis, 2014) on XSEDE using the Cipres portal (Miller et al., 2010) under the GTR-CAT model of evolution with 500 bootstrap pseudo-replicates and Lewis ascertainment bias.
2.6. Pruning of Rogue Taxa
We used RogueNaRok to remove rogue taxa (see Sanderson and Shaffer 2002 for an explanation of this approach). Rogue taxa are defined as taxa that are recovered in multiple places within a phylogeny, therefore, lowering clade (i.e., bootstrap) support mainly due to lack of phylogenetic signal (Aberer et al., 2011), oversampling (Sanderson and Shaffer 2002), and/or samples with a large amount of missing data. In addition to the extensive sampling (> 600 specimens) in this study that likely oversampled common lineages, all samples other than the reference set were derived from the abdomen, which includes DNA of the vector, parasite (if infected), blood meal sources and microbiome. Therefore, the amount of vector genetic information was dependent on the infection status, blood-meal volume and microbiome abundance, making the yield more variable across specimens than would be expected for single-source DNA. Because the method identifying and removing rogue taxa has been tested and verified (Sanderson and Shaffer 2002), we chose this approach to accommodate this variability. RogueNaRok was set to optimize bootstrap support in the “best-known” tree generated by RaxML. Then rogue taxa were pruned from the dataset, RaxML invariant sites were removed and a new phylogeny was inferred following the conditions described above.
2.7. Species delimitation
The ML phylogeny was used as input for the species delimitation analyses. We used The Poisson Tree Process (PTP; Zhang, Kapli, Pavlidis, & Stamatakis, 2013) and the Bayesian implementation of PTP (bPTP) to infer putative species boundaries (http://species.h-its.org/). The reconstructed phylogeny was sampled for 500,000 MCMC (Markov Chain Monte Carlo) generations, where burn-in was set to 25% and convergence was assessed based on the Log-likelihood scores. Lineages delimited with > 0.9 support were deemed as independently evolving, i.e. are distinct species.
3. Results
3.1. Reference catalog construction and quality assessment
Comparison of the three mapping programs revealed that only Bowtie, and not the other two programs, resulted in the correct alignment of the reads (i.e. first nucleotide of the mapped read should be the first nucleotide aligned to the reference tag). For Bowtie2 and BWA alignments an excessive number of reads was observed, including the correct ones. The construction of the reference catalog using Bowtie resulted in a fasta file containing the consensus sequences of the 5,177 retained tags.
This fasta file was then mapped, using Bowtie2 (Langmead and Salzberg, 2012) with default parameters, against all available Archaea, Bacteria, Fungi and Virus genomes, obtained using Taxoner (Pongor et al., 2014) and to the human genome. No contamination from these sources was observed.
3.2. SNP calling and Rogue taxa
SNP calling resulted in a dataset comprising 622 specimens and 25,980 nucleotide sites. After removing the invariant sites using phrynomics, the dataset used for the reconstruction of the preliminary phylogeny contained 17,355 SNPs. Analyses of the best reconstructed tree and bootstrap pseudo-replicates with RogueNarok resulted in the removal of 172 specimens (Table S1). A pruned dataset generated with the 450 retained specimens and 16,202 SNPs was used for subsequent phylogenetic reconstruction in RaxML.
3.3. Species delimitation and Phylogeny
Maximum likelihood phylogenetic reconstruction recovered three well-supported ingroup clades: T. dimidiata s. s. (Groups 1 and 2) + T. hegneri and specimen A10311 from Guatemala; T. sp. aff. dimidiata (Group 3) and T. sp. aff. dimidiata (Group 4 - cave). Out of these clades, only two comprised highly supported species, following species delimitation analysis: T. sp. aff. dimidiata (Group 3; 0.934) and T. sp. aff. dimidiata (Group 4 - cave; 0.992); as well as the outgroup: T. nitida (0.981). T. dimidiata s. s. (Groups 1 and 2) was not recovered as a single species most likely because this group is not a single lineage; however, our sampling was not designed to resolve this.
4. Discussion
4.1. Phylogenetic Inference
Our results show that the group presently referred to as T. dimidiata comprises a species complex of at least three well-defined, monophyletic lineages with T. sp. aff. dimidiata (Group 3) as the basal clade. This species complex was revealed in two ways using the most comprehensive dataset to date, both in terms of sampling >450 T. dimidiata specimens from seven countries) and genome coverage (~16,000 SNPs from across the genome). First, ML phylogenetic inference shows strong support (≥90% bootstrap values) for these three monophyletic clades, T. dimidiata s. s. (Groups 1 and 2), T. sp. aff. dimidiata (Group 4 - cave) and T. sp. aff. dimidiata (Group 3), the latter as the basal clade (Fig. 3). Second, species delimitation supports two [T. sp. aff. dimidiata (Group 4 - cave) and T. sp. aff. dimidiata (Group 3)] out of the three clades as distinct species (>0.9).
Fig. 3.
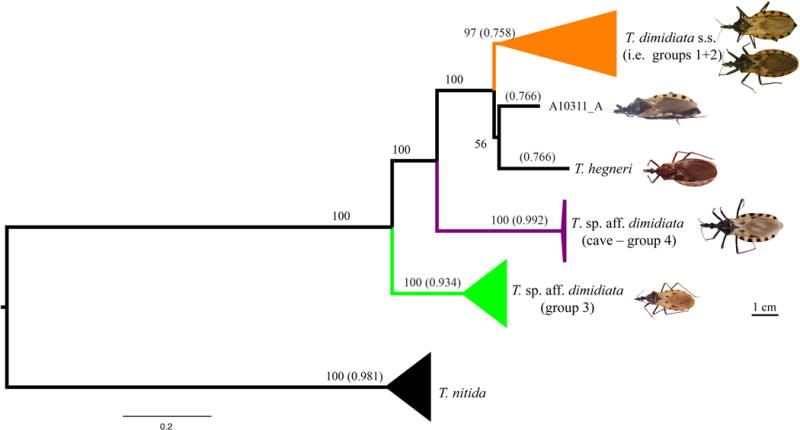
Maximum likelihood phylogeny reconstructed from the 450 T. dimidiata s. l. Numbers outside parenthesis represent bootstrap support > 50, numbers within parenthesis represent the frequency that the clade was recovered as a single species in the species delimitation analysis. Specimen scale :1cm. Photo credits: T. dimidiata s. s. (Groups 1 and 2) and T. hegneri: Carolina Dale; T. sp .aff. dimidiata (Groups 3 and 4 - cave) and A10311: Raquel Lima.
This complex containing at least three lineages is consistent with previous results based on the multi-copy nuclear marker ITS-2 that identified these three taxa within T. dimidiata and this same relationship among the clades (Dorn, 2016). Because of the relative size of the nuclear compared to the mitochondrial genome, the vast majority of the SNPs recovered in this study are expected to be from the nuclear genome. Therefore, these conclusions reflect to the nuclear genome phylogeny.
Overall, the species delimitation analysis supports the same three clades observed in the phylogenetic reconstruction. T. sp. aff. dimidiata (Group 4 – cave) has the highest support, followed by T. nitida (as expected, since it is the outgroup) and then T. sp. aff. dimidiata (Group 3). As previously reported, T. dimidiata Groups 1, 2 and 3 occur in sympatry in Guatemala (Dorn et al., 2009; Monteiro et al., 2013). Despite the three groups occurring in sympatry, studies consistently report T. sp. aff. dimidiata (Group 3) as a distinct lineage both with genetic data (Dorn et al., 2016) and with cross-breeding experiments (García et al., 2013).
The species delimitation results also suggest that the large and diverse T. dimidiata s. s. clade likely includes more than one species. There are two or possibly three distinct groups: T. dimidiata s.s (Groups 1 and 2), T. hegneri and specimen A10311_A were recovered with high bootstrap support (100%) based on the phylogenetic analysis. However, the delimitation support was lower (0.758 – 0.766) suggesting further study is necessary to understand diversification within the T. dimidiata s.s clade.
T. dimidiata s. s. has high diversity and the second highest geographic range of any of the over 150 species of Triatominae; only the tropicopolitan Triatoma rubrofascita is higher. T. dimidiata s. s. also has the lowest support as a single species, perhaps because of the high diversity and the possibility of independently evolving lineages within this clade (Dorn et al., 2016). Groups 1 and 2 as well as T. hegneri were originally described based on geography (Bargues et al., 2008). T. hegneri is restricted to the island of Cozumel, Group 1 from Central America and Mexico, and Group 2 from southern Central America and South America. These groups together form a monophyletic clade but individually have never been shown to be monophyletic (Bargues et al., 2008; Dorn et al., 2016, 2009; Monteiro et al., 2013).
4.2. Conflicting nuclear and mitochondrial phylogeny: Early introgression?
It is interesting that in previous phylogenetic analyses, mitochondrial and nuclear markers recovered conflicting topologies regarding the putative sister taxa of T. dimidiata s. s. (Table 1, Fig. 1). For the mitochondrial genes, T. sp. aff. dimidiata (Group 3) was recovered as sister to T. dimidiata s. s. (Dorn et al., 2016, 2009; Monteiro et al., 2013), while for the nuclear ITS-2 marker (Bargues et al., 2008; Dorn et al., 2016, 2009) and nuclear GBS genome-wide data (presented here), T. sp. aff. dimidiata (Group 4 – cave) was recovered as sister.
Since interspecific mitochondrial recombination has been reported in some insect orders but not for Heteroptera (Hua et al., 2008; Li et al., 2016), we assume the phylogeny recovered for the mitochondrial markers reflects the evolutionary history of the mitochondrial genomes in the Triatoma lineages studied. We speculate that the mitochondrial “swap” in T. dimidiata s. s. sister groups resulted from ancestral hybridization between the lineages (Fig. 4). When speciation occurs in the absence of reinforcement, as is often hypothesized for allopatric speciation, mating barriers might not evolve between the closely related species (Ortiz-Barrientos et al., 2004). Subsequent geographic expansion could lead to a hybrid zone, where with sufficient time, mitochondrial introgression might take place (Mastrantonio et al., 2016).
Fig. 4.

One possible evolutionary history of T. dimidiata s. l. (all groups) based on currently accepted nuclear and mitochondrial phylogenies. Background shaded tree represents the topology observed for the nuclear genome. The single interior line refers to the topology observed for the mitochondrial genome (Orange: T. dimidiata s. s. (Groups 1 and 2); Green: T. sp. aff. dimidiata (Group 3): Purple: T. sp. aff. dimidiata (Group 4 - cave)
Hybridization potentially has a range of distinct consequences, from inviability to the formation of a new hybrid species (Arnold and Martin, 2009). Adaptive alleles in particular may permeate through closely-related species (Baack and Rieseberg, 2007) and could be detected with “gene-by-gene” analysis of the nuclear genome. Complete mitochondrial introgression (i.e. fixation) can occur in the absence of negative consequences of hybridization. This can be observed even if there are a small number of migrants between populations (Takahata and Slatkin, 1984). The observed mitochondrial and nuclear phylogenies are predicted if ancestral populations of Groups 3 and 4 exchanged migrants and the resulting hybrids were either neutral or had some selective advantage. Another possibility is single-direction replacement between T. dimidiata s. s. and T. sp. aff. dimidiata (Group 4 - cave).
It appears that the phylogenetic signal reported here is sufficiently strong that even if there was introgression among nuclear genes it did not obscure overall support for the phylogenetic reconstruction. Although it is unknown if introgressed genes were included in the reduced representation sequencing, if such genes exist it would be interesting to know their ontology or functional role, and especially if they are epidemiologically important.
4.3. Niche divergence
Groups 3 and 4 have very distinct ecological ranges, the former inhabiting palm trees, chultuns (ancient Mayan granaries), and both peridomestic and intradomestic environments (personal observation, Carlota Monroy and Raquel Lima); while the later has only been found in caves (Dorn et al., 2016; Monteiro et al., 2013). The investigation of introgressed alleles within the nuclear genomes would shed light on which genes are responsible for environmental adaptation in the Triatominae and possible drivers of diversification. Comparison of T. sp. aff. dimidiata (Group 3) and T. sp. aff. dimidiata (Group 4 - cave) with T. dimidiata s. s. can shed light on genetic changes involved in domiciliation of these important vector species.
4.4. Morphology versus Molecular Phylogeny
Morphological identification of Triatominae largely relies on the dichotomous keys published by Lent and Wygodzinsky (1979) and subsequent species descriptions. Using their keys all four groups are identified as T. dimidiata s. l. However, there are additional morphological characters that separate T. sp. aff. dimidiata (Group 3) and T. sp. aff. dimidiata (Group 4 - cave) from the “classic” T. dimidiata s. l. description. Such differences are leading our group to describe these new species (Dorn et al. in prep and Lima et al. in prep).
5. Conclusions
In summary, this first genome-wide phylogeny and species delimitation analysis for Chagas disease vectors, based on thousands of SNPs across the nuclear genome of hundreds of specimens from a wide geographic range, reveal that Triatoma dimidiata s. l. is a complex of at least three species. These results combined with previous data on mitochondrial genes show different phylogenies for the nuclear and mitochondrial genomes, suggesting past hybridization and introgression among the lineages. It will be important to identify the introgressed genes and other genetic differences among groups which may reveal the evolution of domestication and epidemiological importance. Furthermore, the distinct ecological preferences observed for each species reinforces the need to incorporate the knowledge that there are three well-defined species in designing vector control strategies.
Supplementary Material
Table S1: Information on the specimens used for this study. Specimens used for the construction of the reference catalog are underlined.
Figure S1: Maximum likelihood tree reconstructed after rogue specimens pruned.
Highlights.
The first phylogeny of a Chagas disease vector based on genome-wide data.
The largest phylogenetic dataset for the Triatominae.
Triatoma dimidiata is one of the most epidemiologically important Chagas disease vectors.
Triatoma dimidiata s. l. is a complex comprised of at least three species.
Nuclear and mitochondrial phylogenies are incongruent, suggesting hybridization and introgression.
Cave specimens form a separate, well-resolved species
Acknowledgments
We thank Andre Elias Soares for helping with part of the analyses, villagers for allowing us to collect in their homes and surrounding areas, Antonieta Rodas for help in collecting insects, Bethany Richards and Meghan Gallaspy for assistance with the database, Carolina Dale for the photographs of T. dimidiata s. s. and T. hegneri (Fig. 3) and Dr. Jürgen Deckert from the Museum fur Naturkunde in Berlin for access to the collection where the photos were taken. This work was funded by NSF grant BCS - 1216193 as part of the joint NSF-NIH-USDA Ecology and Evolution of Infectious Diseases program and NIH grant R03AI26268/1-2. Any opinions, findings, and conclusions or recommendations expressed in this material are those of the author(s) and do not necessarily reflect the views of the National Science Foundation and of the National Institutes of Health. The funders had no role in study design.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Aberer AJ, Krompaß D, Stamatakis A. RogueNaRok : an Efficient and Exact Algorithm for Rogue Taxon Identification. 2011 no ID 12. [Google Scholar]
- Arnold ML, Martin NH. Adaptation by introgression. J Biol. 2009;8:82. doi: 10.1186/jbiol176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baack EJ, Rieseberg LH. NIH Public Access. Curr Opin Genet Dev. 2007;17:513–518. doi: 10.1016/j.gde.2007.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bargues MD, Klisiowicz DR, Gonzalez-Candelas F, Ramsey JM, Monroy C, Ponce C, Salazar-Schettino PM, Panzera F, Abad-Franch F, Sousa OE, Schofield CJ, Dujardin JP, Guhl F, Mas-Coma S. Phylogeography and genetic variation of Triatoma dimidiata, the main Chagas disease vector in Central America, and its position within the genus Triatoma. PLoS Negl Trop Dis. 2008;2:e233. doi: 10.1371/journal.pntd.0000233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Catchen J, Hohenlohe PA, Bassham S, Amores A, Cresko WA. Stacks: an analysis tool set for population genomics. Mol Ecol. 2013;22:3124–3140. doi: 10.1111/mec.12354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Catchen JM, Amores A, Hohenlohe P, Cresko W, Postlethwait JH, De Koning DJ. Stacks: Building and Genotyping Loci De Novo From Short-Read Sequences. G3: Genes|Genomes|Genetics. 2011;1:171–182. doi: 10.1534/g3.111.000240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davey JW, Hohenlohe PA, Etter PD, Boone JQ, Catchen JM, Blaxter ML. Genome-wide genetic marker discovery and genotyping using next generation sequencing. Nat Rev Genet. 2011;12(7):499–510. doi: 10.1038/nrg3012. [DOI] [PubMed] [Google Scholar]
- Dorn PL, Calderon C, Melgar S, Moguel B, Solorzano E, Dumonteil E, Rodas A, de la Rua N, Garnica R, Monroy C. Two distinct Triatoma dimidiata (Latreille, 1811) taxa are found in sympatry in Guatemala and Mexico. PLoS Negl Trop Dis. 2009;3:e393. doi: 10.1371/journal.pntd.0000393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dorn PL, de la Rúa NM, Axen H, Smith N, Richards BR, Charabati J, Suarez J, Woods A, Pessoa R, Monroy C, Kilpatrick CW, Stevens L. Hypothesis testing clarifies the systematics of the main Central American Chagas disease vector, Triatoma dimidiata (Latreille, 1811), across its geographic range. Infect Genet Evol. 2016;44:431–443. doi: 10.1016/j.meegid.2016.07.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dorn PL, Monroy C, Curtis A. Triatoma dimidiata (Latreille, 1811): A review of its diversity across its geographic range and the relationship among populations. Infect Genet Evol. 2007;7:343–352. doi: 10.1016/j.meegid.2006.10.001. [DOI] [PubMed] [Google Scholar]
- Dumonteil E, Ramirez-Sierra Tripet F, Payet MJ, Lanzaro VG, Menu F. Assessment of Triatoma dimidiata dispersal in the Yucatán Peninsula of Mexico by morphometry and microsatellite markers. Am J Human Genet. 2007;76:930–937. [PubMed] [Google Scholar]
- Elshire RJ, Glaubitz JC, Sun Q, Poland JA, Kawamoto K, Buckler ES, Mitchell SE. A robust, simple genotyping-by-sequencing (GBS) approach for high diversity species. PLoS One. 2011;6:1–10. doi: 10.1371/journal.pone.0019379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galvão C, Carcavallo R, Rocha DS, Jurberg J. A Checklist of the current valid species of the subfamily Triatominae Jeannel, 1919 (Hemiptera: Reduviidae) and their geographical distribution, with nomenclatural and taxonomic notes. Zootaxa. 2003;202:1–36. [Google Scholar]
- García M, Menes M, Dorn PL, Monroy C, Richards B, Panzera F, Bustamante DM. Reproductive isolation revealed in preliminary crossbreeding experiments using field collected Triatoma dimidiata (Hemiptera: Reduviidae) from three ITS-2 defined groups. Acta Trop. 2013;128:714–718. doi: 10.1016/j.actatropica.2013.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hatem A, Toland Bozda D, Çatalyürek AE, V Ü. Benchmarking short sequence mapping tools. BMC Bioinformatics. 2013;14:184. doi: 10.1186/1471-2105-14-184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hua J, Li M, Dong P, Cui Y, Xie Q, Bu W. Comparative and phylogenomic studies on the mitochondrial genomes of Pentatomomorpha (Insects: Hemiptera: Heteroptera) BMC Genomics. 2008;9:610. doi: 10.1186/1471-2164-9-610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, Salzberg SL. Fast gapped-read alignment with Bowtie 2. Nat Methods. 2012;9:357–359. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, Trapnell C, Pop M, Salzberg SL. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009;10:R25. doi: 10.1186/gb-2009-10-3-r25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lent H, Wygodzinsky P. Revision of the Triatominae (Hemiptera: reduviidae), and their significance as vectors of Chagas Disease. Bull Am Museum Nat Hist. 1979;163:123–520. [Google Scholar]
- Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li T, Yang J, Li Y, Cui Y, Xie Q, Bu W, Hillis DM, Henry TJ, Dong JZ, Zheng LY, Xie Q, Bu WJ, Zheng LY, Wang Y, Li HM, Tian XX, Cameron SL, Li T, Gao CQ, Cui Y, Xie Q, Bu W, Tang M, Cameron SL, Li T, Yi W, Zhang H, Xie Q, Bu W, Dai X, Zhang DX, Hewitt GM, Lunt DH, Whipple LE, Hyman BC, Vinces MD, Legendre M, Caldara M, Hagihara M, Verstrepen KJ, Huang W, Zheng J, He Y, Luo C, Levinson G, Gutman GA, Fondon JW, Garner HR, Li H, Hua JM, Yuan ML, Zhang QL, Guo ZL, Wang J, Shen YY, Wang Y, Li H, Wang P, Song F, Cai W, Hua JM, Li T, Gao JY, Ojala D, Montoya J, Attardi G, Wang Y, Chen J, Jiang LY, Qiao GX, Hebert PDN, Ratnasingham S, deWaard JR, Roques S, Fox CJ, Villasana MI, Rico C, Schmidt TR, Wu W, Goodman M, Grossman LI, Perna NT, Kocher TD, Hassanin A, Leger N, Deutsch J, Cui Y, Yokobori SI, Pääbo S, Nesnidal MP, Helmkampf M, Bruchhaus I, Hausdorf B, Boore JL, Buckley TR, Simon C, Flook PK, Misof B, Cannone JJ, Niehuis O, Yen SH, Naumann CM, Misof B, Niehuis O, Naumann CM, Misof B, Cameron SL, Whiting MF, Gillespie JJ, Johnston JS, Cannone JJ, Gutell RR, Taanman JW, Zhang DX, Szymura JM, Hewitt GM, Wang X, Li H, Shao R, Barker SC, Cook CE, Song F, Goncalves R, Freitas AI, Jesus J, la Rua PD, Brehm A, Liu J, Bu C, Wipfler B, Liang A, Rokas A, Ladoukakis E, Zouros E, Farabaugh PJ, Schmeissner U, Hofer M, Miller JH, Li H, Schuh RT, Weirauch C, Wheeler WC, Tian Y, Zhu W, Li M, Xie Q, Bu W, Reineke A, Karlovsky P, Zebitz CP, Hall TA, Thompson JD, Gibson TJ, Plewniak F, Jeanmougin F, Higgins DG, Lowe TM, Eddy SR, Tamura K, Stecher G, Peterson D, Filipski A, Kumar S, Benson G, Zuker M, Zhou J, Liu X, Stones DS, Xie Q, Wang G, Silvestro D, Michalak I, Posada D, Crandall KA. A Mitochondrial Genome of Rhyparochromidae (Hemiptera: Heteroptera) and a Comparative Analysis of Related Mitochondrial Genomes. Sci Rep. 2016;6:35175. doi: 10.1038/srep35175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lima-Cordón R, Stevens L, Solorzano-Ortíz E, Rodas G, Castellanos S, Rodas A, Abrego V, Zúñiga C, Monroy MC. In review. Implementation Science: Epidemiology and feeding profiles of the Chagas vector Triatoma dimidiata prior to Ecohealth intervention for three locations in Central America. PLoS Negl Trop Dis. doi: 10.1371/journal.pntd.0006952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcilla A, Bargues MD, Ramsey JM, Magallon-gastelum E, Marı P, Abad-franch F, Dujardin J, Schofield CJ, Mas-Coma S. The ITS-2 of the Nuclear rDNA as a Molecular Marker for Populations, Species, and Phylogenetic Relationships in Triatominae (Hemiptera : Reduviidae), Vectors of Chagas Disease. Mol Phyl Evol. 2001;18:136–142. doi: 10.1006/mpev.2000.0864. [DOI] [PubMed] [Google Scholar]
- Mastrantonio V, Porretta D, Urbanelli S, Crasta G, Nascetti G. Dynamics of mtDNA introgression during species range expansion: insights from an experimental longitudinal study. Sci Rep. 2016;6:30355. doi: 10.1038/srep30355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller MA, Pfeiffer W, Schwartz T. Creating the CIPRES Science Gateway for inference of large phylogenetic trees 2010. Gatew Comput Environ Work. 2010:1–8. doi: 10.1109/GCE.2010.5676129. [DOI] [Google Scholar]
- Monteiro FA, Peretolchina T, Lazoski C, Harris K, Dotson EM, Abad-Franch F, Tamayo E, Pennington PM, Monroy C, Cordon-Rosales C, Salazar-Schettino PM, Gómez-Palacio AM, Grijalva MJ, Beard CB, Marcet PL. Phylogeographic Pattern and Extensive Mitochondrial DNA Divergence Disclose a Species Complex within the Chagas Disease Vector Triatoma dimidiata. PLoS Negl Trop Dis. 2013;8:e70974. doi: 10.1371/journal.pone.0070974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ortiz-Barrientos D, Counterman BA, Noor MAF. The genetics of speciation by reinforcement. PLoS Biol. 2004;2:e416. doi: 10.1371/journal.pbio.0020416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinto C. Valor do rostro e antenas na caracterizacao dos generos de Triatomideos. Hemiptera, Reduvidioidea. Bol Biol Sao Paulo. 1931;19:45–136. [Google Scholar]
- Pongor LS, Vera R, Ligeti B. Fast and Sensitive Alignment of Microbial Whole Genome Sequencing Reads to Large Sequence Datasets on a Desktop PC: Application to Metagenomic Datasets and Pathogen Identification. PLoS One. 2014;9:e103441. doi: 10.1371/journal.pone.0103441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanderson, Shaffer HB. Troubleshooting molecular phylogenetic analyses. Annu Rev Ecol Syst 2002. 2002;33:49–72. doi:0.1146/annurev.ecolsys.33.010802.150509. [Google Scholar]
- Stahl C. Monographie der Gattung Conorhinus und Verwandten. Berl Ent Zeitschr. 1859;3:99–117. [Google Scholar]
- Stamatakis A. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics. 2014;30:1312–3. doi: 10.1093/bioinformatics/btu033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stevens L, Dorn PL. Population Genetics of Triatomines American Trypanosomiasis Chagas Disease – One Hundred Years of Research J Telleria and M Tibayrnec United States. Academic Press; 2017. 844.9780128010297. [Google Scholar]
- Stevens L, Monroy MC, Rodas AG, Hicks RM, Lucero DE, Lyons LA, Dorn PL. Migration and Gene Flow Among Domestic Populations of the Chagas Insect Vector Triatoma dimidiata (Hemiptera: Reduviidae) Detected by Microsatellite Loci. J Med Entomol. 2015;52(3):419–428. doi: 10.1093/jme/tjv002:10.1093/jme/tjv002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahata N, Nei M. Gene genealogy and variance of interpopulational nucleotide differences. Genetics. 1985;110:325–344. doi: 10.1093/genetics/110.2.325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahata N, Slatkin M. Mitochondrial gene flow. Proc Natl Acad Sci U S A. 1984;81(6):1764–7. doi: 10.1073/pnas.81.6.1764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Usinger. Notes and descriptions of neotropicalTriatominae (Hemiptera, Reduviidae) Pan Pacific Entomol. 1941;17:49–57. [Google Scholar]
- Zeledon R, Rabinovich JE. Chagas disease: an ecological appraisal with special emphasis on its insect vectors. Annual Review of Entomology. 1981;26(1):101–33. doi: 10.1146/annurev.en.26.010181.000533. [DOI] [PubMed] [Google Scholar]
- Zhang J, Kapli P, Pavlidis P, Stamatakis A. A general species delimitation method with applications to phylogenetic placements. Bioinformatics. 2013;29:2869–2876. doi: 10.1093/bioinformatics/btt499. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1: Information on the specimens used for this study. Specimens used for the construction of the reference catalog are underlined.
Figure S1: Maximum likelihood tree reconstructed after rogue specimens pruned.