

Abstract
DNA replication occurs in a defined temporal order known as the replication-timing (RT) program. RT is regulated during development in discrete chromosomal units, coordinated with transcriptional activity and 3D genome organization. Here, we derived distinct cell types from F1 hybrid musculus × castaneus mouse crosses and exploited the high single-nucleotide polymorphism (SNP) density to characterize allelic differences in RT (Repli-seq), genome organization (Hi-C and promoter-capture Hi-C), gene expression (total nuclear RNA-seq), and chromatin accessibility (ATAC-seq). We also present HARP, a new computational tool for sorting SNPs in phased genomes to efficiently measure allele-specific genome-wide data. Analysis of six different hybrid mESC clones with different genomes (C57BL/6, 129/sv, and CAST/Ei), parental configurations, and gender revealed significant RT asynchrony between alleles across ∼12% of the autosomal genome linked to subspecies genomes but not to parental origin, growth conditions, or gender. RT asynchrony in mESCs strongly correlated with changes in Hi-C compartments between alleles but not as strongly with SNP density, gene expression, imprinting, or chromatin accessibility. We then tracked mESC RT asynchronous regions during development by analyzing differentiated cell types, including extraembryonic endoderm stem (XEN) cells, four male and female primary mouse embryonic fibroblasts (MEFs), and neural precursor cells (NPCs) differentiated in vitro from mESCs with opposite parental configurations. We found that RT asynchrony and allelic discordance in Hi-C compartments seen in mESCs were largely lost in all differentiated cell types, accompanied by novel sites of allelic asynchrony at a considerably smaller proportion of the genome, suggesting that genome organization of homologs converges to similar folding patterns during cell fate commitment.
Genome duplication in eukaryotes is regulated in coordination with large-scale chromosome organization and transcriptional activity, with discrete chromosome units (replication domains [RDs]) replicating at specific times during S-phase (Hiratani et al. 2010; Rivera-Mulia and Gilbert 2016a,b). Spatiotemporal regulation of RDs is critical for nuclear organization and function (Neelsen et al. 2013; Alver et al. 2014; Donley et al. 2015), and replication timing (RT) alterations are observed in disease (Ryba et al. 2012; Gerhardt et al. 2014a,b; Dixon et al. 2017; Rivera-Mulia et al. 2017a; Sasaki et al. 2017). Early and late RDs segregate to distinct nuclear compartments, with early replicating domains preferentially positioned at the nuclear interior while late RDs are located either at the periphery or close to the nucleolus (Jackson and Pombo 1998; Sadoni et al. 2004). Additionally, RDs correspond to the topologically associating domains (TADs) measured by chromosome conformation capture techniques, such as Hi-C (Ryba et al. 2010; Yaffe et al. 2010; Moindrot et al. 2012; Pope et al. 2014; Rivera-Mulia and Gilbert 2016b). RT is highly conserved in all eukaryotes (Ryba et al. 2010; Yue et al. 2014; Solovei et al. 2016), changes dynamically during development in coordination with changes in nuclear positioning and transcriptional activity (Hiratani et al. 2010; Rivera-Mulia et al. 2015), and can be exploited to characterize complex circuits of gene regulatory networks (Rivera-Mulia et al. 2017b). Hence, RT constitutes a functional readout of genome organization and function.
Chromosome homologs replicate highly synchronously, with very few exceptions that include imprinted genes (with the imprinted allele showing delayed replication) and monoallelically expressed genes, which are also generally replicated earlier when active (Kitsberg et al. 1993; Simon et al. 1999; Mostoslavsky et al. 2001; Singh et al. 2003; Ensminger and Chess 2004; Dutta et al. 2009; Farago et al. 2012). X Chromosome inactivation in female cells is the most impressive example of RT asynchrony linked to large-scale chromosome organization, with the inactive chromosome X (Xi) densely packed into the Barr body at the nuclear periphery and replicating later than the active chromosome X (Avner and Heard 2001; Galupa and Heard 2015). Although most asynchronously replicating loci have been identified by cytogenetic analysis (Selig et al. 1992; Boggs and Chinault 1997), recent studies exploiting single-nucleotide polymorphisms (SNPs) and deep sequencing of phased genomes has allowed measurement of allelic variation in RT genome-wide (Koren et al. 2014; Koren and McCarroll 2014; Mukhopadhyay et al. 2014; Bartholdy et al. 2015). These studies identified RT asynchrony associated with sequence variation and gene imprinting. Although differential efficiency in replication origin firing has been associated with allelic variation in RT (Bartholdy et al. 2015), it is not clear whether the RT asynchrony is linked to differences in 3D genome organization between alleles and if those differences are regulated during development. Here, we took advantage of the high SNP density between genomes of distinct mouse subspecies and measured allelic differences in RT, 3D genome organization, gene expression, and chromatin accessibility. To do so, we developed the algorithm Haplotype-Assisted Read Parsing (HARP), a new computational tool to efficiently sort reads into each genome based on the presence of SNPs. We identified RT asynchrony in ESCs that correlated better with allelic differences in Hi-C compartments than SNP density, gene expression, or chromatin accessibility. Moreover, we found that RT asynchrony was lost during cell fate specification toward distinct cell types in coordination with a decrease in Hi-C compartment differences.
Results
To evaluate allelic RT variation, we exploited the high SNP density between distinct mouse genomes. Mouse crosses of subspecies and strains were generated and ES cell lines were derived from hybrid F1 blastocysts (Fig. 1A) as previously described (Dupont et al. 2016). We characterized a total of six hybrid mESC lines harboring three different genomes (C57BL/6, 129/sv, and CAST/Ei), opposite parental configurations, and gender (Fig. 1B). Genome-wide RT analysis (Fig. 1C) was performed by NGS as described previously (Ryba et al. 2011; Marchal et al. 2017). Allele-specific genome-wide data were measured using our HARP algorithm (see Methods). Chromatin spatial organization (Hi-C) and long-range enhancer-promoter interactions (PC-Hi-C), gene expression (total nuclear RNA-seq), and chromatin accessibility (ATAC-seq) data were also collected (see Methods) to identify possible mechanisms driving the differences between alleles in RT and 3D genome organization (Fig. 1D–F). An exemplary genomic region with allele-specific measurements of RT, Hi-C compartments and interactions, gene expression, and chromatin accessibility is shown in Figure 1G.
Figure 1.
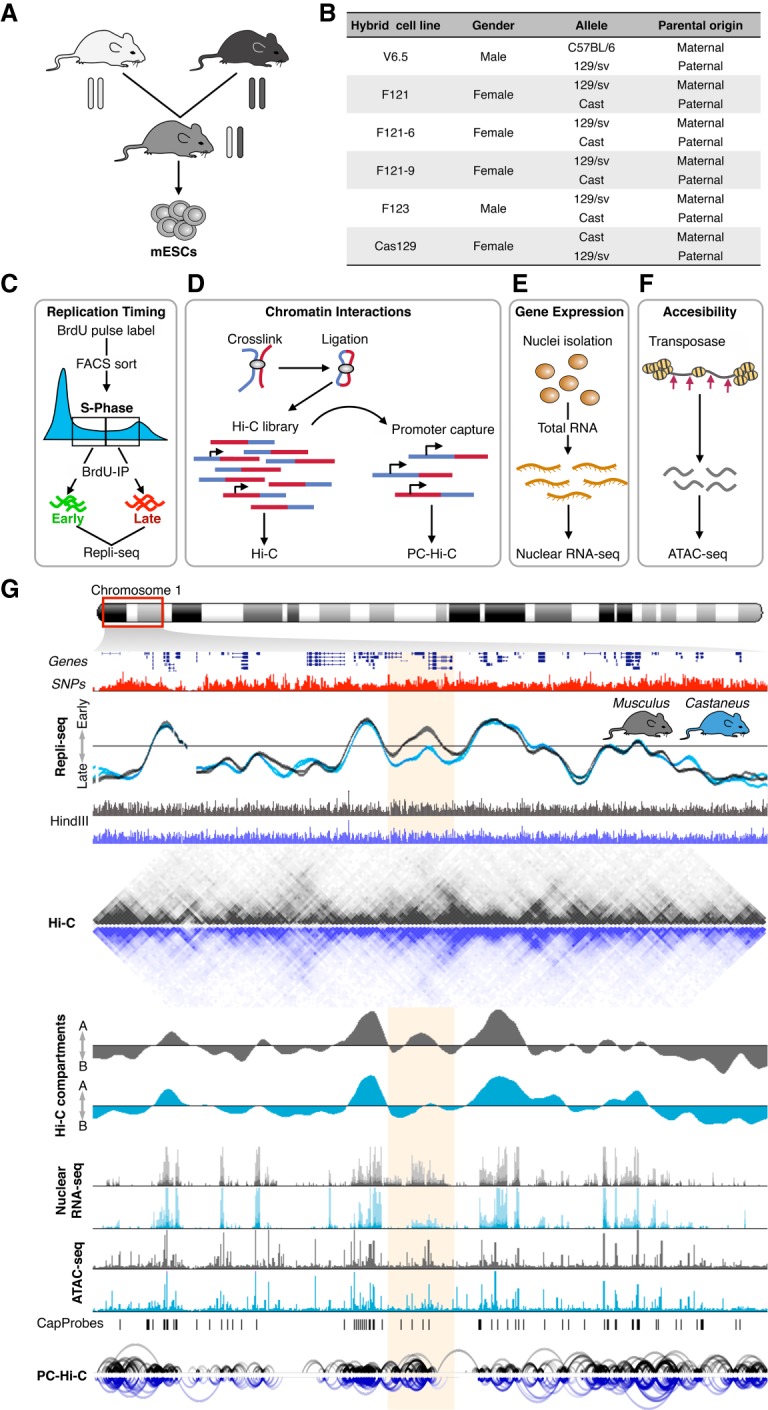
Genome-wide analysis of RT, enhancer-promoter interactions, gene expression, and chromatin accessibility in hybrid mouse ESCs. (A) Mouse ESC lines were derived from hybrid F1 blastocysts from crosses of distinct subspecies and strains. (B) Six distinct hybrid mESC lines harboring three different genomes (C57BL/6, 129/sv, and CAST/Ei), opposite parental configurations, and different genders were analyzed. V6.5, F121, and F123 hybrid cell lines were generated previously (Rideout et al. 2000; Monkhorst et al. 2008). F121-6 and F121-9 are single-cell subclones of F121, and Cas129 was generated in this study from a reciprocal cross between castaneus/musculus mice. (C–F) Genome-wide analysis of RT (C), Hi-C and promoter capture Hi-C (PC-Hi-C) (D), total nuclear RNA-seq (E), and chromatin accessibility measured by Assay for Transposase Accessible Chromatin (ATAC-seq) (F). (G) Representative genomic region on Chromosome 1 showing RT profiles of musculus (129/sv) and castaneus (CAST/Ei) alleles. Two replicates of the F121-9 cell line (two RT profiles of each genome) show the consistency in RT asynchrony. Allele-specific Hi-C matrices and compartments A and B (eigenvectors), RNA-seq, ATAC-seq, and enhancer-promoter interactions are shown. Allele-specific RT, total nuclear RNA-seq, and ATAC-seq were determined based on the SNPs shown in red. Allele-specific Hi-C and PC-Hi-C interactions were obtained using only HindIII fragments containing SNPs that distinguish each genome (HindIII track). Capture probes for PC-Hi-C are shown above promoter-enhancer interactions. Hi-C data were obtained from Giorgetti et al. (2016).
RT asynchrony between alleles in hybrid mouse ESCs
To identify the degree of RT synchrony, we divided the autosomal genome into 43,941 nonoverlapping 50-kb windows. First, we determined the degree of similarity in RT between the two alleles. As expected, we found the strongest genome-wide correlation in the ESC line derived from a mouse inbred cross (genomes C57BL/6 and 129/sv) and slightly lower correlation values for ESCs derived from subspecies Mus musculus musculus (129/sv genome) and M. musculus castaneus (CAST/Ei genome) crosses (Fig. 2A). Although the correlation values were very strong (>0.9) between the alleles from all hybrid mouse ESC lines analyzed, they were segregated by subspecies in a genome-wide correlation matrix, and the highest correlation values were observed between alleles with the same subspecies genome (either M. musculus musculus or M. musculus castaneus) rather than between alleles from the same cell line (Fig. 2B). Consistently, alleles from the V6.5 cell line (derived from a cross of inbreeds from the same subspecies) clustered tightly together with the rest of the M. musculus musculus genomes (Fig. 2B). Next, to identify the RT asynchrony between homologs, we analyzed the magnitude of differences in RT between alleles and between replicates. Considering an average S-phase time of 8 h, we found that the largest differences between replicates of the same allele (same genome) were <80 min (Supplemental Fig. S1), which is consistent with the average technical noise commonly observed in RT measurements (∼10% of the dynamic range). Hence, we considered significant RT asynchrony between alleles to be any difference larger than 80 min (Fig. 2C,D). In agreement with the correlation analysis, we found very few asynchronous regions in the V6.5 cell line (0.7%) with the C57BL/6 and 129/sv genomes (Fig. 2C–E; Supplemental Fig. S2) but found 12% of the genome replicating asynchronously in cell lines derived from M. musculus musculus and M. musculus castaneus crosses (Fig. 2C–E; Supplemental Fig. S3).
Figure 2.

Replication timing asynchrony in hybrid mouse ESCs. (A) Genome-wide correlations of RT between alleles for hybrid ESC lines V6.5 (top panel) and F1219 (bottom panel). RT values of 50-kb windows are displayed as binned scatter plots with a density heat map. (B) Correlation matrix of allele-specific RT separates samples by genome. Data from all cell lines (V6.5, F121, F1216, F1219, F123, Cas129) were analyzed and arranged based on their correlation values. (C,D) RT asynchrony in V6.5 and castaneus × musculus mESC lines. Histogram of RT differences between C57BL/6 and 129/sv (C) and between 129/sv and CAST/Ei genomes (D) are shown. (E) Percentages of autosomal genome showing RT asynchrony in C57BL/6 × 129/sv and musculus × castaneus are shown. Asynchrony was defined as differences >80 min in S-phase. (F) Unsupervised clustering analysis of RT asynchronous chromosomal segments identified specific RT signatures. Heat map shows the RT ratios [=log2(Early/Late)]. Branches of the dendrogram were constructed based on the correlation values (distance = correlation value − 1), and a correlation threshold of 0.9 was used to define two main sample clusters. k-means clustering analysis of RT asynchronous regions defined RT signatures with specific patterns. Specific genomes, parental configuration, gender, and growth conditions are shown at the bottom. (G) RT profiles of exemplary genomic regions showing RT asynchrony between alleles. Cell lines are labeled at the right in gray boxes; SNPs between C57B/6 vs. 129/sv and 129/sv vs. CAST/Ei are shown in black and red peak tracks, respectively. C57B/6 alleles are shown as green lines, 129/sv alleles as gray lines, and castaneus as blue lines. Two replicates of each cell line are shown in each plot, and the gender and parental configuration of each hybrid mESC line are shown.
Next, we extracted all genomic regions with replication asynchrony and classified them into RT signatures (Fig. 2F,G) as previously described (Rivera-Mulia et al. 2015). As expected, the clustering analysis showed that alleles from the V6.5 cell line clustered together and with all other alleles from M. musculus musculus genomes, while M. musculus castaneus alleles formed a separate cluster (Fig. 2F). Unsupervised clustering confirmed that the allelic differences in RT are associated with their respective subspecies genomes but not with parental origin or gender (Fig. 2F). In fact, only 0.10% of the total RT variation across all samples is linked to the gender of mouse ESCs (Supplemental Fig. S3), and no differences were observed linked to parental origin (Fig. 2F). Additional analysis of mouse hybrid ESCs with opposite parental configurations (F121 and F123 compared to Cas129) confirmed that RT differences are associated with musculus and castaneus genomes but not parental origin (Fig. 2G). Exemplary RT profiles show that the differences in RT between distinct alleles span over megabase regions and occur between subspecies genomes (Fig. 2G).
RT asynchrony in hybrid mouse ESCs occurs at developmentally regulated replication domains
We also analyzed whether the RT asynchronous regions identified in hybrid mouse ESCs coincide with replication domains that are regulated during development (i.e., domains that change between early and late compartments during distinct cell specification pathways). To do so, we identified developmentally regulated RT domains (RDs) across 30 mouse cell lines representing cell types from each of the three germ layers (Pope et al. 2014) and measured the overlap with the hybrid mouse ESC asynchronously replicating regions. We found that 67.4% of the RT asynchrony occurred at developmentally RT regulated genomic regions (Supplemental Fig. S3). By using a sequential Monte Carlo multiple testing (MCFDR) algorithm (Sandve et al. 2010, 2011), preserving the RD lengths and positions and randomizing the positions of the RT asynchronous regions, we found a highly significant overlap as compared to what is expected by chance (P-value = 0.003984).
Allelic differences in RT are maintained under different ESC media conditions
RT asynchrony between musculus and castaneus genomes was maintained under different growth conditions, either when cultured on MEFs in serum + LIF or feeder-free in 2i medium (Fig. 2F; Supplemental Fig. S4). In fact, we found that only 0.17% of the autosomal genome showed RT differences that correlated with growth conditions (Supplemental Fig. S3). Overall, our results suggest that allelic RT differences detected in ESCs are associated with the subspecies genomes but not with gender or paternal configuration and are stable in distinct growth conditions that maintain the naive vs. ground states of pluripotency.
RT asynchrony correlates with 3D genome organization but not sequence variation, gene expression, or chromatin accessibility
Previous analysis in human cells suggested that sequence variation is linked to RT asynchrony between alleles (Koren et al. 2014; Mukhopadhyay et al. 2014; Bartholdy et al. 2015). Here, we analyzed the relationship between RT differences and SNP density to determine whether RT asynchrony was related to local SNP density. We found that RT asynchronous regions were not enriched for SNPs, and no significant differences in SNP density were observed between RT constitutive regions and RT variable regions (Fig. 3A). Additionally, since previous studies suggested a link between replication timing and interspersed repetitive sequences (Hiratani et al. 2004), we analyzed whether differences in repetitive sequence content correlates with RT asynchrony. We found that RT asynchrony is not associated with content of LINEs, SINEs, LTRs, or short repetitive sequences (Supplemental Fig. S5).
Figure 3.

RT asynchrony correlates with genome organization. (A) RT synchronous and asynchronous genomic regions contain similar SNP densities. (B) Genome organization, chromatin accessibility, and gene expression of RT synchronous regions that replicate either early or late during S-phase. (C) Spearman correlation values of RT and distinct genomic features per genome. (D) Scatter plots of RT asynchrony vs. SNP density, changes in Hi-C compartments, differential accessibility, and differential expression. Hi-C data were obtained from Giorgetti et al. (2016). (E) Spearman correlation values of RT asynchrony and changes in Hi-C compartments, Hi-C interaction counts and distances, differences in chromatin accessibility, and SNP density. (F) Two exemplary chromosome regions showing the RT asynchrony associated with changes in Hi-C compartments, differential expression, and distinct promoter-enhancer interactions. Two replicates of each cell line are shown in each plot of RT profiles. Allele-specific RT, total nuclear RNA-seq, and ATAC-seq were determined based on the SNPs shown in red. Allele-specific Hi-C and PC-Hi-C data were obtained using HindIII fragments containing SNPs for each genome (HindIII track). Differentially expressed genes measured by total nuclear RNA-seq are shown at the bottom, color- coded according to the allele showing the highest expression value (musculus = black, light blue = castaneus). These two exemplary loci illustrate the strongest differences in RT associated with a measurable difference in gene expression. This association with transcription differences was not seen for most asynchronous regions (see text for details).
Next, we analyzed the global chromatin properties of the genomic regions in which the two alleles replicate synchronously in hybrid mouse ESCs, comparing chromosomal segments that replicate early vs. late during S-phase. Consistent with previous studies (Ryba et al. 2010; Pope et al. 2014; Rivera-Mulia and Gilbert 2016b), we found that synchronously early replicating regions are within Hi-C compartment A, while synchronously late replicating regions are within Hi-C compartment B (Fig. 3B). Moreover, synchronously early replicating allelic regions presented higher densities of long-range chromatin interactions with shorter distances in comparison with the synchronously late replicating allelic regions (Fig. 3B). Similarly, we found that synchronously early replicating regions were more accessible and the genes located within them expressed at higher levels in comparison to synchronously late replicating regions (Fig. 3B). Correlation analysis of RT of each allele focusing on the synchronously replicating portion of the genome with several genomic features confirmed that 3D chromatin organization (Hi-C) is the genomic property with the strongest correlation to RT (Fig. 3C).
We next analyzed the correlation of the asynchronously replicating regions of the genome with all other genomic properties. Scatter plots and Spearman correlation demonstrate that changes in 3D genome organization strongly correlated (correlation >0.6) with RT asynchrony (Fig. 3D,E). This analysis also confirmed that SNP density was not linked to the RT differences between the chromosome homologs (Fig. 3D,E). Differences in chromatin accessibility and gene expression did not correlate as strongly with RT asynchrony (Fig. 3D,E). Exemplary RT profiles of chromosomal regions with RT asynchrony show: (1) the close link between RT and 3D genome organization (Hi-C eigenvectors); (2) the differences in chromatin accessibility and gene expression between the two alleles; and (3) the differences in PC-Hi-C interactions (Fig. 3F). To better determine the correlation strength between RT asynchrony and 3D genome organization, we tested more stringent cutoffs to identify the highest allelic differences in RT. We found that higher allelic RT differences strengthen the correlation of RT asynchrony and Hi-C compartment changes and are associated with a moderate correlation with differential gene expression (Supplemental Fig. S6).
Since we found that RT asynchrony strongly correlated with 3D genome organization, we also performed a reciprocal analysis to determine to what extent differences in Hi-C compartments are linked to RT allelic differences. We found that Hi-C compartment changes correlate with RT asynchrony but not with gene expression or chromatin accessibility (Supplemental Fig. S7). These findings highlight the strong relationship between RT and chromosome architecture.
Although we did not find a correlation between RT asynchrony and differential gene expression, we detected differences in transcription levels for some of the genes present in the RT asynchronous regions (Prex2, A830018L16Rik, and Asph genes in Fig. 3F) and, after increasing the cut-off for allelic RT differences, we found a positive correlation between RT asynchrony and differential gene expression of 0.42 as compared to 0.62 correlation between RT asynchrony and Hi-C (Supplemental Fig. S6). The examples in Figure 3F show two regions with the strongest association between RT and transcription differences, demonstrating a quantitative correlation at a minority of loci. Hence, we took three additional approaches to explore the link between RT asynchrony and allelic differences in transcriptional activity. First, we identified the differentially expressed genes between musculus and castaneus alleles based on the FDR adjusted P-values (q-value) and analyzed their RT differences (the reciprocal of the analysis in Fig. 3). However, we did not find a strong correlation (rs = 0.14) between differential expression and RT asynchrony (Supplemental Fig. S6). Second, we mapped differentially expressed genes into synchronously vs. asynchronously replicated regions and found that only 25% of the differentially expressed genes reside in RT asynchronous regions (Supplemental Fig. S6). Finally, to determine whether RT asynchronous regions have at least one differentially expressed gene, we identified synchronous vs. asynchronous replication domains by merging adjacent windows with constant RT values and quantified the number of differentially expressed genes per RD. We found that most of the RT asynchronous RDs do not contain a differentially expressed gene, and most of these genes reside within synchronously early replicating domains (Supplemental Fig. S6), confirming that allelic differences in transcriptional activity are not necessarily linked to RT asynchrony.
RT asynchrony in hybrid mouse ESCs is not associated with gene imprinting
Early observations of RT asynchrony linked differences in RT to gene imprinting (Reik and Walter 2001), and a recent genome-wide study of RT asynchrony in human adult erythroid cells suggested that allelic differences in RT are enriched in imprinted genes (Mukhopadhyay et al. 2014). Hence, we analyzed whether the RT asynchrony identified in hybrid mouse ESCs is associated with gene imprinting. We obtained RT values at the transcription start sites (TSSs) of all RefSeq genes, identified the genes with RT asynchrony, and measured their overlap with characterized imprinted genes. We found that only 1.5% of the RT asynchronous genes are imprinted (Supplemental Fig. S8). In fact, only a fraction (25.5%) of imprinted genes replicated asynchronously in hybrid mouse ESCs (Supplemental Fig. S8). However, allelic differences in RT at those imprinted genes are not linked to gene imprinting, as identical RT patterns were observed in cell lines with opposite parental configurations. Hence, RT asynchrony in hybrid mouse ESC lines is not due to parental imprinting (Supplemental Fig. S8).
Since we did not find RT asynchrony linked to parental imprinting in the hybrid mouse ESC lines, we analyzed cells from extraembryonic tissue (XEN cells) as a positive control for parental imprinting. In extraembryonic tissues, the paternal X Chromosome is imprinted and inactivated (Takagi and Sasaki 1975; Escamilla-Del-Arenal et al. 2011). Consistently, we detected early replication peaks in both X Chromosomes in hybrid mouse ESCs, but early replication was restricted to the musculus maternal allele in extraembryonic cells (Supplemental Fig. S9). These findings demonstrate that our allele-specific analysis can detect RT asynchrony linked to parental imprinting, confirming that the RT differences observed in hybrid mouse ESCs are not associated with imprinting.
Long-range interactions in RT asynchronous domains are restricted to the early replicating allele
RT and Hi-C eigenvectors correlated strongly and RT asynchrony was associated with differences in Hi-C compartments (Fig. 3), suggesting that differences in chromatin interactions and 3D genome organization are linked to the allelic differences in RT. Hence, to test whether specific interactions are associated with the changes in RT between homologs, we analyzed PC-Hi-C data, which reduces the complexity of Hi-C libraries and allows the identification of significant regulatory interactions (Mifsud et al. 2015; Schoenfelder et al. 2015a). We did not find significant differences when comparing the total number of interactions and average distances between the two homologs at the asynchronous replicating regions. However, we found that discrete long-range enhancer-promoter interactions were restricted to the allele replicating earlier (Fig. 3F; Supplemental Fig. S10). In contrast, PC-Hi-C interactions at the allele replicating later in the asynchronous regions were restricted to short-range distances and the majority were within the replication domain (Fig. 3F; Supplemental Fig. S10). These observations suggest that long-range enhancer-promoter interactions connecting early replicating domains with RT asynchronous domains are restricted to the allele that replicates earlier.
RT asynchrony is lost during cell fate commitment
To determine whether RT asynchrony is maintained or increased during loss of pluripotency, we measured allele-specific RT programs of extraembryonic endoderm stem (XEN) cells (Dupont et al. 2016) and four different primary mouse embryonic fibroblasts (MEFs) derived from musculus × castaneus F1 hybrid embryos, as well as of neural precursors cells (NPCs) differentiated in vitro from hybrid mouse ESC lines with opposite parental configurations (Fig. 4A). Consistent with our previous findings demonstrating that RT is cell-type specific (Hiratani et al. 2010; Rivera-Mulia et al. 2015), genome-wide correlation confirms different RT programs for the distinct hybrid cell types (Supplemental Fig. S11). However, in contrast to ESCs, higher correlations between alleles, replicates, and cell lines were observed for all differentiated hybrid cell types (MEFs, NPCs, and XEN cells), suggesting fewer differences in RT between homologs (Supplemental Fig. S11). Consistently, we found a dramatic decrease in RT asynchrony in all differentiated cell types: 6% in XEN cells, 4% in MEFs, and 1% in NPCs of the autosomal genome. Symmetrical RT asynchrony was observed in all cell types, with a similar number of regions being replicated earlier in musculus or castaneus alleles (Fig. 4B; Supplemental Fig. S12). Very little overlap of RT asynchrony was observed between the distinct hybrid cell types, suggesting that allelic differences in RT are epigenetically regulated during development (Supplemental Fig. S12), although these asynchronous regions were not associated with autosomal gene imprinting (Supplemental Fig. S13). Furthermore, we tracked the RT of hybrid mouse ESC asynchronous regions in MEFs, NPCs, and XEN cells and found that more than 85% of the regions with allelic RT differences in stem cells became synchronously replicated in all differentiated cells (Fig. 4C). Exemplary RT profiles of hybrid mouse ESCs, XEN cells, MEFs, and NPCs confirm that the regions of RT asynchrony observed in pluripotency are lost during cell fate commitment (Fig. 4D).
Figure 4.

RT asynchrony is lost during differentiation. (A) Distinct cell types were derived from hybrid crosses of M. musculus musculus × M. musculus castaneus. ESC and extraembryonic endoderm stem (XEN) cells were derived from hybrid blastocysts, mouse embryonic fibroblasts (MEF) from hybrid mouse embryos (E12–14), and neural precursor cells (NPC) were differentiated in vitro from hybrid ESC lines. (B) Histograms of RT differences between musculus (129/sv) and castaneus (CAST/Ei) genomes. Labeled in red are the changes higher than the differences between replicates of the same genome. (C) RT asynchronous regions in ESCs become synchronous in differentiated cell types. (D) Two exemplary chromosome regions display the loss of RT asynchrony in differentiated cell types. Two replicates of each sample are shown. Four distinct primary MEFs of opposite gender were obtained from different embryos; RT profiles of female MEFs (MEF XX) and male MEFs (MEF XY) are shown. Data from two independently differentiated NPCs derived from distinct ESC lines (F121-9 and Cas129) are shown.
Convergence of RT and 3D genome folding during differentiation
In order to understand the loss of RT asynchrony in differentiated cells, we analyzed the changes in other genomic properties in NPCs, including gene expression, chromatin accessibility, and Hi-C compartments. Consistent with our findings in hybrid mouse ESCS, we found that synchronously early and late replicating domains in NPCs correlate with Hi-C compartment A and B, respectively, and that early synchronously replicating regions have higher densities of chromatin interactions with shorter distances and are more accessible in comparison to synchronously late replicating regions (Supplemental Fig. S14). Similarly, we found that RT correlates strongly with 3D chromatin organization genome-wide (Supplemental Fig. S14). We then tracked the changes in chromatin interactions in the regions that replicate asynchronously in hybrid mESCs after differentiation to NPCs. Consistent with the decrease in RT differences, we found a convergence to similar chromatin organization with a decrease in differences between Hi-C compartments A and B (Supplemental Fig. S14). In fact, we found that the chromosomal regions with allelic differences in RT and Hi-C compartments in stem cells replicate synchronously and were organized within the same A/B compartment in NPCs (Fig. 5).
Figure 5.

RT and genome organization control during development. RT asynchrony loss is coordinated with a decrease in genome organization differences between alleles. Genome tracks of RT (two cell lines with opposite parental configurations), Hi-C eigenvectors, ATAC-seq, and total nuclear RNA-seq are shown for mESCs (blue/gray) and for NPCs (red/orange). Differences in RT and Hi-C compartments are lost during differentiation to NPCs. NPC ATAC-seq, RNA-seq, and Hi-C data were obtained from Giorgetti et al. (2016).
Discussion
In this study, we measured the allelic variation in genome organization and function, exploiting the high SNP density between genomes of distinct mouse subspecies. Cell lines derived from crosses between Mus musculus castaneus and Mus musculus musculus subspecies exhibit a SNP, on average, every 150 bp, rendering informative reads to determine the homolog chromosome of origin. To sort NGS reads into each homolog chromosome, we developed a new algorithm: Haplotype-Assisted Read Parsing (HARP), which can efficiently separate reads per genome based on the presence of SNPs. An advantage of HARP over other allele-parsing methods is the separated alignment of each genome that yields more reads as compared with alignment just to the reference genome. We measured allelic differences in RT, genome organization, gene expression, and chromatin accessibility (Fig. 1), providing a valuable resource of normal variation between homologs that can be leveraged as a reference framework for future studies. Normal variation between homologs would be particularly useful for genome editing studies to characterize gene function and chromatin architecture, where the generation of heterozygous mutants will provide an internal wild-type control for comparison and better assessment of the manipulation effects.
Previous studies found RT variation between homologs in 8%–12% of the human genome (Mukhopadhyay et al. 2014; Bartholdy et al. 2015). Here, despite the 10-fold increase in SNP density, we found significant RT differences between mouse alleles in 12% of the autosomal genome in ESCs (Fig. 2E). Most of the genomic regions with allelic RT differences in mESCs overlap (67.4%) with developmentally regulated replication domains (Supplemental Fig. S3), suggesting that RT asynchrony occurs at loci that change RT dynamically during development. This overlap could be much higher, as the developmentally regulated regions were defined using only cell types with available data and additional genomic regions might change RT during cell fate commitment toward unexplored cell types. Hence, the overlap between RT asynchronous regions and developmentally regulated RDs is an underestimate, but we cannot rule out that RT asynchrony can occur in regions that do not change RT during development.
Consistent with previous studies (Ryba et al. 2010; Pope et al. 2014; Rivera-Mulia and Gilbert 2016b), we found that synchronously early replicating regions on both alleles correlated with Hi-C compartment A, higher gene expression, and accessible chromatin when compared to synchronously late replicating regions (Fig. 3). RT asynchrony also correlated strongly with the 3D chromatin structure but not as strongly with any other genomic property (Fig. 3). Reciprocal analysis of the changes in the Hi-C compartments also indicates that allelic differences in 3D genome organization correlate with RT synchrony but not with differential expression or chromatin accessibility (Supplemental Fig. S7), and higher allelic RT differences further strengthen the correlation of RT asynchrony and Hi-C compartment changes (Supplemental Fig. S6). These results underscore the link between DNA replication and 3D genome organization, supporting the hypothesis that the large genomes in eukaryotes are partitioned into smaller units (TADs/RDs), segregated to different nuclear compartments based on their temporal order of replication (Rivera-Mulia and Gilbert 2016a). Moreover, our findings highlight the measurement of RT programs as a functional readout of the spatial organization of the chromatin in the nucleus.
In contrast with previous studies, where RT asynchrony was found associated with gene imprinting (Mukhopadhyay et al. 2014; Bartholdy et al. 2015), we found that allelic differences in RT did not correlate with gene imprinting (Supplemental Fig. S8). Part of this could be quantitative; here, we have imposed a stringent cut-off for only those regions that differ in RT more than the noise seen between replicate experiments. In addition, a caveat of previous studies was the lack of comparison of individuals with opposite parental configurations to confirm whether the allelic differences were linked to gene imprinting. Here, we took advantage of hybrid cell lines with opposite parental configurations and confirmed that the allelic differences in RT were not associated with parental imprinting (Supplemental Fig. S8). Our study does not, however, address random monoallelic asynchrony. In fact, we have shown previously (Gribnau et al. 2005) that hybrid mESCs show RT asynchrony in the X Chromosome that switches dynamically between alleles; however, detection of dynamic switching random monoallelic asynchrony would require single-cell analyses such as singlet-doublet FISH analysis (Selig et al. 1992; Boggs and Chinault 1997; Singh et al. 2003; Dutta et al. 2009). It is unlikely that stable random allelic asynchrony is occurring in mESCs, however, as several of our mESC lines are clonally derived.
Recent studies report gene expression changes and impaired differentiation capacity linked to culture conditions (MEFs in serum + LIF vs. feeder-free in 2i + LIF) and associated with altered patterns of DNA methylation (Choi et al. 2017; Yagi et al. 2017). However, we found that RT asynchrony in mESCs is stable in all media conditions (Fig. 2F; Supplemental Fig. S4) and that hybrid mESCs maintained in any media conditions can be efficiently differentiated into neural precursors cells.
X Chromosome inactivation (XCI) in female cells occurs post-implantation during development in a random fashion in all embryonic cell types. However, in extraembryonic tissues, the paternal X is imprinted and inactivated (Takagi and Sasaki 1975; Escamilla-Del-Arenal et al. 2011). Consistently, early replication was restricted to the musculus maternal allele in XEN cells but not in mouse hybrid ESCs (Supplemental Fig. S9). However, although XCI occurs randomly in other cell types, we have shown previously that XCI can be detected as a decrease in RT values in differentiated cell types as compared to male cell lines (Hiratani et al. 2010). Here, we found a delay in RT for X Chromosomes of all female differentiated cell lines (Supplemental Fig. S9), with the musculus allele replicated later in all NPC and MEF cell lines, suggesting a subspecies skew in XCI (Supplemental Fig. S9).
Finally, since almost all of the RT asynchrony reported in the literature has been in differentiated cell types, we also evaluated the allelic RT differences in XEN cells, primary MEFs and NPCs. We found that the RT asynchrony observed in mESCs was lost during cell fate commitment (Fig. 4), with more than 85% of the loci showing allelic differences in RT in mESCs replicating synchronously in all differentiated cells. In addition, the few allelic differences in RT were not associated with gene imprinting even in these differentiated cell types (Supplemental Fig. S13). As discussed above, one possible explanation is that differences in RT associated with autosomal gene imprinting are lower than the significant differences considered in this study, and single-cell analysis might identify small degrees of RT variation of autosomal imprinted genes. Similarly, allelic differences in RT have been observed at random monoallelically expressed genes (Donley et al. 2015), but single-cell analysis or subcloning of differentiated cell types would be required to assess any stable RT differences at these random monoallelically expressed loci. Alternatively, RT asynchrony of imprinted genes might be established downstream during differentiation, which would explain the RT asynchrony observed in B lymphocytes and erythroid cells (Koren et al. 2014; Koren and McCarroll 2014; Mukhopadhyay et al. 2014; Bartholdy et al. 2015). Moreover, we found that the loss of RT asynchrony in differentiated cell types was coordinated with a decrease in allelic differences in Hi-C compartments, with the RT asynchronous regions detected in hybrid mESCs becoming synchronously replicating and organized within the same Hi-C compartments after differentiation to NPCs. These results suggest that, during cell fate commitment, there are mechanisms that converge 3D chromatin folding patterns for the chromosome homologs. Future studies, such as genome editing experiments combined with allele-specific genomics, would be required to identify the regulatory factors that control the establishment of the spatial genome organization and its intriguing liaisons to gene function.
Methods
Cell culture
Hybrid mouse ESC lines V6.5, F121, F121-6, F121-9, F123, and Cas129 (Fig. 1B) were grown in two different conditions: (1) on mitomycin-C-inactivated MEFs in ESC medium; and (2) feeder-free in 2i medium (see Supplemental Table S1 for media composition details). For feeder-free/serum-free conditions, cells were grown on 0.1% gelatin-coated dishes in N2B27 media supplemented with 1 µM MEK inhibitor, 4.25 µM GSK3 inhibitor (1-Azakenpaullone), 2 mM glutamine, 1000 U/mL LIF (Cell Guidance Systems GFM200), and 0.15 mM monothioglycerol. Hybrid XEN cells were derived and maintained as previously described (Kunath et al. 2005) in RPMI supplemented with 15% FBS (Life Technologies), 100 mM sodium pyruvate, 10 mM beta-mercaptoethanol, and 200 mM L-glutamine. Hybrid MEFs were grown in DMEM supplemented with 10% FBS, 1× NAAs, 2 mM L-glutamine, and 10 mM beta-mercaptoethanol. NPCs were derived from hybrid mouse ESC lines with opposite parental configurations (F121-9 and Cas129). Hybrid mouse ESCs were grown in a monolayer culture at high density (1.5 × 105 cells/cm2) on 0.1% gelatin-coated six-well plates in serum-free ESGRO Complete Clonal Grade medium (CCGM). After 24 h, cells were dissociated with 0.1% trypsin and were plated onto 0.1% (v/v) gelatin-coated 10-cm dishes at 1 × 104 cells/cm2 in RHB-A medium (Takara). The medium was changed every other day, and NPCs were analyzed at day 11.
RT libraries
Genome-wide RT profiles were constructed as previously described (Ryba et al. 2011; Marchal et al. 2017). Briefly, cells were pulse-labeled with BrdU and separated into early and late S-phase fractions by flow cytometry, followed by DNA immunoprecipitation with anti-BrdU antibody (Sigma-Aldrich, cat. no. B5002). Sequencing libraries of BrdU-substituted DNA from early and late fractions were prepared using the NEBNext Ultra DNA Library Prep kit for Illumina (E7370). Sequencing was performed on an Illumina-HiSeq 2500 by 100-bp single-end sequencing. Approximately 35 million reads per sample were generated.
ATAC-seq libraries
ATAC-seq (Assay for Transposase-Accessible Chromatin using Sequencing) was performed as previously described (Buenrostro et al. 2013), starting with ∼200,000 cells. Lysis was performed for 15 min on ice, and nuclei were collected by spinning at 600g for 10 min at 4°C. Transposase from the Nextera DNA Sample Preparation kit (Illumina) was added, and nuclei were incubated at 37°C for 30 min. Following DNA purification on a MinElute column (Qiagen), libraries were amplified using PCR reagents from the Nextera DNA Sample Preparation kit and index primers from the Nextera Index kit (Illumina). PCR amplification was performed using the following conditions: 72°C for 5 min; 98°C for 30 sec; 10 cycles of 98°C for 10 sec, 63°C for 30 sec, and 72°C for 3 min; and a final extension at 72°C for 5 min. Libraries were purified twice with PCR clean-up kit columns (Qiagen). Library concentration and size distribution were measured using the KAPA Library Quantification kit (KAPA Biosystems) and the Agilent Bioanalyzer 2100. Libraries were sequenced (100-bp paired-end) on the HiSeq 2500 platform (Illumina).
Promoter Capture Hi-C libraries
Promoter Capture Hi-C was performed as previously described (Schoenfelder et al. 2015a), but using an in-nucleus ligation method for Hi-C library preparation (Nagano et al. 2015). Briefly, 3–4 × 107 cells were fixed in 2% formaldehyde for 10 min. Chromatin was digested with HindIII (NEB) overnight at 37°C, and Klenow (NEB) was used to label restriction sites with biotin-14-dATP (Life Technologies). In-nucleus ligation was performed by adding 25 units of T4 DNA ligase (Invitrogen) in T4 ligase buffer (NEB), supplemented with 100 µg/mL BSA, per 5 million cells starting material. The ligation reaction was incubated at 16°C overnight, and crosslinks were reversed by adding Proteinase K and incubating at 65°C overnight. Samples were treated with RNase A, and ligation products were purified by phenol/chloroform extraction. Biotin was removed from nonligated Hi-C library fragment ends with T4 DNA polymerase (NEB), followed by phenol/chloroform extraction and sonication (Covaris E220). Hi-C library DNA was end-repaired with Klenow, T4 DNA polymerase, and T4 polynucleotide kinase (NEB). Library DNA was size-selected with AMPure XP beads (Beckman Coulter), and 3′ ends were adenylated using Klenow exo− (NEB). Biotin-marked ligation products were bound to MyOne Streptavidin C1 Dynabeads (Life Technologies), and PE adapters (Illumina) were ligated. Bead-bound Hi-C DNA was amplified with seven or eight PCR amplification cycles using the PE PCR 1.0 and PE PCR 2.0 primers (Illumina), and amplified libraries were purified using AMPure XP beads (Beckman Coulter).
For the capture step, 500 ng of Hi-C library DNA were denatured by heating to 95°C for 5 min and incubated at 65°C for 24 h with a custom-designed biotinylated RNA bait system targeting the ends of HindIII restriction fragments at 22,225 mouse gene promoters (Schoenfelder et al. 2015a) (Agilent Technologies). The SureSelect Target enrichment protocol (Agilent Technologies) was followed for biotin pull-down and washes, using MyOne Streptavidin T1 Dynabeads (Life Technologies). Captured Hi-C DNA was amplified with four PCR amplification cycles using the PE PCR 1.0 and PE PCR 2.0 primers (Illumina), and amplified libraries were purified using AMPure XP beads (Beckman Coulter). Promoter Capture Hi-C libraries were sequenced (100-bp paired-end) on the HiSeq 1000 platform (Illumina).
Total nuclear RNA-seq data
To measure all transcriptional events (including coding and noncoding transcripts), we performed total nuclear RNA-seq. Previously reported strand-specific nuclear RNA-seq libraries (Schoenfelder et al. 2015b) were resequenced (100-bp paired-end) on the HiSeq 2500 platform (Illumina), and reads were merged with the published data for downstream analysis. Briefly, total nuclear RNA was purified from isolated intact nuclei using TRIsure (Bioline), treated with DNase I (Roche), and repurified using the RNeasy Mini kit (Qiagen). cDNA was made using random primers to include all transcripts (including poly[A]-RNA). Strand-specific RNA-seq libraries were prepared by ligation to TruSeq Illumina adapters with T4 DNA ligase (Enzymatics). BAM files were filtered to remove read-pairs which mapped to ribosomal DNA sequences.
Allele-specific alignment and data analysis
Reads of quality scores above 30 were independently mapped to 129/sv and castaneus reference genomes using Bowtie 2 (Langmead and Salzberg 2012) and assigned to each allele using HARP, a new computational algorithm to filter reads based on the presence of genome-specific SNPs (see Supplemental Methods, Supplemental Scripts, and https://github.com/dvera/harp). Repli-seq analysis was performed in R. RT data sets were normalized using the limma package in R (R Core Team 2017) and rescaled to equivalent ranges by quantile normalization. Correlation analysis was performed using the corrplot package in R. A detailed computational pipeline for genome-wide measurement of RT has been published elsewhere (Ryba et al. 2011; Marchal et al. 2017).
Clustering analysis
Significant RT variable regions were defined as those regions with differences ≥1 in pairwise comparisons between all samples analyzed. Unsupervised hierarchical and k-means clustering analysis were performed using Cluster 3.0 (de Hoon et al. 2004) using uncentered correlation metrics and average linkage. Heat maps and dendrograms were generated in Java Treeview (Saldanha 2004).
Data access
All data sets generated in this study have been submitted to the NCBI Gene Expression Omnibus (GEO; http://www.ncbi.nlm.nih.gov/geo/) under accession number GSE107421, in the 4D Nucleome DCIC portal, and in our laboratory database at http://www.replicationdomain.org. HARP (Haplotype-Assisted Read Parsing) source code is available in Supplemental Scripts and at https://github.com/dvera/harp.
Supplementary Material
Acknowledgments
We thank Ruth A. Didier for assistance with flow cytometry and Ferhat Ay for data processing advice. This work has been supported by National Institutes of Health grants GM083337 and DK107965 to D.M.G.
Author contributions: J.C.R.-M. and D.M.G. conceived and designed the project. C.D. and J.G. generated the F1 hybrid cell lines. A.D. and P.F. generated PC-Hi-C, RNA-seq, and ATAC-seq data sets. J.C.R.-M., T.S., and C.T.-G. performed cell cultures and NPC differentiation. J.C.R.-M., T.S., and C.T.-G. collected the RT data sets. J.C.R.-M., D.V., and J.Z. performed data analysis and interpretation. J.C.R.-M. and D.M.G. wrote the manuscript.
Footnotes
[Supplemental material is available for this article.]
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.232561.117.
References
- Alver RC, Chadha GS, Blow JJ. 2014. The contribution of dormant origins to genome stability: from cell biology to human genetics. DNA Repair (Amst) 19: 182–189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avner P, Heard E. 2001. X-chromosome inactivation: counting, choice and initiation. Nat Rev Genet 2: 59–67. [DOI] [PubMed] [Google Scholar]
- Bartholdy B, Mukhopadhyay R, Lajugie J, Aladjem MI, Bouhassira EE. 2015. Allele-specific analysis of DNA replication origins in mammalian cells. Nat Commun 6: 7051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boggs BA, Chinault AC. 1997. Analysis of DNA replication by fluorescence in situ hybridization. Methods 13: 259–270. [DOI] [PubMed] [Google Scholar]
- Buenrostro JD, Giresi PG, Zaba LC, Chang HY, Greenleaf WJ. 2013. Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position. Nat Methods 10: 1213–1218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi J, Huebner AJ, Clement K, Walsh RM, Savol A, Lin K, Gu H, Di Stefano B, Brumbaugh J, Kim S-Y, et al. 2017. Prolonged Mek1/2 suppression impairs the developmental potential of embryonic stem cells. Nature 548: 219–233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Hoon MJL, Imoto S, Nolan J, Miyano S. 2004. Open source clustering software. Bioinformatics 20: 1453–1454. [DOI] [PubMed] [Google Scholar]
- Dixon J, Xu J, Dileep V, Zhan Y, Song F, Le VT, Yardimci GG, Chakraborty A, Bann DV, Wang Y, et al. 2017. An integrative framework for detecting structural variations in cancer genomes. bioRxiv 10.1101/119651. [DOI]
- Donley N, Smith L, Thayer MJ. 2015. ASAR15, a cis-acting locus that controls chromosome-wide replication timing and stability of human chromosome 15. PLoS Genet 11: e1004923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dupont C, Maduro C, Braanker Den H, Boers R, Kurek D, Gribnau J. 2016. Characterization of histone modifications associated with inactive X-chromosome in trophoblast stem cells, eXtra-embryonic endoderm cells and in in vitro derived undifferentiated and differentiated epiblast like stem cells. PLoS One 11: e0167154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dutta D, Ensminger AW, Zucker JP, Chess A. 2009. Asynchronous replication and autosome-pair non-equivalence in human embryonic stem cells. PLoS One 4: e4970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ensminger AW, Chess A. 2004. Coordinated replication timing of monoallelically expressed genes along human autosomes. Hum Mol Genet 13: 651–658. [DOI] [PubMed] [Google Scholar]
- Escamilla-Del-Arenal M, da Rocha ST, Heard E. 2011. Evolutionary diversity and developmental regulation of X-chromosome inactivation. Hum Genet 130: 307–327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farago M, Rosenbluh C, Tevlin M, Fraenkel S, Schlesinger S, Masika H, Gouzman M, Teng G, Schatz D, Rais Y, et al. 2012. Clonal allelic predetermination of immunoglobulin-κ rearrangement. Nature 490: 561–565. [DOI] [PubMed] [Google Scholar]
- Galupa R, Heard E. 2015. X-chromosome inactivation: new insights into cis and trans regulation. Curr Opin Genet Dev 31: 57–66. [DOI] [PubMed] [Google Scholar]
- Gerhardt J, Tomishima MJ, Zaninovic N, Colak D, Yan Z, Zhan Q, Rosenwaks Z, Jaffrey SR, Schildkraut CL. 2014a. The DNA replication program is altered at the FMR1 locus in fragile X embryonic stem cells. Mol Cell 53: 19–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerhardt J, Zaninovic N, Zhan Q, Madireddy A, Nolin SL, Ersalesi N, Yan Z, Rosenwaks Z, Schildkraut CL. 2014b. Cis-acting DNA sequence at a replication origin promotes repeat expansion to fragile X full mutation. J Cell Biol 206: 599–607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giorgetti L, Lajoie BR, Carter AC, Attia M, Zhan Y, Xu J, Chen CJ, Kaplan N, Chang HY, Heard E, et al. 2016. Structural organization of the inactive X chromosome in the mouse. Nature 535: 575–579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gribnau J, Luikenhuis S, Hochedlinger K, Monkhorst K, Jaenisch R. 2005. X chromosome choice occurs independently of asynchronous replication timing. J Cell Biol 168: 365–373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hiratani I, Leskovar A, Gilbert DM. 2004. Differentiation-induced replication-timing changes are restricted to AT-rich/long interspersed nuclear element (LINE)-rich isochores. Proc Natl Acad Sci 101: 16861–16866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hiratani I, Ryba T, Itoh M, Rathjen J, Kulik M, Papp B, Fussner E, Bazett-Jones DP, Plath K, Dalton S, et al. 2010. Genome-wide dynamics of replication timing revealed by in vitro models of mouse embryogenesis. Genome Res 20: 155–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson DA, Pombo A. 1998. Replicon clusters are stable units of chromosome structure: evidence that nuclear organization contributes to the efficient activation and propagation of S phase in human cells. J Cell Biol 140: 1285–1295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitsberg D, Selig S, Brandeis M, Simon I, Keshet I, Driscoll DJ, Nicholls RD, Cedar H. 1993. Allele-specific replication timing of imprinted gene regions. Nature 364: 459–463. [DOI] [PubMed] [Google Scholar]
- Koren A, McCarroll SA. 2014. Random replication of the inactive X chromosome. Genome Res 24: 64–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koren A, Handsaker RE, Kamitaki N, Karlić R, Ghosh S, Polak P, Eggan K, McCarroll SA. 2014. Genetic variation in human DNA replication timing. Cell 159: 1015–1026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kunath T, Arnaud D, Uy GD, Okamoto I, Chureau C, Yamanaka Y, Heard E, Gardner RL, Avner P, Rossant J. 2005. Imprinted X-inactivation in extra-embryonic endoderm cell lines from mouse blastocysts. Development 132: 1649–1661. [DOI] [PubMed] [Google Scholar]
- Langmead B, Salzberg SL. 2012. Fast gapped-read alignment with Bowtie 2. Nat Methods 9: 357–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marchal C, Sasaki T, Vera D, Wilson K, Sima J, Rivera-Mulia JC, Trevilla-García C, Nogues C, Nafie E, Gilbert DM. 2017. Repli-seq: genome-wide analysis of replication timing by next-generation sequencing. bioRxiv 10.1101/104653. [DOI] [PMC free article] [PubMed]
- Mifsud B, Tavares-Cadete F, Young AN, Sugar R, Schoenfelder S, Ferreira L, Wingett SW, Andrews S, Grey W, Ewels PA, et al. 2015. Mapping long-range promoter contacts in human cells with high-resolution capture Hi-C. Nat Genet 47: 598–606. [DOI] [PubMed] [Google Scholar]
- Moindrot B, Audit B, Klous P, Baker A, Thermes C, de Laat W, Bouvet P, Mongelard F, Arneodo A. 2012. 3D chromatin conformation correlates with replication timing and is conserved in resting cells. Nucleic Acids Res 40: 9470–9481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monkhorst K, Jonkers I, Rentmeester E, Grosveld F, Gribnau J. 2008. X inactivation counting and choice is a stochastic process: evidence for involvement of an X-linked activator. Cell 132: 410–421. [DOI] [PubMed] [Google Scholar]
- Mostoslavsky R, Singh N, Tenzen T, Goldmit M, Gabay C, Elizur S, Qi P, Reubinoff BE, Chess A, Cedar H, et al. 2001. Asynchronous replication and allelic exclusion in the immune system. Nature 414: 221–225. [DOI] [PubMed] [Google Scholar]
- Mukhopadhyay R, Lajugie J, Fourel N, Selzer A, Schizas M, Bartholdy B, Mar J, Lin CM, Martin MM, Ryan M, et al. 2014. Allele-specific genome-wide profiling in human primary erythroblasts reveal replication program organization. PLoS Genet 10: e1004319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagano T, Várnai C, Schoenfelder S, Javierre BM, Wingett SW, Fraser P. 2015. Comparison of Hi-C results using in-solution versus in-nucleus ligation. Genome Biol 16: 175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neelsen KJ, Zanini IMY, Mijic S, Herrador R, Zellweger R, Ray Chaudhuri A, Creavin KD, Blow JJ, Lopes M. 2013. Deregulated origin licensing leads to chromosomal breaks by rereplication of a gapped DNA template. Genes Dev 27: 2537–2542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pope BD, Ryba T, Dileep V, Yue F, Wu W, Denas O, Vera DL, Wang Y, Hansen RS, Canfield TK, et al. 2014. Topologically associating domains are stable units of replication-timing regulation. Nature 515: 402–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- R Core Team. 2017. R: a language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria: https://www.R-project.org/. [Google Scholar]
- Reik W, Walter J. 2001. Genomic imprinting: parental influence on the genome. Nat Rev Genet 2: 21–32. [DOI] [PubMed] [Google Scholar]
- Rideout WM, Wakayama T, Wutz A, Eggan K, Jackson-Grusby L, Dausman J, Yanagimachi R, Jaenisch R. 2000. Generation of mice from wild-type and targeted ES cells by nuclear cloning. Nat Genet 24: 109–110. [DOI] [PubMed] [Google Scholar]
- Rivera-Mulia JC, Gilbert DM. 2016a. Replicating large genomes: divide and conquer. Mol Cell 62: 756–765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rivera-Mulia JC, Gilbert DM. 2016b. Replication timing and transcriptional control: beyond cause and effect—part III. Curr Opin Cell Biol 40: 168–178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rivera-Mulia JC, Buckley Q, Sasaki T, Zimmerman J, Didier RA, Nazor K, Loring JF, Lian Z, Weissman S, Robins AJ, et al. 2015. Dynamic changes in replication timing and gene expression during lineage specification of human pluripotent stem cells. Genome Res 25: 1091–1103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rivera-Mulia JC, Desprat R, Trevilla-García C, Cornacchia D, Schwerer H, Sasaki T, Sima J, Fells T, Studer L, Lemaitre JM, et al. 2017a. DNA replication timing alterations identify common markers between distinct progeroid diseases. Proc Natl Acad Sci 114: E10972–E10980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rivera-Mulia JC, Kim S, Gabr H, Kahveci T, Gilbert DM. 2017b. Replication Timing Networks: a novel class of gene regulatory networks. bioRxiv 10.1101/186866. [DOI] [PMC free article] [PubMed]
- Ryba T, Hiratani I, Lu J, Itoh M, Kulik M, Zhang J, Schulz TC, Robins AJ, Dalton S, Gilbert DM. 2010. Evolutionarily conserved replication timing profiles predict long-range chromatin interactions and distinguish closely related cell types. Genome Res 20: 761–770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryba T, Battaglia D, Pope BD, Hiratani I, Gilbert DM. 2011. Genome-scale analysis of replication timing: from bench to bioinformatics. Nat Protoc 6: 870–895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryba T, Battaglia D, Chang BH, Shirley JW, Buckley Q, Pope BD, Devidas M, Druker BJ, Gilbert DM. 2012. Abnormal developmental control of replication-timing domains in pediatric acute lymphoblastic leukemia. Genome Res 22: 1833–1844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sadoni N, Cardoso MC, Stelzer EHK, Leonhardt H, Zink D. 2004. Stable chromosomal units determine the spatial and temporal organization of DNA replication. J Cell Sci 117: 5353–5365. [DOI] [PubMed] [Google Scholar]
- Saldanha AJ. 2004. Java Treeview—extensible visualization of microarray data. Bioinformatics 20: 3246–3248. [DOI] [PubMed] [Google Scholar]
- Sandve GK, Gundersen S, Rydbeck H, Glad I, Holden L, Holden M, Liestol K, Clancy T, Ferkingstad E, Johansen M, et al. 2010. The Genomic HyperBrowser: inferential genomics at the sequence level. Genome Biol 11: R121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandve GK, Ferkingstad E, Nygård S. 2011. Sequential Monte Carlo multiple testing. Bioinformatics 27: 3235–3241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasaki T, Rivera-Mulia JC, Vera D, Zimmerman J, Das S, Padget M, Nakamichi N, Chang BH, Tyner J, Druker BJ, et al. 2017. Stability of patient-specific features of altered DNA replication timing in xenografts of primary human acute lymphoblastic leukemia. Exp Hematol 51: 71–82.e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schoenfelder S, Furlan-Magaril M, Mifsud B, Tavares-Cadete F, Sugar R, Javierre BM, Nagano T, Katsman Y, Sakthidevi M, Wingett SW, et al. 2015a. The pluripotent regulatory circuitry connecting promoters to their long-range interacting elements. Genome Res 25: 582–597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schoenfelder S, Sugar R, Dimond A, Javierre BM, Armstrong H, Mifsud B, Dimitrova E, Matheson L, Tavares-Cadete F, Furlan-Magaril M, et al. 2015b. Polycomb repressive complex PRC1 spatially constrains the mouse embryonic stem cell genome. Nat Genet 47: 1179–1186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selig S, Okumura K, Ward DC, Cedar H. 1992. Delineation of DNA replication time zones by fluorescence in situ hybridization. EMBO J 11: 1217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simon I, Tenzen T, Reubinoff BE, Hillman D, McCarrey JR, Cedar H. 1999. Asynchronous replication of imprinted genes is established in the gametes and maintained during development. Nature 401: 929–932. [DOI] [PubMed] [Google Scholar]
- Singh N, Ebrahimi FAW, Gimelbrant AA, Ensminger AW, Tackett MR, Qi P, Gribnau J, Chess A. 2003. Coordination of the random asynchronous replication of autosomal loci. Nat Genet 33: 339–341. [DOI] [PubMed] [Google Scholar]
- Solovei I, Thanisch K, Feodorova Y. 2016. How to rule the nucleus: divide et impera. Curr Opin Cell Biol 40: 47–59. [DOI] [PubMed] [Google Scholar]
- Takagi N, Sasaki M. 1975. Preferential inactivation of the paternally derived X chromosome in the extraembryonic membranes of the mouse. Nature 256: 640–642. [DOI] [PubMed] [Google Scholar]
- Yaffe E, Farkash-Amar S, Polten A, Yakhini Z, Tanay A, Simon I. 2010. Comparative analysis of DNA replication timing reveals conserved large-scale chromosomal architecture. PLoS Genet 6: e1001011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yagi M, Kishigami S, Tanaka A, Semi K, Mizutani E, Wakayama S, Wakayama T, Yamamoto T, Yamada Y. 2017. Derivation of ground-state female ES cells maintaining gamete-derived DNA methylation. Nature 548: 224–227. [DOI] [PubMed] [Google Scholar]
- Yue F, Cheng Y, Breschi A, Vierstra J, Wu W, Ryba T, Sandstrom R, Ma Z, Davis C, Pope BD, et al. 2014. A comparative encyclopedia of DNA elements in the mouse genome. Nature 515: 355–364. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.