

Abstract
Pooled CRISPR screens based on lentiviral systems have been widely applied to identify the effect of gene knockout on cellular phenotype. Although many screens were successful, they also have the limitation that genes conferring mild phenotypes or those essential for growth can be overlooked, as every genetic perturbation is incorporated in the same population. Arrayed screens, on the other hand, incorporate a single genetic perturbation in each well and could overcome these limitations. However, arrayed screens based on siRNA-mediated knockdown were recently criticized for low reproducibility caused by incomplete inhibition of gene expression. To overcome these limitations, we developed a novel arrayed CRISPR screen based on a plasmid library expressing a single guide RNA (sgRNA) and disrupted 1514 genes, encoding kinases, proteins related to endocytosis, and Golgi-localized proteins, individually using 4542 sgRNAs (three sgRNAs per gene). This screen revealed host factors required for infection by coxsackievirus B3 (CVB3) from Picornaviridae, which includes human pathogens causing diverse diseases. Many host factors that had been overlooked in a conventional pooled screen were identified for CVB3 infection, including entry-related factors, translational initiation factors, and several replication factors with different functions, demonstrating the advantage of the arrayed screen. This screen was quite reliable and reproducible, as most genes identified in the primary screen were confirmed in secondary screens. Moreover, ACBD3, whose phenotype was not affected by siRNA-mediated knockdown, was reliably identified. We propose that arrayed CRISPR screens based on sgRNA plasmid libraries are powerful tools for arrayed genetic screening and applicable to larger-scale screens.
Functional genetic screens are generally performed using two different methods: pooled or arrayed. Genetic screens using the clustered regularly interspaced repeat (CRISPR)-CRISPR-associated (Cas) system have been widely applied to various systems to reveal relationships between phenotypes and genes (Koike-Yusa et al. 2014; Shalem et al. 2014; Wang et al. 2014). However, most screens are conducted in a pooled format, which uses lentiviruses to deliver a genome-wide library of single guide RNAs (sgRNAs). Pooled screens have some advantages, including easy preparation and reduced labor. Although highly sensitive pooled assays could identify the phenotypes that are mild or that have growth disadvantages (Gilbert et al. 2014), pooled assays, in general, are not compatible with these phenotypes. On the other hand, arrayed screens that are conducted by singular reagents arranged in a multiwell format could overcome these limitations, as each well has individual genetic perturbations. However, small interference RNA (siRNA) library screens, which are the most representative arrayed screen method, are recently being criticized for low reproducibility induced by high off-target effects and incomplete gene knockdown (Buehler et al. 2012; Kaelin 2012). In contrast, genome editing based on CRISPR-Cas9 results in complete knockout of the target gene. Furthermore, off-target effects can be greatly reduced by selecting unique target sites and modifying guide RNAs (Bae et al. 2014b; Cho et al. 2014; Fu et al. 2014). These comparative advantages of the CRISPR system could help overcome the limitations of siRNA library screening. Arrayed CRISPR screens could be a very useful platform to fill the caveats of pooled CRISPR screens and siRNA library screens.
Some groups have previously attempted to develop arrayed CRISPR screens (Hultquist et al. 2016; McCleland et al. 2016; Tan and Martin 2016; Datlinger et al. 2017; Strezoska et al. 2017). These studies were based on guide RNA libraries consisting of individual lentiviral sgRNAs for single genes or synthesized CRISPR RNA (crRNA) in a manner similar to siRNA screens. Although these prior studies have their strengths, they also have some weaknesses. Arrayed lentiviral particles are cumbersome to prepare because each lentiviral vector should be delivered to packaging cell lines individually. Moreover, challenges such as cross-contamination and variable lentiviral titers in the multiwell plate make it difficult to apply lentiviral particles to arrayed screens on a large scale. Furthermore, because of the low expression of sgRNAs and the additional time to select out the untransduced cells with selection marker, it will take more time for screening. This could also be another drawback of the lentiviral method in arrayed screens because a long culture period is not feasible in multiwell plates. Although synthetic crRNA is easier to prepare compared to lentivirus particles, RNA synthesis is cost-inefficient.
In this study, we present a new CRISPR screen method using an arrayed library of sgRNAs to identify essential host genes for coxsackievirus infection. Delivery of Cas9 and sgRNA expression plasmids is the most frequently applied method because of simplicity and high expression levels of both components, which result in high efficiency of genome editing in eukaryotic cells over 2–3 d. With respect to rapid and highly efficient genome editing, this method would reduce the signal-to-noise levels in high-throughput assays, thus improving the reliability and sensitivity of genetic screening.
Enteroviruses from the Picornaviridae family are human pathogens that cause a wide range of illnesses, including aseptic meningitis, encephalitis, and myocarditis (Fields et al. 2007). These viruses depend on host proteins for their life cycle. Determining the role of host factors in viral infection would help with understanding the basic aspects of virus-host interaction and exploring novel therapeutic targets. Host factors that are indispensable for viruses and affect host cells minimally would be ideal targets. We chose coxsackievirus B3 (CVB3), which is a member of human enteroviruses. Some groups have reported that CVB3 exploits kinases, proteins in endocytic pathways, and Golgi-localized proteins for its infection (Coyne and Bergelson 2006; Lanke et al. 2009; Patel et al. 2009). To further investigate the host factors required for CVB3 infection, we selected up to 1514 encoding kinases, proteins related to endocytosis, and Golgi-localized proteins. We constructed a plasmid library expressing sgRNA targeting these 1514 genes (three sgRNAs per gene, 4542 sgRNAs) and performed a high-throughput screen to identify the genes required for CVB3 infection.
Results
Pooled lentiviral CRISPR screen to identify host factors for CVB3 infection
To compare the commonly used pooled screen with the arrayed screen, a lentivirus-based pooled sgRNA screen was conducted to uncover the host genes essential for CVB3. The GeCKOv2 CRISPR knockout pooled library, which is the most widely used, was transduced into HeLa cells at 0.3 multiplicity of infection (MOI), and sgRNA-expressing cells were selected with puromycin. Two weeks later, these cells were subjected to CVB3 infection. We performed targeted deep sequencing for 100× coverage using genomic DNA isolated from hundreds of surviving cells and compared the abundance of sgRNAs from the initial population and the cells surviving after the CVB3 infection, using Model-based Analysis of Genome-wide CRISPR-Cas9 Knockout (MAGeCK) software (Supplemental Fig. S1A; Li et al. 2014). Only one gene, CXADR (Coxsackievirus and adenovirus receptor), which is well known as the receptor of CVB3, was shown as a strong candidate (Supplemental Fig. S1B). Though the lentivirus-based CRISPR screen was easy to perform, we wondered if we had missed other important host factors using this pooled screen. Therefore, we tried to screen each gene one by one in an arrayed format.
Arrayed CRISPR screens
To identify host factors for CVB3 infection in an arrayed format, we developed an image-based assay using sgRNA plasmids. As a proof-of-principle, we chose two genes: CXADR and PI4KB. CXADR is well-known as the receptor of CVB3, which was also identified from the pooled screen, and PI4KB is an essential factor for CVB3 replication (Bergelson et al. 1997; Hsu et al. 2010). We first transfected three sgRNA plasmids targeting CXADR and PI4KB into HeLa cells, along with the Cas9 expression plasmid to induce mutations in each gene. Mutations were induced very efficiently, and 71% and 80% of alleles were disrupted, respectively. As CRISPR knockout generally requires more time to show phenotypes compared to siRNA, experimental conditions like cell number and incubation time after transfection were optimized. To maximize Cas9-mediated mutation and minimize the effect of residual mRNA and proteins, we incubated the transfected cells for 5 d and then infected them with CVB3. The cells were then fixed and stained with the anti-3C rabbit polyclonal antibody and AF-488-conjugated anti-rabbit goat antibody. Images were acquired using the high-content imaging system, Operetta. The CVB3 infection level was significantly reduced when the host factor-targeting sgRNAs were transfected, compared to that using the nontargeting sgRNA (Fig. 1A,B). This result implies that the arrayed sgRNA library, based on plasmid expression, could be useful for screening. Additionally, this image-based assay, which stains viral protein directly, could reduce the artifacts driven by other assays, including reporter cells detecting viral infection (Park et al. 2017). To identify novel host factors for CVB3 infection, we chose three groups of genes encoding 469 kinases, 310 proteins related to endocytosis, and 735 Golgi-localized proteins, which are potential host factors for viral infection. Using a library of individually synthesized oligonucleotides, which encode sgRNAs targeting the human genome that we have described previously (Kim et al. 2017), we prepared an arrayed sgRNA plasmid library in the 96-well format consisting of three target sites per gene for 1514 genes. We carefully designed those target sites to be unique in the human genome and to have a high microhomology score to avoid in-frame mutations as much as possible (Bae et al. 2014a, b). To further reduce off-target effects, we added two extra guanine nucleotides to produce ggX20 sgRNAs (Cho et al. 2014; Kim et al. 2015) and carefully designed three sgRNA target sites that differ from any other site in the human genome by at least 3 nucleotides (nt), which is considered a more important criterion than the microhomology score. To examine the activity of this library, we investigated the gene disruption rate after transfecting sgRNAs targeting 35 genes. Most of the genes were knocked out efficiently, implying that this library is useful and reliable. The average mutation rate for 35 genes was quite high at 72.8% (Fig. 1C). The average out-of-frame ratio was 76.3%.
Figure 1.

Plasmid-based sgRNA constructs as a tool for a genetic perturbation screen in human cells. (A,B) HeLa cells were transfected with sgRNA-expressing plasmids (Control, CXADR, and PI4KB) and infected with CVB3 at an MOI of 5. At 8 h post-infection, cells were fixed and stained with anti-3C antibody (green) and DAPI (blue). (A) Quantification of infection. Mean ± SEM for quadruplicate experiments. (**) P < 0.01. (B) Representative images of infection are shown. (C) Mutation frequencies determined after transient transfection of 35 sgRNAs and Cas9 plasmid.
We performed an arrayed CRISPR screen based on the plasmid sgRNA library for 1514 genes in duplicate (Fig. 2A). We calculated the Z score for CVB3 infection rate reduction for each plate and identified the candidate genes with Z < −1.8 in the duplicate screens (Supplemental Fig. S2A; yellow box indicates the region for candidate genes). From the primary screens, we chose 10 genes as candidates (Supplemental Fig. S2B). To confirm these results, the same sgRNAs used in the screen were transfected again into HeLa cells along with Cas9 and then the infection rate was measured (Fig. 2B). Most of the genes, except two from the primary screens, were confirmed by reduced infectivity. To further verify this result, we transfected newly designed sgRNA sets for each gene and found that CVB3 infection was significantly inhibited (Fig. 2C). Of the 10 genes identified in the primary screen, eight were confirmed in the secondary screens, which are derived by experimental and statistical errors in arrayed screen. This result shows that the arrayed CRISPR screen is quite reliable and reproducible.
Figure 2.

Candidate identification. (A) Schematic presentation of the arrayed CRISPR screen. (B) Quantification of CVB3 infection in HeLa cells transfected with sgRNA-expressing plasmids targeting 10 genes from primary screens. Mean ± SEM for quadruplicate experiments. (**) P < 0.01, (n.s.) nonsignificant. (C) Similar quantification of virus infection in B using newly designed sgRNA. (D) The percentage of CVB3-infected cells (normalized to DMSO control) treated with three known inhibitors of the candidates. Mean ± SD for triplicate experiments. (E) HeLa cells were transfected with ACBD3 and CSDE1 targeting sgRNAs and infected with CVB3. Surviving cells were stained and are shown. Surviving colonies were expanded and the target region was sequenced to confirm the mutations. Red characters indicate the PAM and blue characters indicate insertions. hg19 is the wild-type sequence of each gene. As our target sites have the same sequence in both references (i.e., hg19 and GRCh38), using GRCh38 would not significantly affect our conclusions.
To directly compare arrayed and pooled approaches, we next investigated whether these eight genes could be identified using a pooled library of 4542 sgRNAs targeted to the same 1514 genes. This focused, pooled library was transduced into HeLa cells at 0.3 MOI. Two weeks later, these cells were subjected to CVB3 infection. Targeted deep sequencing revealed that only one gene, CXADR, was identified as a strong candidate (Supplemental Fig. S3). Note that we identified the same gene using the GeCKOv2 library (Supplemental Fig. S1).
DNM2, FASN, OSBP, and SACM1L are known to be important for enterovirus infection
It has been previously reported that some candidates are required for coxsackievirus infection. Dynamin 2 protein encoded by DNM2 is required for CVB3 entry (Patel et al. 2009). Fatty acid synthase encoded by FASN is required for CVB3 replication (Wilsky et al. 2012). OSBP and SACM1L are required for sterol/PI(4)P exchange at the ER-Golgi interface, and rhinovirus and enterovirus, which belong to Picornaviridae like CVB3, use these processes for viral replication (Mesmin et al. 2013; Roulin et al. 2014; Strating et al. 2015). Our screening results are in agreement with these previous reports and give us confidence in our approach. The proteins encoded by DNM2, FASN, and OSBP have known chemical inhibitors: Dynasore, C75, and Itraconazole (ITZ), respectively. As expected, CVB3 infection was dramatically reduced by these chemicals (Fig. 2D). In contrast, cell viability was not affected at the same concentrations (Supplemental Fig. S4).
sgRNAs against CSDE1 and ACBD3 make cells resistant to CVB3
Unlike siRNAs, sgRNAs coupled with Cas9 induce permanent gene knockout at the target sites. We examined whether knocking out the candidate genes could make cells completely resistant to CVB3 infection. Genes that make cells resistant to virus after being knocked out could be important antiviral drug targets because these host factors are absolutely required for virus infection but are not essential for cell viability and proliferation. sgRNAs targeting the cold shock domain containing E1 (CSDE1) and the acyl-CoA binding domain containing 3 (ACBD3) were found to induce CVB3-resistance without preventing cell proliferation (Fig. 2E). As expected, these resistant colonies had mutations at the CSDE1 and ACBD3 target sites (Fig. 2E). However, sgRNAs targeting other genes like DNM2, FASN, and OSBP, although proved to be important host factors by using chemical inhibitors (Fig. 2D), could not generate CVB3-resistant clones (Supplemental Fig. S5). We also tried to isolate KO clones for these genes, but we could not obtain them. This suggests that they are essential factors for cell proliferation, as previously reported (Blomen et al. 2015). Accordingly, we chose CSDE1 and ACBD3 for further study among the candidates.
CSDE1 is required for CVB3 infection, especially in IRES-dependent translation
We isolated a CSDE1 KO cell line for functional study and found that these cells were resistant to CVB3 infection at various MOIs (Fig. 3A). Moreover, they did not permit viral replication, as shown by the CVB3 replicon assay expressing the luciferase reporter in place of the P1 structural region and an immunostaining assay (Fig. 3B; Supplemental Fig. S6). Western blot analysis also showed the absence of CSDE1 protein (Fig. 3C). CVB3 infection and replication was rescued when we transfected the eGFP-CSDE1 plasmid into CSDE1 KO cells (Fig. 3B,C). These results suggest that CSDE1 is a key host factor for CVB3 infection. CSDE1 was previously known to be required for internal initiation of translation of the human rhinovirus internal ribosome entry site (IRES) (Hunt et al. 1999; Anderson et al. 2007). As human rhinovirus and CVB3 both belong to the Enterovirus genus and share similar genomic structure, we hypothesized that CSDE1 might be related to translation initiation directed by the CVB3 IRES as well. A dual luciferase reporter system, which encodes firefly luciferase under the IRES sequence of CVB3 and constitutively expresses Renilla luciferase upstream of them, was used to confirm this hypothesis (Paek et al. 2008; Kang et al. 2015). CVB3 IRES sequences could not initiate translation in CSDE1-depleted cells as efficiently as in wild-type cells (Fig. 3D). To further investigate whether CSDE1 is required for IRES-dependent translation in other viruses, a similar dual luciferase reporter system for four different viruses, Poliovirus 1 (PV1), Enterovirus 71 (EV71), Hepatitis C virus (HCV), and Encephalomyocarditis virus (EMCV), was used (Paek et al. 2008; Kang et al. 2015). Concerning the four IRES sequence origins, PV1 and EV71 belong to the same Enterovirus genus. IRES sequences from these could not initiate translation in CSDE1-depleted cells (Fig. 3D). In contrast, the other two IRES sequences (HCV and EMCV) could initiate high-level translation in both cells, indicating that CSDE1 is an important factor for IRES-dependent translation, especially in the human Enterovirus genus. These results suggest that CSDE1 could be a valuable target for development of broad-spectrum antivirals against diverse enteroviruses that cause numerous diseases in humans.
Figure 3.

CSDE1 is required for CVB3 infection. (A) Viability of wild-type (HeLa) and CSDE1 knockout (KO) cells after CVB3 infection. (B) Viral replication test of wild-type (HeLa) and CSDE1 KO cells using the CVB3 replicon. Transfection with cDNA encoding the eGFP-CSDE1 protein into CSDE1 KO cells rescued CVB3 replication. (C) Transfection with cDNA encoding the eGFP-CSDE1 protein into CSDE1 KO cells rescued CVB3 infection. After 8 h of CVB3 infection in wild-type (HeLa) and CSDE1 KO cells transfected with the plasmid encoding the eGFP-CSDE1 protein, cells were harvested and lysed for western blot analysis using anti-CSDE1 antibody, anti-VP1 antibody, and anti-beta actin antibody. (D) CVB3 IRES-dependent translation assay of wild-type (HeLa) and CSDE1 KO cells. Dual luciferase reporter plasmids containing the IRES sequence from five different viruses were transfected into cells and the activity of firefly luciferase and Renilla luciferase was measured.
ACBD3 is required for CVB3 infection but influences cell growth minimally
Another candidate, ACBD3, was also interesting because some groups have reported that ACBD3 is dispensable for PV1 and CVB3 replication through siRNA knockdown (Teoule et al. 2013; Dorobantu et al. 2014). In contrast, we could obtain CVB3-resistant cells after sgRNA transfection. To further investigate the role of ACBD3 in CVB3 infection, ACBD3 KO clones were isolated. ACBD3 knockout clones were resistant to CVB3 and blocked CVB3 replication completely, as shown by the CVB3 replicon assay (Fig. 4A,B). Western blotting and an immunostaining assay also showed that viral protein expression was significantly inhibited in these cells (Fig. 4C; Supplemental Fig. S7). Ectopic expression of FLAG-tagged ACBD3 protein rescued CVB infection, replication, and viral protein expression (Fig. 4B,C; Supplemental Fig. S8). Sasaki et al. (2012) reported that the Aichi virus of the Picornaviridae family recruits ACBD3 and PI4KB to RNA replication sites and the viral protein 3A interacts with ACBD3 for recruitment. Lei et al. (2017) also reported that ACBD3 facilitates enterovirus 71 replication by interacting with the 3A protein. These reports, combined with our results, suggest that ACBD3 is broadly required for human enterovirus replication. To confirm the interaction between ACBD3 and the human enterovirus 3A protein, we conducted a simple luciferase assay with a mammalian two-hybrid system. Both 3A proteins from CVB3 and PV1 fused with the DNA-binding domain increased luciferase expression only in combination with the ACBD3 protein fused with the transcription activation domain (Fig. 4D). These results indicate that the CVB3 3A protein interacts with ACBD3 and CVB3 uses ACBD3 as an essential host factor similar to that in Aichi virus or enterovirus 71. As ACBD3 is broadly required for enteroviruses infection, this protein might be valuable target for the development of broad spectrum antiviral inhibitors. It is necessary that this host factor be essential for the virus but have a minimal effect on cell proliferation. To determine whether knocking out this protein could influence cell growth, we compared the growth of ACBD3 KO clones with control HeLa cells (Fig. 4E). ACBD3 KO clones showed growth patterns similar to those of HeLa cells. These results suggest that ACBD3 is required for CVB3 infection but minimally influences cellular proliferation.
Figure 4.

ACBD3 is required for CVB3 replication. (A) Viability of wild-type (HeLa) and ACBD3 knockout (KO) cells after CVB3 infection. (B) Viral replication assay of wild-type (HeLa) and ACBD3 KO cells using the CVB3 replicon. Transfection with cDNA encoding FLAG-tagged ACBD3 protein into ACBD3 KO cells rescued CVB3 replication, as shown by a viral replication assay using the CVB3 replicon. (C) Wild-type HeLa cells and ACBD3 KO cells transfected with control and plasmid encoding FLAG-tagged ACBD3 protein were infected with CVB3 at an MOI of 5. At 8 h post-infection, cells were fixed and stained with anti-3C antibody (green) and DAPI (blue). (D) Interaction between ACBD3 and the 3A protein of PV1 and CVB3 was confirmed using a mammalian two-hybrid system. (E) A fixed number of HeLa cells and KO cells were plated into six-well plates and incubated. On each day, live cells were counted and normalized to the initial number of cells.
Arrayed CRISPR screen reliably identifies host factors difficult to find using siRNAs
Since siRNA is the most widely used tool in arrayed screens, we tried to compare it with CRISPR. To do so, we performed CVB3 infection in cells transfected with siRNAs or sgRNAs targeting CSDE1 and ACBD3. Both the knockdown by siRNAs and knockout by sgRNAs of CSDE1 showed reduction in CVB3 infection (Fig. 5A). However, knockdown of ACBD3 could not restrict CVB3 infection in contrast to ACBD3 knockout (Fig. 5A). Moreover, ACBD3 knockdown failed to make HeLa cells resistant to infection and block viral replication (Supplemental Fig. S9), even though knockdown efficiency of siRNA was high for both genes (Fig. 5B,C). This indicates that even a trace amount of ACBD3 protein could be sufficient for the virus, explaining why this gene was disputed in the previous studies using siRNAs. Another candidate, RACK1, also showed a pattern similar to ACBD3. Both siRNA and sgRNA against RACK1 showed good reduction of protein levels (Supplemental Fig. S10B). CVB3 infection was significantly inhibited by sgRNA but was modestly inhibited by siRNA (Supplemental Fig. S10A). This suggests that screening with sgRNAs could reduce the false-negatives induced by the incomplete knockdown events with siRNAs.
Figure 5.
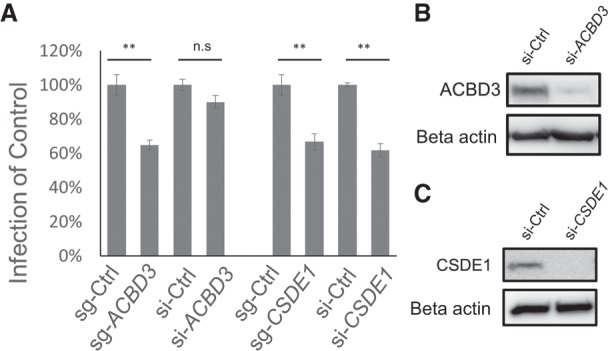
HeLa cells were transiently transfected with siRNAs and sgRNAs targeting ACBD3 or CSDE1. (A) Cells were infected with CVB3, fixed, and stained. The percentage of CVB3-infected cells (normalized to sgRNA or siRNA control). Mean ± SEM for triplicate experiments. (**) P < 0.001, (n.s.) nonsignificant. (B,C) Cells were transfected with control siRNA, ACBD3 siRNA, and CSDE1 siRNA. The expression level of each protein was measured by western blot using the anti-ACBD3 antibody (B) and anti-CSDE1 antibody (C).
Discussion
In this study, we report a novel, high-throughput arrayed CRISPR screen method carried out with an image-based assay. Based on the arrayed plasmid library expressing sgRNA, we extended the library to cover 1514 genes and successfully identified host factors required for CVB3 infection. Compared to pooled screens, arrayed screens can be more expensive and may require automation. However, with arrayed screens, higher hit rates and lower false-positive/-negative ratios are expected. Furthermore, arrayed screens can have broader screen phenotypes because each well has high individual perturbations compared to very low mixed perturbation in the pooled screen (<0.01%). For pooled screens, especially for highly cytopathic viruses like CVB3, a high dose of virus could inhibit survival of virus-resistant cells, which exist in a very small proportion. We could not obtain any hits using a high MOI for the pooled screen with the GeCKOv2 library. Furthermore, as we had to enrich the surviving cells for an additional 14 d post-infection, genes essential for cell proliferation could be missed because of this longer time course. However, in the arrayed screens, we could use a higher MOI, such as 5, and a short time course, such as 8 h, because of the very high proportion of virus-resistant cells (∼80%) in each well. This arrayed screen can be preferred for assays requiring a high dose and short time course, like early entry of viral particles. Notably, numerous arrayed siRNA screens have been performed in many laboratories and automation facilities for siRNA screening have been developed in many research centers. Our arrayed screens are compatible with these screening platforms. Currently, most CRIPSR screens are conducted in a pooled format, using the lentiviral system to identify viral host factors (Ma et al. 2015; Marceau et al. 2016; Savidis et al. 2016; Zhang et al. 2016; Kim et al. 2017; Park et al. 2017). Although these screens have been used successfully, only a few significant factors were selected, even though a genome-wide sgRNA library covering over 10,000 genes was used. In fact, in a previous pooled CRISPR screen that we performed, we were able to identify only a single host factor for enterovirus 68 in the initial screening (Kim et al. 2017). A strong effector in the pooled population could mask other genes that have milder effects. In contrast, we showed that genes with subtle effects or growth disadvantages, which make them difficult to be revealed in a pooled screen, can be identified using the present method when compared with the result of pooled screen as shown in Supplemental Figures S1 and S3. Viral infection is a complex process that involves diverse steps such as receptor binding, endocytosis, translation, replication, morphogenesis, and egress, and each step exploits a variety of host factors required for the process. In our screens, we could identify various host factors for CVB3, including the entry-related factor, the translational initiation factor of viral RNA, and several replication factors with different functions because many physically distinct cells are generated in each well by the knockout of each gene. Furthermore, host factor genes such as DNM2, FASN, and OSBP, which are also important for cell proliferation, could be identified owing to the short assay time in contrast to pooled screens.
There are new CRISPR screens that combine a pooled CRISPR screen and single cell RNA-seq (Dixit et al. 2016; Jaitin et al. 2016; Datlinger et al. 2017). Their major objective is to identify comprehensive effects induced by a single guide RNA, rather than uncovering a specific target gene among a few thousand candidate genes. Those two methods are very valuable when researchers want to discover the effects of gene knockouts at the transcriptome level. However, the methods would be rather inefficient when the goal is to uncover a target gene among thousands of genes. The methods would be more suitable if the aim was to analyze the effect of viral host genes (e.g., ACBD3 or CSDE1) after viral infection at a single-cell level.
So far, arrayed screening formats have mostly relied on siRNA libraries. Arrayed siRNA screens and sgRNA screens were performed with some minor differences. siRNAs were spotted in a multiwell plate and reverse transfected into cells using liposomes. At 48 or 72 h post-transfection, cells were infected by virus and the viral infection was measured using various methods. For sgRNA screens, at 5 d after transfection, cells were infected by virus and the viral infection was measured using 3C-antibody staining and image acquisition with the Operetta system. Although those siRNA screens have advantages over pooled screens, disadvantages of siRNAs sometimes discourage their use in large-scale screens. The biggest issue may be “low reproducibility” across screens, which is promoted by their off-target effects. Target mRNAs can be repressed only by partial matching with the siRNA seed sequence, in contrast to the CRISPR system which requires a near perfect match, not only in the seed sequence. We analyzed off-target effects using sgRNAs specific to ACBD3 and CSDE1 with next-generation sequencing: No off-target mutations were detected at sites that differ by up to 3 nt (Supplemental Fig. S11). Here, our arrayed CRISPR screen identified 10 candidate genes, of which eight genes were confirmed by a secondary screen (Fig. 2C). This high rate (80%) of confirmation by secondary screens and only two false-positives among 1514 genes suggests that this screen was reproducible and reliable and could be useful for large-scale screens. As shown in Figure 5, incomplete suppression of target genes can result in false-negatives during screening. ACBD3, which is essential even in a trace amount of expression, could not be revealed by the traditional siRNA screening, thus demonstrating the value of this screen.
The arrayed CRISPR screen requires different considerations from those of pooled screens. We chose a plasmid-based library, expressing sgRNAs in multiwell plates. Compared with other libraries based on individual lentiviruses or synthetic crRNA (Metzakopian et al. 2017), including commercial sources, our plasmid-based library is scalable. This library was cotransfected with a plasmid expressing Cas9 using lipid-based reagents. As shown in Figure 1C, most genes were knocked out efficiently in HeLa cells within 5 d, demonstrating the strength of the plasmid-based system. However, some cells are difficult to transfect using liposomes. These problems could be solved by electroporation, especially using an electroporator equipped with multiwell modules, as reported by Hultquist et al. (2016). We validated the genes using original and newly designed sgRNAs. Moreover, we validated some candidate genes using known chemical inhibitors (Fig. 2D), analysis of protein expression levels by western blotting (Fig. 3), and rescue of the phenotype by cDNA expression (Figs. 3, 4). Finally, we found clues to how ACBD3 and CSDE1 affect CVB3 infection. Additionally, live-imaging based on GFP-expressing virus could be optimized in future studies.
We identified two host factors, CSDE1 and ACBD3, which are crucial for viral infection. Unlike DNM2, FASN, and OSBP, which are also crucial for cell proliferation, these knocked-out cells could proliferate well to produce resistant colonies (Fig. 2) and completely block viral infection (Figs. 3, 4). We found that three viruses belonging to the human enteroviruses require CSDE1 for RNA translation. Other reports have described that human rhinovirus also uses CSDE1 for IRES-dependent translation (Hunt et al. 1999; Boussadia et al. 2003). Staring et al. (2017) reported that CSDE1 was selected as one of the host factors for poliovirus using haploid genetic screen. These results suggest that CSDE1 is a universal factor for translation initiation in human enteroviruses. Stone et al. (2008) reported that morpholino oligomers targeting IRES inhibited multiple species of picornaviruses. Using a similar approach targeting the IRES region, which interacts with CSDE1, potential broad therapeutic inhibitors against diverse human enteroviruses could be developed. Concerning ACBD3, several reports suggested that human enteroviruses use this factor for replication. In this study, we added that ACBD3 minimally affects cellular proliferation even after complete knockout. Seven acyl-CoA-binding protein domain-containing proteins (ACBD) including ACBD3 have been identified (Fan et al. 2010). Only ACBD3 possesses the GOLD domain at its C terminus, and this domain mediates interaction with the Aichi virus 3A protein (McPhail et al. 2017). The 3A proteins of diverse enteroviruses interact with ACBD3, and inhibitors that block this interaction could be potential broad antiviral therapeutics. In summary, we demonstrate that these two proteins could be interesting targets to develop anti-enteroviral therapeutics.
The two remaining host factors identified in this screen, ADCY8 and RACK1, could also be interesting targets for further study. Coyne et al. (2011) reported that several ADCY family proteins, ADCY1, ADCY4, ADCY6, and ADCY7, were required for CVB3 replication. In our screen, we identified ADCY8 as a host factor for CVB3. This may be due to the fact that different cells were used for the screens or due to the difference in the efficacy of siRNA or sgRNA targeting the ADCY family. This suggests that the two screening methods could be complementary to each other. It has been reported that RACK1 controls IRES-mediated translation of viruses, deactivates IRF3, and limits type I interferon signaling (Long et al. 2014; Majzoub et al. 2014). It is possible that these two functions could be required for CVB3 infection. Determining the molecular details of RACK1 in CVB3 infection could also be intriguing for further study.
Methods
sgRNA oligonucleotide preparation
Oligonucleotides were purchased from Bioneer. Two complementary 24-nt oligonucleotides were mixed at 100 µM in each well in a 96-well plate. Each oligonucleotide pair was diluted to 50 µM in TES buffer (10 mM Tris-HCl [pH7.5], 1 mM EDTA, and 100 mM NaCl), and annealed to form a duplex in a water bath by heating to 80°C and cooling to room temperature.
sgRNA array construction
An empty sgRNA expression vector cleaved using BsaI (New England BioLabs) was ligated with annealed oligonucleotide mixtures (three oligonucleotide pairs for each gene). Ligation products were transformed into DH5α competent cells. Transformed E. coli cells were plated on a 10-cm LB plate with ampicillin and plasmids are purified from them.
Pooled library screen and analysis of target genes
Human GeCKOv2 CRISPR knockout pooled library was a gift from Feng Zhang (Addgene #1000000048) (Sanjana et al. 2014). The oligo pool for the focused library was purchased from CustomArray, Inc. The single-stranded oligos were amplified to double-stranded DNA by PCR and assembled to a lentiviral sgRNA plasmid using NEBuilder HiFi DNA Assembly Master Mix (New England BioLabs); 3 × 107 cells were transduced with a lentiviral sgRNA library at an MOI of 0.3. After 24 h, cells were selected with puromycin. Those cells were cultured for ∼2 wk, and 3 × 107 cells were infected with CVB3 (MOI 0.00001). Genomic DNA was extracted from the initial cell population and surviving cells from CVB3 infected populations. The region containing the gRNA was amplified with primers F-TCTTGTGGAAAGGACGAAACACCG and R-TCTACTATTCTTTCCCCTGCACTGT, using 10 µg of genomic DNA, and sequenced using an Illumina HiSeq 4000 for 100× coverage. After sequencing, reads were aligned to the sgRNA library and counted. Those sgRNA counts are analyzed by MAGeCK version 0.5.5 according to the instructions (Li et al. 2014).
Cell culture and transfection conditions
HeLa (ATCC, CCL-2) cells were maintained in Dulbecco's Modified Eagle's Medium (DMEM) supplemented with 10% fetal bovine serum (FBS). Using 96-well plates (PerkinElmer, #6005550), 25 ng of arrayed sgRNAs and 25 ng of Cas9 expression plasmids were reversely transfected to 5 × 103 HeLa cells using Lipofectamine 2000 in each well. At 5 d post-transfection, cells were used for CVB3 infection. Single-cell-derived knockout cell lines were obtained by limiting dilution.
Virus and plasmids
Coxsackievirus B3 (Nancy; ATCC, VR-30) was expanded by growth in HeLa cells and titered using HeLa cells. The pLuCVB3 plasmid, which contains a coxsackievirus B3 subgenomic replicon carrying the firefly luciferase gene, was provided by Eun-Seok Jeon (Samsung Medical Center, Seoul, Korea) (Lim et al. 2012). A CSDE1-expressing plasmid was purchased form Vigene Bioscience (CH842097) and a plasmid encoding eGFP-CSDE1 was constructed using the eGFP-N1 plasmid (Clontech Laboratories). pCI-FLAG–ACBD3, which contains a FLAG-tagged ACBD3 gene, was obtained from Jun Sasaki (Fujita Health University, Aichi, Japan) (Sasaki et al. 2012). The CheckMate mammalian two-hybrid system was purchased from Promega Corporation. Dual luciferase reporter plasmids containing viral IRES were a gift from Sung Key Jang (POSTECH, Pohang, Korea).
Antibodies and chemicals
Anti-CSDE1 rabbit antibody was obtained from Bethyl Laboratories (catalog no. A303-159A). ACBD3-specific rabbit antibody (catalog no. HPA015594), murine anti-FLAG antibody (catalog no. F3165), and beta actin murine monoclonal antibody (catalog no. A1978) were purchased from Sigma-Aldrich. Murine monoclonal anti-VP1 antibody was obtained from Leica (NCL-ENTERO). Anti-RACK1 mouse antibody was purchased from BD Biosciences (catalog no. 610177). Rabbit polyclonal anti-3C antibody was generated by immunization with recombinant 3C protein. Secondary antibodies conjugated to horseradish peroxidase were purchased from Thermo Fisher Scientific (catalog no. 31466, 31430). Alexa Fluor 488-conjugated goat anti-rabbit antibody was obtained from Life Technologies (catalog no. A11008). Chemical inhibitor Dynasore was purchased from Merck Millipore, and C75 and itraconazole were purchased from Sigma-Aldrich.
Virus infection and screening
Cells transfected with the Cas9 plasmid and sgRNA library in a 96-well plate were infected with coxsackievirus B3 at an MOI of 5 at 37°C for 8 h. Infected cells were fixed and permeabilized with a 3:1 mixture of ice-cold methanol-acetone. Cells were incubated with anti-3C antibody and anti-rabbit secondary antibody conjugated to Alexa Fluor 488 to detect infected cells, and then counterstained with 4′,6-diamidino-2-phenylindole (DAPI) (Thermo Fisher Scientific, #62248). Images were captured with the Operetta system (PerkinElmer). The number of infected cells and the total number of nuclei were quantified using Harmony software installed on the Operetta system. The Z score for CVB3 infection was calculated as previously described (Yasunaga et al. 2014). Candidates genes were selected which are Z < −1.8 in duplicate screens.
Reinfection test and replicon assay
Cells were plated to 96-well plates at 2 × 104 cells per well to test whether knockout cells were resistant to viral infection and whether replication of the CVB subgenomic replicon was inhibited in those cells. For the reinfection assay, 10-fold diluted CVB (MOI 0.1 ∼ 0.0001) was added to each well and the cultures were incubated at 37°C for 2 d. To measure cell viability after virus infection, a modified 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT; Sigma-Aldrich) assay was performed as previously described (Kim et al. 2002). The viability of infected cells was normalized to the viability of mock-infected cells (expressed as 100%). For the replicon assay, the pLuCVB3 plasmid was linearized with ClaI (New England BioLabs) and used as a template for in vitro RNA transcription performed with a MEGAscript T7 kit (Ambion) as suggested by the manufacturer's protocol. The in vitro transcribed replicon RNA was purified using the TRIzol LS reagent (Invitrogen). Transfection was performed with 50 ng of replicon RNA per well with the Lipofectamine 2000 (Thermo Fisher Scientific) reagent. At 4 h post-transfection, cells were washed, resuspended in complete growth medium, and incubated at 37°C or lysed in 20 µL of lysis buffer (0 h). At indicated time points, cells were washed and lysed in lysis buffer. Luciferase activity was measured using a luciferase assay kit (Promega Corporation) according to the manufacturer's protocol and a luminometer (LB960 centro XS3, Berthold Technologies). Luciferase activity was expressed in fold increase as compared with the activity measured at 0 h.
RNA interference
CSDE1 siRNA (L-015834-00) and RACK1 siRNA (L-006876-00) were obtained from Dharmacon. Control and human ACBD3 siRNAs have been described previously (Dorobantu et al. 2014). siRNAs were used at a concentration of 20 nM. Cells were transfected with siRNAs in six-well plates using Lipofectamine 2000 (Thermo Fisher Scientific). After 24 h, cells were trypsinized and replated in 96-well or 12-well plates, and virus infection or replicon RNA transfection was performed 2 d later. Depletion of the target proteins was confirmed by immunoblot analysis. The blots were developed using a LAS 4000 imager (Fujifilm).
IRES activity test
IRES activity was measured as previously described with minor modification (Kang et al. 2015). Wild-type (HeLa) and CSDE1 knockout cells were plated into 96-well plate and transfected with reporter plasmids using Lipofectamine 2000. Forty-eight hours post-transfection, firefly and Renilla luciferase activities were measured.
Isolation of CVB3-resistant colonies
HeLa cells transduced with the GeCKO library or transfected with Cas9 plasmid and candidate sgRNAs were infected with CVB3. Infected cells were washed and changed to complete medium. The culture medium was changed every 2 or 3 d. After 14 d, surviving colonies were fixed and stained by crystal violet (0.05%) in phosphate buffered saline (PBS) solution containing 1% formaldehyde and 25% methanol or isolated and expanded for further analysis.
Data access
The deep sequencing data from this study have been submitted to the NCBI Sequence Read Archive (SRA; https://www.ncbi.nlm.nih.gov/sra) under accession number SRP117996. The plasmids used here have been deposited at Addgene (#110724).
Supplementary Material
Acknowledgments
This research was supported by grants from IBS (IBS-R021-D1) to J.-S.K., from the Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Science, ICT & Future Planning (2016R1C1B2009585), and from the National Research Council of Science & Technology (NST) grant from the Korea government (MSIP) (no. CRC-16-01-KRICT) to C.K.
Author contributions: H.S.K., K.L., S.-J.K., S.C., H.J.S., and C.K. performed the experiments. H.S.K. performed bioinformatics analyses. J.-S.K. and C.K. supervised the research.
Footnotes
[Supplemental material is available for this article.]
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.230250.117.
Freely available online through the Genome Research Open Access option.
References
- Anderson EC, Hunt SL, Jackson RJ. 2007. Internal initiation of translation from the human rhinovirus-2 internal ribosome entry site requires the binding of Unr to two distinct sites on the 5′ untranslated region. J Gen Virol 88: 3043–3052. [DOI] [PubMed] [Google Scholar]
- Bae S, Kweon J, Kim HS, Kim JS. 2014a. Microhomology-based choice of Cas9 nuclease target sites. Nat Methods 11: 705–706. [DOI] [PubMed] [Google Scholar]
- Bae S, Park J, Kim JS. 2014b. Cas-OFFinder: a fast and versatile algorithm that searches for potential off-target sites of Cas9 RNA-guided endonucleases. Bioinformatics 30: 1473–1475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergelson JM, Cunningham JA, Droguett G, Kurt-Jones EA, Krithivas A, Hong JS, Horwitz MS, Crowell RL, Finberg RW. 1997. Isolation of a common receptor for Coxsackie B viruses and adenoviruses 2 and 5. Science 275: 1320–1323. [DOI] [PubMed] [Google Scholar]
- Blomen VA, Majek P, Jae LT, Bigenzahn JW, Nieuwenhuis J, Staring J, Sacco R, van Diemen FR, Olk N, Stukalov A, et al. 2015. Gene essentiality and synthetic lethality in haploid human cells. Science 350: 1092–1096. [DOI] [PubMed] [Google Scholar]
- Boussadia O, Niepmann M, Creancier L, Prats AC, Dautry F, Jacquemin-Sablon H. 2003. Unr is required in vivo for efficient initiation of translation from the internal ribosome entry sites of both rhinovirus and poliovirus. J Virol 77: 3353–3359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buehler E, Chen YC, Martin S. 2012. C911: a bench-level control for sequence specific siRNA off-target effects. PLoS One 7: e51942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho SW, Kim S, Kim Y, Kweon J, Kim HS, Bae S, Kim JS. 2014. Analysis of off-target effects of CRISPR/Cas-derived RNA-guided endonucleases and nickases. Genome Res 24: 132–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coyne CB, Bergelson JM. 2006. Virus-induced Abl and Fyn kinase signals permit coxsackievirus entry through epithelial tight junctions. Cell 124: 119–131. [DOI] [PubMed] [Google Scholar]
- Coyne CB, Bozym R, Morosky SA, Hanna SL, Mukherjee A, Tudor M, Kim KS, Cherry S. 2011. Comparative RNAi screening reveals host factors involved in enterovirus infection of polarized endothelial monolayers. Cell Host Microbe 9: 70–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Datlinger P, Rendeiro AF, Schmidl C, Krausgruber T, Traxler P, Klughammer J, Schuster LC, Kuchler A, Alpar D, Bock C. 2017. Pooled CRISPR screening with single-cell transcriptome readout. Nat Methods 14: 297–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dixit A, Parnas O, Li B, Chen J, Fulco CP, Jerby-Arnon L, Marjanovic ND, Dionne D, Burks T, Raychowdhury R, et al. 2016. Perturb-Seq: dissecting molecular circuits with scalable single-cell RNA profiling of pooled genetic screens. Cell 167: 1853–1866.e17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dorobantu CM, van der Schaar HM, Ford LA, Strating JR, Ulferts R, Fang Y, Belov G, van Kuppeveld FJ. 2014. Recruitment of PI4KIIIβ to coxsackievirus B3 replication organelles is independent of ACBD3, GBF1, and Arf1. J Virol 88: 2725–2736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan J, Liu J, Culty M, Papadopoulos V. 2010. Acyl-coenzyme A binding domain containing 3 (ACBD3; PAP7; GCP60): an emerging signaling molecule. Prog Lipid Res 49: 218–234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fields BN, Knipe DM, Howley PM. 2007. Fields’ virology. Wolters Kluwer Health/Lippincott Williams & Wilkins, Philadelphia, PA. [Google Scholar]
- Fu Y, Sander JD, Reyon D, Cascio VM, Joung JK. 2014. Improving CRISPR-Cas nuclease specificity using truncated guide RNAs. Nat Biotechnol 32: 279–284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilbert LA, Horlbeck MA, Adamson B, Villalta JE, Chen Y, Whitehead EH, Guimaraes C, Panning B, Ploegh HL, Bassik MC, et al. 2014. Genome-scale CRISPR-mediated control of gene repression and activation. Cell 159: 647–661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu NY, Ilnytska O, Belov G, Santiana M, Chen YH, Takvorian PM, Pau C, van der Schaar H, Kaushik-Basu N, Balla T, et al. 2010. Viral reorganization of the secretory pathway generates distinct organelles for RNA replication. Cell 141: 799–811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hultquist JF, Schumann K, Woo JM, Manganaro L, McGregor MJ, Doudna J, Simon V, Krogan NJ, Marson A. 2016. A Cas9 ribonucleoprotein platform for functional genetic studies of HIV-host interactions in primary human T cells. Cell Rep 17: 1438–1452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunt SL, Hsuan JJ, Totty N, Jackson RJ. 1999. unr, a cellular cytoplasmic RNA-binding protein with five cold-shock domains, is required for internal initiation of translation of human rhinovirus RNA. Genes Dev 13: 437–448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaitin DA, Weiner A, Yofe I, Lara-Astiaso D, Keren-Shaul H, David E, Salame TM, Tanay A, van Oudenaarden A, Amit I. 2016. Dissecting immune circuits by linking CRISPR-pooled screens with single-cell RNA-seq. Cell 167: 1883–1896.e15. [DOI] [PubMed] [Google Scholar]
- Kaelin WG Jr. 2012. Molecular biology. Use and abuse of RNAi to study mammalian gene function. Science 337: 421–422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang H, Kim C, Kim DE, Song JH, Choi M, Choi K, Kang M, Lee K, Kim HS, Shin JS, et al. 2015. Synergistic antiviral activity of gemcitabine and ribavirin against enteroviruses. Antiviral Res 124: 1–10. [DOI] [PubMed] [Google Scholar]
- Kim JH, Park JB, Bae PK, Kim HS, Kim DW, Ahn JK, Lee CK. 2002. Establishment and use of a cell line expressing HSV-1 thymidine kinase to characterize viral thymidine kinase-dependent drug-resistance. Antiviral Res 54: 163–174. [DOI] [PubMed] [Google Scholar]
- Kim D, Bae S, Park J, Kim E, Kim S, Yu HR, Hwang J, Kim JI, Kim JS. 2015. Digenome-seq: genome-wide profiling of CRISPR-Cas9 off-target effects in human cells. Nat Methods 12: 237–243. [DOI] [PubMed] [Google Scholar]
- Kim HS, Lee K, Bae S, Park J, Lee CK, Kim M, Kim E, Kim M, Kim S, Kim C, et al. 2017. CRISPR/Cas9-mediated gene knockout screens and target identification via whole-genome sequencing uncover host genes required for picornavirus infection. J Biol Chem 292: 10664–10671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koike-Yusa H, Li Y, Tan EP, Velasco-Herrera MDC, Yusa K. 2014. Genome-wide recessive genetic screening in mammalian cells with a lentiviral CRISPR-guide RNA library. Nat Biotechnol 32: 267–273. [DOI] [PubMed] [Google Scholar]
- Lanke KH, van der Schaar HM, Belov GA, Feng Q, Duijsings D, Jackson CL, Ehrenfeld E, van Kuppeveld FJ. 2009. GBF1, a guanine nucleotide exchange factor for Arf, is crucial for coxsackievirus B3 RNA replication. J Virol 83: 11940–11949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lei X, Xiao X, Zhang Z, Ma Y, Qi J, Wu C, Xiao Y, Zhou Z, He B, Wang J. 2017. The Golgi protein ACBD3 facilitates Enterovirus 71 replication by interacting with 3A. Sci Rep 7: 44592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W, Xu H, Xiao T, Cong L, Love MI, Zhang F, Irizarry RA, Liu JS, Brown M, Liu XS. 2014. MAGeCK enables robust identification of essential genes from genome-scale CRISPR/Cas9 knockout screens. Genome Biol 15: 554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim BK, Yun SH, Gil CO, Ju ES, Choi JO, Kim DK, Jeon ES. 2012. Foreign gene transfer to cardiomyocyte using a replication-defective recombinant coxsackievirus B3 without cytotoxicity. Intervirology 55: 201–209. [DOI] [PubMed] [Google Scholar]
- Long L, Deng Y, Yao F, Guan D, Feng Y, Jiang H, Li X, Hu P, Lu X, Wang H, et al. 2014. Recruitment of phosphatase PP2A by RACK1 adaptor protein deactivates transcription factor IRF3 and limits type I interferon signaling. Immunity 40: 515–529. [DOI] [PubMed] [Google Scholar]
- Ma H, Dang Y, Wu Y, Jia G, Anaya E, Zhang J, Abraham S, Choi JG, Shi G, Qi L, et al. 2015. A CRISPR-based screen identifies genes essential for West-Nile-virus-induced cell death. Cell Rep 12: 673–683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Majzoub K, Hafirassou ML, Meignin C, Goto A, Marzi S, Fedorova A, Verdier Y, Vinh J, Hoffmann JA, Martin F, et al. 2014. RACK1 controls IRES-mediated translation of viruses. Cell 159: 1086–1095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marceau CD, Puschnik AS, Majzoub K, Ooi YS, Brewer SM, Fuchs G, Swaminathan K, Mata MA, Elias JE, Sarnow P, et al. 2016. Genetic dissection of Flaviviridae host factors through genome-scale CRISPR screens. Nature 535: 159–163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCleland ML, Mesh K, Lorenzana E, Chopra VS, Segal E, Watanabe C, Haley B, Mayba O, Yaylaoglu M, Gnad F, et al. 2016. CCAT1 is an enhancer-templated RNA that predicts BET sensitivity in colorectal cancer. J Clin Invest 126: 639–652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McPhail JA, Ottosen EH, Jenkins ML, Burke JE. 2017. The molecular basis of Aichi virus 3A protein activation of phosphatidylinositol 4 kinase IIIβa, PI4KB, through ACBD3. Structure 25: 121–131. [DOI] [PubMed] [Google Scholar]
- Mesmin B, Bigay J, Moser von Filseck J, Lacas-Gervais S, Drin G, Antonny B. 2013. A four-step cycle driven by PI(4)P hydrolysis directs sterol/PI(4)P exchange by the ER-Golgi tether OSBP. Cell 155: 830–843. [DOI] [PubMed] [Google Scholar]
- Metzakopian E, Strong A, Iyer V, Hodgkins A, Tzelepis K, Antunes L, Friedrich MJ, Kang Q, Davidson T, Lamberth J, et al. 2017. Enhancing the genome editing toolbox: genome wide CRISPR arrayed libraries. Sci Rep 7: 2244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paek KY, Kim CS, Park SM, Kim JH, Jang SK. 2008. RNA-binding protein hnRNP D modulates internal ribosome entry site-dependent translation of hepatitis C virus RNA. J Virol 82: 12082–12093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park RJ, Wang T, Koundakjian D, Hultquist JF, Lamothe-Molina P, Monel B, Schumann K, Yu H, Krupzcak KM, Garcia-Beltran W, et al. 2017. A genome-wide CRISPR screen identifies a restricted set of HIV host dependency factors. Nat Genet 49: 193–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel KP, Coyne CB, Bergelson JM. 2009. Dynamin- and lipid raft-dependent entry of decay-accelerating factor (DAF)-binding and non-DAF-binding coxsackieviruses into nonpolarized cells. J Virol 83: 11064–11077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roulin PS, Lotzerich M, Torta F, Tanner LB, van Kuppeveld FJ, Wenk MR, Greber UF. 2014. Rhinovirus uses a phosphatidylinositol 4-phosphate/cholesterol counter-current for the formation of replication compartments at the ER-Golgi interface. Cell Host Microbe 16: 677–690. [DOI] [PubMed] [Google Scholar]
- Sanjana NE, Shalem O, Zhang F. 2014. Improved vectors and genome-wide libraries for CRISPR screening. Nat Methods 11: 783–784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasaki J, Ishikawa K, Arita M, Taniguchi K. 2012. ACBD3-mediated recruitment of PI4KB to picornavirus RNA replication sites. EMBO J 31: 754–766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savidis G, McDougall WM, Meraner P, Perreira JM, Portmann JM, Trincucci G, John SP, Aker AM, Renzette N, Robbins DR, et al. 2016. Identification of Zika virus and dengue virus dependency factors using functional genomics. Cell Rep 16: 232–246. [DOI] [PubMed] [Google Scholar]
- Shalem O, Sanjana NE, Hartenian E, Shi X, Scott DA, Mikkelsen TS, Heckl D, Ebert BL, Root DE, Doench JG, et al. 2014. Genome-scale CRISPR-Cas9 knockout screening in human cells. Science 343: 84–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staring J, von Castelmur E, Blomen VA, van den Hengel LG, Brockmann M, Baggen J, Thibaut HJ, Nieuwenhuis J, Janssen H, van Kuppeveld FJ, et al. 2017. PLA2G16 represents a switch between entry and clearance of Picornaviridae. Nature 541: 412–416. [DOI] [PubMed] [Google Scholar]
- Stone JK, Rijnbrand R, Stein DA, Ma Y, Yang Y, Iversen PL, Andino R. 2008. A morpholino oligomer targeting highly conserved internal ribosome entry site sequence is able to inhibit multiple species of picornavirus. Antimicrob Agents Chemother 52: 1970–1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strating JR, van der Linden L, Albulescu L, Bigay J, Arita M, Delang L, Leyssen P, van der Schaar HM, Lanke KH, Thibaut HJ, et al. 2015. Itraconazole inhibits enterovirus replication by targeting the oxysterol-binding protein. Cell Rep 10: 600–615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strezoska Z, Perkett MR, Chou ET, Maksimova E, Anderson EM, McClelland S, Kelley ML, Vermeulen A, Smith AVB. 2017. High-content analysis screening for cell cycle regulators using arrayed synthetic crRNA libraries. J Biotechnol 251: 189–200. [DOI] [PubMed] [Google Scholar]
- Tan J, Martin SE. 2016. Validation of synthetic CRISPR reagents as a tool for arrayed functional genomic screening. PLoS One 11: e0168968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teoule F, Brisac C, Pelletier I, Vidalain PO, Jegouic S, Mirabelli C, Bessaud M, Combelas N, Autret A, Tangy F, et al. 2013. The Golgi protein ACBD3, an interactor for poliovirus protein 3A, modulates poliovirus replication. J Virol 87: 11031–11046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang T, Wei JJ, Sabatini DM, Lander ES. 2014. Genetic screens in human cells using the CRISPR-Cas9 system. Science 343: 80–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilsky S, Sobotta K, Wiesener N, Pilas J, Althof N, Munder T, Wutzler P, Henke A. 2012. Inhibition of fatty acid synthase by amentoflavone reduces coxsackievirus B3 replication. Arch Virol 157: 259–269. [DOI] [PubMed] [Google Scholar]
- Yasunaga A, Hanna SL, Li J, Cho H, Rose PP, Spiridigliozzi A, Gold B, Diamond MS, Cherry S. 2014. Genome-wide RNAi screen identifies broadly-acting host factors that inhibit arbovirus infection. PLoS Pathog 10: e1003914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang R, Miner JJ, Gorman MJ, Rausch K, Ramage H, White JP, Zuiani A, Zhang P, Fernandez E, Zhang Q, et al. 2016. A CRISPR screen defines a signal peptide processing pathway required by flaviviruses. Nature 535: 164–168. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.