

Abstract
In mouse embryonic stem cells (mESCs), the expression of provirus and endogenous retroelements is epigenetically repressed. Although many cellular factors involved in retroelement silencing have been identified, the complete molecular mechanism remains elusive. In this study, we performed a genome-wide CRISPR screen to advance our understanding of retroelement silencing in mESCs. The Moloney murine leukemia virus (MLV)–based retroviral vector MSCV-GFP, which is repressed by the SETDB1/TRIM28 pathway in mESCs, was used as a reporter provirus, and we identified more than 80 genes involved in this process. In particular, ATF7IP and the BAF complex components are linked with the repression of most of the SETDB1 targets. We characterized two factors, MORC2A and RESF1, of which RESF1 is a novel molecule in retroelement silencing. Although both factors are recruited to repress provirus, their roles in repression are different. MORC2A appears to function dependent on repressive epigenetic modifications, while RESF1 regulates repressive epigenetic modifications associated with SETDB1. Our genome-wide CRISPR screen cataloged genes which function at different levels in silencing of SETDB1-target retroelements and provides a useful resource for further molecular studies.
Transposable element–derived elements comprise <40% of the mouse genome (Waterston et al. 2002). Among these, retroelements including short/long interspersed elements (SINEs/LINEs) and endogenous retroviruses (ERVs) are capable of active retrotransposition (Maksakova et al. 2006; Huang et al. 2012). Although retrotransposition contributes to genome diversification and evolution, highly active retrotransposition can cause genome instability and is commonly deleterious to host species (Kazazian 2004; O'Donnell and Burns 2010; Mager and Stoye 2015). Therefore, evolution has also driven the development of multiple defense mechanisms against retrotransposition. The first line of the defense mechanism is transcriptional silencing of retroelements, using various epigenetic machineries (Goodier 2016). DNA methylation–mediated transcriptional silencing is a well-conserved defense mechanism in various species (Slotkin and Martienssen 2007). Histone H3 lysine 9 trimethylation (H3K9me3) is also a crucial epigenetic modification employed in retroelement silencing. In murine pluripotent stem cells, proviruses (an integrated form of retroviruses) and a subset of the endogenous retroelements, such as ERVs, are transcriptionally repressed by a histone methyltransferase SETDB1 (also known as ESET or KMT1E) (Matsui et al. 2010) and a corepressor complex containing tripartite motif containing 28 (TRIM28 [also known as KAP1 or TIF1B]) (Rowe et al. 2010). This SETDB1/TRIM28 complex is recruited to target retroelements by TRIM28-associating, nucleic acid binding zinc-finger proteins (ZFPs), including KRAB-ZFPs and YY1 (Schlesinger et al. 2013; Wolf et al. 2015). The physical interaction between SETDB1 and TRIM28 is facilitated by SUMOylation of TRIM28 (Ivanov et al. 2007). Therefore, factors involved in TRIM28 SUMOylation as well as regulation of SUMOylation are crucial for SETDB1-mediated retroelement silencing (Thompson et al. 2015; Yang et al. 2015). In addition, chromatin assembly factors such as ATRX, DAXX, and CAF1A/B are also known to be involved in retroelement silencing in early embryos or embryonic stem cells (Elsasser et al. 2015; Hatanaka et al. 2015; Sadic et al. 2015; Voon et al. 2015; Yang et al. 2015).
As exemplified above, multiple cellular factors that are involved in retroelement silencing have been identified. However, the complete mechanism of retroelement silencing via epigenetic modifications, especially downstream from DNA methylation or H3K9me3, is not yet fully understood. Recently, Yang et al. (2015) performed a genome-wide siRNA screen and identified multiple known and previously uncharacterized silencing factors. Since siRNA generally cannot completely down-regulate gene expression, it is possible that there are further unidentified retroelement silencing factors. Gene perturbation by genome editing using the CRISPR-Cas9 technology has now become available as an alternative to the RNAi technology (Cong et al. 2013; Mali et al. 2013). This technology allows us to perform genome-scale gene perturbation, which has recently been developed as CRISPR screens (Koike-Yusa et al. 2014; Shalem et al. 2014; Wang et al. 2014). The CRISPR screening technique has evolved rapidly, and activity-optimized CRISPR libraries are now available (Wang et al. 2015; Doench et al. 2016; Tzelepis et al. 2016).
In order to advance our understanding of retroelement silencing, we performed a CRISPR-gRNA–based genome-wide screen in mESCs harboring MLV-based retroviral vector, murine stem cell virus (MSCV)-GFP as a reporter provirus.
Results
Genome-wide CRISPR screen for genes required for provirus silencing in mESCs
To establish a CRISPR KO screen system, we employed a reporter retrovirus expressing GFP under the control of the long terminal repeat (LTR) enhancer/promoter of MSCV (Matsui et al. 2010). mESCs which were grown in media with serum plus leukemia inhibitory factor (LIF) and were stably expressing hCas9 from the Rosa26 locus (Tzelepis et al. 2016) were infected with the reporter virus, and five cell lines carrying silenced MSCV-GFP provirus were established (for more details, see Methods) (Fig. 1A). The MSCV vector includes an introduced SP1-binding site within the LTR, along with a tRNAGln PBS in place of tRNAPro to be silenced less efficiently in pluripotent stem cells than wild-type (WT) MLV (Hawley et al. 1994) but still silenced by the SETDB1/TRIM28 pathway (Matsui et al. 2010; Maksakova et al. 2011). Transfection of a Setdb1-specific gRNA expression vector to the established cell lines revealed that cell line 7 (clone 7) showed the highest GFP reactivation among the five lines (Supplemental Fig. S1a), but this activation was suppressed by cotransfection with Setdb1 cDNA, indicating that reactivation of proviral GFP occurred via genetic perturbation of Setdb1 (Fig. 1B). Clone 7 grown in culture media containing GSK3 inhibitor CHIR99021 and MEK-1,2 inhibitor PD0325901 (called 2i), which shield pluripotent cells from differentiation triggers (Wray et al. 2011), showed partial reactivation of proviral GFP and DNA hypomethylation in MSCV promoter (Supplemental Fig. S1b, left, c). Furthermore, in the presence of 2i, not only Setdb1 gRNA but also empty control lentivirus infection further enhanced proviral GFP expression by unknown mechanisms. We therefore performed a genome-wide CRISPR KO screen without those two inhibitors.
Figure 1.

Genome-wide genetic screen of provirus silencing factor. (A) Scheme of gRNA vecter (top) and the provirus MSCV-GFP reporter mouse ES cell lines (bottom) for the screen. (B) Validation of provirus silencing by SETDB1 in the reporter cell line. Setdb1 gRNA plasmid only (green) or both Setdb1 gRNA plasmid and transgene (blue) were transfected into clone 7, followed by flow cytometric analysis of GFP expression at 5 d after transfection. (C) Scheme of genome-wide genetic screen of provirus silencing factor by lentiviral gRNA library. (D) Analysis of the screening result by MAGeCK. X- and y-axes indicate rank of genes and –log10(P-value), respectively. P-values of top 222 genes were less than 0.01. All top-ranked genes listed in the red box have been known as a repressor of retroelement. (E) Validation of the top 46 genes by gRNA inactivation. Each gRNA plasmid of the top 46 genes was transfected into clone 7, and GFP expressions were analyzed by flow cytometry at 5 d after transfection. (F) GO term enrichment analysis for biological process of top 100 genes performed by DAVID. (G) Comparison of the top 100 genes between our screen and siRNA screen in Yang et al. (2015). See also Supplemental Figure S1.
The CRISPR library we used was the second-generation mouse library, which contains 89,897 gRNAs targeting 19,150 mouse protein-coding genes (Fig. 1A; Tzelepis et al. 2016). Clone 7 was transduced with the library virus, and 2 d later, BFP+/GFP− cells were collected by cell sorting. These cells were cultured for additional 3 d, and GFP-positive (top 4%–5%) and -negative (the remaining 95%) cells were collected by cell sorting. The collected cells were then analyzed for abundance of the gRNAs by next-generation sequencing, followed by statistical analysis using MAGeCK (Fig. 1C; Li et al. 2014). More than 100 genes were significantly enriched in the GFP-positive cells (P-value <0.005). The top eight out of the 100 genes (Setdb1, Atf7ip [also known as mAM or Mcaf1], Dnmt1, Atrx, Daxx, Smarcb1 [also known as Baf47 or Snf5], Smarcc1 [also known as Baf155], and Smarca4 [also known as Brg1]) have been previously shown to have a role in provirus and/or ERV silencing (Fig. 1D; Supplemental Table S1; Matsui et al. 2010; Rafati et al. 2011; Elsasser et al. 2015; Sadic et al. 2015; Voon et al. 2015; Yang et al. 2015).
We validated the results of our CRISPR screen by transfection of gRNA for the top 95 genes to clone 7. Depletion of gene products was confirmed by Western blot analysis for selected candidate genes (Supplemental Fig. S1d). The majority of the gRNA-based KO genes tested (84/95: 88%) showed enhanced GFP expression, indicating efficient identification of provirus silencing factors in our screen (Fig. 1E; Supplemental Fig. S1e). Gene Ontology (GO) terms enrichment analysis by DAVID 6.7 (Huang da et al. 2009) showed that GO terms related to chromatin organization and localized to heterochromatin were enriched in the top 100 genes (Fig. 1F; Supplemental Fig. S1f). These include components of various protein complexes, such as the BAF, HUSH, ATRX-DAXX, and shelterin complexes (Supplemental Fig. S1g). The HUSH complex is composed of FAM208A [also known as TASOR], MPHOSPH8 [also known as MPP8], and PPHLN1 and functions in regulating H3K9me3 (Tchasovnikarova et al. 2015). All three genes encoding the components of the HUSH complex were relatively highly ranked in our screen (Fam208a, 21; Pphln1, 215; and Mphosph8, 614). Transfection with gRNA specific to each of the three components enhanced proviral GFP expression (Fig. 1E; Supplemental Fig. S1h), indicating that the HUSH complex is also involved in provirus silencing in mouse pluripotent stem cells.
Comparison of the top 100 genes identified in our screen with those identified in an siRNA screen (Yang et al. 2015) showed that only eight genes (Atf7ip, Setdb1, Trim28, Smarca4, Smarcb1, Smarcc1, Sumo2, and Sae1) were common between the two screens (Fig. 1G). Even when the top 650 genes, which were considered as primary candidate genes in the siRNA screen (more than two SD from negative control), were compared with our top 100 genes, only 19 genes were common (Supplemental Table S1). The markedly different outcome could be explained by the differences in reporter retroviruses used (MSCV vs. WT MLV) and cell types used (mESCs vs. EC cells). Nevertheless, our screen identified a number of novel provirus silencing genes such as 2810474O19Rik which we termed Retroelement silencing factor 1 (Resf1).
Roles of the identified provirus silencing factors in retroelement regulation
To investigate the roles of the genes identified by our screen in transcriptional regulation of the retroelements, we performed an RNA-seq analysis of the top 20 genes and the remaining two HUSH complex genes, Pphln1 and Mphosph8. Clone 7 was transfected with gRNA-expressing vector and transiently selected with puromycin. Then, RNA was isolated 5 d post transfection. Cluster analysis of repeat expression showed that genes tended to cluster together according to protein complexes (SETDB1, BAF, NuA4 HAT, ATRX-DAXX, and HUSH complex) (Fig. 2A). Retroelement types and the number repressed by ATF7IP were similar to those repressed by SETDB1 (Fig. 2B). Furthermore, almost all retroelements repressed by SETDB1 (FC > 2, FDR < 0.05) (Supplemental Fig. S2a) were also repressed by ATF7IP (Fig. 2C). Thus, ATF7IP is a pan-repressor of SETDB1 targets. On the other hand, components of other complexes repressed specific types of retroelements although most of these retroelements are repressed by SETDB1 (Fig. 2B,D; Supplemental Fig. S2b). The majority of SETDB1 targets were mildly de-repressed in BAF components Smarcc1 and Smarca4 KO mESCs, but some retroelements such as RLTR4 were not reactivated. HUSH components repressed L1 and some ERVK such as IAPEY. ATRX-DAXX components mainly repressed ERVK, but not ERVL and L1. NuA4 HAT components ACTL6A and DMAP1 repressed ERVL, but not L1 much. These results were validated by RT-qPCR (Supplemental Fig. S2c). We also found that MORC2A and RESF1 showed L1 and ERVK target preference for silencing, respectively (Fig. 2D). Recently, human homolog of MORC2A, MORC2, has been shown to play a crucial role in the HUSH complex–mediated gene silencing (Tchasovnikarova et al. 2017). However, it remains unknown which type of retroelement was targeted by MORC2A. In addition, Resf1 is an uncharacterized gene. Thus, we further characterized these two genes in retroelement silencing and other functions.
Figure 2.

Repeat regulation of top 20 genes and HUSH components. (A) Cluster analysis of the indicated RNA-seq libraries based on log2 fold change of repeats at 5 d after gRNA plasmid transfection. Heatmap color intensity represents Pearson's correlation coefficient. gRNAs are colored by the type of protein complex: (Red) HUSH complex; (green) SETDB1 complex; (blue) BAF complex; (brown) NuA4 HAT complex; (pink) DAXX/ATRX complex. (B) Number of different repeat families up-regulated in the indicated RNA-seq libraries (FC > 1.5). Only repeat elements for which expression levels were more than 100 in the control sample were used in this analysis. (C) Expression change of repeats repressed by SETDB1 in each gRNA transfected mESCs. Fold expression change is classified by color. (D) Heatmap of log2 fold change of retroelements repressed by SETDB1 in the indicated RNA-seq libraries.
MORC2A functions as repressor at H3K9me3 regions
MORC2A is a member of the microchidia (MORC) protein family, which is composed of four MORC proteins (MORC1–4) and SMCHD1 in mice. MORC2A has a GHKL-type ATPase domain, a CW-type zinc finger (CW) domain, and a Chromo-like (CL) domain (Fig. 3A). Proviral GFP expression was reactivated by transient transfection of clone 7 with Morc2a gRNA and three additional mESC reporter cell lines that carry a reporter provirus at a genomic locus different from that of clone 7 (Supplemental Fig. S3a), indicating that provirus silencing by MORC2A is not locus dependent and that MORC2A is a genuine silencing factor. To analyze the function of MORC2A in retroelement silencing, we knocked out Morc2a in Clone 7 using the CRISPR system and isolated a KO clone, which had frameshift mutations in both alleles (c.1303_1324del22, c.1312_1315del4). Expression of Morc2a was significantly reduced in the Morc2a KO cells (Fig. 3B). Reverse-transcription quantitative PCR (RT-qPCR) and flow cytometry analyses showed reactivation of proviral GFP in the Morc2a KO cells (Fig. 3B,C). KO cells complemented with FLAG-MORC2A WT or a FLAG-ΔCL mutant rescued GFP silencing (Fig. 3C). Neither FLAG-ΔATPase nor FLAG-ΔCW mutant MORC2A could rescue GFP silencing (Fig. 3C). The ATP binding defective mutant (DD68/69AA), which abolishes ATPase activity (Li et al. 2012), also could not rescue regardless of similar protein expression levels between WT and the mutant transgene, suggesting the requirement for ATPase activity and CW domain for provirus silencing (Fig. 3C; Supplemental Fig. S3b). These findings are consistent with the human MORC2 studies (Tchasovnikarova et al. 2017).
Figure 3.

Characterization of MORC2A. (A) Scheme of protein domains of MORC2A and MORC2A mutant molecules used in this study. (B) Relative RNA expression of MSCV-GFP and Morc2a in the Morc2a KO cell line. Relative mRNA expression level was examined by RT-qPCR. (C) Flow cytometric analysis of GFP expression in Morc2a KO cell line complemented with WT or mutant transgene vector. (D) ChIP-qPCR confirmation of MORC2A enrichment on MSCV promoter. Anti-FLAG ChIP-qPCR was performed with Morc2a KO cell line stably expressing FLAG-tagged WT or mutant MORC2A. (E) Peptide binding assay using histone peptide (H2A, H2B, H4, H3, and H3K9me3). GST-tagged MORC2A CW domain and biotinylated histone peptide were mixed, and the peptide-bound GST-fusion was collected by avidin beads. (F) Overlap of MORC2A-, SETDB1-, and H3K9me3-enriched regions in entire genome (left) or gene promoter (right). Publicly available SETDB1 and H3K9me3 ChIP-seq data in mESCs were reanalyzed to obtain SETDB1- and H3K9me3-enriched regions (Karimi et al. 2011). (G) Heat map of MORC2A, SETDB1, and H3K9me3 ChIP-seq enrichment across MORC2A binding sites in gene promoter. ChIP-seq enrichment (reads per million [RPM]) is shown by color scale. MORC2A binding sites were classified by overlap with SETDB1- and H3K9me3-enriched regions: All three factors were colocalized (triple positive [TP]); only SETDB1 was colocalized with MORC2A (double positive [DP]), and MORC2A did not overlap with SETDB1 and H3K9me3 (single positive [SP]). (H) Boxplot showing log2 fold change of gene expression in Morc2a KO cells. Only genes with MORC2A binding sites in their promoter were analyzed, and genes were classified by overlap of MORC2A-, SETDB1-, and H3K9me3-enriched regions. MORC2A binding was correlated with gene silencing, when it was colocalized with SETDB1 and H3K9me3. (I–K) Analysis of chromatin status of MSCV promoter in Morc2a KO cells. H3K4me3 (I) and H3K9me3 (K) were analyzed by ChIP-qPCR, and chromatin compaction was analyzed by FAIRE-qPCR (J). (L) DNA methylation status of MSCV promoter in Morc2a KO cells analyzed by bisulfite sequencing analysis. Open and filled circles represent unmethylated or methylated cytosines, respectively. The percentage of total methylated CpGs/CpGs is presented on the left sides of each data set. Data represent mean ± SE (n = 3). (*) P-value <0.01 (t-test). (N.S.) Not significant. See also Supplemental Figure S3.
To investigate whether MORC2A directly binds the reporter provirus, we performed chromatin immunoprecipitation (ChIP)–qPCR assay using Morc2a KO cells expressing FLAG-WT, FLAG-ΔATPase, or FLAG-ΔCW Morc2a cDNA. MORC2A WT and the ΔATPase mutant showed comparable enrichment of the provirus, but the ΔCW mutant did not show significant enrichment (Fig. 3D). Immunofluorescence staining showed that FLAG-MORC2A WT localized to the nucleus and that this localization was not affected by ΔCW domain deletion (Supplemental Fig. S3c). Peptide binding assay using histone peptide (H2A, H2B, H3, and H4) showed that the CW domain bound H3 peptide (1–20) regardless of the methylation status at K9 and bound H4 peptide (2–20) with a lower affinity, but not H2A and H2B peptides (Fig. 3E). Taken together, these results suggest that MORC2A directly binds the provirus locus through the interaction of CW domain with H3 tail and suppresses the provirus.
As the CW domain binds H3 peptide regardless of methylation status at K9, it seems that the H3K9me3 mark may not be a prerequisite for MORC2A binding to chromatin. To investigate it, we performed MORC2A ChIP-seq in mESCs expressing FLAG-MORC2A and identified 2443 MORC2A binding sites. Of these, 570 sites were located in gene promoters. From our ChIP-seq data and previously reported SETDB1 and H3K9me3 ChIP-seq data in mESCs (Yuan et al. 2009), MORC2A enrichment was also detected in regions without H3K9me3 mark or both SETDB1 and H3K9me3 mark, which is consistent with the peptide binding assay (Fig. 3E–G). RNA-seq analysis of the Morc2a KO cells showed that MORC2A binding promoter sites that were simultaneously bound by both SETDB1 and H3K9me3 were strongly associated with gene repression, but the MORC2A binding sites that lack SETDB1 and H3K9me3 enrichment did not show a significant impact on transcription (Fig. 3H). Therefore, MORC2A functions as a transcriptional repressor when it is colocalized with SETDB1 and the H3K9me3 mark. Simultaneous depletion of both SETDB1 and MORC2A by transfection with gRNAs for these two genes did not result in synergistic up-regulation of proviral GFP expression (Supplemental Fig. S3d). Collectively, these results suggest that MORC2A functions in the H3K9me3-dependent silencing pathway of SETDB1, rather than by an independent silencing pathway.
We next analyzed two histone modifications, namely, H3K4me3 and H3K9me3, and chromatin condensation status at the provirus. Enrichment of H3K4me3 and formaldehyde-assisted isolation of regulatory elements (FAIRE) (Giresi et al. 2007) at the 5′ LTR of the provirus were higher in the Morc2a KO cells than those in WT cells (Fig. 3I,J), indicating that the de-repressed provirus formed a more open and transcriptionally active chromatin in the Morc2a KO cells. However, global H3K9me3 and SETDB1 protein levels and localization patterns of H3K9me3 in the nucleus were not affected in the Morc2a KO cells (Supplemental Fig. S3e,f). Furthermore, reduction of H3K9me3 enrichment and the DNA methylation level at the provirus were not significant in the Morc2a KO cells (Fig. 3K,L). Thus, it is possible that MORC2A can function downstream from H3K9me3.
De-repression of L1 in Morc2a KO mESCs
To identify MORC2A targets other than provirus, we compared repeat expressions between WT and the Morc2a KO cells. Consistent with the RNA-seq data from transient Morc2a gRNA transfection (Fig. 2), L1 retrotransposons were commonly silenced by SETDB1 and MORC2A (Fig. 4A). Furthermore, FLAG-MORC2A ChIP-seq analysis showed that the ratio of L1 occupancy among the MORC2A-targeted repeats increases compared with the ratio of L1 occupancy among repeats in the entire mouse genome, especially L1Md_T (L1MdTf_I as Repbase annotation) (Fig. 4B,C). Out of 2632 full-length L1Md_T copies (>6 kb), 843 (32.0%) were overlapped with MORC2A binding sites. Along L1Md_T consensus sequence, FLAG-MORC2A was enriched in the 5′ UTR (Fig. 4D), which was validated with ChIP-qPCR via V5-MORC2A expressing mESCs (Fig. 4E). Unlike MSCV, H3K9me3 in the 5′ UTR of L1Md_T was decreased in Morc2a KO mESCs (Fig. 4F). Thus, the impact of MORC2A for H3K9me3 was different between MSCV and L1, and this difference may be dependent on genomic locus and/or repeat type. Reanalysis of RNA-seq data from human MORC2 KO cells and human MORC2 ChIP-seq data (Tchasovnikarova et al. 2017) showed that human MORC2 also repressed L1s, including retrotransposition defective elements such as L1P and L1M (Fig. 4G), and was enriched in the 5′ terminus of LINE (Fig. 4H), indicating evolutionally conserved role of MORC2/MORC2A in L1 regulation.
Figure 4.

L1 retrotransposons were repressed by MORC2A. (A) Expression change of repetitive elements repressed by SETDB1 and/or MORC2A in Setdb1 and Morc2a KO mESCs. Previously reported RNA-seq data from Setdb1 KO mESC (Karimi et al. 2011) was used for the analysis. (B) Occupancy of repeat class in entire genome (left) or MORC2A binding sites (right). (C) Fraction of MORC2A binding sites overlapping with L1Md_Gf (L1MdTf_II), L1MdA (L1MdA_I), or L1Md_T in all MORC2A binding sites. (D) Enrichment of MORC2A along L1Md_T. MORC2A ChIP-seq reads were aligned to the L1Md_T consensus sequence, and enrichment of MORC2A relative to input for each 100-bp bin was plotted. (E) ChIP-qPCR confirmation of MORC2A binding to 5′ UTR of L1Md_T. MORC2A ChIPs were performed Morc2a KO mESCs stably expressing V5-tagged WT MORC2A. Anti-V5 Ab was used. (F) ChIP-qPCR analysis of H3K9me3 at L1Md_T in Morc2a KO cells. (G) Repression of L1P4a, L1PA2, L1M3a (L1M3B), and L1HS by human MORC2 in HeLa cells. Publicly available RNA-seq data in Morc2 knockout HeLa cells (GSE95452) were used for the analysis. Human MORC2 also repressed L1. (H) Enrichment of human MORC2 in 5′ UTR of L1HS. Publicly available human MORC2 ChIP-seq data (GSE95456) was used for the analysis. Data represent mean ± SE (n = 3). (*) P-value <0.01 (t-test). See also Supplemental Figure S4.
In addition to retroelement, we also focused on gene regulation by MORC2A. From the RNA-seq data of Morc2a KO mESCs, we identified 456 differentially expressed (DE) genes (271 up-regulated and 185 down-regulated genes) (Supplemental Table S2). GO term analysis showed that germ cell–related GO terms were enriched in up-regulated genes (Supplemental Fig. S4a). Consistently, MORC2A binding sites were enriched in germ cell–related genes (Supplemental Fig. S4b,c). Up-regulation of germ cell–related genes (Fkbp6 and Dazl) and L1 was confirmed in another Morc2a KO cell line (Supplemental Fig. S4d). Enrichment of MORC2A in germ cell–related genes was also validated by ChIP-qPCR using anti-FLAG and anti-V5 for Morc2a KO mESCs stably expressing FLAG- and V5-tagged WT MORC2A, respectively (Supplemental Fig. S4e,f). To identify recruiters of MORC2A, we performed motif analysis and found that the MYC and MAX binding motifs were enriched in the MORC2A binding sites (Supplemental Fig. S4g). On the other hand, these motifs (CACGTG) were not found in the L1Md_T full-length consensus or MSCV reporter sequences. Furthermore, MAX was enriched in MORC2A binding sites (Supplemental Fig. S4h), and genes repressed by MORC2A and MAX were significantly overlapped (Supplemental Fig. S4i). We found that enrichment of MORC2A in germ cell–related genes was diminished in conditional Max KO mESCs (Supplemental Fig. S4j,k; Hishida et al. 2011). Although we could not observe physical interaction between MAX and MORC2A (Supplemental Fig. S4l), the MAX complex may recruit MORC2A to specific chromatin loci via an indirect way.
Loss of repressive chromatin modifications in Resf1 KO mESCs
We also characterized novel proviral silencing factor, RESF1, which is a 152-kDa (predicted) protein with no known functional domain and is conserved from chickens to humans. Reactivation of proviral GFP was confirmed in the independent reporter lines (Supplemental Fig. S5a), indicating that the provirus silencing by RESF1 is not locus dependent. Thus, RESF1 functions as a genuine provirus silencing factor. To analyze the molecular function of RESF1, we generated a Resf1 KO cell line, which had frameshift mutations on both alleles (c.446_447del2, c.446_453del8). Proviral GFP expression was reactivated in the Resf1 KO cells and silenced again by re-expressing FLAG-RESF1 (Fig. 5A). The de-repression level of reporter GFP was modest at day 5 after Resf1 gRNA KO (Fig. 1E; Supplemental Fig. S5a) than that in established Resf1 KO ES cells (Fig. 5A), indicating that the full impact of Resf1 KO may take time for the provirus de-repression. DNA methylation analysis showed that Resf1 gRNA did not make clear induction of DNA demethylation on the MSCV promoter at day 5 after transfection (Supplemental Fig. S5b). However, in 2i/LIF medium, Resf1 gRNA could induce relatively efficient reactivation of proviral GFP to a similar extent Setdb1 gRNA did (Supplemental Fig. S5c). Clone 7 grown in 2i/LIF medium showed significant reduction of DNA methylation on the MSCV promoter (Supplemental Fig. S1c), suggesting that unaffected DNA methylation may attenuate full reactivation of MSCV-GFP induced by Resf1 inactivation in a short-term culture. This also indicates that DNA hypomethylation of the MSCV promoter region seen in Resf1 KO ES cells (Fig. 5F) is indirectly induced by Resf1 inactivation.
Figure 5.

RESF1 regulates repressive epigenetic chromatin configurations. (A) Flow cytometric analysis of GFP expression in Resf1 KO cell line and the cell line complemented with Resf1 transgene vector. (B) Localization of RESF1 in DAPI-dense region in mESCs characterized by low DNMT1 expression. Cellular localization of RESF1 and DNMT1 analyzed by immunofluorescent in mESCs stably expressing FLAG-RESF1. In a small cell population, DNMT1 expression was low (DNMT1low) (27/1173), and FLAG signals was enriched in DAPI-dense regions (35/1173). Such cells were indicated by the white arrows. DNMT1low mESCs were significantly overlapped with mESCs showing RESF1 enrichment in DAPI-dense regions (21/27, P-value = 8.5 × 10−31, hypergeometric test). (C–E) Analysis of chromatin status of MSCV promoter in Resf1 KO cells. H3K4me3 (C) and H3K9me3 (E) were analyzed by ChIP-qPCR, and chromatin compaction was analyzed by FAIRE-qPCR (D). (F) DNA methylation status of provirus in Resf1 KO cells analyzed by bisulfite sequencing analysis. In the KO cells, DNA methylation level of MSCV promoter was decreased. (G) Enrichment of SETDB1 in MSCV promoter analyzed by ChIP-qPCR. In Resf1 KO cells, enrichment of SETDB1 on MSCV promoter was reduced. (H) ChIP-qPCR confirmation of RESF1 binding to MSCV promoter. Anti-FLAG ChIPs were performed with Resf1 KO cell line stably expressing FLAG-tagged RESF1. Data represent mean ± SE (n = 3). (*) P-value <0.01 (t-test). See also Supplemental Figure S5.
RESF1 localized in the nucleus and formed foci that overlap with DAPI-dense regions in a small fraction of cells (<2%) that expressed DNMT1 at a markedly low level (Fig. 5B). It has recently been reported that DNMT1 is down-regulated in 2C-like cells, which is a rare group of cells found in mESC culture and express “early-embryonic” transcripts, such as Zscan4 and MERVL ERVs (Eckersley-Maslin et al. 2016). RESF1 also localized around γ-tubulin during M phase (Supplemental Fig. S5d). Thus, subcellular localization of RESF1 dynamically changes with cellular status.
To investigate the role of RESF1 in epigenetic regulation, we performed ChIP-qPCR, FAIRE-qPCR, and bisulfite sequencing analyses at the provirus locus. Enrichment of H3K4me3 and FAIRE in the Resf1 KO cells was higher than that in WT mESCs (Fig. 5C,D), suggesting open and active chromatin configuration. In contrast, H3K9me3 and DNA methylation level of the provirus were substantially diminished in the Resf1 KO cells (Fig. 5E,F). Enrichment of SETDB1 on the provirus also decreased in the Resf1 KO cells (Fig. 5G). On the other hand, Setdb1 knockdown by siRNA did not show significant decrease of RESF1 enrichment in MSCV promoter (Supplemental Fig. S5e–g). ChIP-qPCR assay of the Resf1 KO cells complemented with FLAG-RESF1 showed enrichment of RESF1 at the provirus (Fig. 5H). Transient transfection coimmunoprecipitation experiment showed that RESF1 could interact with SETDB1 (Supplemental Fig. S5h). There were no significant differences between WT and KO cells in SETDB1 expression, as well as the global level and the subnuclear localization of H3K9me3 modification (Supplemental Fig. S5i,j). Taken together, these data indicate that RESF1 physically interacts with SETDB1 and is required for the recruitment or accumulation of SETDB1 to the provirus and maintenance of repressive chromatin configuration.
RESF1 is a repressor of ERV
To identify targets of RESF1 other than provirus, we also performed RNA-seq analysis of the Resf1 KO cell line. We analyzed repeat expression and identified 22 up-regulated repeats in the Resf1 KO cells, most of which were ERVs (19/22, 86.4%) (Supplemental Table S3). Consistent with RNA-seq data from depletion of RESF1 by gRNA transfection, most of the repeats (mostly ERVKs) de-repressed by Resf1 KO were included in the repeats de-repressed by Setdb1 KO (Fig. 6A), suggesting that RESF1 contributes to subset of the SETDB1-dependent ERV repression. Up-regulation of ERV expression was also confirmed in an independent Resf1 KO cell line (Supplemental Fig. S6a). ChIP-seq analysis of H3K9me3 showed reduced H3K9me3 levels of the up-regulated ERVs in the Resf1 KO cells, but those of L1s were not affected (Supplemental Fig. S6b). The DNA methylation level and enrichment of SETDB1 in ETn ERV that is de-repressed by RESF1 depletion were also reduced in the Resf1 KO cells (Supplemental Fig. S6c,d). In addition, we identified 74 RESF1 binding sites by ChIP-seq analysis in mESCs expressing FLAG-RESF1, and ERV retrotransposons were enriched in RESF1 binding sites (Fig. 6B). These data indicate that RESF1 represses ERV by regulating H3K9me3.
Figure 6.

RESF1 regulates imprinting genes and ERVs. (A) Expression change of repetitive elements repressed by SETDB1 and/or RESF1 in Setdb1 and Resf1 KO mESCs. All repetitive elements repressed by both SETDB1 and RESF1 were ERV retrotransposons. Previously reported RNA-seq data from Setdb1 KO mESC (Karimi et al. 2011) was used for the analysis. (B) Length distribution of repeat class in entire genome (left) and RESF1 binding sites (right). ERV retrotransposon (LTR) was enriched in RESF1 binding sites. (C) List of imprinting genes dysregulated in Resf1 KO cells. Direction of expression change of these genes was almost same those in Setdb1 KO mESCs. Six out of 10 dysregulated imprinting genes were located near RESF1 binding sites (<500 kb), and these RESF1 binding sites were occupied by SETDB1. Publicly available RNA-seq data in Setdb1 KO mESCs (GSE29413) (Karimi et al. 2011) and SETDB1 ChIP-seq data in mESCs (GSE17642) (Yuan et al. 2009) were used. (D) Representative view of RESF1 binding site around imprinting genes. Regions in red or blue boxes represent RESF1 binding sites or dysregulated gene in Resf1 KO cells, respectively. See also Supplemental Figure S6.
In addition to repeats, we identified 1038 DE genes (up-regulated, 387; down-regulated, 651). Some imprinted genes, such as Igf2, Peg10, and Dlk1, were dysregulated in the Resf1 KO cells (Supplemental Table S3). Consistent with that, eight of the 11 genes that RESF1 binds in their promoter regions were imprinted genes (Airn, H19, Kcnq1ot1, Peg13, Nap1l5, Nnat, Grb10, and Plagl1). RESF1 was also located in differentially methylated regions (DMRs) of imprinted genes in nonpromoter regions, including Rasgrf1 and Meg3 DMRs. Enrichment of FLAG- and V5-RESF1 in imprinted genes was validated by ChIP-qPCR (Supplemental Fig. S6e,f). Furthermore, RESF1 binding sites were frequently associated with imprinted genes dysregulated in the Resf1 KO cells (Fig. 6C,D; Supplemental Fig. S6g), suggesting a role of RESF1 in regulation of imprinted gene expression.
Discussion
In this study, we conducted a genome-wide CRISPR KO screen and cataloged cellular factors involved in provirus silencing. The top eight genes identified in this screen were previously characterized provirus silencing factors. Approximately 90% of the top 95 genes were successfully validated. These include a number of factors that have not been previously associated with provirus silencing. Functional studies of these novel candidates would provide deeper insights into the mechanisms of retroelement silencing.
Functional interaction of identified provirus silencing factors
The top 100 genes identified in our screen included multiple components of different protein complexes associated with chromatin, such as SETDB1, BAF, ATRX-DAXX, shelterin, and HUSH complexes. ATF7IP interacts with SETDB1 (Wang et al. 2003) and stabilizes SETDB1 by protecting it from proteasomal degradation (Timms et al. 2016). Thus, it is reasonable that Atf7ip and Setdb1 cluster closely together in the retroelement expression profile. In addition to ATF7IP, BAF and HUSH components also repressed majority of SETDB1 targets. It was reported that BAF components such as SMARCA4 and SMARCC1 are involved in the pericentromeric heterochromatin formation and distribution of H3K9me3 (Bourgo et al. 2009; Schaniel et al. 2009) and in the formation of senescence-associated heterochromatin foci (SAHF), which is enriched by H3K9me3 (Tu et al. 2013). It was also reported that the BAF complex represses HIV (Rafati et al. 2011). Although it remains unknown how the BAF complex regulates heterochromatin formation associated with H3K9me3, SMARCC1 may play a key role since it interacts with SETDB1 (Thompson et al. 2015). Components of the HUSH, SETDB1, and BAF complexes were also identified as provirus silencing factors in siRNA screen (Yang et al. 2015). Thus, these protein complexes play more general roles for provirus silencing in murine early embryonic cells. In contrast, ATRX-DAXX components were involved in silencing a part of SETDB1 targets.
Targets of ATRX and DAXX were similar to those of DNMT1. They repress ERVK and ERV1, but not ERVL and L1, which is consistent with previous results of RNA-seq and microarray analyses in Dnmt TKO mESCs (Karimi et al. 2011; Reichmann et al. 2012). It was reported that ATRX-DAXX is responsible for H3.3 deposition at SETDB1-TRIM28–targeted ERV elements (Elsasser et al. 2015). Deposition of H3.3 at the retroelements is also dependent on TRIM28, which is enriched in ERV1 and ERVK rather than ERVL and L1. Furthermore, DAXX interacts with TRIM28 (Elsasser et al. 2015). These reports suggest that TRIM28 may be a determinant of ATRX-DAXX targets.
The shelterin complex is well known for protection of telomeres. The complex and function are highly conserved in different species (de Lange 2005; Palm and de Lange 2008; Xin et al. 2008), but very little is known about the role of the shelterin complex in retroelement silencing. It was recently reported that shelterin components promote facultative heterochromatin assembly at internal chromosomal sites containing late replication origins in fission yeast (Zofall et al. 2016). Therefore, the role of the shelterin complex in gene silencing might have been conserved from yeast to mammal.
MORC2A function
We focused on the analysis of two identified provirus silencing factors, MORC2A and RESF1. Among the MORC family members, mouse MORC1 has been shown to be involved in retroelement silencing in male germ cells (Pastor et al. 2014). AtMORC1 and AtMORC6, homologs of MORC in Arabidopsis thaliana, are known to be involved in heterochromatin condensation and gene and transposon silencing (Moissiard et al. 2012). Furthermore, AtMORC4 and AtMORC7 have been recently shown to form nuclear bodies and repress a large number of protein-coding genes (Harris et al. 2016). Therefore, MORC family proteins seem to have a conserved function as transcriptional repressors. Our CRISPR screen revealed a role of MORC2A in provirus silencing that is consistent with the very recent finding of a human MORC2A counterpart, MORC2 (Tchasovnikarova et al. 2017).
In Morc2a KO mESCs, proviral GFP was de-repressed, which was associated with the open chromatin structure and a transcriptionally active histone mark. To our surprise, we did not detect significant difference in repressive epigenetic marks such as DNA methylation and/or H3K9me3 on provirus between WT and KO cells (Fig. 3K,L). In addition, RNA-seq analysis showed that genes bound simultaneously by MORC2A, SETDB1, and the H3K9me3 mark, but not by MORC2A alone, were up-regulated in Morc2a KO cells (Fig. 3H). These results indicate that MORC2A function is dependent of repressive epigenetic modifications and acts to maintain a transcriptionally repressive state.
The molecular mechanism by which MORC2A represses provirus expression remains to be determined. One potential mechanism is that the repressive function of MORC2A is mediated by histone deacetylases, as human MORC2 was shown to bind to the histone deacetylases HDAC1 and HDAC4 (Shao et al. 2010; Zhang et al. 2015). However, no HDACs were identified either in our CRISPR screen or in the siRNA screen (Yang et al. 2015). It is still possible that multiple HDACs are redundantly involved in the MORC2A-mediated silencing, but we need further investigation. Another possibility and a more likely mechanism is that MORC2A functions as a transcriptional suppressor through its chromatin remodeling activity. Li et al. (2012) reported that MORC2 showed chromatin remodeling activity following DNA damage and that this activity is dependent on the ATPase activity. In fact, a recent study demonstrated that MORC2-mediated gene silencing is ATPase activity dependent (Tchasovnikarova et al. 2017). Our analysis also showed that both the ΔATPase and DD68/69AA (defective for ATP binding) mutants of MORC2A did not show provirus silencing (Fig. 3C), further supporting such a mechanism.
Transcriptome analysis of Morc2a KO mESCs showed that MORC2A represses germ cell–related genes and L1 retrotransposons. ChIP-seq analysis showed that MORC2A directly regulates germ cell–related genes and L1 retrotransposons. L1 retrotransposons are repressed in male germ cells by various mechanisms such as the piRNA pathway (Goodier 2016), and MORC2A is highly expressed in testis (Andrews et al. 2016). These data implicate that MORC2A regulates gene and retroelement expression in germ cell lineages and plays important roles in spermatogenesis and other germ cell functions. While our manuscript was under revision, Liu et al. (2018) reported that MORC2 selectively binds 5′ ends of evolutionarily young, full-length L1s and silences them. Our analysis of MORC2A/human MORC2 ChIP-seq data is partly consistent with their MORC2 data, thus indicating a 5′ binding preference for L1.
Finally, even though MORC2A and MORC2A-associated activity may operate downstream from H3K9m3 in silencing, there are potentially other downstream effectors. H3K9me3 binding proteins, such as HP1, are potential candidates. Although some of them were identified in our screen (ATRX and MPHOSPH8), it is not known whether they function downstream from H3K9me3, especially in provirus silencing. Further analysis of the candidate molecules is necessary for understanding the silencing mechanism via H3K9me3.
RESF1 function
Another and a new provirus silencing factor, RESF1, is an uncharacterized molecule and does not have known protein domains. RESF1 is highly expressed in oocytes and early embryos (Park et al. 2015). We showed that cellular localization of RESF1 overlapped with DAPI-dense regions in small cell populations characterized by low expression of DNMT1 (Fig. 5B). In addition, RESF1 seems to be localized at the centrosome in M phase (Supplemental Fig. S5d). As various heterochromatin-related proteins, such as DNMT1, MBD3 and HDAC1, localize to the centrosome (Chadwick and Willard 2002), it is possible that there is a functional link between the centrosome and heterochromatin.
In Resf1 KO mESCs, reporter provirus was de-repressed and provirus chromatin on provirus became open and active. In addition, repressive modification of chromatin decreased on provirus and ERV in the KO mESCs (Fig. 5E,F; Supplemental Fig. S6b,c). The enrichment of SETDB1 on provirus and ERV was diminished in the Resf1 KO mESCs (Fig. 5G; Supplemental Fig. S6d) Therefore, RESF1 could interact with SETDB1 in the transient transfection experiment (Supplemental Fig. S5h). Thus, it is possible that RESF1 regulates, directly or indirectly, SETDB1 localization and repressive chromatin modifications. It would be interesting to investigate which region of RESF1 is responsible for the SETDB1 interaction and how this region is crucial for the RESF1-mediated ERV silencing. In addition to provirus and ERVs, RESF1 regulated some imprinted genes, and RESF1 and SETDB1 binding sites frequently overlapped with these genes (Fig. 6C; Supplemental Fig. S6g). These imprinted genes were also dysregulated in Setdb1 KO mESCs, suggesting involvement of RESF1 in SETDB1-mediated regulation of imprinted genes as well.
In conclusion, our screen identified various SETDB1-associated provirus silencing factors. We analyzed molecular functions of two uninvestigated silencing factors and provided molecular insights. The possible mechanism of MORC2A- and RESF1-mediated provirus silencing based on our current studies is shown in Figure 7. Further detailed analyses of the provirus silencing factors identified in this study would advance our understanding of provirus silencing and heterochromatin formation.
Figure 7.
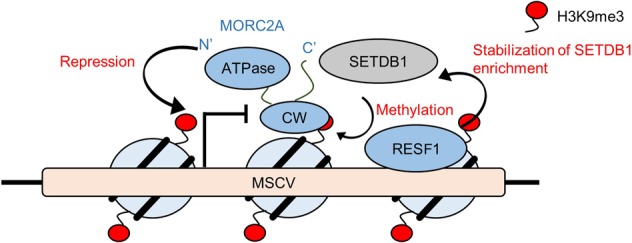
Possible mechanism of MORC2A- and RESF1-mediated provirus silencing. MORC2A interacts with histone H3 tail through its CW domain and functions as transcriptional repressor when it colocalizes with H3K9me3. MORC2A-mediated silencing activity is ATPase activity dependent. RESF1 regulates SETDB1-mediated silencing by stabilizing SETDB1 enrichment on the target provirus loci.
Methods
Generation of MSCV-GFP reporter mESCs lines
Rosa26EF1ahCas9IRESneo/+ mESCs were described previously (Tzelepis et al. 2016). Cells were transduced with MSCV-GFP retrovirus. Transduction efficiency was monitored with GFP expression 4 d after virus infection (>60% GFP-positive). After 14 d of cell culture, GFP-negative cells were collected by cell sorting and plated at clonal density in order to obtain cells in which integrated MSCV-GFP reporter provirus was epigenetically silenced. Colonies were picked and expanded. Ten clones that showed GFP silencing were transfected with a Setdb1-gRNA expressing vector, and GFP reactivation was analyzed by flow cytometry 5 d after transfection. Clone 7 was chosen for further studies.
CRISPR screen
Clone 7 (48 × 106 cells) was transduced with murine genome-wide CRISPR library virus (Tzelepis et al. 2016). We titrated transduction conditions such that ∼30% of input cells were transduced with the virus. Two days later, BFP+/GFP− cells were collected by cell sorting and further cultured. BFP+/GFP+ and BFP+/GFP− cells were collected on the fifth day after transduction. The GFP+ gate was set to isolate the top 4%–5% of GFP-positive cells. The screen was performed in four biological replicates. We collected 1.3 × 106–2.4 × 106 and 20 × 106–46 × 106 cells for BFP+/GFP+ and BFP+/GFP− populations, respectively. Genomic DNA was extracted from the sorted cells by either a DNeasy blood and tissue kit (Qiagen) or a blood and cell culture DNA maxi kit (Qiagen). gRNA amplification, Illumina sequencing, and statistical analysis were conducted as described previously (Tzelepis et al. 2016).
Additional information on cell culture, Native ChIP, crosslinked ChIP, DNA methylation analysis, FAIRE, Western blotting, immunofluorescence analysis, cDNA synthesis, qPCR, RNA-seq analysis, ChIP-seq analysis, knockdown analysis, peptide binding assay, lentivirus infection, antibodies, and oligonucleotides (Supplemental Table S4) is presented in the Supplemental Materials.
Data access
All reads from the RNA-seq and ChIP-seq experiments in this study have been submitted to the Sequence Read Archive (SRA; https://www.ncbi.nlm.nih.gov/sra) under accession number SRP127601.
Supplementary Material
Acknowledgments
We thank Dr. M.C. Lorincz for providing the MSCV-GFP retrovirus vector, Dr. Jason Yu for critically reading the manuscript, and the Sanger Institute Flow Cytometry Core Facility for their technical help. We also thank all of the Shinkai laboratory members for helpful discussions and the Support Unit for Bio-Material Analysis, RIKEN BSI Research Resources Center, with special thanks to Mr. K. Ohtawa for flow cytometric analysis and cell sorting. We also thank the Phyloinformatics Unit, RIKEN Life Science Technologies for NGS analysis, with special thanks to Dr. M. Kadota. This research was supported by KAKENHI (26250039) and a RIKEN internal research fund for Y.S. and the Wellcome Trust (WT098051) for K.Y.
Author contributions: K.F., K.Y., and Y.S. performed experiments. Y.S. established reporter cell lines, K.Y. performed genome-wide CRISPR-gRNA screen, and K.F. performed all the rest of the experiments. K.F. and Y.S. took primary responsibility for writing the manuscript. All authors edited the manuscript.
Footnotes
[Supplemental material is available for this article.]
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.227280.117.
References
- Andrews FH, Tong Q, Sullivan KD, Cornett EM, Zhang Y, Ali M, Ahn J, Pandey A, Guo AH, Strahl BD, et al. 2016. Multivalent chromatin engagement and inter-domain crosstalk regulate MORC3 ATPase. Cell Rep 16: 3195–3207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bourgo RJ, Siddiqui H, Fox S, Solomon D, Sansam CG, Yaniv M, Muchardt C, Metzger D, Chambon P, Roberts CW, et al. 2009. SWI/SNF deficiency results in aberrant chromatin organization, mitotic failure, and diminished proliferative capacity. Mol Biol Cell 20: 3192–3199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chadwick BP, Willard HF. 2002. Cell cycle–dependent localization of macroH2A in chromatin of the inactive X chromosome. J Cell Biol 157: 1113–1123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cong L, Ran FA, Cox D, Lin S, Barretto R, Habib N, Hsu PD, Wu X, Jiang W, Marraffini LA, et al. 2013. Multiplex genome engineering using CRISPR/Cas systems. Science 339: 819–823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Lange T. 2005. Shelterin: the protein complex that shapes and safeguards human telomeres. Genes Dev 19: 2100–2110. [DOI] [PubMed] [Google Scholar]
- Doench JG, Fusi N, Sullender M, Hegde M, Vaimberg EW, Donovan KF, Smith I, Tothova Z, Wilen C, Orchard R, et al. 2016. Optimized sgRNA design to maximize activity and minimize off-target effects of CRISPR-Cas9. Nat Biotechnol 34: 184–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eckersley-Maslin MA, Svensson V, Krueger C, Stubbs TM, Giehr P, Krueger F, Miragaia RJ, Kyriakopoulos C, Berrens RV, Milagre I, et al. 2016. MERVL/Zscan4 network activation results in transient genome-wide DNA demethylation of mESCs. Cell Rep 17: 179–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elsasser SJ, Noh KM, Diaz N, Allis CD, Banaszynski LA. 2015. Histone H3.3 is required for endogenous retroviral element silencing in embryonic stem cells. Nature 522: 240–244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giresi PG, Kim J, McDaniell RM, Iyer VR, Lieb JD. 2007. FAIRE (Formaldehyde-Assisted Isolation of Regulatory Elements) isolates active regulatory elements from human chromatin. Genome Res 17: 877–885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodier JL. 2016. Restricting retrotransposons: a review. Mob DNA 7: 16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris CJ, Husmann D, Liu W, Kasmi FE, Wang H, Papikian A, Pastor WA, Moissiard G, Vashisht AA, Dangl JL, et al. 2016. Arabidopsis AtMORC4 and AtMORC7 form nuclear bodies and repress a large number of protein-coding genes. PLoS Genet 12: e1005998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hatanaka Y, Inoue K, Oikawa M, Kamimura S, Ogonuki N, Kodama EN, Ohkawa Y, Tsukada Y, Ogura A. 2015. Histone chaperone CAF-1 mediates repressive histone modifications to protect preimplantation mouse embryos from endogenous retrotransposons. Proc Natl Acad Sci 112: 14641–14646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hawley RG, Lieu FH, Fong AZ, Hawley TS. 1994. Versatile retroviral vectors for potential use in gene therapy. Gene Ther 1: 136–138. [PubMed] [Google Scholar]
- Hishida T, Nozaki Y, Nakachi Y, Mizuno Y, Okazaki Y, Ema M, Takahashi S, Nishimoto M, Okuda A. 2011. Indefinite self-renewal of ESCs through Myc/Max transcriptional complex-independent mechanisms. Cell Stem Cell 9: 37–49. [DOI] [PubMed] [Google Scholar]
- Huang CR, Burns KH, Boeke JD. 2012. Active transposition in genomes. Annu Rev Genet 46: 651–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang da W, Sherman BT, Lempicki RA. 2009. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc 4: 44–57. [DOI] [PubMed] [Google Scholar]
- Ivanov AV, Peng H, Yurchenko V, Yap KL, Negorev DG, Schultz DC, Psulkowski E, Fredericks WJ, White DE, Maul GG, et al. 2007. PHD domain-mediated E3 ligase activity directs intramolecular sumoylation of an adjacent bromodomain required for gene silencing. Mol Cell 28: 823–837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karimi MM, Goyal P, Maksakova IA, Bilenky M, Leung D, Tang JX, Shinkai Y, Mager DL, Jones S, Hirst M, et al. 2011. DNA methylation and SETDB1/H3K9me3 regulate predominantly distinct sets of genes, retroelements, and chimeric transcripts in mESCs. Cell Stem Cell 8: 676–687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kazazian HH Jr. 2004. Mobile elements: drivers of genome evolution. Science 303: 1626–1632. [DOI] [PubMed] [Google Scholar]
- Koike-Yusa H, Li Y, Tan EP, Velasco-Herrera Mdel C, Yusa K. 2014. Genome-wide recessive genetic screening in mammalian cells with a lentiviral CRISPR-guide RNA library. Nat Biotechnol 32: 267–273. [DOI] [PubMed] [Google Scholar]
- Li DQ, Nair SS, Ohshiro K, Kumar A, Nair VS, Pakala SB, Reddy SD, Gajula RP, Eswaran J, Aravind L, et al. 2012. MORC2 signaling integrates phosphorylation-dependent, ATPase-coupled chromatin remodeling during the DNA damage response. Cell Rep 2: 1657–1669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W, Xu H, Xiao T, Cong L, Love MI, Zhang F, Irizarry RA, Liu JS, Brown M, Liu XS. 2014. MAGeCK enables robust identification of essential genes from genome-scale CRISPR/Cas9 knockout screens. Genome Biol 15: 554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu N, Lee CH, Swigut T, Grow E, Gu B, Bassik M, Wysocka J. 2018. Selective silencing of euchromatic L1s revealed by genome-wide screens for L1 regulators. Nature 553: 228–232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mager DL, Stoye JP. 2015. Mammalian endogenous retroviruses. Microbiol Spectr 3: MDNA3-0009-2014. [DOI] [PubMed] [Google Scholar]
- Maksakova IA, Romanish MT, Gagnier L, Dunn CA, van de Lagemaat LN, Mager DL. 2006. Retroviral elements and their hosts: insertional mutagenesis in the mouse germ line. PLoS Genet 2: e2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maksakova IA, Goyal P, Bullwinkel J, Brown JP, Bilenky M, Mager DL, Singh PB, Lorincz MC. 2011. H3K9me3-binding proteins are dispensable for SETDB1/H3K9me3-dependent retroviral silencing. Epigenetics Chromatin 4: 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mali P, Yang L, Esvelt KM, Aach J, Guell M, DiCarlo JE, Norville JE, Church GM. 2013. RNA-guided human genome engineering via Cas9. Science 339: 823–826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsui T, Leung D, Miyashita H, Maksakova IA, Miyachi H, Kimura H, Tachibana M, Lorincz MC, Shinkai Y. 2010. Proviral silencing in embryonic stem cells requires the histone methyltransferase ESET. Nature 464: 927–931. [DOI] [PubMed] [Google Scholar]
- Moissiard G, Cokus SJ, Cary J, Feng S, Billi AC, Stroud H, Husmann D, Zhan Y, Lajoie BR, McCord RP, et al. 2012. MORC family ATPases required for heterochromatin condensation and gene silencing. Science 336: 1448–1451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Donnell KA, Burns KH. 2010. Mobilizing diversity: transposable element insertions in genetic variation and disease. Mob DNA 1: 21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palm W, de Lange T. 2008. How shelterin protects mammalian telomeres. Annu Rev Genet 42: 301–334. [DOI] [PubMed] [Google Scholar]
- Park SJ, Shirahige K, Ohsugi M, Nakai K. 2015. DBTMEE: a database of transcriptome in mouse early embryos. Nucleic Acids Res 43: D771–D776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pastor WA, Stroud H, Nee K, Liu W, Pezic D, Manakov S, Lee SA, Moissiard G, Zamudio N, Bourc'his D, et al. 2014. MORC1 represses transposable elements in the mouse male germline. Nat Commun 5: 5795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rafati H, Parra M, Hakre S, Moshkin Y, Verdin E, Mahmoudi T. 2011. Repressive LTR nucleosome positioning by the BAF complex is required for HIV latency. PLoS Biol 9: e1001206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reichmann J, Crichton JH, Madej MJ, Taggart M, Gautier P, Garcia-Perez JL, Meehan RR, Adams IR. 2012. Microarray analysis of LTR retrotransposon silencing identifies Hdac1 as a regulator of retrotransposon expression in mouse embryonic stem cells. PLoS Comput Biol 8: e1002486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rowe HM, Jakobsson J, Mesnard D, Rougemont J, Reynard S, Aktas T, Maillard PV, Layard-Liesching H, Verp S, Marquis J, et al. 2010. KAP1 controls endogenous retroviruses in embryonic stem cells. Nature 463: 237–240. [DOI] [PubMed] [Google Scholar]
- Sadic D, Schmidt K, Groh S, Kondofersky I, Ellwart J, Fuchs C, Theis FJ, Schotta G. 2015. Atrx promotes heterochromatin formation at retrotransposons. EMBO Rep 16: 836–850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaniel C, Ang YS, Ratnakumar K, Cormier C, James T, Bernstein E, Lemischka IR, Paddison PJ. 2009. Smarcc1/Baf155 couples self-renewal gene repression with changes in chromatin structure in mouse embryonic stem cells. Stem Cells 27: 2979–2991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlesinger S, Lee AH, Wang GZ, Green L, Goff SP. 2013. Proviral silencing in embryonic cells is regulated by Yin Yang 1. Cell Rep 4: 50–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shalem O, Sanjana NE, Hartenian E, Shi X, Scott DA, Mikkelsen TS, Heckl D, Ebert BL, Root DE, Doench JG, et al. 2014. Genome-scale CRISPR-Cas9 knockout screening in human cells. Science 343: 84–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shao Y, Li Y, Zhang J, Liu D, Liu F, Zhao Y, Shen T, Li F. 2010. Involvement of histone deacetylation in MORC2-mediated down-regulation of carbonic anhydrase IX. Nucleic Acids Res 38: 2813–2824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slotkin RK, Martienssen R. 2007. Transposable elements and the epigenetic regulation of the genome. Nat Rev Genet 8: 272–285. [DOI] [PubMed] [Google Scholar]
- Tchasovnikarova IA, Timms RT, Matheson NJ, Wals K, Antrobus R, Gottgens B, Dougan G, Dawson MA, Lehner PJ. 2015. Gene silencing. Epigenetic silencing by the HUSH complex mediates position-effect variegation in human cells. Science 348: 1481–1485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tchasovnikarova IA, Timms RT, Douse CH, Roberts RC, Dougan G, Kingston RE, Modis Y, Lehner PJ. 2017. Hyperactivation of HUSH complex function by Charcot–Marie–Tooth disease mutation in MORC2. Nat Genet 49: 1035–1044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson PJ, Dulberg V, Moon KM, Foster LJ, Chen C, Karimi MM, Lorincz MC. 2015. hnRNP K coordinates transcriptional silencing by SETDB1 in embryonic stem cells. PLoS Genet 11: e1004933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Timms RT, Tchasovnikarova IA, Antrobus R, Dougan G, Lehner PJ. 2016. ATF7IP-mediated stabilization of the histone methyltransferase SETDB1 is essential for heterochromatin formation by the HUSH complex. Cell Rep 17: 653–659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tu Z, Zhuang X, Yao YG, Zhang R. 2013. BRG1 is required for formation of senescence-associated heterochromatin foci induced by oncogenic RAS or BRCA1 loss. Mol Cell Biol 33: 1819–1829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tzelepis K, Koike-Yusa H, De Braekeleer E, Li Y, Metzakopian E, Dovey OM, Mupo A, Grinkevich V, Li M, Mazan M, et al. 2016. A CRISPR dropout screen identifies genetic vulnerabilities and therapeutic targets in acute myeloid leukemia. Cell Rep 17: 1193–1205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voon HP, Hughes JR, Rode C, De La Rosa-Velazquez IA, Jenuwein T, Feil R, Higgs DR, Gibbons RJ. 2015. ATRX plays a key role in maintaining silencing at interstitial heterochromatic loci and imprinted genes. Cell Rep 11: 405–418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, An W, Cao R, Xia L, Erdjument-Bromage H, Chatton B, Tempst P, Roeder RG, Zhang Y. 2003. mAM facilitates conversion by ESET of dimethyl to trimethyl lysine 9 of histone H3 to cause transcriptional repression. Mol Cell 12: 475–487. [DOI] [PubMed] [Google Scholar]
- Wang T, Wei JJ, Sabatini DM, Lander ES. 2014. Genetic screens in human cells using the CRISPR-Cas9 system. Science 343: 80–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang T, Birsoy K, Hughes NW, Krupczak KM, Post Y, Wei JJ, Lander ES, Sabatini DM. 2015. Identification and characterization of essential genes in the human genome. Science 350: 1096–1101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waterston RH, Lindblad-Toh K, Birney E, Rogers J, Abril JF, Agarwal P, Agarwala R, Ainscough R, Alexandersson M, An P, et al. 2002. Initial sequencing and comparative analysis of the mouse genome. Nature 420: 520–562. [DOI] [PubMed] [Google Scholar]
- Wolf G, Yang P, Fuchtbauer AC, Fuchtbauer EM, Silva AM, Park C, Wu W, Nielsen AL, Pedersen FS, Macfarlan TS. 2015. The KRAB zinc finger protein ZFP809 is required to initiate epigenetic silencing of endogenous retroviruses. Genes Dev 29: 538–554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wray J, Kalkan T, Gomez-Lopez S, Eckardt D, Cook A, Kemler R, Smith A. 2011. Inhibition of glycogen synthase kinase-3 alleviates Tcf3 repression of the pluripotency network and increases embryonic stem cell resistance to differentiation. Nat Cell Biol 13: 838–845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xin H, Liu D, Songyang Z. 2008. The telosome/shelterin complex and its functions. Genome Biol 9: 232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang BX, El Farran CA, Guo HC, Yu T, Fang HT, Wang HF, Schlesinger S, Seah YF, Goh GY, Neo SP, et al. 2015. Systematic identification of factors for provirus silencing in embryonic stem cells. Cell 163: 230–245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan P, Han J, Guo G, Orlov YL, Huss M, Loh YH, Yaw LP, Robson P, Lim B, Ng HH. 2009. Eset partners with Oct4 to restrict extraembryonic trophoblast lineage potential in embryonic stem cells. Genes Dev 23: 2507–2520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Q, Song Y, Chen W, Wang X, Miao Z, Cao L, Li F, Wang G. 2015. By recruiting HDAC1, MORC2 suppresses p21 Waf1/Cip1 in gastric cancer. Oncotarget 6: 16461–16470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zofall M, Smith DR, Mizuguchi T, Dhakshnamoorthy J, Grewal SI. 2016. Taz1-shelterin promotes facultative heterochromatin assembly at chromosome-internal sites containing late replication origins. Mol Cell 62: 862–874. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.