

Abstract
Premise of the Study
Until recently, most phylogenetic studies of ferns were based on chloroplast genes. Evolutionary inferences based on these data can be incomplete because the characters are from a single linkage group and are uniparentally inherited. These limitations are particularly acute in studies of hybridization, which is prevalent in ferns; fern hybrids are common and ferns are able to hybridize across highly diverged lineages, up to 60 million years since divergence in one documented case. However, it not yet clear what effect such hybridization has on fern evolution, in part due to a paucity of available biparentally inherited (nuclear‐encoded) markers.
Methods
We designed oligonucleotide baits to capture 25 targeted, low‐copy nuclear markers from a sample of 24 species spanning extant fern diversity.
Results
Most loci were successfully sequenced from most accessions. Although the baits were designed from exon (transcript) data, we successfully captured intron sequences that should be useful for more focused phylogenetic studies. We present phylogenetic analyses of the new target sequence capture data and integrate these into a previous transcript‐based data set.
Discussion
We make our bait sequences available to the community as a resource for further studies of fern phylogeny.
Keywords: ferns, HybPiper, hybridization, Hyb‐Seq, phylogeny, target sequence capture
Our collective understanding of fern phylogeny has benefited greatly from the explosion of available molecular data since about 1990. Most of this progress has resulted from analyses of chloroplast (plastid) genes (e.g., Schuettpelz and Pryer, 2007; Testo and Sundue, 2016). The preponderance of plastid gene studies in ferns, and in other plant groups, has a biological and historical basis. Whereas diploid individuals typically carry two copies of each nuclear gene per cell, plastid genes are present in as many as thousands of copies of a single haploid allele, providing ample template for amplification and study. Furthermore, most plastid genes are present as single‐copy markers within a genome, thus simplifying the distinction between paralogy and orthology. Plastid genes have few introns, and those that occur tend to be conservative in presence (Plant and Gray, 1988), simplifying alignment of nucleotides for phylogenetic analysis. Finally, the plastid genome is small, typically about 150 kbp, compared to nuclear genomes that are gigabases for most plant taxa. These biological aspects led to the early characterization of plastid genomes in ferns and other plants, which then led to further development of methods for more detailed study. Thus, the first gene for which universal PCR‐amplification primers were developed and broadly applied was the plastid‐encoded gene for the large subunit of RuBisCO (Zurawski et al., 1986; Ritland and Clegg, 1987; Zurawski and Clegg, 1987), subsequently igniting the field of plant molecular systematics. As a result, we now have a dense phylogenetic framework for green plants, and also for certain well‐studied clades therein.
However, phylogenetic estimates resulting from analyses of chloroplast genes can paint an oversimplified picture. In most plants, plastid genomes are inherited uniparentally and, unlike nuclear genomes, do not undergo extensive recombination. Thus, evolutionary events such as hybridization are often missed in plastid phylogenies. On the other hand, evidence from nuclear‐encoded genes can reveal a more complex picture, whereby hybridization events are evident in the patterns of incongruence across genes. Although phylogenetic hypotheses based on nuclear genes can be messy due to incongruence, plastid‐only phylogenies can lead to overconfidence in a phylogenetic framework derived from a single linkage group. As complex evolutionary events increase in frequency, so does the need for phylogenetic estimates based on nuclear‐encoded genes. Ferns are particularly vulnerable to such events because they appear to be capable of hybridization among lineages that are highly diverged. One such example is hybridization across fern genera that shared their last common ancestor approximately 60 million years ago (Rothfels et al., 2015a), and additional, potentially comparable hybridizations have been reported in other groups of ferns (e.g., Wagner et al., 1992; Wagner, 1993; Larsson, 2014b; Engels and Canestraro, 2017). It is hypothesized that organisms that undergo abiotically mediated fertilization may lack the types of pre‐mating barriers that are found in groups such as insect‐pollinated angiosperms; thus, ferns might evolve reproductive barriers more slowly than angiosperms (Ranker and Sundue, 2015; Rothfels et al., 2015a). However, several questions remain: How common are such wide hybridization events in ferns, relative to other plant groups? And are the hybrids evolutionary dead ends? Such hybrids are probably always sterile. However, it is not known whether fertility can be restored via allopolyploidy across such widely divergent genomes, and if so, is there an upper divergence limit to such events? These types of evolutionary processes would be entirely missed with plastid‐based studies, but analyses based on a sufficient number of low‐copy nuclear‐encoded genes should help detect signatures of such complex evolutionary histories.
Published studies using nuclear genes in ferns either focus on one or a few genes (e.g., Ishikawa et al., 2002; Pryer et al., 2004; Ebihara et al., 2005; Adjie et al., 2007; James et al., 2008; Schuettpelz et al., 2008; Chen et al., 2012; Sessa et al., 2012; Schneider et al., 2013; Rothfels and Schuettpelz, 2014; Rothfels et al., 2017) or genes from transcriptome data (Wickett et al., 2014; Li et al., 2015; Rothfels et al., 2015b; Shen et al., 2017). These approaches either scale poorly (PCR amplification of nuclear markers) or are expensive and restrictive with respect to the material that can be used (transcriptomics). Here we present a cost‐effective approach for acquiring multi‐locus nuclear‐encoded gene data in ferns. We apply a target sequence capture approach (Mertes et al., 2011) for 25 low‐copy genes, designed to work across the fern tree of life. This type of approach has been used successfully on other groups of organisms (Lemmon et al., 2012; Hart et al., 2016; Heyduk et al., 2016; Leveille‐Bourret et al., 2018) and can use genomic DNA as a starting material. During the preparation of a genomic library, templates are filtered to enrich for fragments that match certain “bait” oligonucleotides, which are designed from independent data, such as transcriptome sequences. The advantages of this method are that time‐ and labor‐intensive PCR is circumvented, templates can be genomic DNA rather than RNA, template quality conditions are less restrictive than they are for PCR‐based approaches, large contiguous information‐rich homologous sequences (including introns) can be captured, and larger sample sizes can be used. Our goal is to provide the fern systematics community with a cost‐effective tool for generating nucleotide data from nuclear genes. Our approach is focused to exploit data already available for ferns.
METHODS
Bait design
We started with alignments of 25 genes from a previous transcriptome study (Rothfels et al., 2015b) and designed baits from 33 taxa, spanning fern phylogenetic diversity, for which coverage was good (the list of taxa is available on Digital Commons [https://doi.org/10.15142/t3mg95]). We used RepeatMasker version 4.06 (Smit et al., 2013) to flag simple sequence repeats and low‐complexity DNA. From the remaining sequences, we designed 120‐nucleotide baits with ~2× flexible tiling density, for a total of 20,353 unfiltered baits. We then collapsed baits that were 95% identical or higher (with at least 75% sequence overlap) into 19,863 clusters, from which baits were chosen, one from each cluster. We screened these sequences against 38 published fern chloroplast sequences (from GenBank); no baits were flagged as being similar to any of these sequences. Baits were designed and synthesized by Arbor Biosciences (formerly Mycroarray; Ann Arbor, Michigan, USA) and are available on Digital Commons (https://doi.org/10.15142/t3mg95).
Samples, library prep, and sequencing
We chose 24 fern samples to evaluate the efficacy of our approach (Table 1). Taxa were selected from across fern diversity with a slight emphasis on Dennstaedtiaceae, Lindsaeaceae, and some other early diverging Polypodiales, which were underrepresented in the earlier transcriptome‐based work (Rothfels et al., 2015b). Thirteen of the samples were from genera used for bait design, and four were from families absent from the Rothfels et al. (2015b) study (Dicksoniaceae, Cystodiaceae, Saccolomataceae, and Metaxyaceae; Table 1). Samples were extracted from silica‐dried material, except for Osmundastrum C. Presl, which was from a herbarium specimen. We used a cetyltrimethylammonium bromide (CTAB) method (Doyle and Doyle, 1990) to extract DNA, which was then sent to Arbor Biosciences for further processing. Half the volume of each sample was purified using QIAquick PCR Purification columns (QIAGEN, Germantown, Maryland, USA) to remove residual contamination and eluted in 50 μL of EB buffer. Each purified sample was quantified with Quant‐iT PicoGreen dsDNA Assay Kit (Molecular Probes Inc., Eugene, Oregon, USA), and up to 4 μg of total genomic DNA was taken through a sonication and bead‐based size‐selection protocol to achieve modal fragment lengths of approximately 600 nucleotides. Between 99 and 509 ng of processed DNA were then converted to TruSeq‐style libraries and index‐amplified with unique dual eight‐nucleotide indexes for six cycles with KAPA HiFi polymerase (454 Life Sciences, a Roche Company, Branford, Connecticut, USA). Each target enrichment reaction contained two libraries of similar library yield, ranging from 100 to 400 ng of each library per pool. Enrichment reactions followed the standard myBaits version 3 (Arbor Biosciences) protocol (see manual provided on Digital Commons [https://doi.org/10.15142/t3mg95]) using 500‐ng baits, 62°C for 20 h of hybridization, bead‐bait binding, and wash steps. After resuspension of bead‐bound enriched libraries in 30 μL of recommended buffer, 15 μL were amplified for 10 cycles using KAPA HiFi polymerase, and the reactions purified with Solid Phase Reversible Immobilization (SPRI) beads (Applied Biological Materials, Richmond, British Columbia, Canada). The final enriched libraries were then quantified using library qPCR, combined equimolar, and passed to a single lane of MiSeq PE250 (Illumina, San Diego, California, USA). FASTQ files were delivered following standard post‐processing pipelines in BaseSpace Sequence Hub (Illumina). The Culcita C. Presl sample failed on first attempt and therefore was added to a later sequencing run on a HiSeq platform, with 125‐bp paired‐end reads.
Table 1.
Fern species used for target sequence capture in this study
| Taxon | Family | Locality | Voucher (Herbarium)a | Trimmed reads |
|---|---|---|---|---|
| Angiopteris evecta (G. Forst.) Hoffm.b | Marattiaceae | Cultivated | Testo 1475 (VT) | 1,090,305 |
| Asplenium harpeodes Kunzeb | Aspleniaceae | Costa Rica | Testo 666 (CR, VT) | 1,433,417 |
| Azolla caroliniana Willd.b | Salviniaceae | Cultivated | Testo 1472 (VT) | 1,146,104 |
| Blechnum occidentale L.b | Blechnaceae | Costa Rica | Testo 760 (CR, VT) | 1,014,324 |
| Culcita coniifolia (Hook.) Maxonb | Culcitaceae | Mexico | Sundue 4043 (VT) | 15,570,735 |
| Cyrtomium falcatum (L. f.) C. Presl | Dryopteridaceae | Cultivated | Testo 1477 (VT) | 974,984 |
| Cystodium sorbifolium (Sm.) J. Sm. | Cystodiaceae | Papua New Guinea | James 1669 (BISH) | 1,135,945 |
| Dennstaedtia cicutaria (Sw.) T. Mooreb | Dennstaedtiaceae | Costa Rica | Testo 743 (CR, VT) | 682,472 |
| Dicksonia antarctica Labill. | Dicksoniaceae | Cultivated | Testo 1474 (VT) | 807,243 |
| Didymochlaena truncatula (Sw.) J. Sm.b | Didymochlaenaceae | Costa Rica | Testo 1038 (NY, VT) | 1,431,614 |
| Diplazium sanctae‐rosae Christb | Athyriaceae | Costa Rica | Testo 124 (CR, VT) | 898,108 |
| Histiopteris incisa (Thunb.) J. Sm. | Dennstaedtiaceae | Costa Rica | Sundue 3923 (CR, VT) | 1,476,756 |
| Hymenophyllum fragile (Hedw.) C. V. Mortonb | Hymenophyllaceae | Mexico | Testo 911 (MEXU, VT) | 1,081,937 |
| Lindsaea quadrangularis Raddib | Lindsaeaceae | Mexico | Testo 868 (MEXU, VT) | 990,380 |
| Lygodium japonicum (Thunb.) Sw.b | Lygodiaceae | Cultivated | Testo 1476 (VT) | 1,519,904 |
| Metaxya elongata Tuomisto & G. G. Cárdenas | Metaxyaceae | Costa Rica | Testo 783 (CR, VT) | 759,737 |
| Osmundastrum cinnamomeum (L.) C. Preslb | Osmundaceae | USA | Testo 458 (VT) | 1,230,209 |
| Paesia glandulosa (Sw.) Kuhn | Dennstaedtiaceae | Colombia | Testo 1165 (HUA, VT) | 1,982,346 |
| Phlebodium aureum (L.) J. Sm. | Polypodiaceae | Cultivated | Testo 1479 (VT) | 465,304 |
| Polystichum concinnum Lellinger ex Barrington | Dryopteridaceae | Costa Rica | Testo 674 (CR, VT) | 628,777 |
| Saccoloma elegans Kaulf. | Saccolomataceae | Costa Rica | Testo 756 (CR, VT) | 1,034,346 |
| Sphaeropteris cooperi (F. Muell.) R. M. Tryon | Cyatheaceae | Cultivated | Testo 1473 (VT) | 1,100,822 |
| Sticherus palmatus (W. Schaffn. ex E. Fourn.) Copel. | Gleicheniaceae | Costa Rica | Testo 862 (CR, VT) | 791,485 |
| Thelypteris kunthii (Desv.) C. V. Mortonb | Thelypteridaceae | Cultivated | Testo 1480 (VT) | 979,438 |
Herbaria are abbreviated according to Index Herbariorum (http://sweetgum.nybg.org/science/ih/).
Genera also used for bait design.
Data processing
We first trimmed the resulting sequences using Trimmomatic version 0.36 (Bolger et al., 2014) with a quality cutoff of 20, MAXINFO adaptive quality trim with a target length of 10 nucleotides and strictness of 0.2, and discarded any reads trimmed to under 20 nucleotides. We then used HybPiper version 1.3 (Johnson et al., 2016) to extract and sort genes for downstream phylogenetic analyses. This suite of programs uses BLASTX (Altschul et al., 1990) or BWA (Burrows–Wheeler Alignment tool; Li and Durbin, 2009) to assign reads to target genes, and reads are then assembled separately for each target using SPAdes (Bankevich et al., 2012). Our target file was based on the alignments from Rothfels et al. (2015b), with some modifications (e.g., removal of outgroup sequences) to better facilitate the sorting of reads to the appropriate genes. We used 33 taxa, across the fern phylogeny, for which we had most genes. For gene families with duplication events within ferns, one paralog was arbitrarily selected to include the pre‐duplication copies (ApPEFP_AC for ApPEFP_B‐ApPEFP_C; gapCpSh for gapCpSh‐gapCpLg; CRY1 for CRY1‐CRY2‐CRY5; and CRY3 for CRY3‐CRY4 [see Rothfels et al., 2013]) and the reference sequences modified accordingly. We ran HybPiper, using these reference sequences, in nucleotide mode; preliminary runs in amino acid mode were too inclusive in the read‐sorting step: even distantly related paralogs were captured and grouped together. HybPiper annotates intron sequences using Exonerate (Slater and Birney, 2005) via a separate script (intronerate.py). Because our paired‐end reads were approximately 500 bases, we reasoned that we should be able to capture up to that length of intron from the end of each exon (more if unbaited reads are included).
Phylogenetic analyses
The exon‐only “paralog” sequences assembled by HybPiper were aligned for each locus in MUSCLE version 3.8.31 (Edgar, 2004). These alignments were manually refined while viewed as their translated amino acids in AliView version 118 (Larsson, 2014a), with ambiguous regions excluded from subsequent analysis. In several cases, the MUSCLE alignments resulted in long contiguous segments of sequence from individual accessions that were out of alignment compared to the rest of the sequences. BLAST results of these sequences demonstrated that they consisted of exons that were in the incorrect order, due to an Exonerate error that can occur when read coverage is low. The exons of these sequences were manually reassembled in the correct order (based on the BLAST coordinates), and the resulting sequences realigned. Preliminary maximum parsimony trees were inferred from these alignments in PAUP* v4.0 build 159 (Swofford, 2002) and, based on these trees, a small number of mis‐sorted sequences were transferred to their correct locus (e.g., some CRY3 sequences were incorrectly sorted into the CRY4 pool). In the few cases where multiple sequences were recovered for single accessions within a locus, the longest sequence that was resolved in a phylogenetically appropriate location in the maximum parsimony trees was retained and any others erased; these “extra” sequences presumably reflect mis‐assemblies, recent gene duplications, or homeologous sequences.
These 25 individual‐locus alignments were concatenated (while retaining exset and codon position information) into a single alignment with the “seqconcat” command in abioscripts version 0.9.3 (Larsson, 2010). This alignment was subject to maximum likelihood analysis in GARLI version 2.0 (Zwickl, 2006), with the data partitioned by codon position (see Rothfels et al., 2015b) and each data subset given an independent GTR+I+G substitution model. The best‐tree searches were performed from 10 random‐addition‐sequence starting trees, and support was assessed with 720 bootstrap pseudoreplicates (each searched from a single random‐addition‐sequence starting tree). All GARLI analyses were performed on the CIPRES Science Gateway version 3.3 (Miller et al., 2010). The maximum likelihood tree was annotated with support values summarized from the 720 bootstrap trees using the SumTrees version 3.3.1 command in the DendroPy phylogenetic computing library version 3.12.0 (Sukumaran and Holder, 2010).
To further explore the phylogenetic signal in these data and their utility in combination with previously published data, we combined the 25 exon‐only alignments with the corresponding transcript alignment from Rothfels et al. (2015b). We used a custom Python script to profile‐profile align the alignment pairs (each Hyb‐Seq exon‐only sequence paired with its transcriptome‐based partner) for each locus, using MUSCLE version 3.8.31 (Edgar, 2004). These alignments were then manually refined, concatenated, and analyzed as above for the Hyb‐Seq‐only alignments (Swofford, 2002; Zwickl, 2006; Larsson, 2010, 2014a; Miller et al., 2010; Sukumaran and Holder, 2010).
RESULTS
Raw reads, target sequence files, sequence alignments, taxon information, and analysis details are available on Digital Commons (https://doi.org/10.15142/t3mg95). In addition, the intron‐containing “supercontigs” for each sample are deposited in GenBank (MG817735–MG818130), and the exon‐only alignments are deposited in TreeBASE (study S22073 [https://treebase.org/treebase-web/search/study/anyObjectAsRDF.rdf?namespacedGUID=TB2:S22073]). We averaged 1,071,998 reads per sample for the MiSeq samples; the one HiSeq sample returned 15,570,735 reads (Table 1). We measured success of the baits by the proportion of the average gene length in the target file represented in the recovered exon‐only sequences. By this measure, we recovered an average of 51% coverage (across all genes and all accessions; Fig. 1). However, there was considerable variation across genes and taxa. Recovery efficiency was poorest for Phlebodium (R. Br.) J. Sm., Hymenophyllum Sm., Saccoloma Kaulf., and Metaxya C. Presl and the genes NDUFS6 and COP9. We did not see a clear pattern of taxon success relative to genera used for baits, although the results suggest a combination of bait‐related and taxon‐specific factors. For example, Metaxya and Saccoloma are both phylogenetically distant from any sample included in the bait set, whereas Hymenophyllaceae species, while included in the bait set, have sequenced poorly in other broad analyses (e.g., Rothfels et al., 2015b). The mean per‐base read depth for exon regions was 31.2. However, this includes regions that failed to get captured or were absent in a particular taxon; mean depth excluding samples with zero depth was 50.1.
Figure 1.
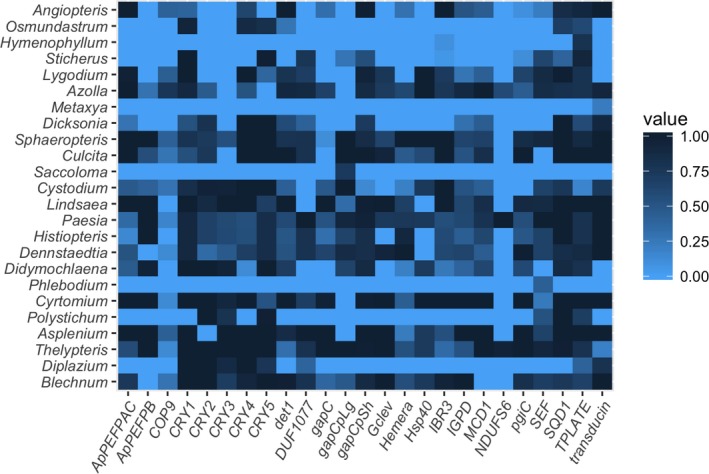
Heat map showing recovery efficiency for 25 genes targeted in 24 fern taxa. Each column corresponds to a gene and each row is a sample. The shading of each box represents the proportion of the target gene length recovered; darker colors indicate a higher percentage of the gene was recovered.
Average supercontig lengths varied from 700 (NDUFS6) to 6356 (IBR3) bp (mean = 3426; Table 2). The mean length of (partial) intron sequences captured was 267 bp. For all introns detected, including those with no coverage, the mean was 135 bp. On average, sequences of introns and intron fragments made up 41% of the supercontig data; these intron sequences could be useful in future studies with more focused phylogenetic samples, but were effectively unalignable at the phylogenetic depth of this study.
Table 2.
Supercontig and intron fragment lengths for each gene
| Gene | Estimated no. of introns | Mean length (zero‐length introns excluded) | Mean length (zero‐length introns included) | Mean length of supercontigs (ignoring zero lengths) | Alignment length (exons only) |
|---|---|---|---|---|---|
| ApPEFPAC | 13 | 295.52 | 131.66 | 5445.0 | 1800 |
| ApPEFPB | 12 | 182.55 | 79.23 | 3627.3 | 1626 |
| COP9 | 1 | 331.92 | 165.96 | 1225.0 | 417 |
| CRY1 | 3 | 335.44 | 232.94 | 4127.4 | 2163 |
| CRY2 | 3 | 400.95 | 233.89 | 3376.0 | 2070 |
| CRY3 | 4 | 568.93 | 242.98 | 4472.3 | 2277 |
| CRY4 | 3 | 440.19 | 293.46 | 6353.3 | 2244 |
| CRY5 | 3 | 314.81 | 227.36 | 2980.0 | 1488 |
| det1 | 7 | 378.99 | 207.54 | 5576.5 | 2919 |
| DUF1077 | 4 | 181.31 | 101.99 | 2198.6 | 570 |
| gapC | 8 | 255.12 | 87.70 | 3455.0 | 1041 |
| gapCpLg | 7 | 118.53 | 42.33 | 3790.0 | 1228 |
| gapCpSh | 9 | 134.63 | 83.52 | 3684.7 | 1356 |
| Gclev | 4 | 339.46 | 162.66 | 1723.6 | 540 |
| Hemera | 4 | 226.41 | 96.70 | 2490.0 | 1707 |
| Hsp40 | 1 | 119.00 | 54.54 | 1150.9 | 525 |
| IBR3 | 15 | 211.07 | 120.78 | 6356.5 | 2544 |
| IGPD | 5 | 249.62 | 141.45 | 2355.0 | 1122 |
| MCD1 | 2 | 467.17 | 282.25 | 2192.0 | 1056 |
| NDUFS6 | 0 | 0.00 | 0.00 | 700.0 | 342 |
| pgiC | 20 | 193.17 | 80.08 | 5456.0 | 1733 |
| SEF | 2 | 378.61 | 244.52 | 1492.0 | 549 |
| SQD1 | 1 | 532.95 | 488.54 | 2469.5 | 1533 |
| TPLATE | 5 | 447.08 | 242.17 | 4517.1 | 3555 |
| transducin | 10 | 232.62 | 136.66 | 4683.8 | 2729 |
| Mean | 5.84 | 266.59 | 134.97 | 3435.9 | 1565.4 |
Phylogeny estimates from our Hyb‐Seq data were generally well supported (Fig. 2) and are broadly consistent with previous studies based on both nuclear and chloroplast genes (e.g., Schuettpelz and Pryer, 2007; Lehtonen, 2011; Rothfels et al., 2015b; Testo and Sundue, 2016). The analyses of our data combined with those from Rothfels et al. (2015b) find the same major relationships as in the earlier study, with some refinements, and with novel inferences for taxa not previously included in broad nuclear phylogenies (Fig. 3 and Discussion).
Figure 2.
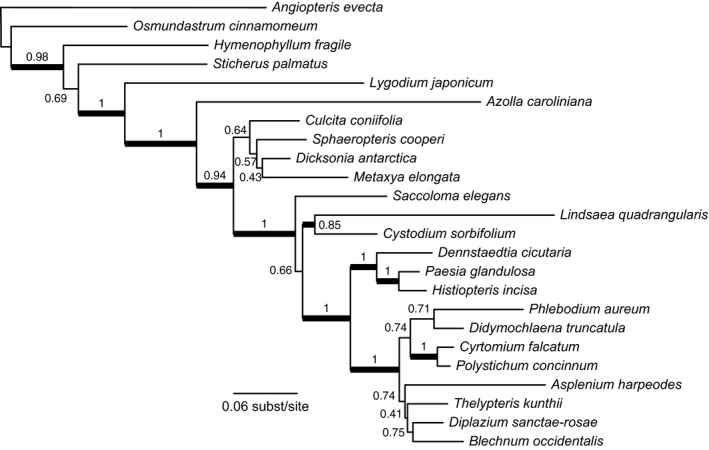
Maximum likelihood phylogeny from our exon‐only sequence‐capture data set. Thickened branches indicate bootstrap support greater than 80%.
Figure 3.
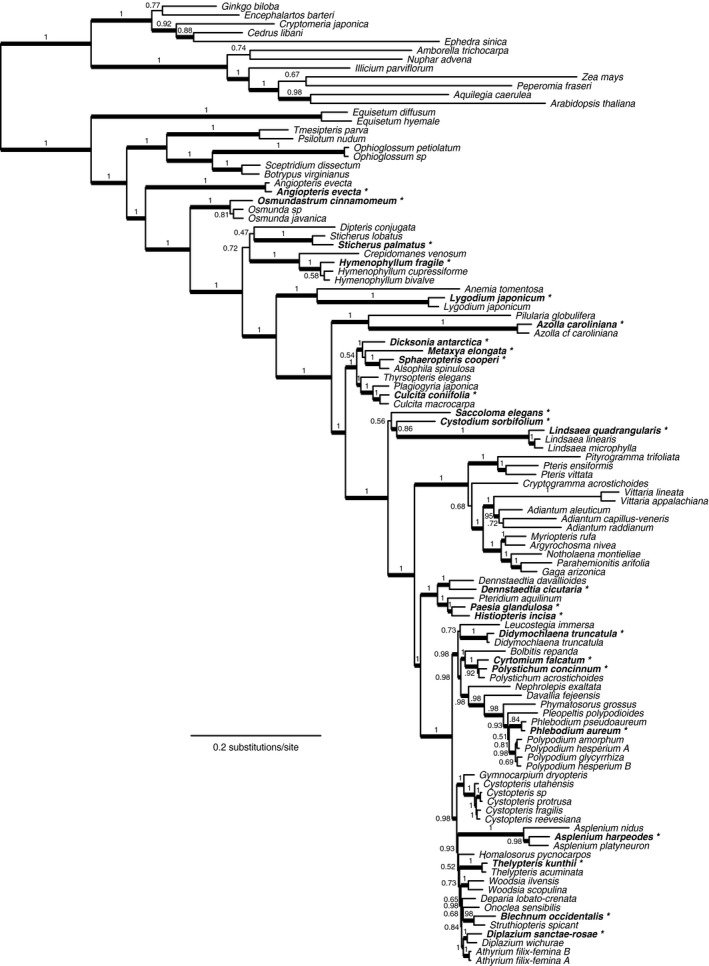
Maximum likelihood phylogeny from our sequence‐capture data set combined with the transcriptome‐derived data from Rothfels et al. (2015b). Thickened branches indicate bootstrap support greater than 80%, and bold taxon names followed by an asterisk indicate accessions from the sequence‐capture data set (see Fig. 1).
DISCUSSION
Our results demonstrate that our target bait collection is effective on a broad selection of fern taxa across the tree of life. We did not see a clear pattern of capture success based on relationships between taxa used in bait design and taxa sampled for capture. Nor is there a clear pattern with respect to genome size, for those few taxa for which we have estimates. We suspect that the main factors that affect success are DNA quantity and quality. Because the baits are designed from exon regions, they are more effective at capturing exon regions. However, the long read lengths enabled us to capture considerable (41% of the supercontig sequence) intron data. We were unable to align the intron regions because of extensive divergence across the taxa sampled. However, these regions can potentially be useful for more focused phylogenetic studies, and it should be possible to use information on exon/intron boundaries in our supercontig files to design more intron‐aware baits for more phylogenetically targeted studies.
We were able to get approximately 1 million reads per sample for the 25 genes by loading 24 samples on a single Illumina MiSeq lane. This appears to be sufficient for target sequence capture in a project of this scale. Using the Illumina HiSeq platform, it may be possible to load about 100 samples per lane, further reducing costs.
In comparison with the phylogenetic results of Rothfels et al. (2015b), we find the same broad relationships, including Equisetum L. sister to the remainder of ferns, Marattiales sister to leptosporangiate ferns, Cyatheales sister to Polypodiales, and Dennstaedtiaceae sister to the eupolypods (for the latter three relationships, we find increased maximum likelihood bootstrap support: 100% vs. 79%, 100% vs. 57%, and 100% vs. 89%, respectively). In addition, we resolve (but do not support) a monophyletic Gleicheniales and we find marginal support (72%) for a Gleicheniales + Hymenophyllales clade; neither relationship was resolved by Rothfels et al. (2015b) but has been reported in other previous studies (Pryer et al., 2004; Shen et al., 2017). The novel taxa sampled add further insights: Dicksonia L’Hér. and Metaxya, representing two previously unsampled families, are well supported in a clade with Cyatheaceae, although the relationships among the three families are uncertain; Cystodiaceae (Cystodium J. Sm.) is resolved as sister to Lindsaeaceae with 86% bootstrap support; Saccolomataceae (Saccoloma) is weakly resolved as sister to the Cystodiaceae + Lindsaeaceae clade; and within Dennstaedtiaceae Paesia A. St.‐Hil. is sister to Histiopteris (J. Agardh) J. Sm. and they, together, are sister to Pteridium Gled. ex Scop. These relationships are all consistent with earlier plastid‐based inferences (Korall et al., 2006a, 2006b; Lehtonen et al., 2012; PPG, 2016). Taxon sampling from Dennstaedtiaceae and Lindsaeaceae remains sparse and more work is required in this area to robustly resolve the early evolutionary history of the Polypodiales. Nevertheless, results from phylogenetic analyses suggest that our approach is capable of capturing DNA sequences with phylogenetic signal. The genes we chose were specifically selected because they have phylogenetic utility across ferns. Not all of these genes may necessarily be useful at shallow levels, e.g., comparing closely related species. Furthermore, we have not specifically tested how effective these genes are at detecting hybrids, because we did not include any suspected hybrids in the study. However, by choosing genes for which nucleotide sequences are already available, researchers will more likely be able to detect hybrids in future studies using a growing collection of available data. The main challenge in analyzing nuclear gene data for analysis of hybrids is phasing of alleles and homeologs, especially in young hybrids. This is an issue with all such data and can be minimized by favoring long‐read sequencing of MiSeq PE250 over the shorter reads from HiSeq. It should also be possible to combine the target sequence capture approach with sequencing on the PacBio platform for even longer reads, which has been shown to be effective for resolving polyploid and hybrid origins (Rothfels et al., 2017).
As evolutionary studies increasingly switch to nuclear gene data, our bait collection should provide a useful tool for a range of projects across ferns. Design of baits for additional genomic regions should soon be possible using accumulating transcriptome data (Matasci et al., 2014) as well as sequences from two fern genome projects (Azolla and Salvinia Genomes Consortium, unpublished data). Our approach to gathering data for ferns is complementary to other projects in progress at a broader scale, including the Genealogy of Flagellate Plants (GoFlag) project (http://flagellateplants.group.ufl.edu/) and the Plant and Fungal Trees of Life (PAFTOL) project (https://www.kew.org/science/who-we-are-and-what-we-do/strategic-outputs-2020/plant-and-fungal-trees-life). Our hope is that the accumulation of nuclear gene data will enhance our understanding of fern phylogeny and contribute to an increased understanding of the role of hybridization to fern evolution, across both deep and shallow phylogenetic depths. Although we are not yet able to evaluate the level of overlap between the regions captured in the method described here and those captured in the broader‐scale studies mentioned previously, we believe there is value in data sets that differ, as well as in those that have sufficient overlap to be combined. Just as phylogenies generated with sequence data from different regions of chloroplast DNA in ferns provided us with a better understanding of the general pattern of fern evolution (Hasebe et al., 1995; Schuettpelz and Pryer, 2007; Kuo et al., 2011) and identified challenging taxa (Pryer et al., 2004; Rothfels et al., 2012; Testo and Sundue, 2016; Wei et al., 2017), those generated from differing subsets of the nuclear genome should further elucidate the evolutionary history of the group. As a diversity of methods become available over the coming years, researchers will be able to choose an approach that is best suited for their needs.
DATA ACCESSIBILITY
Raw reads, target sequence files, sequence alignments, taxon information, and analysis details are available on Digital Commons (https://doi.org/10.15142/t3mg95). Intron‐containing “supercontigs” for each sample are deposited in GenBank (MG817735–MG818130), and the exon‐only alignments are deposited in TreeBASE (study S22073; https://treebase.org/treebase-web/search/study/anyObjectAsRDF.rdf?namespacedGUID=TB2:S22073).
ACKNOWLEDGMENTS
The authors thank three anonymous reviewers for suggestions that improved the manuscript.
Wolf, P. G. , Robison T. A., Johnson M. G., Sundue M. A., Testo W. L., and Rothfels C. J.. 2018. Target sequence capture of nuclear‐encoded genes for phylogenetic analysis in ferns. Applications in Plant Sciences 6(5): e1148.
LITERATURE CITED
- Adjie, B. , Masuyama S., Ishikawa H., and Watano Y.. 2007. Independent origins of tetraploid cryptic species in the fern Ceratopteris thalictroides . Journal of Plant Research 120: 129–138. [DOI] [PubMed] [Google Scholar]
- Altschul, S. F. , Gish W., Miller W., Myers E. W., and Lipman D. J.. 1990. Basic local alignment search tool. Journal of Molecular Biology 215: 403–410. [DOI] [PubMed] [Google Scholar]
- Bankevich, A. , Nurk S., Antipov D., Gurevich A., Dvorkin M., Kulikov A., Lesin V., et al. 2012. SPAdes: A new genome assembly algorithm and its applications to single‐cell sequencing. Journal of Computational Biology 19: 455–477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolger, A. M. , Lohse M., and Usadel B.. 2014. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 30: 2114–2120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, C. W. , Kuo L. Y., Wang C. N., and Chiou W. L.. 2012. Development of PCR primer sets for intron 1 of the low‐copy gene LEAFY in Davalliaceae. American Journal of Botany 99: E223–E225. [DOI] [PubMed] [Google Scholar]
- Doyle, L. L. , and Doyle J. L.. 1990. Isolation of plant DNA from fresh tissue. Focus 12: 13–15. [Google Scholar]
- Ebihara, A. , Ishikawa H., Matsumoto S., Lin S. J., Iwatsuki K., Takamiya N., Watano Y., and Ito M.. 2005. Nuclear DNA, chloroplast DNA, and ploidy analysis clarified biological complexity of the Vandenboschia radicans complex (Hymenophyllaceae) in Japan and adjacent areas. American Journal of Botany 92: 1535–1547. [DOI] [PubMed] [Google Scholar]
- Edgar, R. C. 2004. MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Research 19: 1792–1797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engels, M. E. , and Canestraro B. K.. 2017. ×Cyclobotrya: A new hybrid genus between Cyclodium and Polybotrya (Dryopteridaceae) from the Brazilian Amazon. Brittonia 69: 307–312. [Google Scholar]
- Hart, M. L. , Forrest L. L., Nicholls J. A., and Kidner C. A.. 2016. Retrieval of hundreds of nuclear loci from herbarium specimens. Taxon 65: 1081–1092. [Google Scholar]
- Hasebe, M. , Wolf P. G., Pryer K. M., Ueda K., Ito M., Sano R., Gastony G. J., et al. 1995. Fern phylogeny based on rbcL nucleotide sequences. American Fern Journal 85: 134–181. [Google Scholar]
- Heyduk, K. , Trapnell D. W., Barrett C. F., and Leebens‐Mack J.. 2016. Phylogenomic analyses of species relationships in the genus Sabal (Arecaceae) using targeted sequence capture. Biological Journal of the Linnean Society 117: 106–120. [Google Scholar]
- Ishikawa, H. , Watano Y., Kano K., Ito M., and Kurita S.. 2002. Development of primer sets for PCR amplification of the PgiC gene in ferns. Journal of Plant Research 115: 65–70. [DOI] [PubMed] [Google Scholar]
- James, K. E. , Schneider H., Ansell S. W., Evers M., Robba L., Uszynski G., Pedersen N., et al. 2008. Diversity Arrays Technology (DArT) for pan‐genomic evolutionary studies of non‐model organisms. PLoS ONE 3: e1682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson, M. G. , Gardner E. M., Liu Y., Medina R., Goffinet B., Shaw A. J., Zerega N. J. C., and Wickett N. J.. 2016. HybPiper: Extracting coding sequence and introns for phylogenetics from high‐throughput sequencing reads using target enrichment. Applications in Plant Sciences 4: 1600016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korall, P. , Pryer K. M., Metzgar J. S., Schneider H., and Conant D. S.. 2006a. Tree ferns: Monophyletic groups and their relationships as revealed by four protein‐coding plastid loci. Molecular Phylogenetics and Evolution 39: 830–845. [DOI] [PubMed] [Google Scholar]
- Korall, P. , Conant D. S., Schneider H., Ueda K., Nishida H., and Pryer K. M.. 2006b. On the phylogenetic position of Cystodium: It’s not a tree fern—It’s a polypod! American Fern Journal 96: 45–53. [Google Scholar]
- Kuo, L. Y. , Li F. W., Chiou W. L., and Wang C. N.. 2011. First insights into fern matK phylogeny. Molecular Phylogenetics and Evolution 59: 556–566. [DOI] [PubMed] [Google Scholar]
- Larsson, A. 2010. abioscripts, version 0.9.3. Available at: http://ormbunkar.se/phylogeny/abioscripts/ [accessed 23 April 2018].
- Larsson, A. 2014a. AliView: A fast and lightweight alignment viewer and editor for large datasets. Bioinformatics 30: 3276–3278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larsson, A. 2014b. Systematics of Woodsia: Ferns, bioinformatics and more. PhD thesis, Uppsala University, Uppsala, Sweden.
- Lehtonen, S. 2011. Towards resolving the complete fern tree of life. PLoS ONE 6: e24851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehtonen, S. , Wahlberg N., and Christenhusz M. J. M.. 2012. Diversification of lindsaeoid ferns and phylogenetic uncertainty of early polypod relationships. Botanical Journal of the Linnean Society 170: 489–503. [Google Scholar]
- Lemmon, A. R. , Emme S. A., and Lemmon E. M.. 2012. Anchored hybrid enrichment for massively high‐throughput phylogenomics. Systematic Biology 61: 727–744. [DOI] [PubMed] [Google Scholar]
- Leveille‐Bourret, E. , Starr J. R., Ford B. A., Lemmon E. M., and Lemmon A. R.. 2018. Resolving rapid radiations within angiosperm families using anchored phylogenomics. Systematic Biology 67: 94–112. [DOI] [PubMed] [Google Scholar]
- Li, F. W. , Melkonian M., Rothfels C. J., Villarreal J. C., Stevenson D. W., Graham S. W., Wong G. K. S., et al. 2015. Phytochrome diversity in green plants and the origin of canonical plant phytochromes. Nature Communications 6: 7852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, H. , and Durbin R.. 2009. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics 25: 1754–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matasci, N. , Hung L.‐H., Yan Z., Carpenter E. J., Wickett N. J., Mirarab S., Nguyen N., et al. 2014. Data access for the 1,000 Plants (1KP) project. GigaScience 3: 17–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mertes, F. , ElSharawy A., Sauer S., van Helvoort J. M. L. M., van der Zaag P. J., Franke A., Nilsson M., et al. 2011. Targeted enrichment of genomic DNA regions for next‐generation sequencing. Briefings in Functional Genomics 10: 374–386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller, M. A. , Pfeiffer W., and Schwartz T.. 2010. Creating the CIPRES Science Gateway for inference of large phylogenetic trees. Proceedings of the Gateway Computing Environments Workshop (GCE), New Orleans, Louisiana, USA, 2010, 1–8.
- Plant, A. L. , and Gray J. C.. 1988. Introns in chloroplast protein‐coding genes of land plants. Photosynthesis Research 16: 23–39. [DOI] [PubMed] [Google Scholar]
- PPG . 2016. A community‐derived classification for extant lycophytes and ferns. Journal of Systematics and Evolution 54: 563–603. [Google Scholar]
- Pryer, K. M. , Schuettpelz E., Wolf P. G., Schneider H., Smith A. R., and Cranfill R.. 2004. Phylogeny and evolution of ferns (monilophytes) with a focus on the early leptosporangiate divergences. American Journal of Botany 91: 1582–1598. [DOI] [PubMed] [Google Scholar]
- Ranker, T. A. , and Sundue M. A.. 2015. Why are there so few species of ferns? Trends in Plant Science 20: 402–403. [DOI] [PubMed] [Google Scholar]
- Ritland, K. , and Clegg M. T.. 1987. Evolutionary analysis of plant DNA sequences. American Naturalist 130: 74–100. [Google Scholar]
- Rothfels, C. J. , and Schuettpelz E.. 2014. Accelerated rate of molecular evolution for vittarioid ferns is strong and not driven by selection. Systematic Biology 63: 31–54. [DOI] [PubMed] [Google Scholar]
- Rothfels, C. J. , Larsson A., Kuo L. Y., Korall P., Chiou W. L., and Pryer K. M.. 2012. Overcoming deep roots, fast rates, and short internodes to resolve the ancient rapid radiation of eupolypod II ferns. Systematic Biology 61: 490–509. [DOI] [PubMed] [Google Scholar]
- Rothfels, C. J. , Larsson A., Li F. W., Sigel E. M., Huiet L., Burge D. O., Ruhsam M., et al. 2013. Transcriptome‐mining for single‐copy nuclear markers in ferns. PLoS ONE 8: e76957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothfels, C. J. , Johnson A. K., Hovenkamp P. H., Swofford D. L., Roskam H. C., Fraser‐Jenkins C. R., Windham M. D., and Pryer K. M.. 2015a. Natural hybridization between genera that diverged from each other approximately 60 million years ago. American Naturalist 185: 433–442. [DOI] [PubMed] [Google Scholar]
- Rothfels, C. J. , Li F.‐W., Sigel E. M., Huiet L., Larsson A., Burge D. O., Ruhsam M., et al. 2015b. The evolutionary history of ferns inferred from 25 low‐copy nuclear genes. American Journal of Botany 102: 1089–1107. [DOI] [PubMed] [Google Scholar]
- Rothfels, C. J. , Pryer K. M., and Li F. W.. 2017. Next‐generation polyploid phylogenetics: Rapid resolution of hybrid polyploid complexes using PacBio single‐molecule sequencing. New Phytologist 213: 413–429. [DOI] [PubMed] [Google Scholar]
- Schneider, H. , Navarro‐Gomez A., Russell S. J., Ansell S., Grundmann M., and Vogel J.. 2013. Exploring the utility of three nuclear regions to reconstruct reticulate evolution in the fern genus Asplenium . Journal of Systematics and Evolution 51: 142–153. [Google Scholar]
- Schuettpelz, E. , and Pryer K. M.. 2007. Fern phylogeny inferred from 400 leptosporangiate species and three plastid genes. Taxon 56: 1037–1050. [Google Scholar]
- Schuettpelz, E. , Grusz A. L., Windham M. D., and Pryer K. M.. 2008. The utility of nuclear gapCp in resolving polyploid fern origins. Systematic Botany 33: 621–629. [Google Scholar]
- Sessa, E. B. , Zimmer E. A., and Givnish T. J.. 2012. Reticulate evolution on a global scale: A nuclear phylogeny for New World Dryopteris (Dryopteridaceae). Molecular Phylogenetics and Evolution 64: 563–581. [DOI] [PubMed] [Google Scholar]
- Shen, H. , Jin D., Shu J.‐P., Zhou X.‐L., Lei M., Wei R., Shang H., et al. 2017. Large‐scale phylogenomic analysis resolves a backbone phylogeny in ferns. GigaScience 7: gix116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slater, G. S. , and Birney E.. 2005. Automated generation of heuristics for biological sequence comparison. BMC Bioinformatics 6: 31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smit, A. F. A. , Hubley R., and Green P.. 2013. RepeatMasker, version open‐4.0.6. Available at: http://www.repeatmasker.org [accessed 23 April 2018].
- Sukumaran, J. , and Holder M. T.. 2010. DendroPy: A Python library for phylogenetic computing. Bioinformatics 26: 1569–1571. [DOI] [PubMed] [Google Scholar]
- Swofford, D. L. 2002. PAUP*: Phylogenetic analysis using parsimony (*and other methods) version 4. Sinauer Associates, Sunderland, Massachusetts, USA. [Google Scholar]
- Testo, W. , and Sundue M.. 2016. A 4000‐species dataset provides new insight into the evolution of ferns. Molecular Phylogenetics and Evolution 105: 200–211. [DOI] [PubMed] [Google Scholar]
- Wagner, W. H. J. 1993. New species of Hawaiian pteridophytes. Contributions to University of Michigan Herbarium 19: 63–82. [Google Scholar]
- Wagner, W. H. J. , Wagner F. S., Reznicek A. A., and Werth C. R.. 1992. ×Dryostichum singulare (Dryopteridaceae), a new fern nothogenus from Ontario. Canadian Journal of Botany 70: 245–253. [Google Scholar]
- Wei, R. , Yan Y. H., Harris A. J., Kang J. S., Shen H., Xiang Q. P., and Zhang X. C.. 2017. Plastid phylogenomics resolve deep relationships among Eupolypod II ferns with rapid radiation and rate heterogeneity. Genome Biology and Evolution 9: 1646–1657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wickett, N. J. , Mirarab S., Nam N., Warnow T., Carpenter E., Matasci N., Ayyampalayam S., et al. 2014. Phylotranscriptomic analysis of the origin and early diversification of land plants. Proceedings of the National Academy of Sciences, USA 111: E4859–E4868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zurawski, G. , and Clegg M. T.. 1987. Evolution of higher‐plant chloroplast DNA‐encoded genes: Implications for structure‐function and phylogenetic studies. Annual Review of Plant Physiology 38: 391–418. [Google Scholar]
- Zurawski, G. , Whitfeld P. R., and Bottomley W.. 1986. Sequence of the gene for the large subunit of ribulose 1,5‐bisphosphate carboxylase from pea chloroplasts. Nucleic Acids Research 14: 3975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zwickl, D. J. 2006. GARLI: Genetic algorithm approaches for the phylogenetic analysis of large biological sequence datasets under the maximum likelihood criterion. PhD thesis, University of Texas, Austin, Texas, USA.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Raw reads, target sequence files, sequence alignments, taxon information, and analysis details are available on Digital Commons (https://doi.org/10.15142/t3mg95). Intron‐containing “supercontigs” for each sample are deposited in GenBank (MG817735–MG818130), and the exon‐only alignments are deposited in TreeBASE (study S22073; https://treebase.org/treebase-web/search/study/anyObjectAsRDF.rdf?namespacedGUID=TB2:S22073).