

Abstract
Droplet microfluidics can identify and sort cells using digital reverse transcription polymerase chain reaction (RT-PCR) signals from individual cells. However, current methods require multiple microfabricated devices for enzymatic cell lysis and PCR reagent addition, making the process complex and prone to failure. Here, we describe a new approach that integrates all components into a single device. The method enables controlled exposure of isolated single cells to a high pH buffer, which lyses cells and inactivates reaction inhibitors but can be instantly neutralized with RT-PCR buffer. Using our chemical lysis approach, we distinguish individual cells’ gene expression with data quality equivalent to more complex two-step workflows. Our system accepts cells and produces droplets ready for amplification, making single-cell droplet RT-PCR faster and more reliable.
Introduction
Cells are the elementary unit of biology, but rarely exist in isolation. More often, biological systems are composed of millions of cells with different phenotypes. This complexity means that analysis of bulk samples can mask the importance of subpopulations. Single-cell analysis using well-plates and robotics can interrogate individual cells, but normally analyze a small fraction of the sample. Ultrahigh-throughput methods are necessary to fully characterize millions of cells, especially when important cell types are present at low levels.
Transcriptional analysis with RNA-seq has become the gold standard for understanding cell state because it provides genome-wide characterization of RNA expression.1,2 When applied to single cells, transcriptome analysis can identify the molecular underpinnings of many biological phenotypes3–10 including the functional properties of tissues and the dysregulated gene expression of disease.11–13 However, single-cell transcriptome sequencing on millions of cells is currently prohibitively expensive—the best methods capable of analyzing just thousands of cells. Often, a single transcript biomarker is sufficient for determining the cell state of interest, for example, when seeking cells expressing cancer-specific or viral transcripts. Imaging technologies with in situ hybridization can differentiate between cells based on specific mRNAs, are capable of high throughput, and provide valuable spatial information.14,15 However, these methods have limited sensitivity for detecting lowly-expressed genes, and downstream sequencing of sorted cells can be challenging due to nucleic acid degradation from the requisite chemical fixation.
Microfluidic techniques allow specific RNA sequences in single cells to be detected via reverse transcription polymerase chain reaction (RT-PCR), providing an alternative route to single-cell gene expression analysis. They are high throughput, capable of analyzing thousands (chambers, wells) to millions (microdroplets) of single cells.16–19 Moreover, droplets can be analyzed in flow and sorted, affording a capability reminiscent of flow cytometric analysis but in which cell interrogation is based on nucleic acid content. Since nucleic acids are the principal information-carrying medium of cells, the ability to robustly distinguish between and sort cells based on them is a significant advance. Moreover, sorted cells can be subjected to additional analyses, such as genomic and transcriptomic sequencing.20–22
Performing RT-PCR on single cells in microfluidic droplets is difficult because cells must be lysed to release RNA. Lysis agents and lysate can inhibit the RT-PCR reactions required to identify target sequences. This is especially true in droplet microfluidics, where a single cell can represent an appreciable fraction of the droplet volume. Several strategies have been employed to reduce inhibition including lysate digestion with proteases,20,21 which must be inactivated by heating prior to addition of reverse transcriptase and DNA polymerase. This necessitates separate microfluidic devices for cell lysis and reagent addition. Moreover, the droplets from the lysis device must be collected off chip, heated, and reinjected into the next device, a labor-intensive process that is error-prone due to emulsion coalescence during heating and handling. To enable more reliable high-throughput single-cell droplet RT-PCR, an optimal method would integrate cell lysis and reagent addition into one device, obviating the complicated steps of collecting, heating, and reinjecting emulsions.
In this paper, we describe a novel single-cell RT-PCR method that integrates lysis and reagent addition into one device. The method is based on alkaline lysis, which ruptures cell membranes and rapidly denatures endogenous nucleases and inhibitory proteins in a high pH buffer. In addition, the lysing agent can be instantly neutralized with RT-PCR buffer. Our system comprises one device that accepts cells and produces droplets ready for amplification on a PCR machine, making high-throughput single-cell droplet RT-PCR faster and more reliable than previous methods.
Materials and methods
Microfabrication
The microfluidic device is made by multilayer soft lithography.23 Photomasks for channel layers of different heights are drawn with a vector graphics software (AutoCAD) and printed on a transparency film (CAD/Art Services). Layer one (6.5-μm tall) is composed of thin narrow oil extractor channels (Fig. 2d), which enable oil removal while keeping droplets in the main channel.24 Layer two (35-μm tall) consists of cell encapsulation and coflow channels (Fig. 2b,c). Layer three (80-μm tall) consists of the delay line, RT-PCR drop maker, merger and saltwater electrode/moat channels (Fig. 2e,f). The master mold is fabricated by spincoating (SU-8 3005 for layer one and SU-8 3025 for layers two and three) on a 3-inch silicon wafer (University Wafers) at recommended speeds. Mask alignments and UV irradiations are performed after each spin coat, and the final wafer is developed in propylene glycol monomethyl ether acetate (PGMEA). If first layer alignment marks are difficult to see due to their shallow depth, it is possible to spin, expose, and develop guide alignment marks at higher depth (~50 μm). All subsequent layers can be aligned to these guide marks. Poly(dimethylsiloxane) (PDMS) (Dow Corning, Sylgard 184) base and curing agent are mixed at a weight ratio of 10:1, degassed under vacuum and poured on the master mold placed in a petri dish. The clear PDMS mixture is cured at 65°C for 4 h and cut out with a blade. Access holes of 750-μm diameter are punched to match polyethylene tubing (Scientific Commodities, PE/2, OD 1.09 mm) for injecting liquid into microchannels. The channel side of a punched PDMS slab is cleaned with a Scotch tape and plasma-treated for 60 s at 1 mbar of oxygen (Harrick Plasma, PDC-001) for bonding to a glass slide. After baking at 65°C for 1 h, the channels are treated with Aquapel to convert inner surface to be hydrophobic to facilitate droplet generation.
Figure 2.
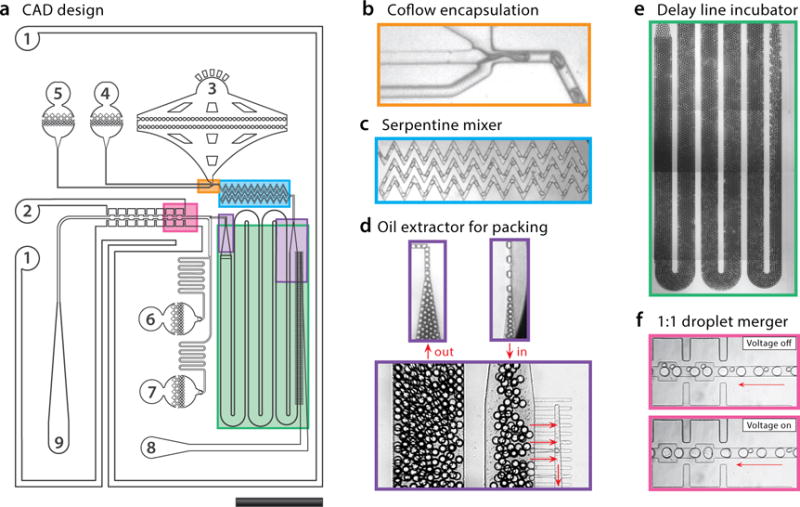
CAD design of the device with important regions indicated by colored boxes, and inlets and outlets numbered (a). Ports #1 and #2 are electrodes, generated by filling the channels with salt water. Ports #3–#7 accept oil or aqueous reagents for device operation. Ports #8 (extractor) and #9 (droplets) are outlets. Cell encapsulator consisting of a modified T-junction with co-flow inlets for cell suspension and lysis buffer (b). The serpentine mixer homogenizes droplet contents in under a second (c). The oil extractor packs droplets together so that they flow at uniform velocity (d). To increase incubation time, the delay line incubator length is set according to the droplet velocity (e). Lysis droplets are paired with PCR droplets produced by a parallel T-junction (not shown) and fused via electrocoalescence (f). The droplets are ~80 μm after coalescence and the entire device is 10 × 15 mm.
Cells & chemicals
Jurkat and MCF7 cells are cultured in RPMI-1640 medium supplemented with 5% fetal bovine serum (FBS) and antibiotics at 37°C in the presence of 5% CO2. 0.8–1 million Jurkat/MCF7 cells are collected and washed with cold PBS supplemented with 2% FBS (PBS-F). Cells are centrifuged at 300 RCF for 3 minutes, resuspended in 100 μL of 10 μM calcein violet (Thermo Fisher, C34858) or 25 μM calcein red (C34851) in PBS-F, and incubated on ice for 30 minutes. After cell staining, cells are pelleted, resuspended in 100 μL of PBS-F, mixed together and supplemented with 40 μL (0.2× volume) of Optiprep (Sigma, D1556) for density matching. The alkaline lysis buffer (ALB) contains 200 mM NaOH, 60%(v/v) PEG-200 and 2%(v/v) Triton X-100. It is noteworthy that the addition of PEG-200 increases pH significantly,25 thus enabling the use of a lower NaOH concentration during the lysis step. This is important so that the buffer capacity of RT-PCR reagents is sufficient to effectively neutralize the alkaline after the merge step. Water-in-oil droplets are generated in a fluorinated oil (3M, HFE Novec 7500) supplemented with 2%(w/w) 008-FluoroSurfactant (RAN Biotechnologies). The oil phase of collected droplets is swapped with the FC-40 oil (Sigma, F9755) supplemented with 5%(w/w) 008-FluoroSurfactant immediately before thermocycling droplets for RT-PCR. The RT-PCR mix is composed of 1× Reaction Mix and 1× Enzyme Mix (Thermo Fisher, 12574030, SuperScript III One-Step) supplemented with 2.5%(v/v) Tween 20 and 2.5%(v/v) PEG 6,000. The FAM-labeled TaqMan probe specific to CD45 protein (IDT PrimeTime, Assay ID: Hs.PT.53a.2558434) is added to the RT-PCR mix.
Device operation & RT-PCR
Computer-controlled syringe pumps (New Era Pump Systems, NE-501) are used to inject liquids into the device at the flow rates as specified below (the numbers match those on Fig. 2): #3 (cell) = 40 μL/hr, #4 (ALB) = 40 μL/hr, #5 (oil) = 250 μL/hr, #6 (oil extractor) = −220 μL/hr, #7 (RT-PCR mix) = 400 μL/hr, and #8 (oil) = 900 μL/hr. The oil extractor uses a syringe pump operating in the withdrawing mode. Droplet generation is monitored on an inverted microscope equipped with a fast-shutter camera (Unibrain, Fire-i 530b). When 1:1 pairing of cell lysate drops and RT-PCR mix drops is achieved, a voltage is applied through the saltwater electrode filled with 1 M NaCl to induce droplet merging via electrocoalescence. The droplets are collected in multiple PCR tubes by collecting for 2 minutes each, the oil phase is exchanged with 5%(w/w) surfactant oil, and the emulsions are subjected to the recommended RT-PCR thermal cycles: 55°C for 15 minutes, 94°C for 2 minutes, 40 cycles of denature (94°C, 15 s), anneal (55°C, 30 s) and extend (68°C, 60 s) steps, and a final extension (68°C, 5 minutes).
Fluorescence imaging & analysis
After RT-PCR thermal cycles, the droplets are loaded into a microscope slide (Thermo Fisher, C10312) and imaged with an inverted epi-fluorescence microscope (Nikon, Ti microscope, DS-Qi2 camera and Plan Apo λ 4× objective lens) using the following filter settings: brightfield, DAPI (for calcein violet; 440/40m), FITC (for FAM; 525/30m) and CY3 (for calcein red; 607/36m). The exposure times are optimized by the camera control software. Droplet parameters and fluorescence signals are determined with Fiji’s Analyze Particles routine (centroidSize = 350–1500, circularity = 0.7–1.0).26 Obtained fluorescence signals are analyzed and plotted with R.
Results and discussion
A single cell contained in a 100 pL droplet has the equivalent concentration is 10 million cells/mL. At this concentration, endogenous molecules release by a cell during lysis can inhibit many biological reactions. A common strategy for overcoming inhibition is to purify molecules of interest (DNA, RNA, protein) from the lysate, and then analyze them. However, purifications are challenging in microfluidic droplets because droplets are self-contained reactors that cannot be subjected to the requisite extraction columns, capillary electrophoresis, and chemical precipitation techniques.
Under such circumstances, efficient RT-PCR can be achieved by digesting concentrated lysates with proteases, which non-specifically degrade cellular proteins, including endogenous nucleases and proteases that can inhibit biochemical reactions. However, these proteases are incompatible with RT-PCR reagents because they degrade the essential reaction enzymes. Consequently, methods employing protease digestion require two steps to prepare droplet reactions; a first in which the cells are encapsulated and lysed, and a second in which the lysis droplets are merged with reagent droplets after the protease has been deactivated by heating. While effective, these workflows are difficult, requiring careful emulsion handling, off-chip thermal incubation, and droplet reinjection into a second device. Droplet reinjection, in particular, is error-prone due to emulsion coalescence. Performing lysis and RT-PCR reagent addition on a single chip would, thus, increase the speed and reliability of droplet single-cell RT-PCR analysis.
Our integrated device uses alkaline lysis, a chemical strategy that denatures inhibitory proteins but is instantly and conveniently neutralized by buffer. Moreover, alkaline lysis is fast and effective at room temperature, requiring only seconds to lyse cells for RT-PCR. This allows integration of all steps into one device and obviates thermal control (Fig. 1), which simplifies the workflow significantly. Our device encapsulates live cells and alkaline buffer in droplets (Fig. 1a). The droplets pass through a micromixer (Fig. 1b) and are incubated in a long channel for 30 s (Fig. 1c). After incubation, the alkaline lysis solution is neutralized and the reagent added in a merger (Fig. 1d), yielding droplets ready for amplification (Figs. 1e,f). In addition to streamlining the workflow and increasing reliability, alkaline lysis enables efficient RT-PCR at smaller droplet sizes relative to reported two-step workflows. This boosts throughput from 168 droplets/s to 411 droplets/s and reduces RT-PCR reagent volume to analyze 100,000 cells from 660 μL to 270 μL at 10% cell loading ratio. The result is that reported two-step workflows require 120 minutes to prepare 47,078 single-cell RT-PCR droplets,20 including proteinase treatment (55°C, 15 min), heat inactivation (90°C, 10 min), and reinjection of cell lysate droplets into the second microfluidic device, while our integrated approach does this in 19 min with one device.
Figure 1.
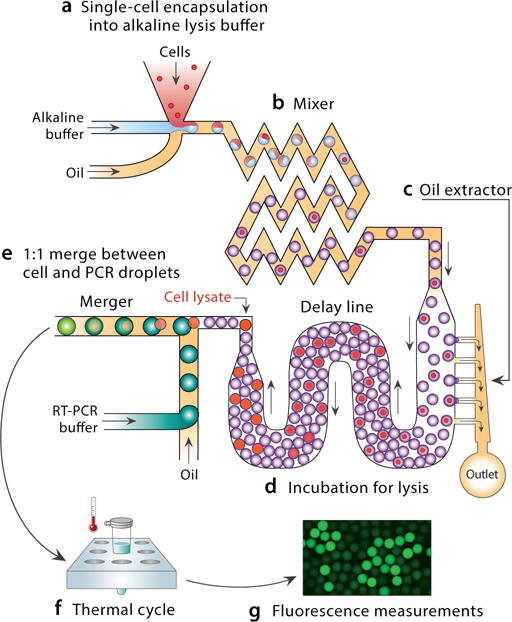
Schematic of single-cell RT-PCR in a device integrating lysis and reagent addition. The device accepts cells and produces droplets ready for amplification. Cells are encapsulated with lysis buffer (a) and flowed through a mixer (b). Excess oil is removed from the emulsion (c), thereby packing the droplets and yielding uniform flow that allows them to be incubated for a controlled time in a delay line (d). Lysed-cell containing droplets are merged with PCR droplets, neutralizing the lysis buffer and readying them for amplification (e). Cells expressing the target mRNA amplify by RT-PCR, yielding TaqMan-positive droplets (f).
A detailed schematic of our device is shown in Fig. 2a. Single-cell encapsulation is achieved with a co-flow geometry in which the cell stream (#3) merges with the alkaline lysis stream (#4) from a parallel channel; the oil (#5) required for droplet generation is added from a third channel (Fig. 2b). To ensure that droplet contents immediately mix and the cells bathe in lysis buffer, we pass the droplets through a serpentine mixer (Fig. 2c).27,28 While high pH is effective for cell lysis and protein denaturation, it also results in hydrolysis of mRNA molecules, reducing RT-PCR sensitivity. Thus, alkaline incubation must be controlled tightly to ensure full lysis and protect RNA quality, which is accomplished by packing droplets so that they move with uniform velocity to control lysis time. Droplet packing is achieved by removing oil from the emulsion with an oil extractor (Fig. 2d).24 The droplets then move through the incubation line with length selected to achieve the optimal alkaline incubation for 30 s (Fig. 2e). The alkaline buffer is neutralized and RT-PCR reagent added via droplet merger. A parallel T-junction (inlets #6 and #7, Fig. 2a) generates 80-μm reagent droplets at 411 droplets/s. Packing of the droplets also allows controlled, periodic spacing, to synchronize cell lysate and reagent droplet coalescence (Fig. 2f).24
To illustrate the effectiveness of alkaline lysis for mammalian cells, we use the co-flow device to encapsulate calcein-stained cells in 30-μm droplets. When we omit the alkaline lysing agent, the cells remain intact and appear as bright puncta (Fig. 3a). By contrast, when it is included, the cells lyse, yielding homogeneously fluorescent droplets distributed with lysate (Fig. 3b).
Figure 3.

Alkaline lysis of cells. When stained cells are encapsulated in PBS buffer droplets, they remain fluorescent (a). When they are encapsulated in alkaline lysis buffer droplets, they quickly lyse, yielding homogenously fluorescent droplets containing their lysate (b). The scale bar is 400 μm.
Our streamlined approach allows cells to be identified with RT-PCR of specific genes that are expressed. To demonstrate this, we analyze a sample of Jurkat cells using the live-cell stain calcein (red fluorescence), to identify cell positive droplets, and RT-PCR TaqMan (green fluorescence) targeting CD45 to identify a specific transcript produced by these cells. We process the cells through the workflow and image the resultant droplets with microscopy. The cells are loaded at a limiting dilution per droplet of 5.8%; consequently, when viewed under the red channel, we observe rare instances of red-fluorescent droplets containing lysates (Fig. 4a). The dark droplets, which cannot be easily seen, are devoid of cells. Because Jurkat cells express CD45, droplets containing these cells should be TaqMan-positive, exhibiting fluorescence in the FAM channel (Fig. 4b). Indeed, we find that calcein-positive droplets tend to also be TaqMan-positive, appearing orange in the composite image (Fig. 4c). However, while nearly all cell-containing droplets are TaqMan-positive, many TaqMan-positive droplets are devoid of cell stain; this is also evident in the scatter plot at high FAM fluorescence but low calcein (Fig 4d). This is commonly observed when performing single-cell RT-PCR in droplets and is thought to result from release of mRNAs from cells prior to encapsulation.20 A single lysed cell can release thousands of mRNA targets and since droplet RT-PCR is single-molecule sensitive, this can lead to thousands of TaqMan-positive droplets devoid of cells. Such “digital background” is an important issue that is best addressed by staining cells so that all cell-negative droplets can be omitted from the analysis.21 These results demonstrate that our integrated device behaves equivalently to reported two-step devices and allows detection of cells based on expressed genes.
Figure 4.
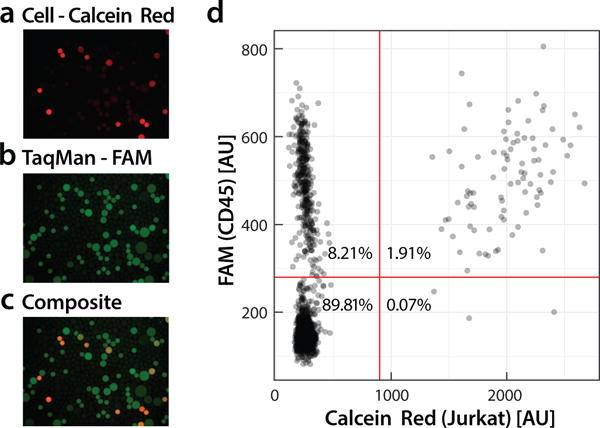
Fluorescence analysis of single-cell RT-PCR droplets. Jurkat cells are stained with calcein red dye (a) and analyzed for expression of CD45 mRNA using a TaqMan probe (b). Colocalization of calcein red and FAM indicates droplets containing cells expressing the target (c). Droplets with FAM fluorescence but no calcein red represent “digital background” in which positive amplification has resulted due to presence of single mRNA molecules of CD45. Using image analysis, droplet fluorescence is measured and the results plotted (d). Double-positive droplets appear on the upper-right and represent cells detected to express the target gene.
To show that our streamlined workflow can identify specific cells based on unique gene expression signatures, we analyze a mixed population of MCF7 and Jurkat cells. Prior to combining these cells, we stain both types with different calcein dyes (red for Jurkat and violet for MCF7), which act as internal controls to evaluate the specificity of the gene expression-based determination. For detection, we again target CD45, a gene expressed in Jurkat but not MCF7 cells. We process the mixed population through the workflow and assess the results via fluorescence microscopy. When viewed under the relevant fluorescence channels, many droplets exhibit red, violet, or green fluorescence, corresponding to Jurkat, MCF7, or TaqMan-positive droplets, respectively (Figs. 5a–c). When the images are overlaid, we observe a mosaicity of fluorescence colors (Fig. 5d). To determine whether this is consistent with the known gene expression of these cells, we plot droplet fluorescence for the different colors (Fig. 5e). Droplets positive for Jurkat dye also tend to be positive for CD45, while ones positive for MCF7 dye tend to be negative for this gene, as expected. These differences are also clear when projecting the results on the three relevant 2D scatter plots (Figs. 5f–h), demonstrating that the TaqMan assay is specific and can detect a target cell in a mixed population, just as described two-step workflows.20,21 By combining detection with droplet sorting, as has been demonstrated previously, a specific subpopulation of cell lysates can be recovered and analyzed further.
Figure 5.

Detection of Jurkat cells spiked into MCF7 cells. Jurkat (a) and MCF7 (b) cells are stained with calcein red and calcein violet, respectively. Mixed cells are analyzed using the workflow with a FAM-labeled TaqMan probe specific to CD45 (c), expressed in Jurkat but not in MCF7 cells. An overlay of all channels reveals that FAM-positive droplets colocalize with calcein red-positive droplets, appearing yellow-orange (d). Using image analysis, droplet fluorescence is measured for all channels and the results plotted (e-h). The total number of analyzed droplets is 14,249. The scatter plots between FAM and cell stain channels show that most calcein red-positive droplets are also FAM-positive (g), while most calcein violet-positive droplets are FAM-negative (h), as expected based on the differing gene expression of these cells.
Conclusion
Flow cytometry is an immensely important tool in biology but is largely limited to identifying cells based on proteomic markers detectable with specific antibodies. Nucleic acids are the primary information encoding medium of cells and many traits of interest are easiest detected at the nucleic acid level, such as genetic mutations, mRNA splicing variants, or unique gene expression signatures.29–31 The inability to reliably detect and sort cells based on nucleic acid content is, thus, a major limitation of current flow cytometry.32 New droplet microfluidic techniques allow this using single-cell PCR, filling a major need, but are challenging to implement due to the requisite complex, multi-step and multi-device workflows.33 Our results demonstrate an integrated on-chip solution for cell lysis and reagent merger, providing a faster and more reliable method for single-cell droplet RT-PCR.
The ability to rapidly interrogate single cells with RT-PCR is valuable for a broad variety of applications in biology, from identifying specific cell types in tissue and blood to deconvoluting the dysregulated gene expression of cancer cells. Previous microfluidic techniques have used proteases for cell lysis that are incompatible with RT-PCR reagents; this necessitates multiple steps of lysis, protease deactivation, and reagent addition that results in long and error-prone workflows. Using our alkaline strategy, lysis and reagent addition are accomplished with an integrated device accepting a cell suspension and producing droplets ready for RT-PCR analysis, each cell spending less than 45 s in the device. Integration of all components and the ability to use smaller droplets boosts throughput; this shortens workflow duration, increases the number of cells that can be analyzed, and circumvents the error-prone steps of off-chip emulsion handling, heating, and reinjection required for previous methods. These attributes should make our integrated approach the method of choice for single-cell RT-PCR in microfluidic droplets.
Acknowledgments
We are grateful to J. Haliburton and L. Liu for helpful discussions. We thank the Nikon Imaging Center at UCSF for the fluorescence microscopy. This work was supported by the National Science Foundation through a CAREER Award (grant number DBI-1253293); the National Institutes of Health (NIH) (grant numbers HG007233-01, R01-EB019453-01, 1R21HG007233, DP2-AR068129-01, R01-HG008978, R21AI116218); and the Defense Advanced Research Projects Agency Living Foundries Program (contract numbers HR0011-12-C-0065, N66001-12-C-4211, HR0011-12-C-0066) and Fold F(x) Program (Contract No. DE-AC02-05CH11231). Funding for open access charge: NIH grant number DP2-AR068129-01.
References
- 1.Wang Z, Gerstein M, Snyder M. Nat Rev Genet. 2009;10:57–63. doi: 10.1038/nrg2484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wagner A, Regev A, Yosef N. Nat Biotech. 2016;34:1145–1160. doi: 10.1038/nbt.3711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Tang F, Lao K, Surani MA. Nat Meth. 2011;8:S6–S11. doi: 10.1038/nmeth.1557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Shapiro E, Biezuner T, Linnarsson S. Nat Rev Genet. 2013;14:618–630. doi: 10.1038/nrg3542. [DOI] [PubMed] [Google Scholar]
- 5.Jaitin DA, Kenigsberg E, Keren-Shaul H, Elefant N, Paul F, Zaretsky I, Mildner A, Cohen N, Jung S, Tanay A, Amit I. Science. 2014;343:776–779. doi: 10.1126/science.1247651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Klein AM, Mazutis L, Akartuna I, Tallapragada N, Veres A, Li V, Peshkin L, Weitz DA, Kirschner MW. Cell. 2015;161:1187–1201. doi: 10.1016/j.cell.2015.04.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Macosko E, et al. Cell. 2015;161:1202–1214. doi: 10.1016/j.cell.2015.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fan HC, Fu GK, Fodor SPA. Science. 2015;347:1258367. doi: 10.1126/science.1258367. [DOI] [PubMed] [Google Scholar]
- 9.Zilionis R, Nainys J, Veres A, Savova V, Zemmour D, Klein AM, Mazutis L. Nat Protocols. 2017;12:44–73. doi: 10.1038/nprot.2016.154. [DOI] [PubMed] [Google Scholar]
- 10.Gierahn TM, Wadsworth MH, II, Hughes TK, Bryson BD, Butler A, Satija R, Fortune S, Love JC, Shalek AK. Nat Meth. 2017;14:395–398. doi: 10.1038/nmeth.4179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Melé M, et al. Science. 2015;348:660–665. doi: 10.1126/science.aaa0355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Scotti MM, Swanson MS. Nat Rev Genet. 2016;17:19–32. doi: 10.1038/nrg.2015.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gupta S, Ellis SE, Ashar FN, Moes A, Bader JS, Zhan J, West AB, Arking DE. Nature Communications. 2014;5:5748. doi: 10.1038/ncomms6748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Baker M. Nat Meth. 2012;9:787–790. [Google Scholar]
- 15.Moffitt JR, Hao J, Wang G, Chen KH, Babcock HP, Zhuang X. PNAS. 2016;113:11046–11051. doi: 10.1073/pnas.1612826113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.White AK, VanInsberghe M, Petriv OI, Hamidi M, Sikorski D, Marra MA, Piret J, Aparicio S, Hansen CL. PNAS. 2011:13999–14004. doi: 10.1073/pnas.1019446108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhang Y, Zhu Y, Yao B, Fang Q. Lab on a Chip. 2011;11:1545–1549. doi: 10.1039/c0lc00502a. [DOI] [PubMed] [Google Scholar]
- 18.Sanchez-Freire V, Ebert AD, Kalisky T, Quake SR, Wu JC. Nat Protocols. 2012;7:829–838. doi: 10.1038/nprot.2012.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhang H, Jenkins G, Zou Y, Zhu Z, Yang CJ. Anal Chem. 2012;84:3599–3606. doi: 10.1021/ac2033084. [DOI] [PubMed] [Google Scholar]
- 20.Eastburn DJ, Sciambi A, Abate AR. Anal Chem. 2013;85:8016–8021. doi: 10.1021/ac402057q. [DOI] [PubMed] [Google Scholar]
- 21.Eastburn DJ, Sciambi A, Abate AR. Nucl Acids Res. 2014;42:e128. doi: 10.1093/nar/gku606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhu Y, Zhang Y-X, Liu W-W, Ma Y, Fang Q, Yao B. Scientific Reports. 2015;5:9551. doi: 10.1038/srep09551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Unger MA, Chou HP, Thorsen T, Scherer A, Quake SR. Science. 2000;288:113–116. doi: 10.1126/science.288.5463.113. [DOI] [PubMed] [Google Scholar]
- 24.Haliburton JR, Kim SC, Clark IC, Sperling RA, Weitz DA, Abate AR. Biomicrofluidics. 2017;11:034111. doi: 10.1063/1.4984035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chomczynski P, Rymaszewski M. BioTechniques. 2006;40:454, 456, 458. doi: 10.2144/000112149. [DOI] [PubMed] [Google Scholar]
- 26.Schindelin J, et al. Nat Meth. 2012;9:676–682. doi: 10.1038/nmeth.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Song H, Tice JD, Ismagilov RF. Angewandte Chemie International Edition. 2003;42:768–772. doi: 10.1002/anie.200390203. [DOI] [PubMed] [Google Scholar]
- 28.Wang J, Wang J, Feng L, Lin T. RSC Adv. 2015;5:104138–104144. [Google Scholar]
- 29.Pekin D, Skhiri Y, Baret J-C, Corre DL, Mazutis L, Salem CB, Millot F, Harrak AE, Hutchison JB, WLarson J, RLink D, Laurent-Puig P, DGrif-fiths A, Taly V. Lab on a Chip. 2011;11:2156–2166. doi: 10.1039/c1lc20128j. [DOI] [PubMed] [Google Scholar]
- 30.Manning KS, Cooper TA. Nat Rev Mol Cell Biol. 2017;18:102–114. doi: 10.1038/nrm.2016.139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Krebs MG, Metcalf RL, Carter L, Brady G, Blackhall FH, Dive C. Nat Rev Clin Oncol. 2014;11:129–144. doi: 10.1038/nrclinonc.2013.253. [DOI] [PubMed] [Google Scholar]
- 32.Clark IC, Abate AR. Lab Chip. 2017;17:2032–2045. doi: 10.1039/c7lc00241f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pellegrino M, Sciambi A, Yates JL, Mast JD, Silver C, Eastburn DJ. BMC Genomics. 2016;17:361. doi: 10.1186/s12864-016-2694-2. [DOI] [PMC free article] [PubMed] [Google Scholar]