

Summary
Background
Dabrafenib plus trametinib (D+T) improves outcomes in BRAF V600–mutant metastatic melanoma without brain metastases; however, activity of D+T has not been studied in active melanoma brain metastases (MBM). Here, we report results from the phase 2 trial COMBI-MB. Our aim was to build upon the current body of evidence of targeted therapy in melanoma brain metastases through an evaluation of D+T in patients with BRAF V600–mutant melanoma brain metastases.
Methods
This ongoing open-label, phase 2 study (NCT02039947) evaluated dabrafenib 150 mg twice daily plus trametinib 2 mg once daily in four melanoma patient cohorts: (A) BRAF V600E, asymptomatic MBM, no prior local brain therapy; (B) BRAF V600E, asymptomatic MBM, prior local brain therapy; (C) BRAF V600D/K/R, asymptomatic MBM, with or without prior local brain therapy; and (D) BRAF V600D/E/K/R, symptomatic MBM, with or without prior local brain therapy. The primary objective was to assess intracranial response rate (IRR) in cohort A in the all-treated-subjects population. Secondary endpoints included IRR in cohorts B–D; extracranial and overall response rates; disease control rates; duration of intracranial, extracranial, and overall response; progression-free survival; overall survival; and safety.
Findings
A total of 125 patients were enrolled (A: n=76; B: n=16; C: n=16; D: n=17). At the data cutoff (November 28, 2016; median follow-up 8·5 months) investigator-assessed IRR was 58% (n=44/76) in cohort A. Intracranial response by investigator assessment was also achieved in 9 (56%) of 16 patients in cohort B, 7 (44%) of 16 patients in cohort C, and 10 (59%) of 17 patients in cohort D. Safety results were consistent with prior D+T studies, with 60 (48%) of 125 patients across cohorts experiencing grade 3/4 adverse events. The most common serious adverse events across cohorts were pyrexia (n=9/125; 7%) and ejection fraction decreased (n=5/125; 4%).
Interpretation
D+T was active with a manageable safety profile in patients with BRAF V600–mutant MBMs, but the median duration of response was relatively short. These results provide evidence of clinical benefit with D+T and support the need for additional research to further improve outcomes in patients with MBMs.
Funding
Novartis.
Introduction
Among common cancers, metastatic melanoma has the highest risk of spreading to the central nervous system.1,2 The development of brain metastases in patients with melanoma has been observed at an incidence of up to 43% and 75% in clinical and autopsy studies, respectively.2,3 Historically, brain metastases in patients with metastatic melanoma have been associated with poor overall survival (median 4–5 months), and the poorest outcomes are observed in those presenting with neurological symptoms and leptomeningeal involvement.4,5 Multiple targeted therapies (ie, BRAF and MEK inhibitors) and checkpoint inhibitor immunotherapies (ie, anti–cytotoxic T-lymphocyte–associated protein 4 [CTLA-4], anti–programmed death-1 [PD-1], alone or combined) available for the treatment of BRAF V600–mutant melanoma have demonstrated significant improvements in clinical outcomes in patients with metastatic disease.6–10 However, patients with active brain metastases have typically been excluded from large trials to date and treatments specifically indicated for the treatment of melanoma brain metastases remain an unmet need.
In the phase 2 BREAK-MB trial (NCT01266967), dabrafenib monotherapy exhibited clinical activity and had a manageable safety profile in patients with BRAF V600E–mutant melanoma brain metastases (n=139), including patients with or without prior local treatment for brain metastases.11 In BRAF V600E patients without prior local treatment (n=74), the overall intracranial response rate was 39% and the overall response rate was 38% by investigator assessment. In BRAF V600E patients with prior local treatment (n=65), both the investigator-assessed overall intracranial response rate and overall response rate were 31%. In both BRAF V600E groups, 6-month overall survival was 61%. Patients with BRAF V600K–mutant disease included in this study (n=33) had lower response rates regardless of whether they had received prior local treatment (overall intracranial response rate, 22% and 7%, respectively). Response has also been observed in prospective clinical trials of small cohorts of molecularly-unselected patients with asymptomatic brain metastases treated with ipilimumab (n=51; brain metastasis response, 16%) or pembrolizumab (n=18; brain metastasis response, 22%).12,13
The combination of dabrafenib and trametinib has demonstrated improved progression-free and overall survival compared with BRAF inhibitor monotherapy with a manageable safety profile in phase 2 and phase 3 trials of patients with BRAF V600E/K–mutant stage IIIC unresectable or stage IV metastatic melanoma without brain metastases.6,7,14–19 However, this combination targeted therapy has not been previously evaluated prospectively in patients with BRAF V600–mutant melanoma brain metastases.
Here, we report the primary analysis of the phase 2 COMBI-MB trial (NCT02039947) evaluating dabrafenib plus trametinib in patients with active BRAF V600–mutant melanoma brain metastases. Findings described include investigator- and independently assessed best-confirmed intracranial, extracranial, and overall response; progression-free survival; duration of response; and safety.
Methods
Study design and patients
This open-label, multicohort, phase 2 trial evaluated the activity and safety of dabrafenib plus trametinib in four patient cohorts: (A) BRAF V600E–mutant, asymptomatic melanoma brain metastases, without prior local brain-directed therapy, Eastern Cooperative Oncology Group (ECOG) performance status ≤1; (B) BRAF V600E–mutant, asymptomatic melanoma brain metastases, with prior local therapy, ECOG performance status ≤1; (C) BRAF V600D/K/R–mutant, asymptomatic melanoma brain metastases, with or without prior local therapy, ECOG performance status ≤1; and (D) BRAF V600D/E/K/R–mutant, symptomatic melanoma brain metastases, with or without prior local therapy ECOG performance status ≤2 (appendix p 2).
Patients ≥18 years old with histologically-confirmed stage IV metastatic BRAF V600E/K/D/R–mutant cutaneous melanoma, determined using the THxID BRAF Assay (investigational use only; bioMerieux, Marcy-l’Étoile, France) at a central reference laboratory, were eligible for enrolment. Target lesions could be ≥0·5 to ≤4·0 cm in diameter; we excluded patients with the presence of any leptomeningeal disease or parenchymal brain metastasis measuring >4·0 cm in diameter. Patients with any RAS-mutant positive malignancy, history of malignancy other than the disease under study within 3 years (except completely resected non-melanoma skin cancer or patients with indolent malignancies), history of hepatitis B virus or hepatitis C virus without laboratory evidence of clearance, and any other serious or unstable pre-existing medical conditions or psychiatric disorders that could interfere with patient safety, consent, or compliance to study procedures were not eligible. Adequate organ function was also required for eligibility. Patients could have received up to two previous systemic therapies for metastatic melanoma, except for BRAF or MEK inhibitors. Prior temozolomide for brain metastases and adjuvant interferon were permitted and did not count towards the maximum of two previous systemic treatments. Prior systemic anti-cancer treatment within the last 3 weeks or chemotherapy without delayed toxicity within the last 2 weeks preceding the first dose of the combination was not permitted. Prior systemic treatment in the adjuvant setting was permitted; however, ipilimumab treatment must have ended ≥8 weeks prior to enrolment. In cohorts including patients who had received prior local therapies, previous treatments could have included, but were not limited to, craniotomy, whole-brain radiotherapy, and stereotactic radiosurgery. Treatment with stereotactic radiosurgery or whole-brain radiation must have occurred ≥14 and ≥28 days prior to start of study treatment, respectively. Eligible patients with prior local therapy to all brain lesions must have demonstrated progression of pre-existing target lesions per Response Evaluation Criteria in Solid Tumours (RECIST) v1·1 criteria. For cohorts A, B, and C (but not D), patients who were receiving concomitant corticosteroids must have been on a stable or decreasing dose for ≥1 month prior to study treatment initiation, and no prophylactic or preventative antiepileptic therapy was permitted.
The study protocol was approved by the institutional review board or human research ethics committee at each participating institution. Furthermore, the study was conducted in accordance with both the Declaration of Helsinki and International Conference of Harmonisation Good Clinical Practice.
Procedures
Patients in each cohort received dabrafenib 150 mg twice daily plus trametinib 2 mg once daily orally until evidence of disease progression, death, or unacceptable toxicity. Dose interruptions and/or reductions down to 75 mg twice daily for dabrafenib and 1 mg once daily for trametinib were permitted to manage adverse events. If a dose reduction of both below 75 mg twice daily for dabrafenib and below 1 mg once daily for trametinib was required, the combination study treatment was discontinued. If a dose reduction below 75 mg twice daily for dabrafenib was required, dabrafenib was permanently discontinued but trametinib could be continued. If a dose reduction below 1 mg once daily for trametinib was required, trametinib was permanently discontinued, but dabrafenib could be continued.
While patients were on study treatment, palliative radiation therapy was permitted for nontarget lesions that were either new or present at baseline; however, patients were censored from progression assessment at the time of initiating a new anticancer therapy (either alone or in combination with dabrafenib plus trametinib). All participants provided written informed consent.
Intracranial disease was assessed by a neuroradiologist/appropriately qualified radiologist/neurosurgeon at baseline, week 4, week 8, and every 8 weeks thereafter until week 40, using gadolinium contrast-enhanced magnetic resonance imaging (MRI)—the only method accepted for assigning intracranial lesions. After week 40, disease assessments were to be performed every 12 weeks. Extracranial disease was assessed at the same time points using contrast-enhanced computed tomography or MRI. An independent assessment of tumour response and progression was provided by Bioclinica, Inc (Doylestown, PA, USA). Adverse events were graded throughout the study by the investigator per the National Cancer Institute Common Terminology Criteria for Adverse Events v4·0, from the first study dose until 30 days following discontinuation of study treatment. Laboratory assessments evaluating chemistry and haematology parameters, were performed at screening, upon initiating study treatment, every 4 weeks through week 36, followed by monthly thereafter, and at the time of study discontinuation.
Outcomes
The primary endpoint was intracranial response rate in cohort A, defined as the percentage of patients in the all-treated-subjects population (patients who received at least 1 dose of study medication) with a confirmed intracranial complete or partial response assessed by the investigator using modified RECIST v1·1 criteria. RECIST was extended to include up to five intracranial and up to five extracranial target lesions, intracranial target lesions 5 to 40 mm in diameter were permitted. Intracranial disease could be assessed only by contrast-enhanced MRI, and MRI scan slices of 1 mm were required for brain metastases ≥5 mm but <10 mm. Secondary endpoints were intracranial response in cohorts B, C, and D; intracranial disease control, defined as the percentage of patients with a complete or partial response or stable disease; extracranial response rate, defined as the percentage of patients with a confirmed extracranial complete or partial response assessed by the investigator using modified RECIST v1·1 criteria; overall response rate, defined as the percentage of patients with a confirmed complete or partial response by investigator assessment; duration of intracranial, extracranial, and overall response, defined as the time from first documented complete or partial response until the time of disease progression; progression-free survival, defined as the interval between the first dose of study treatment and the earliest date of disease progression or death from any cause; overall survival, defined as the time from first dose until death due to any cause; and safety, measured by the frequency and severity of adverse events per skin, laboratory, vital-sign, cardiac function, and neurological assessment data. An independent data monitoring committee assessed safety periodically until the primary analysis was performed. The data sets for assessing all safety endpoints included all safety data collected on patients in the all-treated-subjects population.
Statistical analysis
This study was designed to assess the null hypothesis of intracranial response rate of ≤35% in cohort A and to provide 82% power to detect an intracranial response rate of ≥50% for patients in cohort A. Sample sizes were determined to fit the purpose of exploratory analyses and hypothesis generating. The cohort A sample size was based on the hypothesized improvement in intracranial response. Assessments of intracranial response in other cohorts were considered exploratory analyses; thus, there were no sample size calculations for cohorts B-D.
The study was designed to have a formal interim analysis for cohort A with a statistical decision rule for futility only, without p-value adjustment. The interim analysis was to take place after 22 patients were treated and had the opportunity for ≥2 disease assessments. The responses used in this interim analysis did not require confirmation. At least 8 of the 22 patients must have had an intracranial response (intracranial complete response or partial response) for the trial to continue. If 7 or fewer subjects had an intracranial response, this would have been considered as evidence that the null hypothesis was true. The interim analysis was conducted (cutoff date: January 30, 2015), and 10 of the 22 subjects had an intracranial response; thus, the decision was made to continue the trial. The results presented here are from the primary analysis of activity, which was performed when all patients in cohort A had the opportunity for 3 postbaseline disease assessments. The current analysis is considered as an interim analysis of progression-free and overall survival. The final analysis of progression-free survival, overall survival, and safety will occur when 70% of the total enrolled population has died or is lost to follow-up.
Response outcomes were summarised using point estimates and two-sided 95% confidence intervals (CI) calculated using the unconditional exact method. Duration of response outcomes, progression-free survival, and overall survival were summarised using Kaplan-Meier estimates along with two-sided 95% CI calculated using the Brookmeyer Crowley method. Additional endpoint definitions and censoring methods are described in the appendix (p 1). Adverse events were summarised by the frequency and proportion of total subjects, by system organ class and preferred term, with separate summaries provided for all, drug-related, and serious adverse events and adverse events leading to study treatment discontinuation. Statistical analyses were performed using SAS 9.3 software. This study is registered with ClinicalTrials.gov, NCT01584648.
Role of the funding source
The study was designed by the authors and sponsor. Data were collected by the study site staff and monitored by the sponsor, and the sponsor was involved in the data analysis, data interpretation, and writing of the report. All authors had full access to all data in the study and had final responsibility for the decision to submit for publication.
Results
From February 28, 2014 to August 5, 2016, 125 patients were enrolled, including 76 in cohort A, 16 in cohort B, 16 in cohort C, and 17 in cohort D (figure 1). Lactate dehydrogenase was elevated in 28 (37%) of 76 patients in cohort A, 3 (19%) of 16 patients in cohort B, 6 (38%) of 16 patients in cohort C, and 5 (29%) of 17 patients in cohort D, and extracranial metastases were present at a frequency of 89% (n=68/76), 75% (n=12/16), 100% (n=16/16), and 71% (n=12/17), respectively (table 1). Prior to starting dabrafenib plus trametinib, steroid treatment was received by 3 (4%) of 76 patients in cohort A, 1 (6%) of 16 patients in cohort B, 0 patients in cohort C, and 5 (29%) of 17 patients in cohort D. In cohort A, 59 (78%) of 76 patients were naive to any systemic anticancer treatment. At the data analysis cutoff, November 28, 2016, 14 (18%) of 76 patients in cohort A and 2 (13%) of 16 patients in cohort B remained on study treatment, whereas all patients in cohorts C and D had discontinued study treatment (appendix p 2). Median follow-up times at the data cutoff (ie, intervals between the date of first study treatment dose to the last patient contact date), which varied across cohorts due to differences in timing of enrolment dates for each cohort, were 8·5 months (IQR 5·5–14·0) in cohort A, 20·0 months (IQR 8·5–23·5) in cohort B, 9·5 months (IQR 4·5–17·5) in cohort C, and 11·0 months (95% CI 6·0–20·0) in cohort D. At the data cutoff, 8 (50%) of 16 patients in cohort B were still in follow-up, a proportion that was higher than those remaining in follow-up in cohort C (n=3/16; 19%) and cohort D (n=2/17; 12%; appendix p 2).
Figure 1. Trial profile.
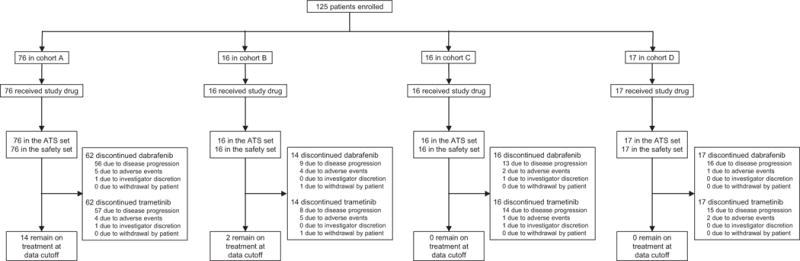
ATS=all-treated subjects.
Table 1.
Baseline characteristics
| Cohort A (n=76) |
Cohort B (n=16) |
Cohort C (n=16) |
Cohort D (n=17) |
|
|---|---|---|---|---|
|
| ||||
| Age (range), years | 52·0 (23–84) | 54·5 (36–74) | 63·0 (44–84) | 46·0 (23–68) |
|
| ||||
| Age category, n (%) | ||||
| <65 years | 60 (79) | 12 (75) | 9 (56) | 16 (94) |
| ≥65 years | 16 (21) | 4 (25) | 7 (44) | 1 (6) |
|
| ||||
| Sex, n (%) | ||||
| Male | 40 (53) | 10 (63) | 11 (69) | 11 (65) |
| Female | 36 (47) | 6 (38) | 5 (31) | 6 (35) |
|
| ||||
| ECOG performance status, n (%) | ||||
| 0 | 50 (66) | 11 (69) | 12 (75) | 9 (53) |
| 1 | 25 (33) | 5 (31) | 4 (25) | 6 (35) |
| 2 | 1 (1) | 0 | 0 | 2 (12) |
|
| ||||
| BRAF genotype, n (%) | ||||
| V600E | 73 (96) | 16 (100) | 0 | 15 (88) |
| V600K | 3 (4) | 0 | 14 (88) | 1 (6) |
| V600R | 0 | 0 | 2 (13) | 1 (6) |
| V600D | 0 | 0 | 0 | 0 |
|
| ||||
| Target brain metastases, n (%) | ||||
| 1 | 41 (54) | 7 (44) | 7 (44) | 7 (41) |
| 2 | 20 (26) | 7 (44) | 6 (38) | 7 (41) |
| 3 | 7 (9) | 2 (13) | 2 (13) | 1 (6) |
| 4 | 4 (5) | 0 | 0 | 1 (6) |
| 5 | 4 (5) | 0 | 1 (6) | 1 (6) |
|
| ||||
| Median SLD of target intracranial lesions (range), mm | 19·5 (6–117) | 14·0 (5–40) | 20·0 (5–61) | 33·0 (10–84) |
|
| ||||
| Extracranial metastases, n (%) | ||||
| No | 8 (11) | 4 (25) | 0 | 5 (29) |
| Yes | 68 (89) | 12 (75) | 16 (100) | 12 (71) |
|
| ||||
| Lactate dehydrogenase level, n (%) | ||||
| Normal (≤ULN) | 48 (63) | 13 (81) | 10 (63) | 12 (71) |
| Elevated (>ULN) | 28 (37) | 3 (19) | 6 (38) | 5 (29) |
|
| ||||
| Receiving steroid therapy, n (%) | ||||
| Prior treatment | 3 (4) | 1 (6) | 0 | 5 (29) |
| On-treatment or post-treatment | 38 (50) | 8 (50) | 9 (56) | 15 (88) |
|
| ||||
| Previous systemic anticancer treatment, n (%) | ||||
| No | 59 (78) | 11 (69) | 13 (81) | 10 (59) |
| Yes | 17 (22) | 5 (31) | 3 (19) | 7 (41) |
ECOG=Eastern Cooperative Oncology Group. SLD=sum of lesion diameters. ULN=upper limit of normal.
The primary endpoint was met, as 44 of 76 patients in cohort A (58%; 95% CI 46%–69%) had an investigator-assessed intracranial response, including 3 (4%) of 76 patients who achieved a complete response (table 2; figure 2A). Median duration of investigator-assessed intracranial response was 6·5 months (95% CI 4·9–10·3) in cohort A (figure 3A), which was supported by independent review (appendix p 6). Intracranial response was also observed in 9 (56%) of 16 patients in cohort B, 7 (44%) of 16 patients in cohort C, and 10 (59%) of 17 patients in cohort D (table 2; figure 2B-D). Extracranial responses were observed in 42 (55%) of 76 patients in cohort A, which lasted a median of 10·2 months (95% CI 5·8–not estimable), and in 7 (44%) of 16 patients in cohort B, 12 (75%) of 16 patients in cohort C, and 7 (41%) of 17 patients in cohort D (table 2). Overall responses were achieved by 44 (58%) of 76 patients in cohort A, which lasted a median of 6·5 months (95% CI 4·9–10·3), and in 9 (56%) of 16 patients in cohort B, 7 (44%) of 16 patients in cohort C, and 11 (65%) of 17 patients in cohort D (table 2). Most of the intracranial, extracranial, and overall responses observed in cohort A occurred by week 4 of study treatment, and by week 4 or week 8 in cohorts B-D (appendix p 2).
Table 2.
Investigator-assessed disease response and survival outcomes
| Cohort A (n=76) |
Cohort B (n=16) |
Cohort C (n=16) |
Cohort D (n=17) |
|
|---|---|---|---|---|
|
| ||||
| Intracranial response, n (%) [95% CI] | ||||
| Overall intracranial response (CR+PR) | 44 (58) [46–69] | 9 (56) [30–80] | 7 (44) [20–70] | 10 (59) [33–82] |
| Intracranial disease control (CR+PR+SD) | 59 (78) | 14 (88) | 12 (75) | 14 (82) |
| Intracranial CR | 3 (4) | 1 (6) | 0 | 1 (6) |
| Intracranial PR | 41 (54) | 8 (50) | 7 (44) | 9 (53) |
| Intracranial SD | 15 (20) | 5 (31) | 5 (31) | 4 (24) |
| Intracranial PD | 14 (18) | 1 (6) | 4 (25) | 3 (18) |
| Not evaluable | 3 (4) | 1 (6) | 0 | 0 |
|
| ||||
| Intracranial duration of response | ||||
| Events, n/N (%) | 29/44 (66) | 6/9 (67) | 4/7 (57) | 8/10 (80) |
| Median (95% CI), months | 6·5 (4·9–10·3) | 7·3 (3·6–12·6) | 8·3 (1·3–15·0) | 4·5 (2·8–5·9) |
| 6-month rate (95% CI), % | 63% (45–76) | 73% (28–93) | 67% (19–90) | 13% (1–43) |
|
| ||||
| Extracranial response, n (%) [95% CI] | ||||
| Overall extracranial response (CR+PR) | 42 (55) [43–67] | 7 (44) [20–70] | 12 (75) [48–93] | 7 (41) [18–67] |
| Extracranial disease control (CR+PR+SD) | 60 (79) | 11 (69) | 15 (94) | 11 (65) |
| Extracranial CR | 3 (4) | 1 (6) | 0 | 0 |
| Extracranial PR | 39 (51) | 6 (38) | 12 (75) | 7 (41) |
| Extracranial SD | 15 (20) | 2 (13) | 2 (13) | 3 (18) |
| Extracranial non-CR/non-PD | 3 (4) | 2 (13) | 1 (6) | 1 (6) |
| Extracranial PD | 6 (8) | 0 | 0 | 1 (6) |
| Not evaluable | 10 (13) | 5 (31) | 1 (6) | 5 (29) |
|
| ||||
| Extracranial duration of response | ||||
| Events, n/N (%) | 16/42 (38) | 0/7 | 6/12 (50) | 4/7 (57) |
| Median (95% CI), months | 10·2 (5·8–NE) | NE (NE–NE) | 4·9 (3·0–NE) | 5·9 (1·8–NE) |
| 6-month rate (95% CI), % | 69% (50–82) | 100% (100–100)* | 40% (10–70) | 48% (8–81) |
|
| ||||
| Overall response, n (%) [95% CI] | ||||
| Overall response (CR+PR) | 44 (58) [46–69] | 9 (56) [30–80] | 7 (44) [20–70] | 11 (65) [38–86] |
| Overall disease control (CR+PR+SD) | 60 (79) | 14 (88) | 12 (75) | 14 (82) |
| Overall CR | 1 (1) | 0 | 0 | 0 |
| Overall PR | 43 (57) | 9 (56) | 7 (44) | 11 (65) |
| Overall SD | 16 (21) | 5 (31) | 5 (31) | 3 (18) |
| Overall PD | 14 (18) | 1 (6) | 4 (25) | 3 (18) |
| Not evaluable | 2 (3) | 1 (6) | 0 | 0 |
|
| ||||
| Overall duration of response | ||||
| Events, n/N (%) | 32/44 (73) | 5/9 (56) | 6/7 (86) | 9/11 (82) |
| Median (95% CI), months | 6·5 (4·9–10·3) | 12·5 (5·3–NE) | 6·6 (1·3–16·3) | 4·5 (2·8–11·2) |
| 6-month rate (95% CI), % | 57% (41–71) | 86% (33–98) | 50% (11–80) | 23% (3–52) |
|
| ||||
| Progression-free survival | ||||
| Events, n (%) | 58 (76) | 10 (63) | 14 (88) | 15 (88) |
| Median (95% CI), months | 5·6 (5·3–7·4) | 7·2 (4·7–14·6) | 4·2 (1·7–6·5) | 5·5 (2·8–7·3) |
| 6-month rate (95% CI), % | 44% (32–56) | 71% (40–88) | 31% (10–55) | 46% (21–67) |
| 12-month rate (95% CI), % | 19% (10–31) | 47% (20–71) | 16% (3–39) | 8% (1–30) |
|
| ||||
| Overall survival (preliminary) | ||||
| Events, n (%) | 44 (58) | 7 (44) | 13 (81) | 13 (76) |
| Median (95% CI), months | 10·8 (8·7–19·6) | 24·3 (7·9–NE) | 10·1 (4·6–17·6) | 11·5 (6·8–22·4) |
| 6-month rate (95% CI), % | 79% (68–87) | 81% (52–94) | 69% (40–86) | 88% (61–97) |
| 12-month rate (95% CI), % | 46% (33–58) | 69% (40–86) | 44% (20–66) | 44% (20–66) |
CR=complete response. NE=not estimable. n/N=number with event/number with confirmed response. PD=progressive disease. PR=partial response. SD=stable disease.
No progression event occurred up to the data cutoff date.
Figure 2. Confirmed maxiumum reduction in intracranial target lesion in cohort A (A), cohort B (B), cohort C (C), and cohort D (D).
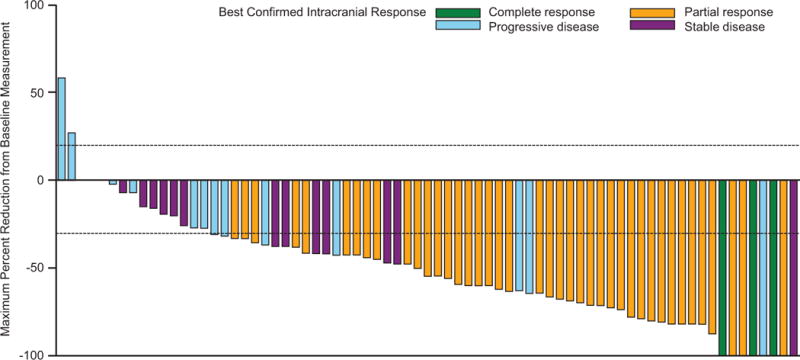
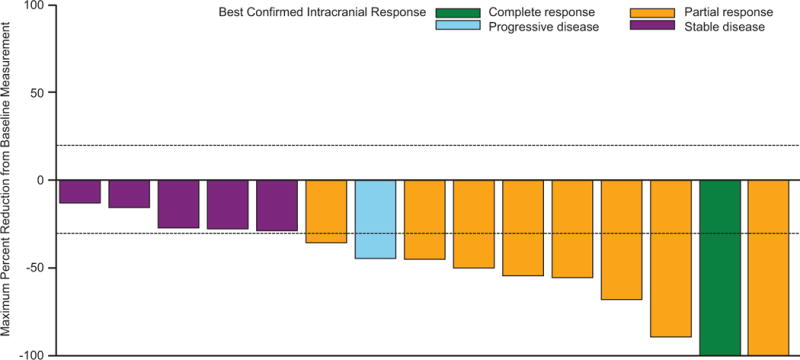
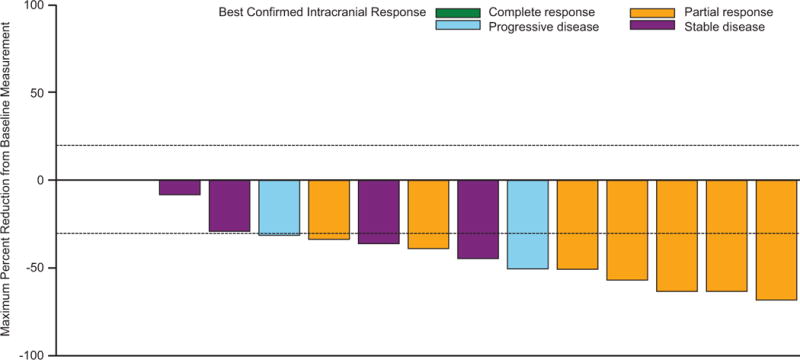
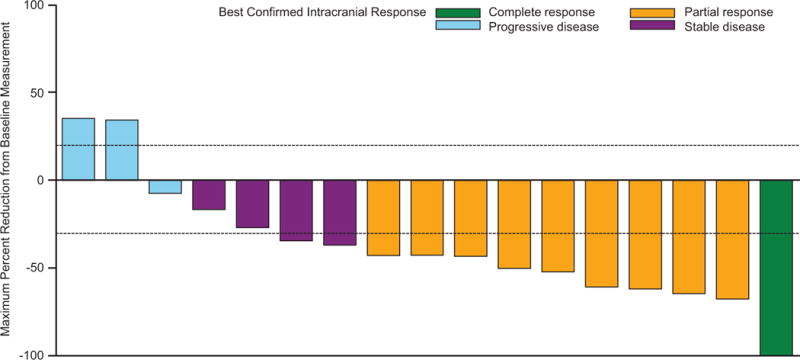
*Patient had a complete response in the target lesion but the best confirmed response was determined to be progressive disease due to development of an unequivocal new lesion. †Patient had an unconfirmed complete response but a best confirmed response was stable disease.
Figure 3. Investigator-assessed duration of intracranial response (A) progression-free survival (B) and preliminary overall survival (C) in cohort A.
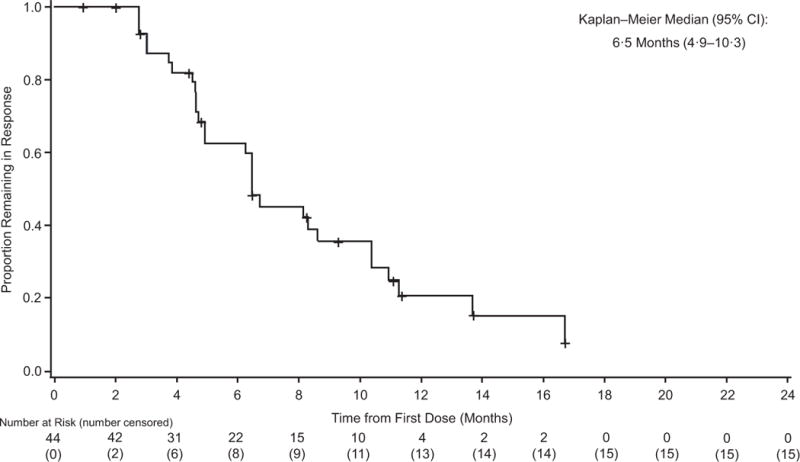
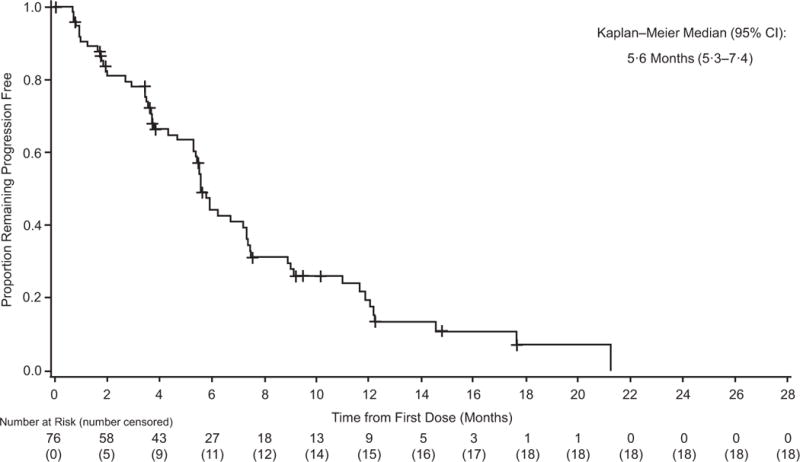
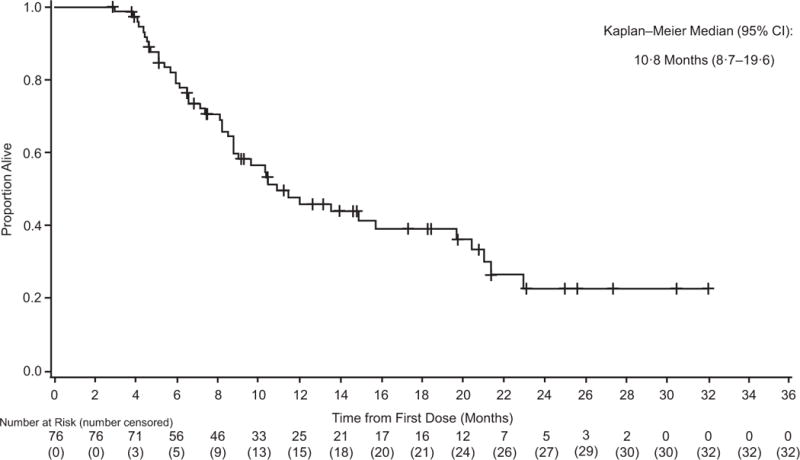
+=censored.
At the time of analysis, 58 (76%) of 76 patients in cohort A experienced progression-free survival events, with a median investigator-assessed progression-free survival of 5·6 months (95% CI 5·3–7·4) (table 2; figure 3B), which was supported by independent review (appendix p 6); 6- and 12-month progression-free survival rates were 44% and 19%, respectively. Median progression-free survival was 7·2 months (95% CI 4·7–14·6) in cohort B, 4·2 months (95% CI 1·7–6·5) in cohort C, and 5·5 months (95% CI 2·8–7·3) in cohort D (table 2; appendix p 7). Most patients across cohorts had progressive disease in intracranial lesions (n=66/125; 53%) or both intracranial and extracranial lesions (n=28/125; 22%) (appendix p 2). The most common type of systemic subsequent therapy received in patients who progressed across cohorts was immunotherapy, including anti–PD-1 and anti–CTLA-4 regimens (appendix p 3). Some patients also received on- or post-treatment anticancer surgery or radiotherapy (appendix p 3).
At the time of analysis, median time on study treatment across cohorts was 6·0 months (range, 0–24·0), with 24 (19%) of 125 patients treated with dabrafenib plus trametinib for >12 months. With follow-up ongoing for 44 (35%) of 125 patients at the time of analysis, preliminary median overall survival was 10·8 months (95% CI 8·7–19·6) in cohort A, with 6- and 12-month overall survival rates of 79% and 46%, respectively (table 2; figure 3C; appendix p 8).
Adverse events of any grade, regardless of study drug relationship, were observed in 123 (98%) of 125 patients, with 60 (48%) of 125 patients experiencing one or more grade 3/4 events (table 3) and 44 (35%) of 125 patients experiencing serious adverse events (appendix p 4). A total of 108 (86%) of 125 patients had adverse events considered to be related to study treatment (appendix p 4); the most common treatment-related serious adverse event related to study treatment was pyrexia for dabrafenib (n=8/124; 6%) and ejection fraction increased (n=5/125; 4%) for trametinib (appendix p 5). The most common adverse events in this study with dabrafenib plus trametinib, regardless of study drug relationship, included pyrexia (n=34/125; 54%), headache (n=46/125; 37%), asthenia (n=40/125; 32%), diarrhoea (n=40/125; 32%), nausea (n=40/125; 32%), and chills (n=37/125; 30%). Dose interruptions and reductions due to adverse events were necessary in 62 (50%) and 28 (22%) of 125 patients, respectively (appendix p 4). Discontinuations due to adverse events occurred in 12 (10%) of 125 patients (appendix p 4), mostly due to ejection fraction decreased (n=4/125; 3%) and pyrexia (n=3/125; 2%). One patient experienced a fatal serious adverse event of intracranial tumour haemorrhage, which was not considered related to study treatment. The primary cause of death in all cases across cohorts was considered to be related to cancer, disease progression, or complications due to melanoma.
Table 3.
Any-cause adverse events (grade ≤2 events occurring in ≥10% of patients across cohorts)
| Adverse event, n (%) |
Cohort A (n=76) | Cohort B (n=16) | Cohort C (n=16) | Cohort D (n=17) | Total (N=125) | |||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Grade 1-2 |
Grade 3 |
Grade 4 |
Grade 5 |
Grade 1-2 |
Grade 3 |
Grade 4 |
Grade 5 |
Grade 1-2 |
Grade 3 |
Grade 4 |
Grade 5 |
Grade 1-2 |
Grade 3 |
Grade 4 |
Grade 5 |
Grade 1-2 |
Grade 3 |
Grade 4 |
Grade 5 |
|
| Any | 39 (51) | 30 (39) | 4 (5) | 1 (1)* | 7 (44) | 8 (50) | 1 (6) | 0 | 7 (44) | 8 (50) | 1 (6) | 0 | 9 (53) | 7 (41) | 1 (6) | 0 | 62 (50) | 53 (42) | 7 (6) | 1 (1)* |
| Pyrexia | 42 (55) | 2 (3) | 0 | 0 | 7 (44) | 0 | 0 | 0 | 7 (44) | 1 (6) | 0 | 0 | 7 (41) | 1 (6) | 0 | 0 | 63 (50) | 4 (3) | 0 | 0 |
| Asthenia | 27 (36) | 0 | 0 | 0 | 5 (31) | 0 | 0 | 0 | 2 (13) | 1 (6) | 0 | 0 | 5 (29) | 0 | 0 | 0 | 39 (31) | 1 (1) | 0 | 0 |
| Headache | 26 (34) | 1 (1) | 0 | 0 | 4 (25) | 1 (6) | 0 | 0 | 6 (38) | 0 | 0 | 0 | 7 (41) | 1 (6) | 0 | 0 | 43 (34) | 3 (2) | 0 | 0 |
| Nausea | 23 (30) | 0 | 0 | 0 | 7 (44) | 0 | 0 | 0 | 4 (25) | 0 | 0 | 0 | 6 (35) | 0 | 0 | 0 | 40 (32) | 0 | 0 | 0 |
| Diarrhoea | 22 (29) | 0 | 0 | 0 | 8 (50) | 0 | 0 | 0 | 3 (19) | 0 | 0 | 0 | 7 (41) | 0 | 0 | 0 | 40 (32) | 0 | 0 | 0 |
| Vomiting | 21 (28) | 1 (1) | 0 | 0 | 2 (13) | 0 | 0 | 0 | 2 (13) | 0 | 0 | 0 | 6 (35) | 0 | 0 | 0 | 31 (25) | 1 (1) | 0 | 0 |
| Chills | 16 (21) | 0 | 0 | 0 | 6 (38) | 0 | 0 | 0 | 8 (50) | 0 | 0 | 0 | 5 (29) | 2 (12) | 0 | 0 | 35 (28) | 2 (2) | 0 | 0 |
| Arthralgia | 15 (20) | 0 | 0 | 0 | 3 (19) | 0 | 0 | 0 | 2 (13) | 0 | 0 | 0 | 6 (35) | 0 | 0 | 0 | 26 (21) | 0 | 0 | 0 |
| Myalgia | 13 (17) | 0 | 0 | 0 | 5 (31) | 0 | 0 | 0 | 1 (6) | 1 (6) | 0 | 0 | 5 (29) | 1 (6) | 0 | 0 | 24 (19) | 2 (2) | 0 | 0 |
| Aspartate aminotransferase increase | 11 (14) | 0 | 0 | 0 | 3 (19) | 0 | 0 | 0 | 2 (13) | 0 | 0 | 0 | 3 (18) | 0 | 0 | 0 | 19 (15) | 0 | 0 | 0 |
| Cough | 10 (13) | 0 | 0 | 0 | 2 (13) | 0 | 0 | 0 | 1 (6) | 0 | 0 | 0 | 3 (18) | 0 | 0 | 0 | 16 (13) | 0 | 0 | 0 |
| Oedema peripheral | 10 (13) | 0 | 0 | 0 | 2 (13) | 0 | 0 | 0 | 2 (13) | 0 | 0 | 0 | 3 (18) | 0 | 0 | 0 | 17 (14) | 0 | 0 | 0 |
| Back pain | 9 (12) | 0 | 0 | 0 | 1 (6) | 0 | 0 | 0 | 3 (19) | 0 | 0 | 0 | 2 (12) | 0 | 0 | 0 | 15 (12) | 0 | 0 | 0 |
| Rash | 9 (12) | 0 | 0 | 0 | 6 (38) | 1 (6) | 0 | 0 | 3 (19) | 0 | 0 | 0 | 3 (18) | 0 | 0 | 0 | 21 (17) | 1 (1) | 0 | 0 |
| Constipation | 8 (11) | 0 | 0 | 0 | 2 (13) | 0 | 0 | 0 | 4 (25) | 0 | 0 | 0 | 6 (35) | 0 | 0 | 0 | 20 (16) | 0 | 0 | 0 |
| Decreased appetite | 8 (11) | 0 | 0 | 0 | 4 (25) | 0 | 0 | 0 | 7 (44) | 1 (6) | 0 | 0 | 3 (18) | 0 | 0 | 0 | 22 (18) | 1 (1) | 0 | 0 |
| Pain in extremity | 8 (11) | 0 | 0 | 0 | 3 (19) | 0 | 0 | 0 | 3 (19) | 0 | 0 | 0 | 2 (12) | 0 | 0 | 0 | 16 (13) | 0 | 0 | 0 |
| Dizziness | 5 (7) | 0 | 0 | 0 | 5 (31) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 3 (18) | 0 | 0 | 0 | 13 (10) | 0 | 0 | 0 |
| Fatigue | 7 (9) | 0 | 0 | 0 | 4 (25) | 0 | 0 | 0 | 7 (44) | 1 (6) | 0 | 0 | 3 (18) | 0 | 0 | 0 | 21 (17) | 1 (1) | 0 | 0 |
| Abdominal pain | 6 (8) | 0 | 0 | 0 | 2 (13) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 5 (29) | 0 | 0 | 0 | 13 (10) | 0 | 0 | 0 |
| Alanine aminotransferase increased | 7 (9) | 0 | 0 | 0 | 2 (13) | 0 | 0 | 0 | 1 (6) | 0 | 0 | 0 | 2 (12) | 1 (6) | 0 | 0 | 13 (10) | 0 | 0 | 0 |
| Dry skin | 6 (8) | 0 | 0 | 0 | 2 (13) | 0 | 0 | 0 | 3 (19) | 0 | 0 | 0 | 3 (18) | 0 | 0 | 0 | 14 (11) | 0 | 0 | 0 |
Intracranial tumour haemorrhage, considered to be unrelated to study treatment.
Discussion
This primary analysis of the COMBI-MB study, representing the first report of a phase 2 trial evaluating BRAF and MEK inhibitor combination therapy in patients with BRAF V600E–mutant melanoma brain metastases, provides evidence of activity of dabrafenib plus trametinib in active melanoma brain metastases. The primary study endpoint of investigator-assessed intracranial response in cohort A was met (intracranial response rate, 58%). Intracranial responses were also observed in cohorts B, C, and D (intracranial response rates, 56%, 44%, and 59%, respectively); however, due to the sample sizes of these cohorts, these findings are considered exploratory and hypothesis generating. Furthermore, dabrafenib and trametinib had a manageable safety profile in this population. The benefits experienced by patients treated with dabrafenib plus trametinib in this study were improved over historic outcomes of patients with melanoma brain metastases treated with local therapies (eg, whole-brain radiotherapy: median overall survival, 3·4 months).20 However, the durability of clinical responses and disease control was relatively short compared with that in previous trials in patients without melanoma brain metastases.
The findings of this study suggest that intracranial response rates of patients with BRAF V600–mutant melanoma brain metastases were improved with dabrafenib and trametinib combination therapy compared with previously reported analyses of BRAF inhibitor monotherapy in this setting. Single-agent vemurafenib was associated with intracranial responses in 16% of patients with symptomatic brain metastases and prior central nervous system–directed therapy, and single-agent dabrafenib has been associated with intracranial response rates in 31% and 39% of patients with asymptomatic active brain metastases with and without prior therapy, respectively.11,21
In a phase 2 study of ipilimumab in patients with melanoma brain metastases, 16% (n=8/51) of asymptomatic patients not requiring steroids and 5% (n=1/21) of symptomatic patients receiving steroids to control neurological symptoms or perilesional oedema achieved a central nervous system response, with a median overall survival of 7·0 and 3·7 months, respectively.12 In a phase 2 study of pembrolizumab, which excluded patients receiving steroids to control neurological symptoms or perilesional oedema, 22% (n=4/18) of patients with untreated or progressive melanoma brain metastases experienced a brain metastasis response lasting ≥4–10 months.13 In part 4 of the phase 1 CheckMate 038 study, in patients with active melanoma brain metastases, nivolumab elicited an objective response rate per RECIST v1·1 of 50% alone (n=5/10) or in combination with ipilimumab (n=5/10), with a median progression-free survival of 10·8 months for the combination and not reached with nivolumab monotherapy.22 Of note, patients enrolled in part 4 of CheckMate 038 generally had more favourable baseline clinical features (eg, elevated lactate dehydrogenase level, 20%) than those included in the current study. The activity of nivolumab plus ipilimumab continues to be evaluated in phase 2 studies, including the anti–PD-1 Brain Collaboration Trial (NCT02374242) and the CheckMate 204 study of the combination in patients with melanoma brain metastases.23,24
The safety profile of dabrafenib plus trametinib in this study was similar to that reported in previous studies, including in patients with metastatic melanoma without brain metastases,6,7,14–19 in which pyrexia and gastrointestinal issues remain common adverse events.6,7,14–16,18 Thus, the combination also has a manageable safety profile in patients with melanoma brain metastases.
While the initial response rates and safety data observed in this population are reassuring, the duration of overall response and progression-free survival were short across cohorts in this study (eg, median of 6·5 and 5·6 months in cohort A, respectively) compared with what has been observed for these outcomes in randomised trials evaluating dabrafenib and trametinib in patients with metastatic melanoma without brain metastases (approximately 12–14 and 11–12 months, respectively in phase 3 trials).6,17,19 Outcomes for patients treated with the combination in this study were similar to those observed in patients with poor clinical features in previous randomised studies evaluating first-line dabrafenib plus trametinib in patients with metastatic melanoma without brain metastases, such as those who had baseline lactate dehydrogenase levels more than two-fold higher than the upper limit of normal (overall response rate, 51%; median progression-free survival, 5·5 months).25 Other studies support that melanoma brain metastases may have distinct molecular features, such as increased activation of the PI3K-AKT signalling pathway, which has also been associated with resistance to BRAF and MEK inhibitors.26–28 Altogether, these findings support the need for additional research in patients with melanoma brain metastases. Additional clinical trials with dabrafenib and trametinib in these patients are also needed, including combinatorial approaches with brain-directed treatments such as stereotactic radiosurgery, and/or with other systemic therapies.
This study clearly demonstrates intracranial activity of dabrafenib plus trametinib; however, from a practical viewpoint, cerebral disease is not controlled in nearly half of patients, which represents an unmet medical need. Most complete responses were achieved in patients with 1-2 brain metastases, which are effectively managed with surgery or stereotactic radiosurgery. Despite these findings, brain metastases are rarely isolated and BRAF and MEK inhibitor combination therapies are efficient in controlling extra-cerebral disease. Together, these data strongly support multidisciplinary combination strategies incorporating dabrafenib plus trametinib for the management of patients with brain metastases.
We acknowledge that the non-randomised one-arm design of COMBI-MB was a limitation for this study, as results can therefore not be compared directly to other current treatments. The small sample sizes for cohorts B-D limited the extent of interpretation of results for these patient subsets. Additionally, due to differences in the prevalence of patients with very different eligibility and enrolment numbers required in each cohort, patients in cohorts B-D were enrolled in the study at an earlier date than patients in cohort A, which resulted in differences in median follow-up time between these patient groups. Analyses incorporating information on patients who received surgery or radiosurgery (eg, gamma-knife) during or after study treatment may have also provided further insights on the appropriate role of dabrafenib plus trametinib in the current treatment landscape of patients with melanoma brain metastases.
To our knowledge, this analysis of COMBI-MB is the first report of a phase 2 trial evaluating a BRAF and MEK inhibitor combination therapy in patients with melanoma that has metastasised to the brain. These preliminary results provide evidence that clinical benefit and tolerability is achievable with dabrafenib plus trametinib in a subset of patients with BRAF V600–mutant metastatic melanoma. Continued follow-up is needed to assess the full impact of dabrafenib plus trametinib on overall survival in these cohorts of patients. Nevertheless, these findings serve as a framework for future studies in this setting, in which effective treatments remain a critical unmet medical need.
Supplementary Material
Research in context.
Evidence before this study
We searched PubMed up to March 7, 2017 for clinical studies with the terms “BRAF,” “melanoma,” and “brain metastases,” and identified 144 articles, of which 4 were primary analyses of phase 1 or 2 clinical trials of BRAF inhibitor regimens in patients with BRAF V600–mutant melanoma brain metastases. BRAF inhibitor monotherapy has previously been shown to exhibit clinical activity and tolerability in patients with BRAF-mutant melanoma who developed metastases in the brain. Although the BRAF and MEK inhibitor combination therapy has demonstrated superiority over BRAF inhibitor monotherapy in patients with BRAF V600–mutant metastatic melanoma without brain metastases, the clinical effect of this regimen on melanoma brain metastases has not been characterised.
Added value of this study
We report here findings from a phase 2 trial evaluating the activity and safety of dabrafenib plus trametinib in patients with BRAF V600–mutant melanoma brain metastases. Intracranial outcomes demonstrated that dabrafenib plus trametinib was active in brain metastases in patients with BRAF V600–mutant melanoma and the primary study endpoint was met; however, responses were less durable than those previously observed for the combination in patients with melanoma without brain metastases. No unexpected safety issues were observed with dabrafenib plus trametinib in this setting.
Implications of all the available evidence
Our findings represent the first report of a phase 2 trial evaluating a BRAF and MEK inhibitor combination therapy in patients with melanoma brain metastases and provide evidence that clinical benefit and tolerability is achievable with dabrafenib plus trametinib in a subset of patients with BRAF V600–mutant melanoma that has metastasised to the brain. Continued follow-up is necessary to determine the full impact of dabrafenib plus trametinib on overall survival in this setting; however, these preliminary results support the use of this targeted therapy combination as a treatment option for these patients, in which effective treatments remain a critical unmet medical need.
Acknowledgments
Medical writing assistance in the form of collating author comments, copyediting, and editorial assistance was provided by Amanda L. Kauffman, PhD (ArticulateScience LLC), and funded by Novartis Pharmaceuticals Corporation. This study was sponsored by GlaxoSmithKline; dabrafenib and trametinib are assets of Novartis AG as of March 2, 2015.
Footnotes
Contributors
MAD, KTF, and GVL contributed to the study design. MAD, PS, CR, J-JG, KTF, AA, VC-S, LT, TL, LM, SJM, DH, IMR, MDV, CL, NM, YH, and GVL recruited the patients and/or collected data. YZ conducted the statistical analyses. All authors analysed and interpreted the data, drafted the manuscript, and approved the final version.
Declaration of interests
MAD has been a principal investigator grants to his institution received from AstraZeneca, Merck, Roche/Genentech, and Sanofi; and has received personal fees from Novartis, Bristol-Myers Squibb, Sanofi, and Merck for advisory board participation. PS has received personal fees from Amgen, Bristol-Myers Squibb, MSD, Merck-Serono, Pfizer, Roche/Genentech, Pierre Fabre, and Novartis; has received nonfinancial support from Bristol-Myers Squibb, MSD, Roche/Genentech, and Novartis; and has received funding grant from Roche/Genentech. CR has received personal fees for advisory board participation from Bristol-Myers Squibb, GlaxoSmithKline, Roche, Merck, Amgen, and Novartis. J-JG has received personal fees for participating in advisory boards for Novartis, Roche, GlaxoSmithKline, Bristol-Myers Squibb, Merck, Amgen, and Pierre Fabre. KTF has received personal fees and grant support from Novartis. AA has received personal fees from Novartis, Roche, Merck/MSD, and Bristol-Myers Squibb and financial support for the study from Novartis. VC-S has participated in advisory boards for Merck/MSD, Merck-Serono, Roche, Bristol-Myers Squibb, and Novartis. LT has nothing to disclose. TL has received grants and personal fees for clinical trials and advisory board participation from Bristol-Myers Squibb, Roche, Novartis, and MSD; has participated in speakers’ bureaus for Roche, MSD, and Novartis; and has received travel/accommodations expenses from Roche. LM has nothing to disclose. SJM has received personal fees for consultancy or travel from Merck, Amgen, Novartis and and Bristol-Myers Squibb; and has received grants from Merck, Amgen, and Pharmacyclics. DH has received personal fees for advisory board participation or lectures from Roche, Bristol-Myers Squibb, EMD Serono, Merck, Amgen, and Novartis. IM-R has received personal fees from Novartis, Roche, MSD, Amgen, Merck-Serono, Pierre Fabre, Bioncotech, GlaxoSmithKline, and Bristol-Myers Squibb and has received travel/accommodation expenses from MSD, Bristol-Myers Squibb, and Amgen. MDV has consulted or had an advisory role for and received honoraria from Bristol-Myers Squibb, Roche, Novartis, and Merck. CL has received grants from Roche and Bristol-Myers Squibb and personal fees for consultancy, advisory roles, speaker’s bureaus, and/or travel/accommodation expenses from Roche, Bristol-Myers Squibb, Novartis, Merck/MSD, Amgen, and GlaxoSmithKline. NM has received personal fees from Roche, Bristol-Myers Squibb, MSD, Novartis, GlaxoSmithKline, Pierre Fabre, and grants from Bristol-Myers Squibb, MSD, and Pierre Fabre. YZ, YH, and BM are employees of Novartis. GVL received personal fees for her role as a consultant advisor to Amgen, Bristol-Myers Squibb, Merck/MSD, Novartis, Pierre Fabre, Array Biopharma, and Roche.
References
- 1.Cohen JV, Tawbi H, Margolin KA, et al. Melanoma central nervous system metastases: current approaches, challenges, and opportunities. Pigment Cell Melanoma Res. 2016;29:627–42. doi: 10.1111/pcmr.12538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sampson JH, Carter JH, Jr, Friedman AH, Seigler HF. Demographics, prognosis, and therapy in 702 patients with brain metastases from malignant melanoma. J Neurosurg. 1998;88:11–20. doi: 10.3171/jns.1998.88.1.0011. [DOI] [PubMed] [Google Scholar]
- 3.Long GV, Margolin KA. Multidisciplinary approach to brain metastasis from melanoma: the emerging role of systemic therapies. Am Soc Clin Oncol Educ Book. 2013:393–8. doi: 10.14694/EdBook_AM.2013.33.393. [DOI] [PubMed] [Google Scholar]
- 4.Davies MA, Liu P, McIntyre S, et al. Prognostic factors for survival in melanoma patients with brain metastases. Cancer. 2011;117:1687–96. doi: 10.1002/cncr.25634. [DOI] [PubMed] [Google Scholar]
- 5.Raizer JJ, Hwu WJ, Panageas KS, et al. Brain and leptomeningeal metastases from cutaneous melanoma: survival outcomes based on clinical features. Neuro Oncol. 2008;10:199–207. doi: 10.1215/15228517-2007-058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Long GV, Stroyakovskiy D, Gogas H, et al. Dabrafenib and trametinib versus dabrafenib and placebo for Val600 BRAF-mutant melanoma: a multicentre, double-blind, phase 3 randomised controlled trial. Lancet. 2015;386:444–51. doi: 10.1016/S0140-6736(15)60898-4. [DOI] [PubMed] [Google Scholar]
- 7.Robert C, Karaszewska B, Schachter J, et al. Improved overall survival in melanoma with combined dabrafenib and trametinib. N Engl J Med. 2015;372:30–9. doi: 10.1056/NEJMoa1412690. [DOI] [PubMed] [Google Scholar]
- 8.Larkin J, Chiarion-Sileni V, Gonzalez R, et al. Combined nivolumab and ipilimumab or monotherapy in untreated melanoma. N Engl J Med. 2015;373:23–34. doi: 10.1056/NEJMoa1504030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Robert C, Schachter J, Long GV, et al. Pembrolizumab versus ipilimumab in advanced melanoma. N Engl J Med. 2015;372:2521–32. doi: 10.1056/NEJMoa1503093. [DOI] [PubMed] [Google Scholar]
- 10.Ascierto PA, McArthur GA, Dreno B, et al. Cobimetinib combined with vemurafenib in advanced BRAFV600-mutant melanoma (coBRIM): updated efficacy results from a randomised, double-blind, phase 3 trial. Lancet Oncol. 2016;17:1248–60. doi: 10.1016/S1470-2045(16)30122-X. [DOI] [PubMed] [Google Scholar]
- 11.Long GV, Trefzer U, Davies MA, et al. Dabrafenib in patients with Val600Glu or Val600Lys BRAF-mutant melanoma metastatic to the brain (BREAK-MB): a multicentre, open-label, phase 2 trial. Lancet Oncol. 2012;13:1087–95. doi: 10.1016/S1470-2045(12)70431-X. [DOI] [PubMed] [Google Scholar]
- 12.Margolin K, Ernstoff MS, Hamid O, et al. Ipilimumab in patients with melanoma and brain metastases: an open-label, phase 2 trial. Lancet Oncol. 2012;13:459–65. doi: 10.1016/S1470-2045(12)70090-6. [DOI] [PubMed] [Google Scholar]
- 13.Goldberg SB, Gettinger SN, Mahajan A, et al. Pembrolizumab for patients with melanoma or non-small-cell lung cancer and untreated brain metastases: early analysis of a non-randomised, open-label, phase 2 trial. Lancet Oncol. 2016;17:976–83. doi: 10.1016/S1470-2045(16)30053-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Flaherty KT, Infante JR, Daud A, et al. Combined BRAF and MEK inhibition in melanoma with BRAF V600 mutations. N Engl J Med. 2012;367:1694–703. doi: 10.1056/NEJMoa1210093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Long GV, Weber JS, Infante JR, et al. Overall survival and durable responses in patients with BRAF V600–mutant metastatic melanoma receiving dabrafenib combined with trametinib. J Clin Oncol. 2016;34:871–8. doi: 10.1200/JCO.2015.62.9345. [DOI] [PubMed] [Google Scholar]
- 16.Long GV, Stroyakovskiy D, Gogas H, et al. Combined BRAF and MEK inhibition versus BRAF inhibition alone in melanoma. N Engl J Med. 2014;371:1877–88. doi: 10.1056/NEJMoa1406037. [DOI] [PubMed] [Google Scholar]
- 17.Long GV, Flaherty KT, Stroyakovskiy D, et al. Dabrafenib plus trametinib versus dabrafenib monotherapy in patients with metastatic BRAF V600E/K–mutant melanoma: long-term survival and safety analysis of a phase 3 study. Ann Oncol. doi: 10.1093/annonc/mdz221. (in press) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Robert C, Karaszewska B, Schachter J, et al. Two year estimate of overall survival in COMBI-v, a randomized, open-label, phase III study comparing the combination of dabrafenib (D) and trametinib (T) with vemurafenib (Vem) as first-line therapy in patients (pts) with unresectable or metastatic BRAF V600E/K mutation-positive cutaneous melanoma. Eur J Cancer. 2015;51(suppl 3) abstr 3301. [Google Scholar]
- 19.Robert C, Karaszewska B, Schachter J, et al. Three-year estimate of overall survival in COMBI-v, a randomized phase 3 study evaluating first-line dabrafenib (D) + trametinib (T) in patients (pts) with unresectable or metastatic BRAF V600E/K–mutant cutaneous melanoma. Ann Oncol. 2016;27(suppl 6) abstr LBA40. [Google Scholar]
- 20.Fife KM, Colman MH, Stevens GN, et al. Determinants of outcome in melanoma patients with cerebral metastases. J Clin Oncol. 2004;22(7):1293–300. doi: 10.1200/JCO.2004.08.140. [DOI] [PubMed] [Google Scholar]
- 21.Dummer R, Goldinger SM, Turtschi CP, et al. Vemurafenib in patients with BRAF(V600) mutation-positive melanoma with symptomatic brain metastases: final results of an open-label pilot study. Eur J Cancer. 2014;50:611–21. doi: 10.1016/j.ejca.2013.11.002. [DOI] [PubMed] [Google Scholar]
- 22.Haanen J, Hwu W, Martín-Algarra S, et al. Efficacy and safety of nivolumab (NIVO) alone or combined with ipilimumab (IPI) in patients with melanoma (MEL) metastatic to the brain in a phase 1 study; Presented at: Society for Melanoma Research. 2016. [Google Scholar]
- 23.Tawbi H, Algazi A, Forsyth P, et al. Safety of nivolumab (NIVO) plus ipilimumab (IPI) in patients with advanced melanoma (MEL) metastatic to the brain: initial results from phase 2 CheckMate 204; Presented at: Society for Melanoma Research. 2016. [Google Scholar]
- 24.Long GV, Atkinson V, Menzies AM, et al. A randomized phase 2 study of nivolumab and nivolumab combined with ipilimumab in patients (pts) with melanoma brain metastases: the Anti-PD1 Brain Collaboration (ABC Study) J Clin Oncol. 2016;34(suppl) abstr TPS9591. [Google Scholar]
- 25.Long GV, Grob J, Nathan P, et al. Factors predictive of response, disease progression, and overall survival after dabrafenib and trametinib combination treatment: a pooled analysis of individual patient data from randomised trials. Lancet Oncol. 2016;17:1743–54. doi: 10.1016/S1470-2045(16)30578-2. [DOI] [PubMed] [Google Scholar]
- 26.Chen G, Chakravarti N, Aardalen K, et al. Molecular profiling of patient-matched brain and extracranial melanoma metastases implicates the PI3K pathway as a therapeutic target. Clin Cancer Res. 2014;20:5537–46. doi: 10.1158/1078-0432.CCR-13-3003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Niessner H, Forschner A, Klumpp B, et al. Targeting hyperactivation of the AKT survival pathway to overcome therapy resistance of melanoma brain metastases. Cancer Med. 2013;2:76–85. doi: 10.1002/cam4.50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Amaral T, Sinnberg T, Meier F, et al. The mitogen-activated protein kinase pathway in melanoma part I - activation and primary resistance mechanisms to BRAF inhibition. Eur J Cancer. 2017;73:85–92. doi: 10.1016/j.ejca.2016.12.010. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.