

Abstract
Capsaicin, the ingredient responsible for the pungent taste of hot chili peppers, is widely used in the study and management of pain. Recently, its neuroprotective effect has been described in multiple studies. Herein, we investigated the underlying mechanisms for the neuroprotective effect of capsaicin. Direct injection of capsaicin (1 or 3 nmol) into the peri-infarct area reduced the infarct volume and improved neurological behavioral scoring and motor coordination function in the middle cerebral artery occlusion (MCAO)/reperfusion model in rats. The time window of the protective effect of capsaicin was within 1 h after reperfusion, when excitotoxicity is the main reason of cell death. In cultured cortical neurons, administration of capsaicin attenuated glutamate-induced excitotoxic injury. With respect to the mechanisms of the neuroprotective effect of capsaicin, reduced calcium influx after glutamate stimulation was observed following capsaicin pretreatment in cortical neurons. Trpv1 knockout abolished the inhibitory effect of capsaicin on glutamate-induced calcium influx and subsequent neuronal death. Reduced expression of GluN1 and GluN2B, subunits of NMDA receptor, was examined after capsaicin treatment in cortical neurons. In summary, our studies reveal that the neuroprotective effect of capsaicin in cortical neurons is TRPV1-dependent and down-regulation of the expression and function of NMDA receptors contributes to the protection afforded by capsaicin.
Keywords: Capsaicin, NMDA receptor, Cerebral ischemia, MCAO, Excitotoxicity, Neuroprotection
1. Introduction
Capsaicin (PubChem CID: 1548943), a phenolic compound (8-methyl-N-vanillyl-6-nonenamide), is the principle component that gives chili peppers their pungent taste. However, the effects of capsaicin go well beyond taste. It can selectively activate the unmyelinated C-fiber and is widely used in the study and management of pain (Szolcsanyi, 2014). Capsaicin can also limit energy intake and increase energy expenditure, induce the secretion of insulin, lower blood pressure, and decrease lipid storage and atherosclerotic lesions. In addition, the anti-tumorigenic activity of capsaicin and its effects in reducing inflammation of the allergic airway and relieving the symptoms of neurogenic bladder have also been demonstrated (Fattori et al., 2016). Currently, low- and high-concentration creams, lotions, patches, capsules and nasal sprays of capsaicin are clinically available.
The receptor for capsaicin, transient receptor potential vanilloid 1 (TRPV1), was first cloned in 1997 (Caterina et al., 1997). It is a Ca2+-permeable, non-selective cation channel with a PCa/PNa of 9.6. TRPV1 is highly expressed in primary sensory neurons and serves as a signal integrator in pathological pain. In addition to capsaicin, TRPV1 can be activated by heat, low pH, and endovanilloids such as arachidonylethanolamide (anandamide) and N-arachidonoyl-dopamine (Julius, 2013). One of the functional features of TRPV1 is desensitization, a refractory state in which the receptor function is inhibited after exposure to a high or repeated dose of capsaicin. Therefore, capsaicin, the most specific known agonist of TRPV1, can be used to relieve pain (Brederson et al., 2013). Certainly, other mechanisms also contribute to the analgesic effect of capsaicin.
In recent years, accumulating evidence has shown the neuroprotective effect of capsaicin. Capsaicin can provide protection against ouabain-induced excitotoxicity in the brain (Veldhuis et al., 2003), global cerebral ischemia in Mongolian gerbils (Pegorini et al., 2005), N-methyl-D-aspartic acid (NMDA)-induced retinal ganglion cell loss (Sakamoto et al., 2014), hypoxia/reoxygenation-induced apoptosis in hippocampal neurons (Guo et al., 2008), and glutamate-induced cortical neuron injury (J.G. Lee et al., 2012). These studies further extend the potential clinical application of capsaicin. However, there has been a lack of study on the underlying mechanisms for the neuroprotective effect of capsaicin. It is proposed that capsaicin-induced TRPV1 desensitization or the hypothermic effect following systemic application of capsaicin might contribute to its neuroprotective action (Pegorini et al., 2005; Veldhuis et al., 2003). In addition, a recent study indicated that activation of TRPV1 can play a neuroprotective action through enhancing neuronal activity for axonal signaling (Ward et al., 2014). They found that elevated ocular pressure-induced axonopathy of the optic projection was accelerated by trpv1 knock-out (KO) or pharmacological antagonism of TRPV1.
In the present studies, we used the in vivo micro-injection approach combined with the in vitro neuronal cultures to examine the direct effect of capsaicin on neuronal ischemia/reperfusion injury and attempted to elucidate the underlying molecular mechanisms.
2. Materials and methods
2.1. Drugs and antibodies
All drugs and antibodies were purchased from Sigma-Aldrich (St Louis, MO, USA) unless otherwise stated. Capsaicin (Cat # BML-EI125, Enzo Life Sciences, Farmingdale, NY, USA) was dissolved in ethanol as a 200 mM stock solution. For stereotaxic injection into the cerebral cortex, the capsaicin stock solution was diluted with PBS to the suitable concentration, for example, 1 mM for 1 nmol/μl injection. For in vitro application, capsaicin was diluted with PBS and immediately added to the medium. Resiniferatoxin (RTX, Cat # 1137, Tocris Bioscience, MO, USA) was dissolved in ethanol as a 50 mM stock solution and diluted with PBS just before use. Fura-2 AM ester (Cat # 50033, Biotium, Hayward, CA, USA) was dissolved in dimethyl sulphoxide (DMSO) to produce a 1 mM stock solution and diluted as indicated below just before use.
Antibodies against TRPV1 (Cat # sc-12498), GluN1 (Cat # sc-1467) and GluN2B (Cat # sc-1469) were purchased from Santa Cruz Biotechnology (CA, USA). An antibody against the transferrin receptor was purchased from Invitrogen (Cat # 13-6800, Carlsbad, CA, USA).
2.2. Animals
Male Sprague Dawley (SD) rats were purchased from the animal center of Peking University Health Science Center (Beijing, China). The rats were housed under a 12/12 h dark/light cycle. All of the experimental procedures were approved by the Animal Care and Welfare Committee in Peking University Health Science Center and conformed to the “Guide for the Care and Use of Laboratory Animals” of Peking University Health Science Center. All efforts were made to minimize pain and suffering of the animals.
Gene deletion of trpv1 in C57BL/6J mice was generated by the Biocytogen Company (Beijing, China) using transcription activator-like effector nuclease (TALEN) technology (Fig. 6A). Genotypes of mice were determined using PCR amplification (94 °C for 5 min; 30 cycles of 94 °C for 50 s, 57 °C for 50 s, 72 °C for 50 s; 72 °C for 10 min). The following primer pairs were used: wild type (WT) (470 bp): forward, 5′-TCTTTAGAGGGAGTACCAAGACCCT-3′; reverse, 5′-CTGAGCTGAGCTACAAGGAAAGC-3′. trpv1 KO (350 bp): forward, 5′-CTGCCAGGGACTACACTTGTGAAG-3′; reverse, 5′-CTGAGCTGAGCTACAAGGAAAGC-3′.
Fig. 6.
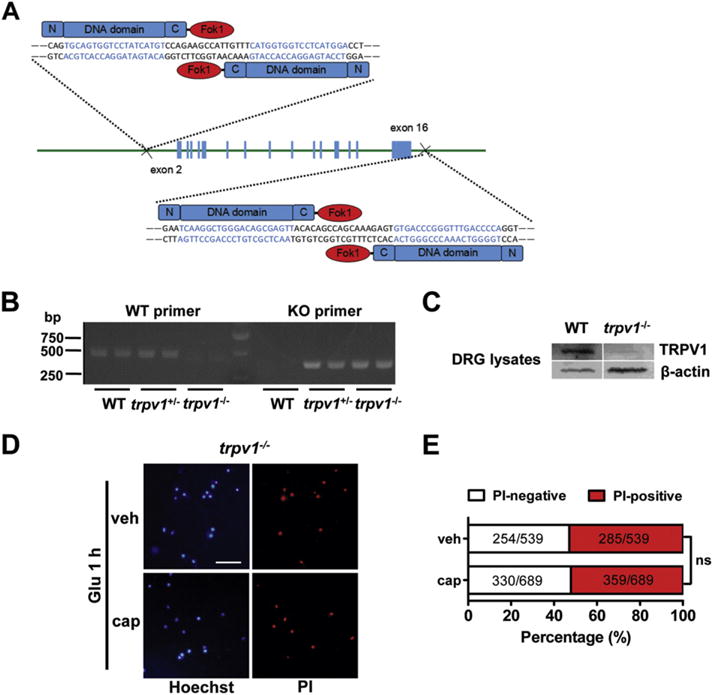
Trpv1 knock-out abolishes the neuroprotective effect of capsaicin. (A) Schematic diagram of the generation of trpv1 knock-out (KO) mice targeting the sequence located upstream of exon 2 and downstream of exon 16 of the trpv1 gene. (B) PCR genotyping of trpv1 knock-out mice. (C) Western blot analysis of TRPV1 protein expression in the dorsal root ganglion tissue of wild type (WT) and trpv1 knock-out mice. (D) and (E) Trpv1 knock-out abolished the effect of capsaicin to reduce glutamate-induced neuronal death in the cortical neurons from KO mice. Scale bar = 100 μm. At least three parallel experiments were repeated. (D) Representative images show absence of the effect of capsaicin on glutamate-induced neuronal death after trpv1 knock-out. (E) Quantification of the PI-positive and PI-negative neurons as shown in (D). ns, no statistical significance. Fisher’s exact test of one representative group.
2.3. Middle cerebral artery occlusion (MCAO)
Focal cerebral ischemia was induced in 280–320 g rats by middle cerebral artery occlusion as described by Longa et al. (1989). In brief, under chloral hydrate anesthesia (350 mg/kg, i.p.), a surgical silica gel monofilament with a rounded tip was introduced into the right internal carotid artery through the external carotid stump and advanced approximately 20 mm past the carotid bifurcation until a slight resistance was felt. Regional cerebral blood flow was measured by laser Doppler flowmetry (Moor Instruments, Axminster, UK). After 2 h, the filament was withdrawn for reperfusion. In the sham rats, the filament was inserted only 7 mm from the carotid bifurcation.
During the surgery, a homemade heating incubator was used to keep the animals warm. The rectal temperature, blood pressure and blood oxygen saturation were monitored using the multifunctional monitoring equipment for animal studies (RWD life science, Shenzhen, China).
2.4. Behavioral test
The neurological deficit was assessed 22 h after reperfusion blinded to the observer. Behavioral score was evaluated by the following criteria: 0 = no deficit, 1 = failure to extend left forepaw, 2 = spontaneous circling, 3 = falling to one side, and 4 = coma. Then, the rat was placed on an accelerated rotating rod (Panlab, Barcelona, Spain). The latency to fall off the rod was recorded three times, and the average time was calculated.
2.5. Measurement of infarct volume
After reperfusion for 24 h, the rats were sacrificed under anesthesia. The brains were rapidly removed, coronally sectioned at 2 mm, and stained with 2% 2, 3, 5-triphenyltetrazolium chloride (TTC, in 0.9% saline) for 10 min at 37 °C. Infarct size was determined by digital planimetry of the slices using ImageJ software and normalized for edema. The infarct volume for each brain was calculated as follows: Percentage of infarct volume = Σ (area of contralateral − normalized area of ipsilateral)/Σ area of contralateral.
2.6. Stereotaxic injection into the cerebral cortex
Cortical micro-injection was performed under anesthesia in a stereotaxic instrument using a microsyringe pump. A scalp incision was made, and a hole was opened in the right parietal skull, 4.6 mm lateral and 0.3 mm posterior to Bregma. A syringe was inserted into the brain to a depth of 3.0 mm below the skull surface. Capsaicin (1 μl) was injected slowly (0.25 μl/min). The syringe was kept in place for 5 min prior to removal. PBS (containing ethanol at a volume equal to the stock solution of capsaicin) was used as a vehicle control.
2.7. Primary cultured cortical neurons
Cerebral cortex isolated from embryonic day 18 rat (or embryonic day 16 mouse) embryos was digested with 0.25% trypsin for 30 min at 37 °C, followed by triturating with a pipette in plating medium (DMEM with 10% fetal bovine serum). Then, cells were plated onto 35-mm dishes coated with poly-D-lysine. After culturing for 4 h, the medium was changed to neurobasal medium (Invitrogen) supplemented with 2% B27 (Invitrogen) and 0.5 mM GlutaMAX-I (Invitrogen). To inhibit the growth of glial cells, 10 μM cytosine-1-β-D-arabinofuranoside was added to the medium at day 3 until the culture was used.
2.8. Glutamate-induced excitotoxicity
Cortical neurons at 7–9 DIV were stimulated with glutamate (50 μM) plus glycine (10 μM) in Mg2+-free extracellular solution (Mg2+-free ECS, 140 mM NaCl, 5.4 mM KCl, 1.3 mM CaCl2, 33 mM D-glucose, and 25 mM HEPES, pH 7.35, osmotic pressure 320–330 mOsm) for 1 h at 37 °C. Then, neurons were washed with ECS and incubated in normal culture medium for an additional 20 (for lactate dehydrogenase assay) or 24 h (for propidium iodide – Hoechst double staining).
2.9. Lactate dehydrogenase (LDH) assay
Cell viability was assessed by measuring the release of LDH into the culture medium using the CytoTox 96 Non-Radioactive Cytotoxicity Assay kit (Promega, Madison, WI, USA) according to the protocol of the manufacturer. Briefly, 50 of medium was transferred from culture wells to 96-well plates, mixed with 50μl of reaction solution and incubated for 30 min at room temperature. Stop solution was then added, and the optical density was measured at 490 nm using a microplate reader (Bio-Rad Laboratories, Hercules, CA, USA).
2.10. Propidium iodide – Hoechst double staining
Hoechst 33342 (5 μg/ml in PBS) was added to the culture for 10 min. Propidium iodide (PI, 15 μg/ml) was added later for an additional 10 min. The numbers of PI-positive and Hoechst-positive neurons were counted, and the percentage of PI-positive cells was calculated as the ratio of PI-positive neurons to Hoechst-positive neurons.
2.11. Calcium imaging
Fura-2 AM was applied to cortical neurons at a 5 μM dilution with the extracellular solution (ECS, 140 mM NaCl, 5.4 mM KCl, 1.3 mM CaCl2, 1 mM MgCl2, 33 mM D-glucose, and 25 mM HEPES, pH 7.35, osmotic pressure 320–330 mOsm). Neurons were washed with ECS three times and incubated in dye solution for 30 min at 37 °C. Then, the cells were washed twice with ECS and allowed to recover in ECS for 1 h. Changes in the intracellular Ca2+ concentration upon capsaicin or glutamate application were measured using the Live Cell Imaging System (Olympus, Japan). Fluorescence intensity at an excitation wavelength of 340 nm compared to that at an excitation wavelength of 380 nm (F340/F380) was calculated.
2.12. Western blot analysis
Tissues were homogenized in ice-cold lysis buffer (50 mM Tris-HCl, pH 7.4, 150 mM NaCl, 1.5 mM MgCl2, 10% glycerol, 1% Triton X-100, 5 mM EGTA, 1 μg/ml leupeptin, 1 mM PMSF, 1 mM Na3VO4, 10 mM NaF and proteinase inhibitor cocktail) and rotated at 4 °C for 1 h. Cell cultures were washed twice with ice-cold PBS and lysed with the lysis buffer indicated above. The lysates were centrifuged at 12,000g for 5 min to yield the total protein extract in the supernatant. The concentration of protein was measured with a BCA protein assay kit (Thermo Scientific, MA, USA). Equal amounts of samples (50 μg) were denatured and subjected to 10% SDS-PAGE. After separation, proteins were transferred to nitrocellulose membranes (Bio-Rad Laboratories). The membranes were blocked with 5% non-fat milk in TBST (25 mM Tris-HCl, pH 7.4, 137 mM NaCl, 2.7 mM KCl and 0.05% Tween 20) for 1 h at room temperature and incubated with primary antibody overnight at 4 °C. After washing three times with TBST, the membranes were incubated with HRP-labeled secondary antibody (Jackson Immunoresearch, West Grove, PA, USA) for 1 h at room temperature, then washed again and developed with ECL solutions (Santa Cruz). The immunoreactive bands were scanned and analyzed quantitatively by densitometry with ImageJ software.
2.13. Statistical analysis
The data are expressed as the mean ± SEM unless otherwise stated. Statistical analyses were performed using Prism 5.0 software. Differences between groups were compared using Fisher’s exact test, Student’s t-test, and one-way ANOVA followed by Dunnett’s post hoc test or two-way repeated measures ANOVA followed by Bonferroni’s post hoc test. Statistical significance was set at p < 0.05. Outliers were detected by Grubb’s test and excluded before further analysis.
3. Results
3.1. Topical administration of capsaicin in the peri-infarct area attenuates cerebral ischemia/reperfusion injury in the rat MCAO/reperfusion model
The neuroprotective effect of capsaicin following systemic administration (no more than the toxic dose: 1 mg/kg) is well described (Park et al., 2012; Pegorini et al., 2005; Veldhuis et al., 2003). However, whether capsaicin can protect neuronal ischemic injury via directly acting on cortical neurons independent of its vasodilation or hypothermic effects remains unknown. Thus, we injected capsaicin into the peri-infarct area to observe its effect on neuronal ischemic/reperfusion injury in the rat MCAO/reperfusion model. The procedures for MCAO/reperfusion surgery, capsaicin injection (1 μl), behavioral tests and TTC staining are shown in Fig. 1A. An equal volume of ethanol was dissolved in PBS and injected into the same site as a vehicle control.
Fig. 1.
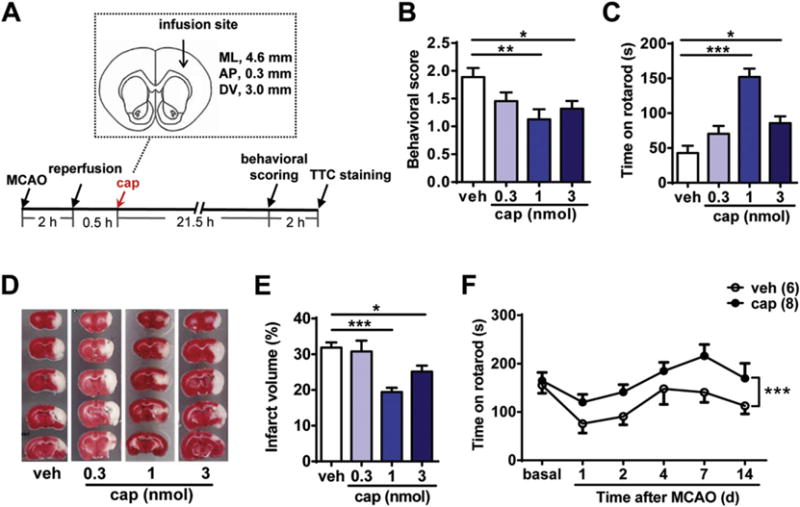
Topical application of capsaicin in the peri-infarct area alleviates the ischemic brain injury in the rat MCAO/reperfusion model. (A) Schematic diagram of rat MCAO/reperfusion procedure and capsaicin (cap) infusion site (relative to Bregma). Capsaicin was infused into the S1 area of the cortex. ML, mediolateral; AP, anteroposterior; DV, dorsoventral. (B) Intracortical injection of capsaicin (1 or 3 nmol) improved the neurological scoring of MCAO rats 22 h after reperfusion. n = 8–11, *p < 0.05, **p < 0.01, one-way ANOVA with Dunn’s post hoc tests. veh, vehicle. (C) Intracortical injection of capsaicin (1 or 3 nmol) increased the latency to fall from the accelerated rotarod of MCAO rats 22 h after reperfusion. n = 7–11, *p < 0.05, ***p < 0.001, one-way ANOVA with Dunnett’s post hoc tests. (D) and (E) Intracortical injection of capsaicin (1 or 3 nmol) 0.5 h after reperfusion significantly reduced the cerebral infarct volume. (D) Representative images of TTC-stained brain sections of MCAO/reperfusion rats with injection of different doses of capsaicin (0.3, 1, and 3 nmol) or vehicle. (E) Quantification of the infarct volume in the vehicle- or capsaicin-injected groups. n = 9–11, *p < 0.05, ***p < 0.001, one-way ANOVA with Dunn’s post hoc tests. (F) The improvement of motor coordination function of MCAO rats by capsaicin (1 nmol) persisted for at least 2 weeks. Open circles indicate the vehicle group and solid circles indicate the capsaicin group. ***p < 0.001, two-way ANOVA.
The behavioral tests of the MCAO/reperfusion rats were performed 22 h after reperfusion. The neurological scoring of MCAO rats was significantly improved in the capsaicin (1, or 3 nmol)-injected groups compared with that in the vehicle-injected group (p < 0.01, for 1 nmol; p < 0.05, for 3 nmol, compared to the vehicle group, Fig. 1B). The latency to fall from the accelerating rotarod of MCAO rats was also elongated in the capsaicin (1, or 3 nmol)-injected groups (p < 0.001, for 1 nmol; p < 0.05, for 3 nmol, compared to the vehicle group, Fig. 1C). At 24 h after reperfusion, the rats were sacrificed, and the volume of the brain infarct was assessed using TTC staining. Pale regions that failed to be stained with TTC represent non-viable infarcted brain regions. The quantification results showed that the infarct volume was significantly decreased following the injection of 1 or 3 nmol capsaicin 0.5 h after reperfusion (vehicle, 31.84 ± 1.43%; 0.3 nmol, 30.74 ± 3.05%; 1 nmol, 19.42 ± 1.21%; 3 nmol, 25.15 ± 1.67%) (p < 0.001, for 1 nmol; p < 0.05, for 3 nmol, compared to the vehicle control, Fig. 1D and E). Altogether, the above data demonstrated the protective effect of capsaicin following injection in the peri-infarct area.
We also tested the long-term effects on the cerebral MCAO/reperfusion injury after single capsaicin injection. During the 14 days after MCAO, the riding time on the rotarod of the capsaicin (1 nmol)-injected group was significantly longer than that in the vehicle-injected group (p < 0.001, Fig. 1F). This result suggested the long-term protective effect of capsaicin.
To further examine the therapeutic time window, we injected 1 nmol capsaicin into the MCAO rats at different time points after reperfusion. The results showed that capsaicin injected 0.5 or 1 h after reperfusion significantly decreased the infarct volume (0.5 h + vehicle, 31.84 ± 1.43%; 0.5 h + capsaicin, 19.42 ± 1.21%; 1 h + vehicle, 33.06 ± 3.16%; 1 h + capsaicin, 19.40 ± 2.65%) (p < 0.001, at 0.5 h; p < 0.01, at 1 h, compared to the vehicle control, Fig. 2A). Consistently, behavioral scoring showed that capsaicin (1 nmol) injected 1 h after reperfusion alleviated the stroke-related behaviors (p < 0.01, compared to the vehicle control, Fig. 2B). These data showed that the therapeutic time window of capsaicin injected in the peri-infarct area is within 1 h after reperfusion, when excitotoxicity is the main reason of cell death (Lai et al., 2014). Therefore, we inferred that capsaicin might alleviate neuronal ischemic injury through inhibiting excitotoxicity.
Fig. 2.
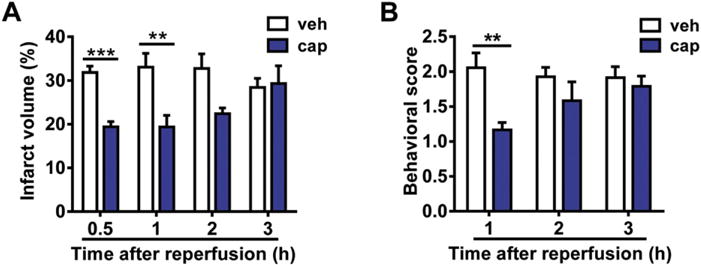
Time window of the neuroprotective effect of capsaicin. (A) Intracortical injection of capsaicin (cap, 1 nmol) 0.5 or 1 h after reperfusion reduced the cerebral infarct volume of MCAO/reperfusion rats. n = 5–11. veh, vehicle. **p < 0.01, ***p < 0.001, two-way ANOVA with Bonferroni post hoc tests. (B) Intracortical injection of capsaicin (1 nmol) 1 h after reperfusion improved the neurological scoring of MCAO/reperfusion rats. n = 6–12. **p < 0.01, two-way ANOVA with Bonferroni post hoc tests.
During the MCAO surgery, blood flow was measured using a laser Doppler flowmetry. Approximately 30% of blood flow remained after the insertion of the filament in the different doses of capsaicin- or vehicle-injected groups (Fig. 3A). No significant difference was detected among these groups. In addition, rectal temperature, blood pressure and blood oxygen saturation were monitored using the multifunctional monitoring equipment during the procedure of drug injection. At 30 min and 1 h after capsaicin injection, there was a decreasing trend in rectal temperature both in the capsaicin (1 nmol)- and vehicle-injected groups (Fig. 3B) without the use of heating incubator. However, no significant difference was detected between the two groups. Thus, the hypothermia is probably caused by anesthesia or surgery per se but not the action of capsaicin. To prevent the decline of body temperature, the rats were put into the homemade heating incubator for 2 h after surgery. Monitoring of blood pressure (Fig. 3C) and blood oxygen saturation (Fig. 3D) showed no significant difference between the capsaicin (1 nmol)- and vehicle-injected groups. Altogether, our data demonstrate that local administration of capsaicin in the peri-infarct area can alleviate neuronal injury in the rat MCAO/reperfusion model. Moreover, the protective effect of capsaicin is independent of its hypothermic effect.
Fig. 3.
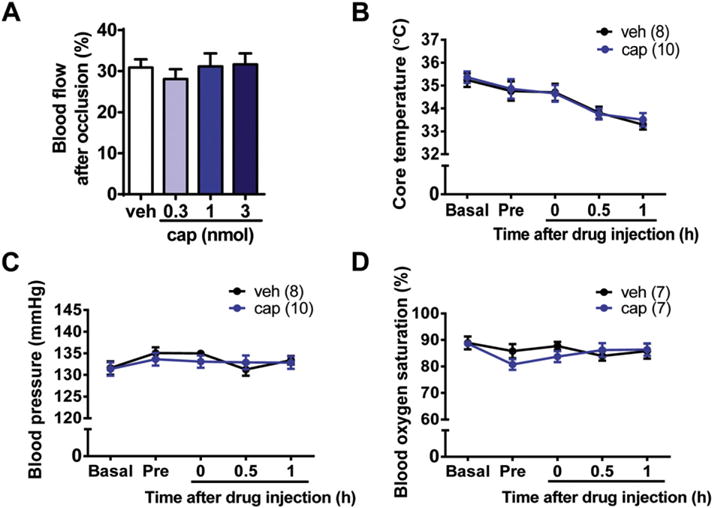
Monitoring of the blood flow, core temperature, blood pressure, and blood oxygen saturation of MCAO rats before and after drug administration. (A) Approximately 30% of blood flow remained after the artery occlusion both in the vehicle (veh)- and capsaicin (cap)-injected groups. n = 6–14. (B–D) No significant differences in the core temperature (B), blood pressure (C), and blood oxygen saturation (D) were detected between capsaicin (1 nmol)- and vehicle-injected groups. Pre, pre-injection.
3.2. Pre- or post-treatment with capsaicin attenuates glutamate-induced excitotoxic injury in cultured cortical neurons
The above in vivo data could not rule out the possible action of capsaicin on glia. To directly observe the effects of capsaicin on the neuronal excitotoxic injury, we used the glutamate-induced excitotoxicity model in cultured rat cortical neurons. Different doses of capsaicin (1, 3 or 10 μM) were added to the culture medium 30 min before glutamate stimulation (Fig. 4A) or together with glutamate (Fig. 4B).
Fig. 4.
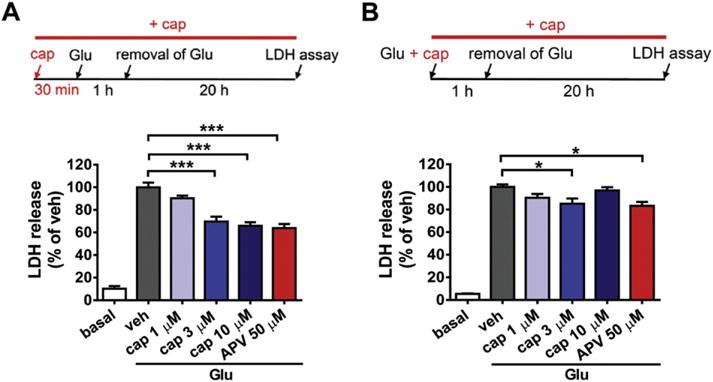
Capsaicin treatment attenuates glutamate-induced excitotoxic injury in cultured cortical neurons. (A) Capsaicin (cap, 3 and 10 μM) pretreatment for 30 min significantly reduced glutamate (Glu, 50 μM, plus 10 μM glycine)-induced LDH release in cultured cortical neurons. Amount of LDH released in the vehicle (veh) group was normalized to 100%. Top panel: Timeline of the drug treatment and LDH assay. n = 6–8, ***p < 0.001, one-way ANOVA with Bonferroni’s post hoc test. (B) Capsaicin (3 μM) treatment together with glutamate reduced the amount of LDH released in the culture medium. Amount of LDH released in the vehicle group was normalized to 100%. n = 6–8, *p < 0.05, one-way ANOVA with Bonferroni’s post hoc test.
Results from the LDH release assay showed that 50 μM glutamate (plus 10 μM glycine, 1 h) induced a remarkable increase of LDH in the culture medium at 20 h after glutamate stimulation compared to the normal control. Pretreatment with 3 or 10 μM capsaicin significantly decreased the amount of LDH released (veh, 100 ± 4.17%; 3 μM, 69.65 ± 4.34%; 10 μM, 65.81 ± 3.26%) (p < 0.001, both for 3 and 10 μM groups compared to the vehicle control, Fig. 4A), suggesting an attenuation of neuronal injury by capsaicin pretreatment. The NMDA receptor antagonist, APV (2-amino-5-phosphonopentanoic acid, 50 μM), was used as a positive control, which reduced the amount of LDH release to 63.85 ± 3.62% (p < 0.001, compared to the vehicle control). No significant differences were detected between the 3 or 10 μM capsaicin and APV groups, suggesting that the neuroprotective effect of capsaicin was comparable to that of APV.
The possible protective effect of capsaicin applied together with glutamate was also examined (Fig. 4B). The results showed that treatment with 3 μM capsaicin reduced the amount of LDH released in the medium (veh, 100 ± 2.20%; 3 μM, 85.19 ± 4.56%) (p < 0.05, compared to the vehicle control). Moreover, its protective effect was comparable to that of 50 μM APV (83.25 ± 3.47%) (p < 0.05, compared to the vehicle control). However, both the protective effects of capsaicin and APV were less than that seen in the pre-treatment procedure. Based on the above results, we chose 3 μM as the dose for in vitro application of capsaicin in the following studies.
PI-Hoechst double staining was also used to examine glutamate-induced neuronal death. PI-positive cells are considered late apoptotic or necrotic cells, i.e., dead cells. The proportion of cells that underwent glutamate-induced cell death was analyzed. Capsaicin treatment decreased glutamate-induced cell death in cortical neurons (PI-positive: vehicle, 148/269 = 55.02%; capsaicin, 117/270 = 43.33%) (p < 0.01, Fig. 5B and C). Similarly, RTX (10 nM), a potent TRPV1 agonist, also reduced glutamate-induced neuronal death (PI-positive: vehicle, 148/269 = 55.02%; RTX, 128/283 = 45.23%) (p < 0.05). Additionally, the protective effect of APV was observed (PI-positive: vehicle, 148/269 = 55.02%; APV, 93/234 = 39.74%) (p < 0.001). However, capsaicin combined with APV treatment could not further augment their protective effect, implying that the two drugs might act on the same pathways that involved in the neuronal injury. Altogether, the in vitro studies indicate that capsaicin protects cortical neurons from glutamate-induced excitotoxic injury.
Fig. 5.
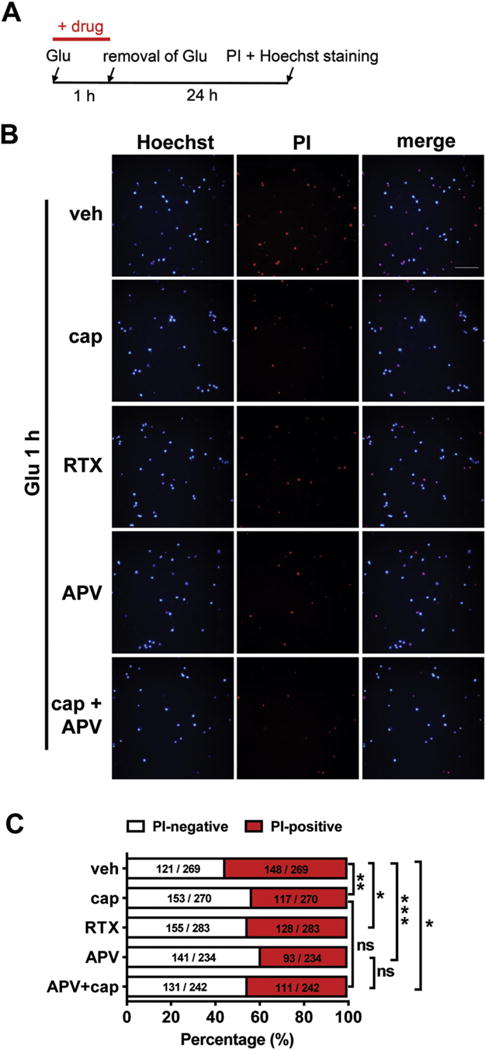
Capsaicin treatment attenuates glutamate-induced neuronal death in cultured mouse cortical neurons. (A) Timeline of the drug treatment and propidium iodide (PI)/Hoechst staining. (B) and (C) Attenuation of glutamate-induced neuronal death by capsaicin (cap, 3 pM), a potent TRPV1 agonist RTX (10 nM), NMDA antagonist APV (50 μM), or APV combined with capsaicin treatment in cultured cortical neurons. ns, no statistical significance. *p < 0.05, **p < 0.01, ***p < 0.001, Fisher’s exact test of one representative group. At least three parallel experiments were repeated. (B) Representative images show the protective effects of capsaicin, RTX, APV or combined treatment of APV and capsaicin on glutamate-induced neuronal death. Scale bar = 50 pm. (C) Quantification of the glutamate-induced neuronal death with the different drug treatment as shown in (B).
33. Trpv1 knock-out abolished the neuroprotective effect of capsaicin
To elucidate the role of TRPV1 in the neuroprotective effect of capsaicin, we generated trpv1 KO mice using TALEN technology (Fig. 6A). PCR genotyping was performed using the WT and KO primer pairs (Fig. 6B). As TRPV1 is highly expressed in DRG, we examined the content of TRPV1 protein in the DRG lysates of WT and KO mice. A faint expression of TPRV1 was examined in the KO mice (Fig. 6C), suggesting the efficient deletion of trpv1 gene. Importantly, trpv1 gene deletion abolished the protective effect of capsaicin on glutamate-induced neuronal death (PI-positive: vehicle, 285/539 = 52.88%; capsaicin, 359/689 = 52.10%) (p = 0.82, Fig. 6D and E), suggesting that the neuroprotective effect of capsaicin is TRPV1-dependent.
3.4. Capsaicin reduced glutamate-induced calcium influx in cultured cortical neurons
Based on the above result that capsaicin alleviates the glutamate-induced neuronal injury, we inferred that capsaicin might reduce the glutamate-induced cellular response. Calcium imaging assay was used to measure the neuronal response to glutamate stimulation. In rat cortical neurons, capsaicin pretreatment (3 μM, 1 h, Fig. 7A) significantly reduced glutamate-induced calcium influx (time = 100 s, vehicle, Ratio340/380 = 4.96 ± 0.70; capsaicin, Ratio340/380 = 2.80 ± 0.24) (p < 0.05, Fig. 7B and C). This result indicates that capsaicin can inhibit glutamate-induced calcium influx.
Fig. 7.
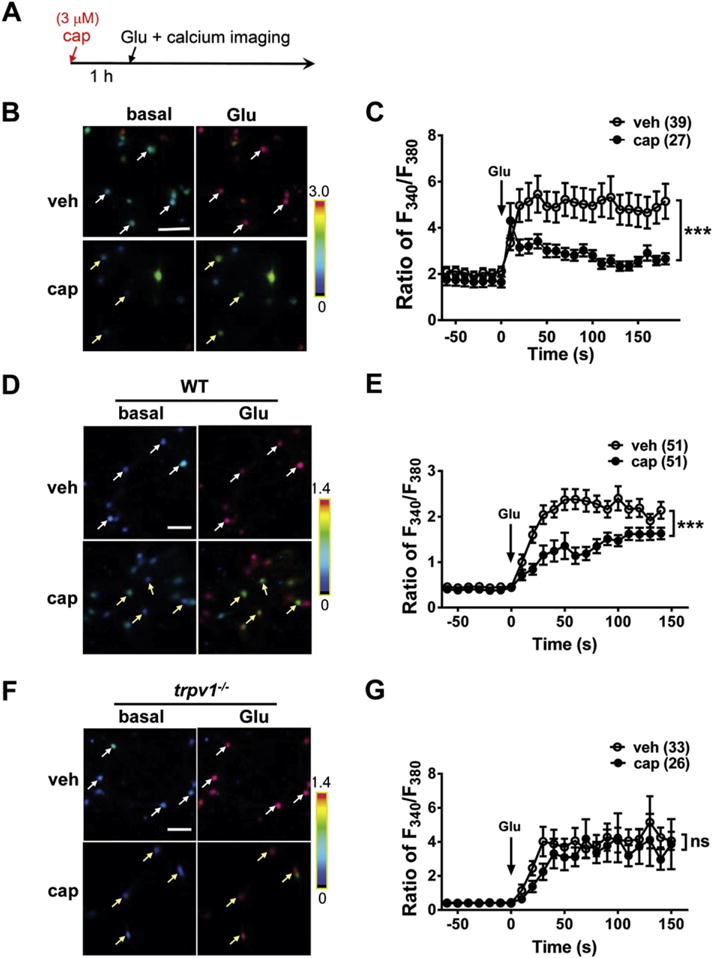
Trpv1 knock-out abolishes the inhibitory effect of capsaicin on glutamate-induced calcium influx in cortical neurons. (A) Timeline of the drug treatment and calcium imaging assay. (B) and (C) Capsaicin (cap, 3 μM) pretreatment for 1 h reduces glutamate (Glu, 100 μM)-induced calcium influx in cultured cortical neurons. (B) Representative images show glutamate-induced calcium influx in rat cortical neurons with vehicle (veh) or capsaicin pretreatment. (C) Quantification of the glutamate-induced calcium influx as shown in (B). ***p < 0.001, twoway ANOVA. (D) and (E) Capsaicin pretreatment significantly reduced glutamate-induced calcium influx in cultured cortical neurons from wild-type (WT) mice. (D) Representative images show glutamate-induced calcium influx in cortical neurons from wild-type mice with vehicle or capsaicin pretreatment. (E) Quantification of glutamate-induced calcium influx as shown in (D). ***p < 0.001, two-way ANOVA. (F) and (G) Capsaicin pretreatment had no effect on glutamate-induced calcium influx in the cortical neurons from trpv1 knock-out (KO) mice. (F) Representative images show the glutamate-induced calcium influx in cortical neurons from trpv1 knock-out mice with vehicle or capsaicin pretreatment. (G) Quantification of glutamate-induced calcium influx as shown in (F). ns, no statistical significance. Arrows in (B), (D) and (F) indicate the representative cells. Scale bar = 50 μm.
Similarly, a reduced response to glutamate was observed after capsaicin pretreatment in the cortical neurons from WT mice (time = 100 s, vehicle, Ratio340/380 = 2.40 ± 0.26; capsaicin, Ratio340/380 = 1.47 ± 0.12) (p < 0.001, Fig. 7D and E). However, the inhibitory effect of capsaicin on glutamate-induced calcium influx was absent in the cortical neurons from trpv1 KO mice (time = 100 s, vehicle, Ratio340/380 = 4.10 ± 0.72; capsaicin, Ratio340/380 = 4.24 ± 1.44) (p > 0.05, Fig. 7F and G). These data indicate that the inhibitory effect of capsaicin on glutamate-induced calcium influx is TRPV1-dependent.
3.5. Capsaicin down-regulates the amount of NMDA receptors
As NMDA receptor is a major contributor of glutamate-induced calcium influx and excitotoxic injury, we examined the expression of NMDA receptors after capsaicin treatment. Western blot analysis showed that the amount of GluN1 subunit, the obligatory subunit of NMDA receptor, in the peri-infarct area of MCAO rats was significantly reduced after capsaicin treatment for 0.5 h (p < 0.001, for the 1 nmol group; p < 0.001, for the 3 nmol group, compared to the vehicle control, Fig. 8A). In the cultured cortical neurons, capsaicin (3 μM) triggered a time-dependent reduction in the amount of the GluN1 subunit (p < 0.05, for the 30 min group; p < 0.01, for the 60 min group; p < 0.01, for the 120 min group, compared to the vehicle control, Fig. 8B). In addition, the expression of GluN2B subunit was also markedly reduced 60 min after capsaicin treatment (p < 0.01, Fig. 8C). Taken together, the above data indicate that down-regulation of the expression level of NMDA receptors might contribute to the protective effect of capsaicin on neuronal ischemic injury.
Fig. 8.
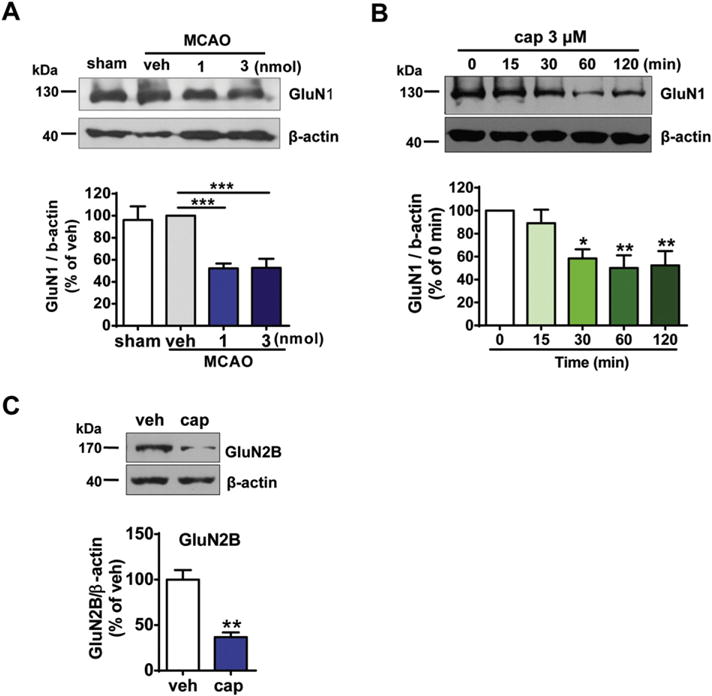
Capsaicin treatment down-regulates the amount of NMDA receptors. (A) The amount of GluN1 subunit was significantly reduced after capsaicin injection into the peri-infarct area of MCAO/reperfusion rats. Peri-infarct tissue was collected 1 h after reperfusion following 2 h MCAO. Capsaicin was injected 0.5 h after reperfusion. n = 3–4, ***p < 0.001, one-way ANOVA with Dunnett’s post hoc tests. (B) Time-dependent decrease in the GluN1 protein after capsaicin (cap, 3 μM) treatment in the rat cortical neurons. n = 5, *p < 0.05, **p < 0.01, one-way ANOVA with Dunnett’s post hoc tests. (C) The expression of GluN2B subunit of NMDA receptors was decreased after capsaicin (3 μM) treatment for 1 h in rat cortical neurons. n = 6, **p < 0.01, paired t-test
4. Discussion
Multiple lines of evidence indicate the neuroprotective effect of capsaicin (Guo et al., 2008; J.G. Lee et al., 2012; Pegorini et al., 2005; Sakamoto et al., 2014; Veldhuis et al., 2003). In the present work, we investigated the molecular mechanisms of the protection afforded by capsaicin on neuronal ischemic injury. As both morphological and functional studies indicate that TRPV1 is expressed in cortical neurons (Aguiar et al., 2009; de Novellis et al., 2011; Giordano et al., 2012; Goswami et al., 2010; Huang et al., 2015; Mezey et al., 2000; Pezzoli et al., 2014; Roberts et al., 2004; Shirakawa et al., 2008), investigation of the effects of capsaicin on neuronal survival via its direct action on neuronal TRPV1 is worth noting. Our in vivo combined with in vitro data, particularly the data from trpv1 KO mice, provide evidence for the involvement of neuronal TRPV1 in the protective effect of capsaicin on neuronal ischemic injury. With respect to the neuroprotective mechanisms of capsaicin, reduced expression of NMDA receptors and suppression of glutamate-induced calcium influx were observed following capsaicin treatment in cortical neurons.
4.1. Neurotoxic versus neuroprotective effects of capsaicin
Capsaicin is well-known for its ability to selectively damage primary sensory neurons with small-diameter unmyelinated C fibers. When 50 mg/kg capsaicin is given to neonatal rats subcutaneously, about half of the DRG neurons are rapidly killed (Jancso et al., 1977). Moreover, the neurotoxicity of capsaicin is not limited to DRG neurons and is more widespread in the nervous system. Degeneration of cell bodies, axons and nerve terminals can also be observed in retinal ganglion, bipolar cells, and some regions in the brain with administration of high doses of capsaicin. Activation of calcium-activated protease, apoptosis and blockade of axoplasmic transport of nerve growth factor all contribute to the neurotoxic effect of capsaicin after systemic administration (Caterina et al., 1997; Chard et al., 1995; Hail, 2003; Song et al., 2013).
On the other hand, the protective effect of capsaicin against ischemia/reperfusion injury in the heart (Wang and Wang, 2005), lung (Wang et al., 2012) and kidney (Ueda et al., 2008) has been reported. In spontaneously hypertensive rats, dietary intake of capsaicin (0.02%) increases activation and expression of endothelia nitric oxide synthase in the cerebrovasculature, an effect associated with a reduction of arteriolar hypertrophy and a delay in the onset of stroke (Xu et al., 2011). The direct evidence for the neuroprotective effect of capsaicin includes its protection from ouabain-induced excitotoxic injury in the brain (Veldhuis et al., 2003), transient global cerebral ischemia (Pegorini et al., 2005), NMDA-induced retinal ganglion cell loss (Sakamoto et al., 2014), hypoxia/reoxygenation-induced apoptosis of hippocampal neurons (Guo et al., 2008), and 1-methyl-4-phenylpyridinium (MPP+)-induced dopaminergic neuronal death (Park et al., 2012). Different mechanisms were proposed to account for the protection afforded by capsaicin. As both capsaicin and capsazepine show protection against ouabain-induced brain injury, it is proposed that the protective effect of capsaicin might be attributed to TRPV1 desensitization (Veldhuis et al., 2003). In the global cerebral ischemia model in Mongolian gerbils, hypothermic effect caused by systemic administration of capsaicin might contribute to its protective effect (Pegorini et al., 2005). Consistently, another TRPV1 agonist dihydrocapsaicin was reported to provide neuroprotection following ischemic stroke through promoting hypothermia (Cao et al., 2014). In addition, capsaicin-induced neuronal activity and anti-oxidative stress effect of capsaicin on microglia were proposed to contribute to the neuroprotective effect of capsaicin (Park et al., 2012; Sakamoto et al., 2014).
In terms of the discrepancy between the neurotoxic and neuroprotective effects of capsaicin, we infer that the outcome might depend on the dosage of capsaicin. Usually, the dosage used to ablate small-diameter unmyelinated C-fibers following systemic administration in neonatal or adult animals is ~50 mg/kg, whereas the maximal nontoxic dose of capsaicin in rats is 1 mg/kg (Di Marzo et al., 2001). Consistently, the neuroprotective effect of capsaicin via systemic administration has been observed at a dose no > 1 mg/kg (Park et al., 2012; Pegorini et al., 2005; Veldhuis et al., 2003). Herein, 0.3–3 nmol capsaicin was locally injected into the rat cerebral cortex. The dosage is in accordance with that used via intravitreal injection (Sakamoto et al., 2014). On the other hand, 3 μM capsaicin was applied in the in vitro studies, which is lower than the toxic dose of capsaicin normally used (Kim et al., 2005). Therefore, capsaicin shows a neuroprotective effect when it is used in a low dose.
In fact, the biphasic action of capsaicin could also be observed in its effect on gastric ulcer formation. Higher doses of capsaicin induced mucosal damage, whereas lower doses of capsaicin promoted gastric protection against ulcer or did not affect the mucosa (Abdel Salam et al., 1995). Thus, high concentrations of capsaicin are likely to evoke deleterious effects, suggesting that capsaicin activates different pathways at different concentrations. Effective dose showing the beneficial action of capsaicin should be investigated carefully.
4.2. Mechanism of the neuroprotective effect of capsaicin: involvement of neuronal TRPV1 and down-regulation of NMDA receptors
Our studies indicate that either direct injection of capsaicin into the peri-infarct area or application of capsaicin in the cultured cortical neurons protect neurons against ischemic injury. Although TRPV1-independent actions of capsaicin in non-neuronal cells have been reported (Gebhardt et al., 2016; Yang et al., 2014), it is very likely that neuronal TRPV1 mediates the neuroprotective effect of capsaicin. Our results of trpv1 knock-out mice provide direct evidence for the involvement of TRPV1 in the neuroprotective effect of capsaicin.
In terms of the molecular mechanisms of the neuroprotective effect of capsaicin, we observed down-regulation of the expression level and function of NMDA receptors after capsaicin treatment. As NMDA receptors are thought of as the major mediators of excitotoxicity, capsaicin may attenuate neuronal injury through inhibiting excitotoxicity. Our data from both in vivo and in vitro studies supported this notion. In the rat MCAO/reperfusion model, the time window of protective action afforded by capsaicin is within 1 h after reperfusion, when excitotoxicity is the main reason of cell death (Lai et al., 2014). The in vitro studies provided direct evidence for the attenuation of glutamate-induced excitotoxic injury by capsaicin.
With respect to the functional interaction between TRPV1 and NMDA receptors, previous studies have revealed the regulation of TRPV1 by NMDA receptor-initiated downstream signaling cascades such as protein kinase C and calcium/calmodulin-dependent kinase II in sensory neurons (Lee et al., 2012a, 2012b). Herein, we demonstrated a reciprocal regulation of NMDA receptors by TRPV1. This is in accordance with a recent study that showed that down-regulation of NMDA receptor function partially contributed to the antidepressantlike effects of capsaicin intracerebroventricular injection (Amiri et al., 2016). On the contrary, another study indicated that anandamide, an endogenous agonist of TRPV1, potentiated NMDA receptor-mediated current in a TRPV1-dependent manner in hippocampal neurons (K. Yang et al., 2014). Thus, TPPV1 activity has a complex modulatory role on NMDA receptors. Activation of TRPV1 by differential ligands or in differential conditions (physiological or pathological) might exert distinct effects on NMDA receptors. This is accordance with the reports that both TRPV1 agonists and antagonists show the antidepressant effects partly through the inhibition of NMDA receptors (Abdelhamid et al., 2014; Amiri et al., 2016; Manna and Umathe, 2012; Sartim et al., 2017).
4.3. Other possible mechanisms for the neuroprotective effect of capsaicin
Our studies demonstrated one new mechanism for the neuroprotective effect of capsaicin: down-regulation of NMDA receptors. However, we could not exclude the involvement of other possible mechanisms, for example, TRPV1 desensitization and anti-oxidant and anti-inflammatory actions of capsaicin. During brain injury, N-acyl-ethanolamines (including anandamide) and other membrane lipid derivatives accumulate in the brain (Hansen et al., 2002). These substances may act as the endogenous activators of TRPV1 and exacerbate Ca2+-overload through TRPV1-mediated calcium influx. However, capsaicin applied exogenously may quickly desensitize TRPV1, thus preventing TRPV1 activation through the endogenous pathway. This hypothesis is compatible with a recent study in trpv1 KO mice (Miyanohara et al., 2015). They found that either gene deletion of trpv1 or application of capsazepine reduced neuronal injury in MCAO/reperfusion model in mice and concluded that TRPV1 had a pathological role in cerebral ischemia/reperfusion. Combined with our results, we infer that activation of TRPV1 might activate beneficial or harmful pathways in different conditions. In addition, the anti-oxidant and anti-inflammatory effects of capsaicin should not be overlooked. Capsaicin represses reactive oxygen species generation and caspase-3 activation induced by oxidized low-density lipoprotein in human umbilical vein endothelial cells (Chen et al., 2015). Capsaicin can also inhibit the release of pro-inflammatory cytokine, interleukin-8, by H. pylori-infected gastric epithelial cells through the modulation of the IκB, NF-κB signaling pathway (Lee et al., 2007). This ability is related to the protective effect of capsaicin against gastro ulcers. Recently, it was reported that capsaicin-nanoparticles showed synergism with TNF-α-siRNA to decrease skin inflammation in mouse models (Desai et al., 2013).
On the other hand, our in vivo results could not rule out the possible action of capsaicin on astrocytes or microglia. A recent study showed that systemic administration of capsaicin protected nigral dopamine neurons in Parkinson’s disease, which was attributed to the activation of TRPV1 on astrocytes followed by the ciliary neurotrophic factor release (Nam et al., 2015). TRPV1 on astrocytes also contributes to the decrease of blood-brain barrier (BBB) permeability after the application of endocannabinoids in the in vitro BBB model (Hind et al., 2015). Additionally, TRPV1 in microglia is involved in cell migration, phagocytic activity, secretion of inflammatory cytokines and production of reactive oxygen species (Kong et al., 2017). Altogether, lines of evidence indicate the functional roles of TRPV1 in astrocytes and microglia. The possible effect of capsaicin on glia needs further investigation.
However, we can rule out the contribution of the hypothermic and vasodilation effects of capsaicin on its protection against brain ischemia. It was shown that the hypothermic effect of capsaicin is most prominent 30 min after systemic administration (Pegorini et al., 2005). We indeed observed a decreasing trend in rectal temperature at 30 min and 1 h after capsaicin injection in MCAO rats (Fig. 3B). To prevent this decline of body temperature, we placed the rats into the homemade heating incubator for 2 h after surgery. Thus, our work ruled out the contribution of the hypothermia to the neuroprotective effect of capsaicin. In terms of the vasodilation action of capsaicin, there is a low probability that it is involved in our studies. In spite of the fact that capsaicin can exert vasodilation effects through promoting the release of substance P and calcitonin gene-related peptide in sensory afferents (Burks et al., 1985; Wang, 2005), the brain parenchyma is void of the innervation of these sensory fibers. Thus, the route of drug administration in our studies, intracortical injection, avoided the possible effect of capsaicin to induce dilation of cerebral blood vessel through promoting the release of vasodilator neuropeptides.
5. Conclusions
Our work reveals a new mechanism for the neuroprotective effect of capsaicin. The contribution of NMDA receptor down-regulation was demonstrated. We found that topical administration of capsaicin, which avoids the possible side effects associated with systemic administration of capsaicin such as hypothermia, hypotension and bronchoconstriction, can attenuate neuronal ischemia/reperfusion injury. In contrast to the intracortical injection in the present studies, the available pharmaceutical formulations of capsaicin, including creams, lotions and patches, might be more practicable for clinical use. Better skin permeability and long-acting capsaicin formulations are helpful for its topical application. Capsaicin, an old molecule, might become a new promising drug for use in the treatment of brain ischemia.
Acknowledgments
This work was supported by grants from the National Natural Science Foundation of China (31371143 to Y.Z., 31530028, 91332119, 81161120497 to Y.W.), the Beijing Higher Education Young Elite Teacher Project (YEPT0051 to Y.Z.) and the Ministry of Science and Technology of China (973 Program: 2014CB542204 to Y.W.).
Abbreviations
- BBB
blood-brain barrier
- DIV
days in vitro
- DMSO
dimethyl sulphoxide
- DRG
dorsal root ganglion
- KO
knock-out
- LDH
lactate dehydrogenase
- MCAO
middle cerebral artery occlusion
- MPP+
1-methyl-4-phenylpyridinium
- NMDA
N-methyl-D-aspartic acid
- PI
propidium iodide
- RTX
resiniferatoxin
- TNF-α
tumor necrosis factor-α
- TRPV1
transient receptor potential vanilloid 1
- TTC
2, 3, 5-triphenyltetrazolium chloride
- WT
wild type
Footnotes
Conflict of interest statement
The authors declare no conflict of interests.
References
- Abdel Salam OM, Mozsik G, Szolcsanyi J. Studies on the effect of intragastric capsaicin on gastric ulcer and on the prostacyclin-induced cytoprotection in rats. Pharmacol Res. 1995;32:209–215. doi: 10.1016/s1043-6618(05)80024-6. [DOI] [PubMed] [Google Scholar]
- Abdelhamid RE, Kovács KJ, Nunez MG, Larson AA. Depressive behavior in the forced swim test can be induced by TRPV1 receptor activity and is dependent on NMDA receptors. Pharmacol Res. 2014;79:21–27. doi: 10.1016/j.phrs.2013.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aguiar DC, Terzian AL, Guimaraes FS, Moreira FA. Anxiolytic-like effects induced by blockade of transient receptor potential vanilloid type 1 (TRPV1) channels in the medial prefrontal cortex of rats. Psychopharmacology. 2009;205:217–225. doi: 10.1007/s00213-009-1532-5. [DOI] [PubMed] [Google Scholar]
- Amiri S, Alijanpour S, Tirgar F, Haj-Mirzaian A, Amini-Khoei H, Rahimi-Balaei M, Rastegar M, Ghaderi M, Ghazi-Khansari M, Zarrindast MR. NMDA receptors are involved in the antidepressant-like effects of capsaicin following amphetamine withdrawal in male mice. Neuroscience. 2016;329:122–133. doi: 10.1016/j.neuroscience.2016.05.003. [DOI] [PubMed] [Google Scholar]
- Brederson JD, Kym PR, Szallasi A. Targeting TRP channels for pain relief. Eur J Pharmacol. 2013;716:61–76. doi: 10.1016/j.ejphar.2013.03.003. [DOI] [PubMed] [Google Scholar]
- Burks TF, Buck SH, Miller MS. Mechanisms of depletion of substance P by capsaicin. Fed Proc. 1985;44:2531–2534. [PubMed] [Google Scholar]
- Cao Z, Balasubramanian A, Marrelli SP. Pharmacologically induced hypothermia via TRPV1 channel agonism provides neuroprotection following ischemic stroke when initiated 90 min after reperfusion. Am J Phys Regul Integr Comp Phys. 2014;306:R149–R156. doi: 10.1152/ajpregu.00329.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caterina MJ, Schumacher MA, Tominaga M, Rosen TA, Levine JD, Julius D. The capsaicin receptor: a heat-activated ion channel in the pain pathway. Nature. 1997;389:816–824. doi: 10.1038/39807. [DOI] [PubMed] [Google Scholar]
- Chard PS, Bleakman D, Savidge JR, Miller RJ. Capsaicin-induced neurotoxicity in cultured dorsal root ganglion neurons: involvement of calcium-activated proteases. Neuroscience. 1995;65:1099–1108. doi: 10.1016/0306-4522(94)00548-j. [DOI] [PubMed] [Google Scholar]
- Chen KS, Chen PN, Hsieh YS, Lin CY, Lee YH, Chu SC. Capsaicin protects endothelial cells and macrophage against oxidized low-density lipoprotein-induced injury by direct antioxidant action. Chem Biol Interact. 2015;228:35–45. doi: 10.1016/j.cbi.2015.01.007. [DOI] [PubMed] [Google Scholar]
- Desai PR, Marepally S, Patel AR, Voshavar C, Chaudhuri A, Singh M. Topical delivery of anti-TNFalpha siRNA and capsaicin via novel lipid-polymer hybrid nanoparticles efficiently inhibits skin inflammation in vivo. J Control Release. 2013;170:51–63. doi: 10.1016/j.jconrel.2013.04.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Marzo V, Lastres-Becker I, Bisogno T, De Petrocellis L, Milone A, Davis JB, Fernandez-Ruiz JJ. Hypolocomotor effects in rats of capsaicin and two long chain capsaicin homologues. Eur J Pharmacol. 2001;420:123–131. doi: 10.1016/s0014-2999(01)01012-3. [DOI] [PubMed] [Google Scholar]
- Fattori V, Hohmann MS, Rossaneis AC, Pinho-Ribeiro FA, Verri WA. Capsaicin: current understanding of its mechanisms and therapy of pain and other pre-clinical and clinical uses. Molecules. 2016;21 doi: 10.3390/molecules21070844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gebhardt C, von Bohlen Und Halbach O, Hadler MD, Harteneck C, Albrecht D. A novel form of capsaicin-modified amygdala LTD mediated by TRPM1. Neurobiol Learn Mem. 2016;136:1–12. doi: 10.1016/j.nlm.2016.09.005. [DOI] [PubMed] [Google Scholar]
- Giordano C, Cristino L, Luongo L, Siniscalco D, Petrosino S, Piscitelli F, Marabese I, Gatta L, Rossi F, Imperatore R, Palazzo E, de Novellis V, Di Marzo V, Maione S. TRPV1-dependent and -independent alterations in the limbic cortex of neuropathic mice: impact on glial caspases and pain perception. Cereb Cortex. 2012;22:2495–2518. doi: 10.1093/cercor/bhr328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goswami C, Rademacher N, Smalla KH, Kalscheuer V, Ropers HH, Gundelfinger ED, Hucho T. TRPV1 acts as a synaptic protein and regulates vesicle recycling. J Cell Sci. 2010;123:2045–2057. doi: 10.1242/jcs.065144. [DOI] [PubMed] [Google Scholar]
- Guo SY, Yang GP, Jiang DJ, Wang F, Song T, Tan XH, Sun ZQ. Protection of capsaicin against hypoxia-reoxygenation-induced apoptosis of rat hippocampal neurons. Can J Physiol Pharmacol. 2008;86:785–792. doi: 10.1139/Y08-083. [DOI] [PubMed] [Google Scholar]
- Hail N., Jr Mechanisms of vanilloid-induced apoptosis. Apoptosis. 2003;8:251–262. doi: 10.1023/a:1023620821878. [DOI] [PubMed] [Google Scholar]
- Hansen HS, Moesgaard B, Petersen G, Hansen HH. Putative neuroprotective actions of N-acyl-ethanolamines. Pharmacol Ther. 2002;95:119–126. doi: 10.1016/s0163-7258(02)00251-6. [DOI] [PubMed] [Google Scholar]
- Hind WH, Tufarelli C, Neophytou M, Anderson SI, England TJ, O’Sullivan SE. Endocannabinoids modulate human blood-brain barrier permeability in vitro. Br J Pharmacol. 2015;172:3015–3027. doi: 10.1111/bph.13106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang WX, Yu F, Sanchez RM, Liu YQ, Min JW, Hu JJ, Bsoul NB, Han S, Yin J, Liu WH, He XH, Peng BW. TRPV1 promotes repetitive febrile seizures by proinflammatory cytokines in immature brain. Brain Behav Immun. 2015;48:68–77. doi: 10.1016/j.bbi.2015.01.017. [DOI] [PubMed] [Google Scholar]
- Jancso G, Kiraly E, Jancso-Gabor A. Pharmacologically induced selective degeneration of chemosensitive primary sensory neurones. Nature. 1977;270:741–743. doi: 10.1038/270741a0. [DOI] [PubMed] [Google Scholar]
- Julius D. TRP channels and pain. Annu Rev Cell Dev Biol. 2013;29:355–384. doi: 10.1146/annurev-cellbio-101011-155833. [DOI] [PubMed] [Google Scholar]
- Kim SR, Lee DY, Chung ES, Oh UT, Kim SU, Jin BK. Transient receptor potential vanilloid subtype 1 mediates cell death of mesencephalic dopaminergic neurons in vivo and in vitro. J Neurosci. 2005;25:662–671. doi: 10.1523/JNEUROSCI.4166-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kong WL, Peng YY, Peng BW. Modulation of neuroinflammation: Role and therapeutic potential of TRPV1 in the neuro-immune axis. Brain Behav Immun. 2017 doi: 10.1016/j.bbi.2017.03.007. Epub ahead of print. http://dx.doi.org/10.10167j.bbi.2017.03.007. [DOI] [PubMed]
- Lai TW, Zhang S, Wang YT. Excitotoxicity and stroke: identifying novel targets for neuroprotection. Prog Neurobiol. 2014;115:157–188. doi: 10.1016/j.pneurobio.2013.11.006. [DOI] [PubMed] [Google Scholar]
- Lee IO, Lee KH, Pyo JH, Kim JH, Choi YJ, Lee YC. Anti-inflammatory effect of capsaicin in Helicobacter pylori-infected gastric epithelial cells. Helicobacter. 2007;12:510–517. doi: 10.1111/j.1523-5378.2007.00521.x. [DOI] [PubMed] [Google Scholar]
- Lee J, Chung MK, Ro JY. Activation of NMDA receptors leads to phosphorylation of TRPV1 S800 by protein kinase C and A-kinase anchoring protein 150 in rat trigeminal ganglia. Biochem Biophys Res Commun. 2012b;424:358–363. doi: 10.1016/j.bbrc.2012.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J, Saloman JL, Weiland G, Auh QS, Chung MK, Ro JY. Functional interactions between NMDA receptors and TRPV1 in trigeminal sensory neurons mediate mechanical hyperalgesia in the rat masseter muscle. Pain. 2012c;153:1514–1524. doi: 10.1016/j.pain.2012.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JG, Yon JM, Lin C, Jung AY, Jung KY, Nam SY. Combined treatment with capsaicin and resveratrol enhances neuroprotection against glutamate-induced toxicity in mouse cerebral cortical neurons. Food Chem Toxicol. 2012a;50:3877–3885. doi: 10.1016/j.fct.2012.08.040. [DOI] [PubMed] [Google Scholar]
- Longa EZ, Weinstein PR, Carlson S, Cummins R. Reversible middle cerebral artery occlusion without craniectomy in rats. Stroke. 1989;20:84–91. doi: 10.1161/01.str.20.1.84. [DOI] [PubMed] [Google Scholar]
- Manna SS, Umathe SN. A possible participation of transient receptor potential vanilloid type 1 channels in the antidepressant effect of fluoxetine. Eur J Pharmacol. 2012;685:81–90. doi: 10.1016/j.ejphar.2012.04.023. [DOI] [PubMed] [Google Scholar]
- Mezey E, Toth ZE, Cortright DN, Arzubi MK, Krause JE, Elde R, Guo A, Blumberg PM, Szallasi A. Distribution of mRNA for vanilloid receptor subtype 1 (VR1), and VR1-like immunoreactivity, in the central nervous system of the rat and human. Proc Natl Acad Sci U S A. 2000;97:3655–3660. doi: 10.1073/pnas.060496197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyanohara J, Shirakawa H, Sanpei K, Nakagawa T, Kaneko S. A pathophysiological role of TRPV1 in ischemic injury after transient focal cerebral ischemia in mice. Biochem Biophys Res Commun. 2015;467:478–483. doi: 10.1016/j.bbrc.2015.10.027. [DOI] [PubMed] [Google Scholar]
- Nam JH, Park ES, Won SY, Lee YA, Kim KI, Jeong JY, Baek JY, Cho EJ, Jin M, Chung YC, Lee BD, Kim SH, Kim EG, Byun K, Lee B, Woo DH, Lee CJ, Kim SR, Bok E, Kim YS, Ahn TB, Ko HW, Brahmachari S, Pletinkova O, Troconso JC, Dawson VL, Dawson TM, Jin BK. TRPV1 on astrocytes rescues nigral dopamine neurons in Parkinson’s disease via CNTF. Brain. 2015;138:3610–3622. doi: 10.1093/brain/awv297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Novellis V, Vita D, Gatta L, Luongo L, Bellini G, De Chiaro M, Marabese I, Siniscalco D, Boccella S, Piscitelli F, Di Marzo V, Palazzo E, Rossi F, Maione S. The blockade of the transient receptor potential vanilloid type 1 and fatty acid amide hydrolase decreases symptoms and central sequelae in the medial prefrontal cortex of neuropathic rats. Mol Pain. 2011;7:7. doi: 10.1186/1744-8069-7-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park ES, Kim SR, Jin BK. Transient receptor potential vanilloid subtype 1 contributes to mesencephalic dopaminergic neuronal survival by inhibiting microglia-originated oxidative stress. Brain Res Bull. 2012;89:92–96. doi: 10.1016/j.brainresbull.2012.07.001. [DOI] [PubMed] [Google Scholar]
- Pegorini S, Braida D, Verzoni C, Guerini-Rocco C, Consalez GG, Croci L, Sala M. Capsaicin exhibits neuroprotective effects in a model of transient global cerebral ischemia in Mongolian gerbils. Br J Pharmacol. 2005;144:727–735. doi: 10.1038/sj.bjp.0706115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pezzoli M, Elhamdani A, Camacho S, Meystre J, Gonzalez SM, le Coutre J, Markram H. Dampened neural activity and abolition of epileptic-like activity in cortical slices by active ingredients of spices. Sci Rep. 2014;4:6825. doi: 10.1038/srep06825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts JC, Davis JB, Benham CD. [3H]Resiniferatoxin autoradiography in the CNS of wild-type and TRPV1 null mice defines TRPV1 (VR-1) protein distribution. Brain Res. 2004;995:176–183. doi: 10.1016/j.brainres.2003.10.001. [DOI] [PubMed] [Google Scholar]
- Sakamoto K, Kuroki T, Okuno Y, Sekiya H, Watanabe A, Sagawa T, Ito H, Mizuta A, Mori A, Nakahara T, Ishii K. Activation of the TRPV1 channel attenuates N-methyl-D-aspartic acid-induced neuronal injury in the rat retina. Eur J Pharmacol. 2014;733:13–22. doi: 10.1016/j.ejphar.2014.03.035. [DOI] [PubMed] [Google Scholar]
- Sartim AG, Moreira FA, Joca SR. Involvement of CB1 and TRPV1 receptors located in the ventral medial prefrontal cortex in the modulation of stress coping behavior. Neuroscience. 2017;340:126–134. doi: 10.1016/j.neuroscience.2016.10.031. [DOI] [PubMed] [Google Scholar]
- Shirakawa H, Yamaoka T, Sanpei K, Sasaoka H, Nakagawa T, Kaneko S. TRPV1 stimulation triggers apoptotic cell death of rat cortical neurons. Biochem Biophys Res Commun. 2008;377:1211–1215. doi: 10.1016/j.bbrc.2008.10.152. [DOI] [PubMed] [Google Scholar]
- Song J, Lee JH, Lee SH, Park KA, Lee WT, Lee JE. TRPV1 activation in primary cortical neurons induces calcium-dependent programmed cell death. Exp Neurobiol. 2013;22:51–57. doi: 10.5607/en.2013.22.1.51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szolcsanyi J. Capsaicin and sensory neurones: A historical perspective. Prog Drug Res. 2014;68:1–37. doi: 10.1007/978-3-0348-0828-6_1. [DOI] [PubMed] [Google Scholar]
- Ueda K, Tsuji F, Hirata T, Takaoka M, Matsumura Y. Preventive effect of TRPV1 agonists capsaicin and resiniferatoxin on ischemia/reperfusion-induced renal injury in rats. J Cardiovasc Pharmacol. 2008;51:513–520. doi: 10.1097/FJC.0b013e31816f6884. [DOI] [PubMed] [Google Scholar]
- Veldhuis WB, van der Stelt M, Wadman MW, van Zadelhoff G, Maccarrone M, Fezza F, Veldink GA, Vliegenthart JF, Bar PR, Nicolay K, Di Marzo V. Neuroprotection by the endogenous cannabinoid anandamide and arvanil against in vivo excitotoxicity in the rat: role of vanilloid receptors and lipoxygenases. J Neurosci. 2003;23:4127–133. doi: 10.1523/JNEUROSCI.23-10-04127.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang DH. The vanilloid receptor and hypertension. Acta Pharmacol Sin. 2005;26:286–294. doi: 10.1111/j.1745-7254.2005.00057.x. [DOI] [PubMed] [Google Scholar]
- Wang L, Wang DH. TRPV1 gene knockout impairs postischemic recovery in isolated perfused heart in mice. Circulation. 2005;112:3617–3623. doi: 10.1161/CIRCULATIONAHA.105.556274. [DOI] [PubMed] [Google Scholar]
- Wang M, Ji P, Wang R, Zhao L, Xia Z. TRPV1 agonist capsaicin attenuates lung ischemia-reperfusion injury in rabbits. J Surg Res. 2012;173:153–160. doi: 10.1016/j.jss.2010.08.053. [DOI] [PubMed] [Google Scholar]
- Ward NJ, Ho KW, Lambert WS, Weitlauf C, Calkins DJ. Absence of transient receptor potential vanilloid-1 accelerates stress-induced axonopathy in the optic projection. J Neurosci. 2014;34:3161–3170. doi: 10.1523/JNEUROSCI.4089-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu X, Wang P, Zhao Z, Cao T, He H, Luo Z, Zhong J, Gao F, Zhu Z, Li L, Yan Z, Chen J, Ni Y, Liu D, Zhu Z. Activation of transient receptor potential vanilloid 1 by dietary capsaicin delays the onset of stroke in stroke-prone spontaneously hypertensive rats. Stroke. 2011;42:3245–3251. doi: 10.1161/STROKEAHA.111.618306. [DOI] [PubMed] [Google Scholar]
- Yang K, Lei G, Xie YF, MacDonald JF, Jackson MF. Differential regulation of NMDAR and NMDAR-mediated metaplasticity by anandamide and 2-AG in the hippocampus. Hippocampus. 2014a;24:1601–1614. doi: 10.1002/hipo.22339. [DOI] [PubMed] [Google Scholar]
- Yang R, Xiong Z, Liu C, Liu L. Inhibitory effects of capsaicin on voltage-gated potassium channels by TRPV1-independent pathway. Cell Mol Neurobiol. 2014b;34:565–576. doi: 10.1007/s10571-014-0041-1. [DOI] [PMC free article] [PubMed] [Google Scholar]