

Abstract
Aims
Following an acute myocardial infarction (MI) the extracellular matrix (ECM) undergoes remodeling in order to prevent dilation of the infarct area and maintain cardiac output. Excessive and prolonged inflammation following an MI exacerbates adverse ventricular remodeling. Macrophages are an integral part of the inflammatory response that contribute to this remodeling. Treatment with histone deacetylase (HDAC) inhibitors preserves LV function and myocardial remodeling in the post-MI heart. This study tested whether inhibition of HDAC activity resulted in preserving post-MI LV function through the regulation of macrophage phenotype and early resolution of inflammation.
Methods and Results
HDAC inhibition does not affect the recruitment of CD45+ leukocytes, CD45+/CD11b+ inflammatory monocytes or CD45+/CD11b+CD86+ inflammatory macrophages for the first 3 days following infarct. Further, HDAC inhibition does not change the high expression level of the inflammatory cytokines in the first days following MI. However, by day 7, there was a significant reduction in the levels of CD45+/Cd11b+ and CD45+/CD11b+/CD86+ cells with HDAC inhibition. Remarkably, HDAC inhibition resulted in the dramatic increase in the recruitment of CD45+/CD11b+/CD206+ alternatively activated macrophages as early as 1 Day which remained significantly elevated until 5 days post-MI. qRT-PCR revealed that HDAC inhibitor treatment shifts the cytokine and chemokine environment towards an M2 phenotype with upregulation of M2 markers at 1 and 5 days post-MI. Importantly, HDAC inhibition correlates with significant preservation of both LV ejection fraction and end-diastolic volume and is associated with a significant increase in micro-vessel density in the border zone at 14 days post-MI.
Conclusion
Inhibition of HDAC activity result in the early recruitment of reparative CD45+/CD11b+/CD206+ macrophages in the post-MI heart and correlates with improved ventricular function and remodeling. This work identifies a very promising therapeutic opportunity to manage macrophage phenotype and enhance resolution of inflammation in the post-MI heart.
Keywords: Myocardial Infarction, Histone Deacetylases, Macrophage polarization
1. Introduction
Coronary Artery Disease with resultant myocardial infarction (MI) is one of the leading causes of progression to Heart Failure (HF). One of the major complications of MI leading to HF and ultimately death is remodeling of the left ventricle1. Macroscopically, these changes include: 1) expansion and extension of the infarct into the non-ischemic or remote zone, and 2) hypertrophy and dilation of the left ventricle which alters LV loading leading to LV dysfunction and ultimately HF2. The heart undergoes three phases as part of the healing process post-MI: inflammation, proliferation, and scar maturation. However; these macroscopic changes are affected by molecular abnormalities including: excessive extracellular matrix (ECM) degradation by matrix metalloproteinases (MMPs), aberrant initiation and prolongation of inflammation, disproportionate fibrosis, and cardiomyocyte hypertrophy3–6. Successfully targeting one or more of these abnormalities has been challenging. In 2007 Lee et al. showed in a rat model of MI, HDAC inhibition resulted in significant decreases in cardiomyocyte hypertrophy and reduced collagen formation in the border zone and remote zone along with an improvement in ejection fraction 4 weeks following MI7. Treatment with the class I and IIb HDAC inhibitor, SAHA, has been shown to reduce myocardial infarct size in rabbits when delivered at the time of reperfusion8. Recently, we demonstrated that inhibition of class I HDAC activity repressed MMP-9 expression and preserved LV function in post myocardial infarction remodeling in mice9.
Interestingly, a significant source of MMP-9 post MI-injury is inflammatory cells, especially macrophages. Many studies have shown post-MI healing is a complex process and that the quality of infarct healing determines the prognosis10, 11. Ischemic injury activates the recruitment of large numbers of inflammatory monocytes which give rise to classically activated (stimulated by TLR-ligands and IFN-γ), inflammatory (M1) macrophages which are responsible for removal of cell debris and necrotic cell clearance and are a significant source of MMP-9 for degradation and remodeling of the ECM. Between day 3 and 7 the inflammatory monocytes give rise to less inflammatory, alternatively activated (stimulated by IL-4/IL-13), reparative (M2) macrophages12. These reparative macrophages promote collagen deposition, scar maturation and angiogenesis13. Clearly, macrophage populations in the post-MI ventricle are heterogeneous and more complex than characterized by the M1/M2 model14, but until a more accurate paradigm is established and with the understanding that it is limiting, this work will use the M1 and M2 designations. The balance between inflammatory pathways and reparative pathways determines the mechanical properties of the infarct tissue and sets the stage for either infarct healing or heart failure development15. Prolonged inflammation accentuates matrix degradation leading to deleterious remodeling and instability of scar13. Several studies have shown that influencing the earlier transition of macrophage phenotypes from inflammatory to reparative is potentially cardioprotective. siRNA targeting of the IRF5 transcription factor reduced the expression of inflammatory M1 markers and showed an increase in the presence of M2 macrophages within the heart following ischemia reperfusion. Depletion of M2-like macrophages in Trib1−/− mice results in impaired fibroblast-mediated repair of the infarcted myocardium 16. This resulted in accelerated myocardial healing17. HDAC inhibition has also been shown to induce M2 macrophage polarization in vitro18 but the role of HDAC inhibition on M2 macrophage polarization in the heart is not known. Development of therapies which increase the number of cardiac M2 macrophages appears to be a promising approach to improve healing following MI19. Therefore, the objective of this study was to determine the impact of HDAC inhibition on MI induced macrophage recruitment and polarization and how that influences LV remodeling and dysfunction.
2. Materials and Methods
2.1. Animal Models
Briefly, the left anterior descending coronary artery was permanently ligated on 12–15 week old CD1 male mice. Mice were anesthetized for all procedures (isoflurane, 2 %; O2 2 L/min), intubated and ventilated. Left thoracotomy was performed in the fourth intercostal space and left anterior descending coronary artery was visualized and ligated just distal to its main bifurcation using 8.0 Neuriolon. MI was confirmed by LV blanching and ST-segment elevation on the electrocardiogram. Mice were randomly assigned to treatment cohort. The first dose of the class I/IIb inhibitor, SAHA was administered by IP injection at 100mg/kg immediately after surgery and subsequent doses were delivered via drinking water as described previously.20 At termination the heart was excised then perfused with ice cold saline, separated into infarct/border zone and remote zone for flow cytometry, Western analysis, qRT-PCR or whole hearts used for immunostaining. All of the animal experiments were performed according to protocols approved by the Medical University of South Carolina Institutional Animal Care and Use Committee and in accordance with U.S. National Institute of Health (NIH) guidelines.
2.2. Functional Analysis
Echocardiography was performed on mice before MI surgery and just prior to sacrifice. A 15-MHz transducer (Sonos 5500; Agilent Technologies, Andover, MA, USA) was placed on a layer of acoustic coupling gel applied to the hemithorax. Standard Doppler techniques were used to collect data. LV dimensions and wall thickness were made at end-systole and end-diastole using the American Society of Echocardiography criteria. Mean wall thickness will be calculated as the average of the end diastolic intraventricular septal wall thickness and left ventricular posterior wall thickness. Relative wall thickness was calculated as: mean LV thickness divided by the LV internal dimension at end-diastole. LV mass was calculated using the standard formula and LV mass was normalized to body weight. LV end diastolic volume and end systolic volume was determined using Simpson’s method of disks and used to compute ejection fraction.
2.3. Flowcytometry
Hearts were harvested, and infarct tissue was minced, and digested in 1 mg/ml collagenase type II (Worthington, Lakewood, NJ, USA) in 30mM Taurine-10 mM HEPES-HBSS buffer, (10 min., 5 times, 37°C). Tissues were triturated and filtered with 40 μm cell strainer (Denville, USA). Isolated cells were then stained with primary antibodies in FACS buffer (5% FBS, 0.1% NaN3 sodium azide in PBS buffer) for 30 min. at 4°C, Table 1 before staining with Live/Dead Liability Dye in PBS buffer (Thermofisher Scientific, Waltham, MA, USA) for 45 min. at 4°C. Flowcytometry analysis was performed on a BD LSR Fortessa X-20/FACS using Diva 6 Software (BD Biosciences, San Jose, CA, USA). Results were analyzed using FlowJo Software V10.2 (TreeStar, Inc., Ashland, OR, USA), and gates were set based on Fluorescence Minus One controls.
Table 1.
| Antibody | Clone | Source(s) | Location | Catalog Number | Host Species | Concentration |
|---|---|---|---|---|---|---|
| CD11b | CBRM1/5 | Biolegend | San Diego, CA | 301410 | Mouse | 2μg/mL |
| CD45 | 30-F11 | Biolegend | San Diego, CA | 103114 | Rat | 2μg/mL |
| CD206 | C068C2 | Biolegend | San Diego, CA | 141717 | Rat | 2μg/mL |
| CD86 | GL-1 | Biolegend | San Diego, CA | 105008 | Rat | 2μg/mL |
2.4. Culture of RAW 264.7 macrophages
The murine macrophage cell line, RAW264.7 (ATCC) were maintained in Macrophage Complete Media [Dulbecco’s Modified Eagle Medium (DMEM; Invitrogen) supplemented with 10 mM L-glutamine, 100 IU/ml penicillin, and 100 μg/ml streptomycin, 10% (v/v) fetal bovine serum (FBS; Atlanta Biological), 100μg/ ml Primocin (invivogen), 1X Fungizone (Gibco), incubated at 5% CO2 and 95% oxygen. Cells will be used for experimentation between 2 and 5 passages. Experimentation was carried out in macrophage complete media lacking FBS. To induce M1 macrophage polarization the cells were treated with 100ng/ml of lipopolysaccharide (Sigma). To induce M2 macrophage polarization cells were treated with IL-4 at 30ng/ml and IL-10 at 20ng/ml (BioAbChem) based on dose response curve validation. Cells were treated with SAHA at 5μM and TSA at 100nM concentrations.
2.5. Isolation of murine Bone marrow macrophages
Murine bone marrow macrophages were isolated from CD1 mice between ages 8–10 weeks. The femur and tibia of male mice were amputated and washed in 1X Hanks balanced salt solution without phenol red (Gibco) following sacrifice. Legs were cleaned of muscle and tissue; ends clipped and placed in sterile 1.5 ml eppendorf tubes. Tubes containing legs were then centrifuged at 6000 RCF for 30 seconds to remove bone marrow from bones. Red blood cells were then lysed using ACK lysing buffer (Gibco) for 3 minutes. Remaining cells are then washed 3 times in HBSS for 10 minutes each and centrifuged at 1200 RPM. Bone marrow cells were then re-suspended in Macrophage complete media [Dulbecco’s Modified Eagle Medium (DMEM; Invitrogen) supplemented with 10 mM L-glutamine, 100 IU/ml penicillin, and 100 μg/ml streptomycin, 10% (v/v) fetal bovine serum (FBS; Atlanta Biological), 100μg/ ml Primocin (invivogen), 1X Fungizone (Gibco), and 100 U/ml recombinant mouse macrophage colony stimulating factor (M-CSF; BioAbChem).] Re-suspended cells were then plated at a density of 3.0 × 106 and grown in macrophage complete media for 5 days with media changes on days 3 and 5. On day five cells were polarized to M1 with interferon-gamma [IFN-γ (20ng/ml)] and lipopolysacharide [LPS (100ng/ml)] for 24 hours before experimentation. Experimentation is then carried out on day 6.
2.6. Protein Isolation
For tissue, 50–100mg sections of both infarct and remote zone were homogenized in lysis buffer (20mM Tris, 150mM NaCl, 1mM EDTA, 1mM EGTA, 1mM β-glycerol, 2.5 mM Na pyrophosphate, and 1% Triton x-100) in varying volumes according to weight of sections supplemented with phosphatase and protease inhibitor cocktails (Roche). Protein concentration was measured using BioRad BCA assay (Bio-Rad, Hercules, CA USA) and samples were prepared in 2X SDS at 15μg/15μl.
For cells, media was removed and immediately used for zymography or stored at −80°C until usage with no addition of protease or phosphatase inhibitors. Cells were washed with ice cold PBS, then lysed with lysis buffer (20mM Tris, 150mM NaCl, 1mM EDTA, 1mM EGTA, 1mM β-glycerol, 2.5 mM Na pyrophosphate, and 1% Triton x-100) supplemented with phosphatase and protease inhibitor cocktails (Roche). The cells were then frozen at −80 for 1 hours, removed and allowed to thaw on ice and then insoluble material pelleted by centrifugation in a tabletop microcentrifuge at 4°C. Protein concentration was measured using BCA assay (Bio-Rad, Hercules, CA USA) and samples prepared with 4X SDS at 15ug/15ul.
2.7. Quantitative RT-PCR
Total messenger RNA (mRNA) was extracted using the RNeasy Fibrous Tissue Kit (Qiagen) for tissue or RNeasy Mini Kit (Qiagen) for cells according to manufacturer’s instructions. One microgram of mRNA was reverse transcribed using the high capacity BioRAD qRT-PCR cDNA synthesis kit. BioRad syber green primer probe expression assays were used to quantify gene targets. The date is represented as fold changes in gene expression normalized to GAPDH mRNA relative treated controls using the Pfaffl Method and standard curves.
2.8. Zymography
Abundance of the active form of MMP-9 was evaluated using gelatin zymography. Briefly, 25 ul of concentrated cell media containing secreted protein was separated out on 10% SDS-PAGE gels copolymerized with 0.1% gelatin (Sigma) and analyzed using invitrogen manufactures protocol. Gels were soaked in renaturing buffer (Invitrogen) at RT and then incubated for 18 hours at 37°C in developing buffer (Invitrogen). Following incubation gels were stained with Coomassie blue staining (Sigma) and destained (Methanol:Acetic acid:Water, 50:10:40) until clear bands were noticeable ~15 minutes, then washed in DDW water. Areas of MMP activity were interpreted as clear bands and then analyzed using densitometry.
Immunostaining
Hearts were perfused with ice-cold PBS after mice were euthanized. Hearts were removed, rinsed in saline and immediately embedded in O.C.T. compound (Sakura Finetek) at −80°C or placed at 4°C overnight in 10% formalin for fixation followed by dehydration and paraffin embedding. Sections were taken at 5μm on a cryostat for O.C.T embedded hearts or at 7.5–10μm for paraffin embedded formalin fixed hearts.
2.9. Fluorescence
O.C.T embedded 5μm sections were fixed with 4% paraformaldehyde for 10 minutes and permeabilized with 0.1% Triton for 5 minutes at room temperature (RT). Blocking was done at RT for 1 hour with 1% BSA and serial sections were stained overnight at 4°C with anti-MAC-3-FITC (MAC-3, CL8943A, Cederlane), anti-F4/80-FITC (invitrogen), anti-CD11c (Abcam, ab11029), and CH3L3 (Ym1) (Abcam, ab930304). Sections were washed the following day and mounted using DAPI-gold (Invitrogen).
2.10. Immunohistochemistry
Masson trichrome staining was performed on paraffin embedded tissue. Briefly, slides containing 7.5μm sections were warmed in a 60°C oven for 1 hours. Slides were de-paraffinized and rehydrated and stained for Masson trichrome following the manufactures protocol using Thermo Scientifics Masson Trichrome kit (VWR, #84000-286). Images were taken using a Nikon 90i Eclipse. Hematoxyline & Eosin (H&E) staining was performed on paraffin embedded tissue. Briefly, slides containing 7.5μm sections were warmed in a 60°C oven for 1 hours. Slides were de-paraffinized and rehydrated at RT. Mayer’s hematoxylin solution (Sigma) was then applied for 15m then rinsed in distilled water and 95% ethanol. Next the slides were incubated with Eosin solution (Sigma) for 30s, cleared, dehydrated and mounted. Images were taken using a Nikon 90i Eclipse.
2.11. Western Blotting
In tissue, samples of 15 μg were loaded on 4–12% Bis-Tris SDS-PAGE (Invitrogen) and transferred onto PVDF membranes (Millipore). Membranes were blocked with 5% non-fat dry milk in PBS-Tween 0.1% and incubated against MMP-9 (Millipore, ab19016) at a concentration of 1:1000 overnight at 4°C. GAPDH or β-tubulin was used as loading controls. Detection was carried out using peroxidase-coupled anti-rabbit (Vector) at 1:10000 for 1 hour at room temperature. Protein bands were visualized using enhanced chemiluminescence (ECL). Cell media was thawed to room temperature and mixed with 4x SDS at a 1:3 ratio. Samples of 15 μl were loaded on 4–12% Bis-Tris SDS-PAGE (Invitrogen) and transferred onto PVDF membranes (Millipore). Membranes were blocked with 5% non-fat dry milk in PBS-Tween 0.1% and incubated against MMP-9 (Millipore ab19016) at a concentration of 1:1000 overnight at 4°C. Cell count was used for normalization. Detection was carried out using peroxidase-coupled anti-rabbit (Vector) at 1:10000 for 1 hour at room temperature. Protein bands were visualized using enhanced chemiluminescence (ECL).
2.12. Statistical Analysis
Data are expressed as mean ± sem. Analyses were performed using Prism 5.0a (GraphPad Software Inc.). The group means were compared using two-way ANOVA followed by t-test (for 2 groups) or Bonferroni post-tests (for > 2 groups). P values of <0.05 indicate statistical significance.
3. Results
3.1. HDAC inhibition promotes the early and robust appearance of reparative macrophages
SAHA treatment has been shown to decrease MMP-9 expression9, and as macrophages are a significant source of MMP-9, we examined whether in-vivo SAHA administration influenced expression of inflammatory cytokines or reduced the numbers of CD68+ inflammatory macrophages recruited to the ischemic heart. As expected, flow cytometric analysis showed that the numbers of CD45+ leukocytes and CD45+/CD11b+ monocytes were dramatically increased with infarct. Notably, their recruitment was unaffected by treatment with SAHA through day 5 (Fig 1A–B). However, SAHA treatment significantly reduced the CD45+/CD11b+ monocyte population in the infarct zone at day 7 post-MI.
Figure 1. HDAC inhibitor treatment does not affect initial recruitment of monocytes and macrophages to the ischemic myocardium.
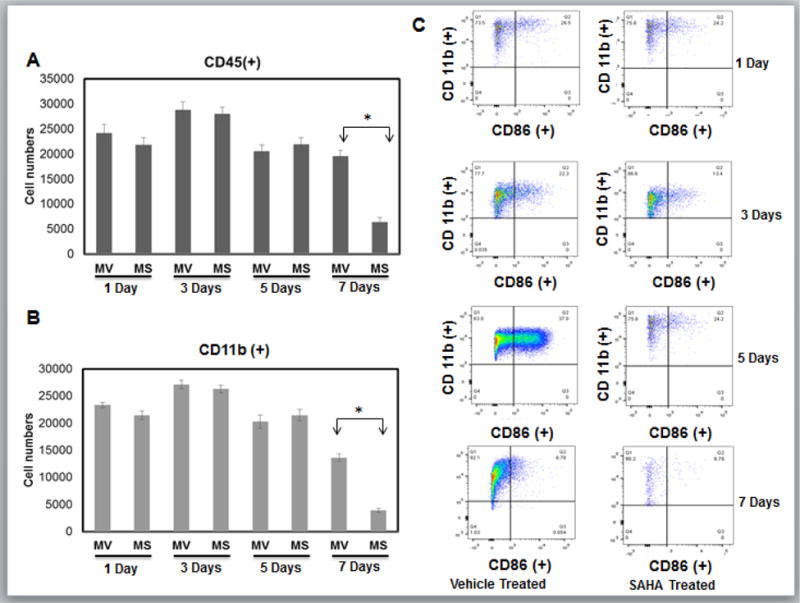
Cell suspensions from infarct zone of vehicle (MV) and SAHA (MS) treated CD1 mice post MI were stained with anti-CD45, CD11b and CD86 mAbs. Results were first processed with live/dead assay and gated with live cells normalized to 200,000 cells. Relative cell numbers are shown as the mean +/− the SEM. A) Live cells were then gated with CD45 positive population to isolate leukocytes. B) Monocytes were then gated with CD11b positive population to identify monocytes [CD45(+)/CD11b(+)]. Any population lower than 103 for either antibody will not be recognized as valid results. C) Original flow cytometry dot plots for infarct tissue in vehicle and SAHA treated mice 1, 3, and 5 and 7 day post-MI. Macrophage were then gated into CD86 positive population that represents classical inflammatory M1 macrophages and will be in region Q2 [CD11b (+)/CD86 (+)] that are greater than 103 for either antibody. (n= 6 for 1, 3, and 5 day groups, n=5 for 7 day group.). *p<0.05 by one-way ANOVA and Bonferroni post-test.
The infarct tissue of both SAHA and vehicle treated mice accumulated similar numbers of CD86+ inflammatory macrophages on both day 1, and day 3. CD86+ cell numbers continued to climb in vehicle treated hearts but they dropped at day 5 and were dramatically lower at day 7 in the SAHA treated hearts (Fig 1C). Consistent with these findings, MCP-1, the primary cytokine responsible for monocyte recruitment to the infarct, had similar expression with SAHA treatment compared to vehicle within the infarct region from day 1 through day 3 post-MI (Fig 2AB). MCP-1 expression drops significantly with SAHA treatment at 5 days post-MI (Fig 2C). Further, RT-PCR revealed that the M1 markers, TNF-α, and IL-1Beta were not significantly changed with SAHA treatment at day 1 through Day 3 (Fig 3AB). IL-6 is unchanged with SAHA treatment at Day 1 but has significantly less expression at day 3 (Fig 3C). At day 5 when there was a significant drop in the CD86+ cell numbers with SAHA treatment, TNF-α, IL-1β and IL-6 expression were downregulated in both MI and MI + SAHA treatment compared to Day 3. But Importantly, TNF-α, IL-1β and IL-6 expression was significantly less with SAHA treatment compared to MI alone.
Figure 2. There is no change in the monocyte chemoattractant protein 1 (MCP-1) in the first 3 days but expression drops with SAHA treatment at day 5 post-MI.
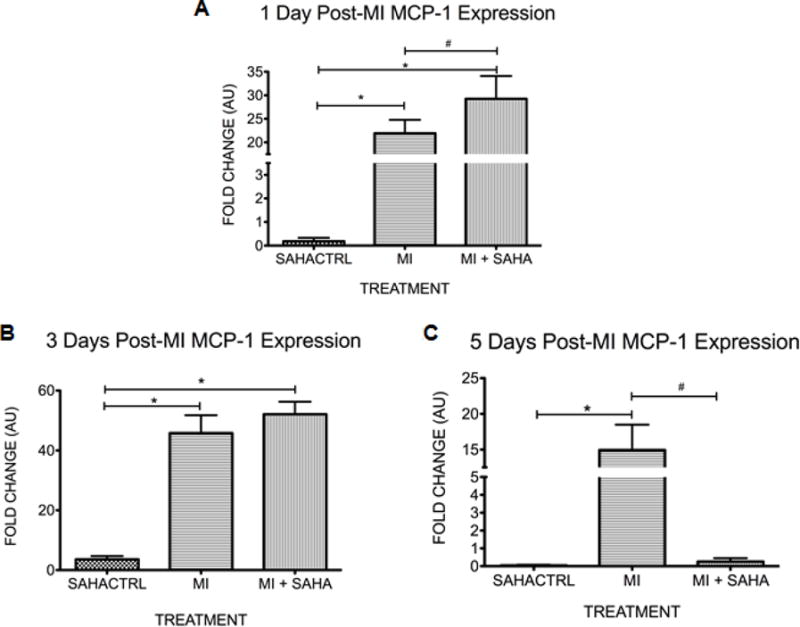
qRT-PCR analysis of monocyte chemoattractant protein 1 (MCP-1) within the infarct zone at A)1 day, B) 3 days and C) 5 days post-MI. The fold change in mRNA value are shown as fold change over sham SAHA treated animals. Values of all qRT-PCR data are normalized to GAPDH. Each bar represents the fold change +/− SEM of three independent experiments with a group of at least n=3 animals per treatment. #p<0.05 vs control, *p<0.05 vs MI, by one-way ANOVA and Bonferroni post-test.
Figure 3. M1 macrophage markers are unchanged at day 1 but drop dramatically at day 5 post-MI with SAHA treatment.
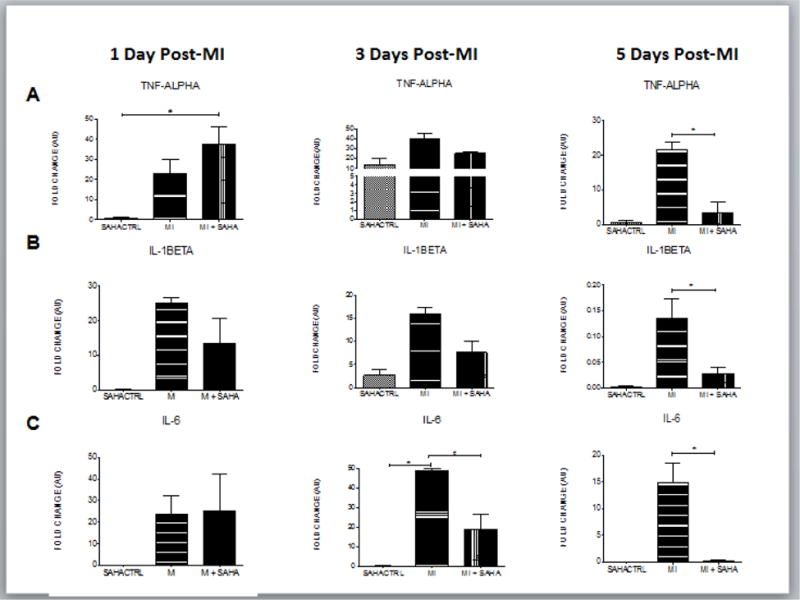
qRT-PCR analysis of mRNA fold change of the M1 associated genes, A) TNF-alpha, B) IL-1beta and C) IL-6 at 1, 3 and 5 days post-MI. Values of all qRT-PCR data are normalized to GAPDH. Each bar represents the fold change +/− SEM of three independent experiments with a group of at least n=3 animals per treatment. #p<0.05 vs control, *p<0.05 vs MI, by one-way ANOVA and Bonferroni post-test.
As seen in previous studies11, 21, 22, very few reparative CD206+ macrophages were present at day 1 post-MI but accumulated from day 3 through day 7 in the vehicle treated mice. Remarkably, SAHA treated hearts had dramatically higher numbers of CD206+ reparative macrophages as early as day 1 and numbers remained very high through day 5 in the infarct region when compared to mice treated with vehicle (Fig. 4A). Further, at day 7 post-MI, very few CD206+ (or CD86+) cells were present in the SAHA treated mice, showing that SAHA treatment effected both the ratio of CD86+/CD206+ cells and the kinetics of their recruitment to the infarct region (Fig. 4B and 4C).
Figure 4. HDAC inhibitor treatment promotes the early and robust recruitment of reparative macrophages.
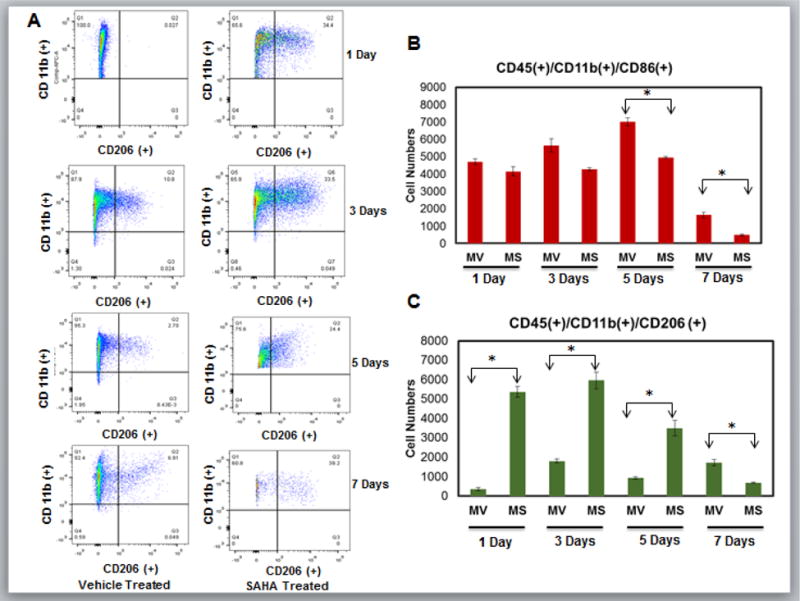
Cell suspensions from infarct zone of vehicle (MV) and SAHA (MS) treated CD1 mice post MI were stained with anti-CD45, CD11b and CD206 mAbs. Results were first processed with live/dead assay and gated with live cells normalized to 200,000 cells. Relative cell numbers are shown as the mean +/− the SEM. A) Original flow cytometry dot plots for infarct tissue in vehicle and SAHA treated mice 1, 3, 5 and 7 days post-MI. Live cells were then gated with CD45 positive population to isolate leukocytes. were then gated with CD11b positive population to identify Monocytes/macrophages [CD45+/CD11b+]. Macrophage were then gated into CD206 positive population that represents reparative M2 macrophages and will be in region Q2 [CD11b(+)/CD206(+)] that are greater than 103 for either antibody. Any population lower than 103 for any antibody will not be recognized as valid results. B) Relative numbers of CD45+/CD11b+/CD86+ cells at 1, 3, 5 and 7 days post-MI. C) Relative numbers of CD45+/CD11b+/CD206+ cells at 1, 3, 5 and 7 days post-MI. n= 6 for 1, 3, and 5 day groups, n=5 for 7 day group. *p<0.05 by one-way ANOVA and Bonferroni post-test.
To validate the findings of SAHA treatment promoting the early and robust presence of CD206+ reparative macrophages we examined the expression pattern of other M2 polarization associated markers in the 1 and 5 day post-MI hearts. Treatment with SAHA resulted in the significant upregulation of M2 markers: CD163, chitinase 3 like 3 (CH3L3), arginase 1, mannose receptor (CD206), and the cytokines IL-4 and IL-10 at one and five days post-MI compared to MI with vehicle, respectively (Fig 5 A–F).
Figure 5. M2 macrophage markers are dramatically upregulated at post-MI day 1, 3 and 5 with SAHA treatment.
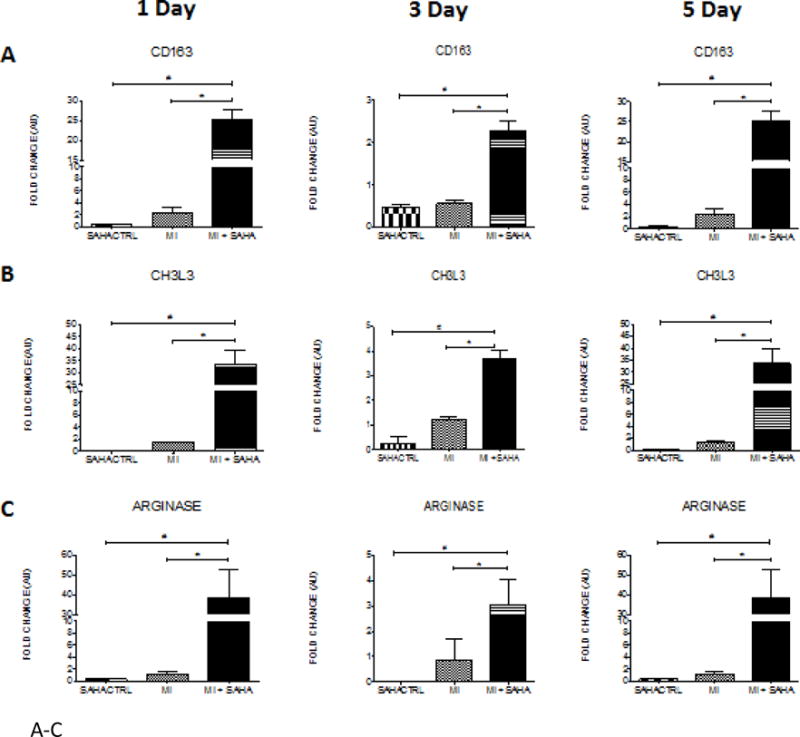
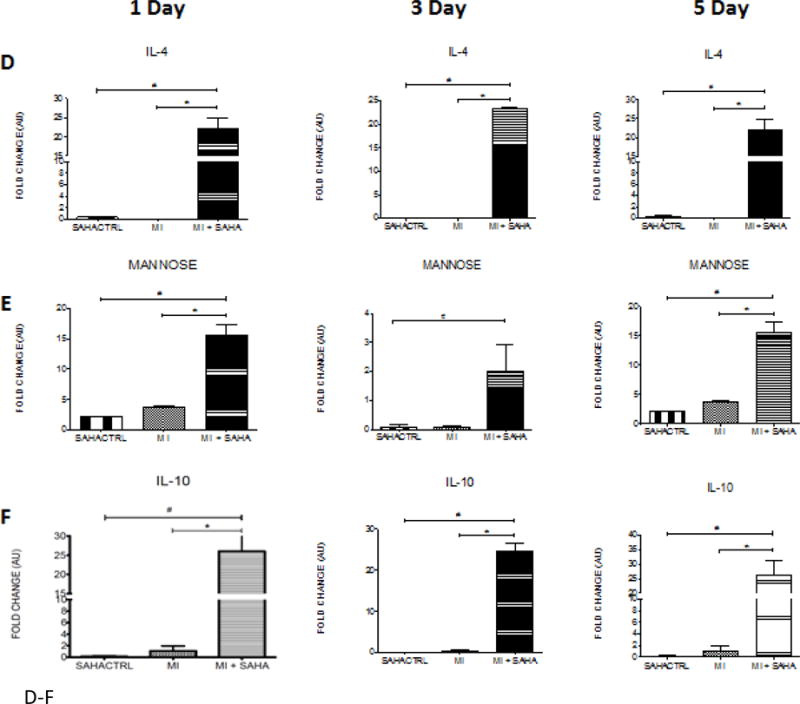
qRT-PCR analysis of mRNA fold change of the M2 associated genes, A) CD163, B) CH3L3, C) Arginase, D) IL-4, E) Mannose receptor and F) IL-10 at 1, 3 and 5 days post-MI. Values of all qRT-PCR data normalized to GAPDH. Each bar represents the fold change ± SEM of three independent experiments with a group of at least n=3 animals per treatment. #p<0.05 vs control, *p<0.05 vs MI, by one-way ANOVA and Bonferroni post-test.
3.2. HDAC inhibition promotes angiogenesis, limits LV dilation and preserves function in the post-MI ventricle
Timely progression and resolution of both the inflammatory and reparative phase is critical for healing. Therefore, we examined some of the possible structural and functional aspects of the early and robust recruitment of CD206+ macrophages to the infarct. M2 macrophages promote tissue remodeling and angiogenesis in part via elevated secretion of VEGF-A which is linked to stimulation of cardiac angiogenesis in the post-MI heart23–25. Interestingly, the early robust recruitment of M2 macrophages with SAHA treatment correlates with a significant increase in micro-vessel density in the border zone at 2 weeks post-MI (Fig. 6A, B). We previously reported that SAHA treatment preserves ejection fraction and LV dilation after 7 days post MI 26. We examined cardiac function and remodeling to see if they are still preserved after 14 days with two treatment strategies. In the first we used daily treatment with SAHA (as the previous experiments) but we end treatment at day 7 and examine if the beneficial effect of SAHA treatment is retained when the function and remodeling are assessed at 14 days post-MI. In the second we decreased the frequency of SAHA treatment to every other day but treated the mice for the entire 14 days. Treatment with SAHA every other day still preserved ejection fraction and LV dilation after 14 days post MI (Fig. 7A). Both ejection fraction and end-diastolic volume was preserved to nearly the same extent when SAHA was administered for the first 7 days and then mice were followed with-out treatment to 14 days post-MI (Fig. 7B).
Figure 6. SAHA treatment enhances microvascular density at 14 days post MI injury.
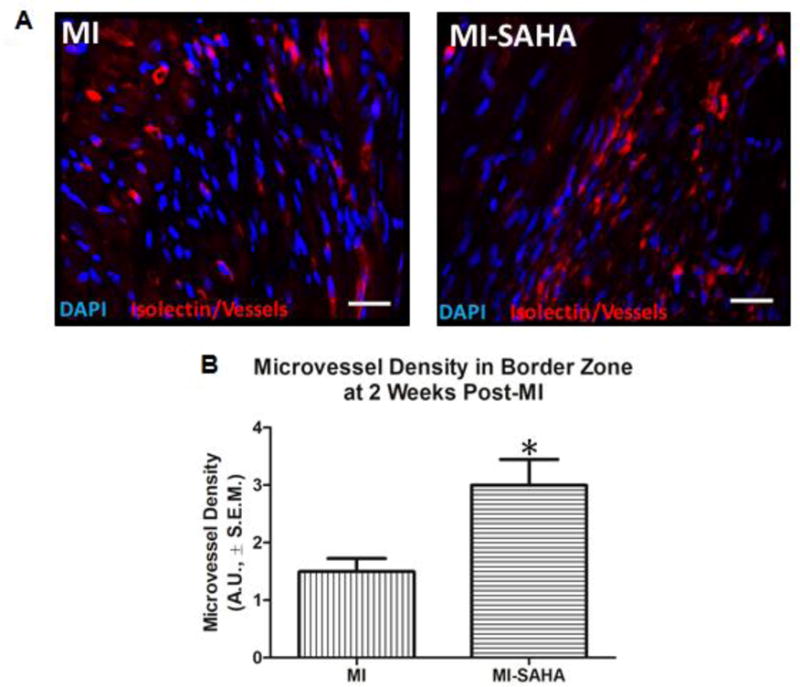
10-week old male CD1 mice were subjected to ligation of the coronary artery surgery and received I.P. injections of DMSO (vehicle-control) or the HDAC inhibitor SAHA (5mg/kg) every 48 hours for 2 weeks. At 2 weeks post-MI, mice were euthanized. Hearts were extracted upon euthanasia and processed for immunofluorescent chemical analyses of the left ventricle border zone of infarction using immunostaining and microscopy at 20X magnification, white bar=25μm. Microvessel density was assessed via immunofluorescent labelling of lectin, a blood vessel marker. Tissue was imaged using an Olympus 1×71 Slide Book-Inverted fluorescent microscope (5 images per heart/sample). A) Images are representative of repeated studies. B) Graphs show quantitated arbitrary fluorescence intensities ±S.E.M., n=5 per group. P-value was obtained using Student’s T-test. *p-value ≤ 0.05
Figure 7. SAHA treatment improves cardiac function at 14 days post-MI injury.
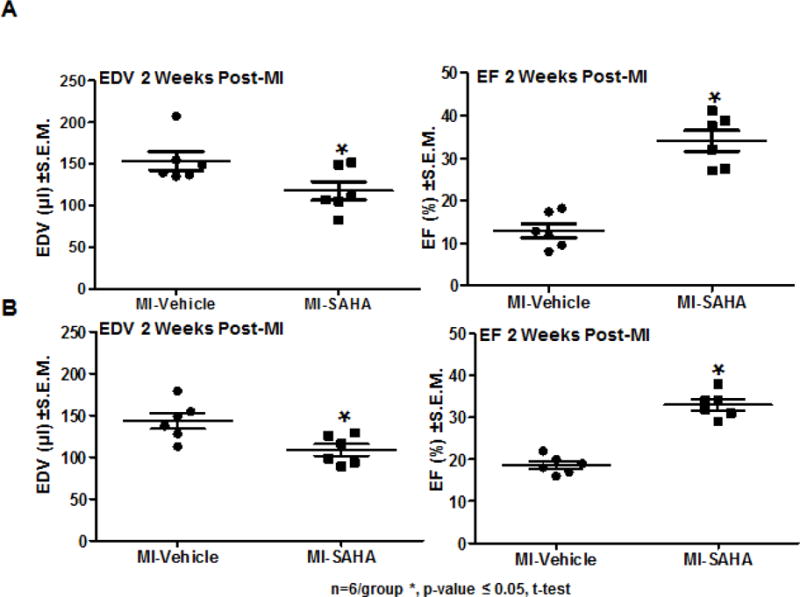
10-week old male CD1 mice were subjected to ligation of the coronary artery surgery and received I.P. injections of DMSO (vehicle-control) or the HDAC inhibitor SAHA (25mg/kg) either A) every 48 hours for 2 weeks or B) SAHA was administered daily for the first week and then mice received no drug until 2 weeks post-MI. At 2 weeks post-MI, mice were subjected to echocardiogram to assess cardiac function as measured by end diastolic volume and ejection fraction. n=6 per group *p-value ≤ 0.05. P-value was obtained using Student’s T-test.
3.3. HDAC inhibition results in M1 to M2 polarization in cultured RAW 264.7 and bone marrow derived macrophages
The data shown above were collected from the infarct zone where multiple cells including macrophages could have contributed to M1 and M2 marker expression. In order to determine if HDAC inhibition shifted M1/M2 marker expression in macrophages we utilized an in vitro model using the murine RAW 264.7 macrophage cell line. Morphologically inflammatory M1 macrophages (stimulated with lipid polysaccharide (LPS)) adopt a circular “pancake like” shape comparable to monocytes, while reparative M2 macrophages (stimulated with IL-4) adopt an elongated neuronal or fibroblastic appearance27. Compared to control or control + LPS stimulated cells, about 50% of treated with the pan HDAC inhibitor TSA adopted an IL-4 morphology suggesting polarization to an M2 phenotype (Fig. 8A and B). Importantly, LPS stimulated cells when treated with the class I and II HDAC inhibitor, TSA >60% of the cells adopt a M2 phenotype. In addition, cells treated with TSA or SAHA exhibited increased expression of the M2 markers, arginase 1, IL-10, and CD206 compared to vehicle. Notably, the M2 markers were increased with SAHA even in the presence of LPS (Fig. 8 C–E). As expected, inflammatory treatment of RAW 264.7 macrophages with LPS resulted in robust transcription and secretion of MMP-9. SAHA treated M2 macrophages transcribed and secreted significantly lower levels of MMP-9 even when stimulated with LPS. (Fig 9 E–F). Lastly, in order to examine the role of HDACs in macrophage polarization in a more relevant in vitro model we looked at murine bone marrow derived macrophages. Similar to the RAW 264.7 macrophages, about 70% of the murine bone marrow derived macrophages displayed the inflammatory circular “pancake like” (M1) shape under control media and media + LPS conditions (Fig 10 A and B). Treatment with SAHA or TSA resulted in over 90% of the cells adopting an elongated neuronal or fibroblastic (M2) appearance. Treatment with LPS + SAHA resulted in over 70% of the cells with a M2 phenotype and LPS + TSA over 80% of the cells adopted a 2 phenotype.
Figure 8. HDAC inhibition induces morphologic changes indicative of M2 polarization and activation of M2 associated gene markers.
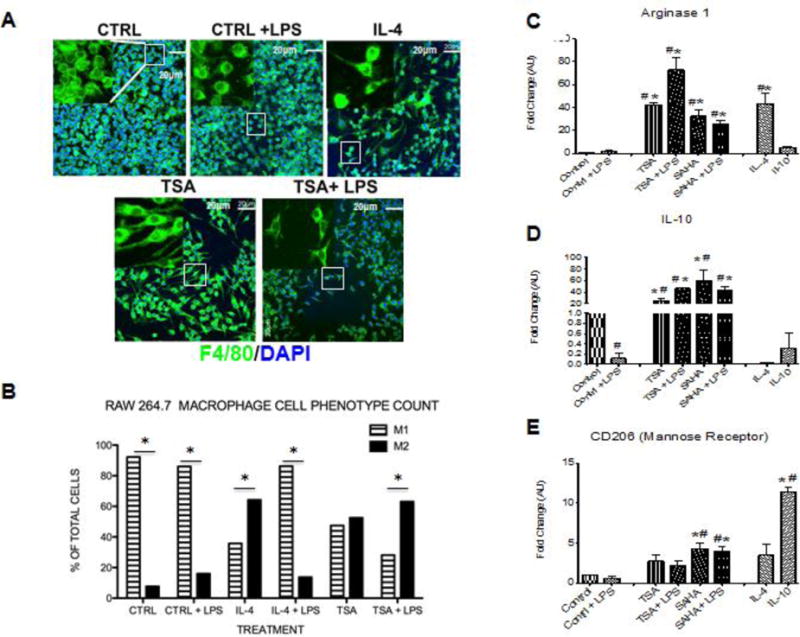
A) RAW264.7 macrophages were treated with medium alone (CTRL), LPS (100ng/ml), IL-4 (30ng/ml), IL-10 (20ng/ml) treated with the pan HDAC inhibitor TSA (100nM) or SAHA (5μM) +/−LPS (100ng/ml) and incubated for 6 hours after treatment. Cells were stained for F4/80 (macrophage marker) and DAPI (nuclear marker). Images were obtained using an Olympus Confocal microscope at 40x objective and inset represents a 60X zoom. B) Quantification of M1 and M2 phenotype was assessed for multiple fields from each condition by 3 investigators who were blinded to what conditions they were assessing. *p<0.05 M1 vs M2 phenotype. C–E) Cells were isolated 6 h after treatment and qRT-PCR analysis was performed on mRNA for Arginase 1, IL-10, and CD206 (mannose receptor) and normalized to GAPDH. The fold change mRNA values are shown as fold change of medium alone cells (D and E) or cells treated with LPS (C). For all the data shown in C–E, each bar represents the fold change ± SEM of three independent experiments. #p<0.05 vs control, *p<0.05 vs control + LPS.
Figure 9.
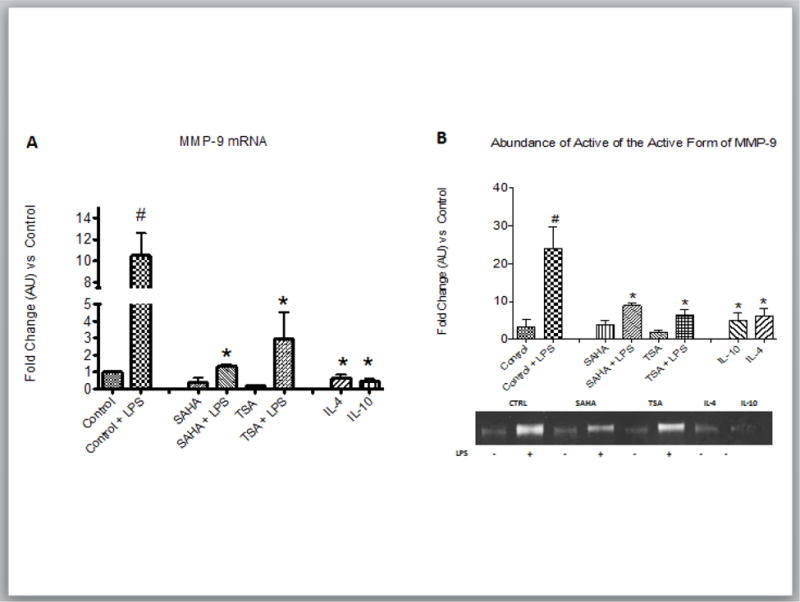
RAW264.7 macrophages were treated with medium alone (CTRL), LPS (100ng/ml), IL-4 (30ng/ml), IL-10 (20ng/ml) treated with the pan HDAC inhibitor TSA (100nM) or SAHA (5μM) +/−LPS (100ng/ml) and incubated for 6 hours after treatment. A) Cells were isolated 6 h after treatment and qRT-PCR analysis was performed on mRNA for MMP-9 and normalized to GAPDH. The fold change mRNA values are shown as fold change of medium alone cells or cells treated with LPS. B) Representative zymogram and quantification of the abundance of the active form of MMP-9 (86 kDa) secreted from RAW264.7 cells after indicated treatment for 12 hr. For all the data shown in A and B, each bar represents the fold change ± SEM of three independent experiments. #p<0.05 vs control, *p<0.05 vs control + LPS.
Figure 10. HDAC inhibition induces morphologic changes indicative of M2 polarization in murine bone marrow derived macrophages.
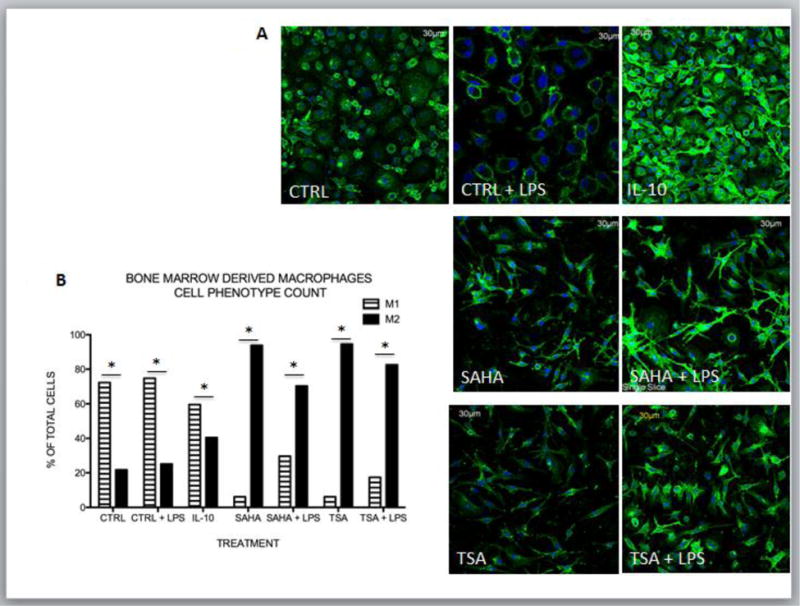
A) Murine bone marrow derived macrophages were treated with medium alone (CTRL), LPS (100ng/ml), IL-4 (30ng/ml), IL-10 (20ng/ml) treated with the pan HDAC inhibitor TSA (100nM) or SAHA (5μM) +/−LPS (100ng/ml) and incubated for 8 hours after treatment. Cells were stained for F4/80 (macrophage marker) and DAPI (nuclear marker). Images were obtained using an Olympus Confocal microscope at 40x objective and inset represents a 60X zoom. B) Quantification of M1 and M2 phenotype was assessed for multiple fields from each condition by 3 investigators who were blinded to what conditions they were assessing. *p<0.05 M1 vs M2 phenotype.
4. Discussion
The extent and quality of post MI remodeling impacts long-term outcomes. Inflammation is critical during the early time points post-MI but timely resolution is key to reparative remodeling and healing. Prolonged inflammation results in increased LV dilation and excessive scar formation28. Macrophages play a critical role in this process by clearing the wound of dead cells and debris, promoting collagen deposition, scar maturation and angiogenesis. The object of this study was to determine the effect of HDAC inhibition on MI induced macrophage recruitment and polarization and how that influences LV remodeling and dysfunction. Remarkably, our data show that inhibition of HDAC activity immediately following MI, results in the robust recruitment of CD45+/CD11b+/CD206+, M2 reparative macrophages during the very early inflammatory phase of healing (30 h). Further, HDAC inhibitor treatment resulted in the robust expression of M2 markers in the first 24 hours post-MI. HDAC inhibitor treatment does not diminish the initial recruitment of inflammatory bone-marrow derived monocytes that give rise to CD45+/CD11b+/CD86+, M1 inflammatory macrophages in the first 3 days post injury. CD86+ cell numbers were lower at 5 days and dropped dramatically at day 7 with HDAC inhibition. Importantly, treatment with HDAC inhibitor for only the first 7 days post-MI resulted in improved ventricle function and remodeling at 14 days post-MI.
Ischemic injury activates the recruitment of large numbers of inflammatory monocytes which give rise to M1, inflammatory macrophages that are responsible for removal of cell debris and necrotic cell clearance. M1 macrophages are a significant source of inflammatory cytokines and matrix metalloproteinases such as MMP-9 for degradation and remodeling of the ECM. Between day 3 and 7 reparative M2 macrophages infiltrate the myocardium29. These reparative macrophages promote collagen deposition, scar maturation and angiogenesis. The balance and timing of appearance of classically activated M1 and alternatively activated M2 macrophages in the post-MI heart are critical. Although it is clear that macrophages do not fall into distinct subsets but are present in a wide spectrum of phenotypes, we will use the conventional and simplified M1 and M2 designation for this discussion. Multiple studies have shown that M2 macrophage polarization is beneficial for the improvement of wound healing and for improving LV function following myocardial ischemic injury17, 30–35. In fact, complete depletion of M2-like macrophages using Trib1−/− mice resulted in reduced collagen fibril formation in infarct tissue resulting in frequent cardiac rupture16. This could be rescued by either IL-4 administration, increasing the number of M2 macrophages or injection of external M2-like macrophages. Further, delay of M1 to M2 polarization as a result of prolonged recruitment of inflammatory monocytes impairs infarct healing36. Yet, therapeutic strategies to increase M2 polarization and aid inflammatory resolution within the infarct are not yet available.
Our results present the possibility of HDAC inhibition as a novel therapy for inducing M2 macrophage polarization following myocardial ischemic injury. Other possible approaches include siRNA which was used to knockdown the interferon regulatory factor 5 (IRF5) in cardiac macrophages resulting in improved post MI healing37. IRF5 is a transcription factor which regulates the expression of genes associated with M1 macrophages. IRF5 silencing reduced expression of M1 macrophage markers at 4 days post-MI but did not appear to affect the expression of M2 markers. Unfortunately, the M2 macrophage population in post-infarct tissue was not assessed. In contrast, HDAC inhibition, in our study, resulted in significantly reduced expression of IL-1β, TNF-α and IL-6 at day 5 and resulted in the very early (Day 1) expression of M2 markers and recruitment of CD45+/CD11b+/CD206+ reparative macrophages. It is important to note that the Courties et al. study37 was carried out in an atherosclerotic mouse model (ApoE−/−) and our study was done in WT mice. Additional approaches to promote inflammatory resolution includes treatment with the anti-inflammatory cytokine IL-10 which resulted in significantly improved cardiac function and post-MI remodeling38. Il-10 treatment resulted in elevated M2 marker gene expression at post-MI day 7, again long after HDAC inhibitor treatment elicits an increase in M2 marker expression. A aging mouse model of macrophage overexpression of MMP-9 shows increased macrophage numbers and anti-inflammatory polarization at 7 days post MI39. Interestingly, the increased macrophage MMP-9 expression contributes to post-MI healing and improved diastolic function. Nahrendorf’s group has used nanoparticles to knockdown monocyte CCR-2 receptor by RNAi to reduce recruitment of monocytes and limit infarct inflammation40. CCR2 silencing reduced expression of several inflammatory genes and increased IL-10 expression, but M1/M2 macrophage levels were not addressed in this study. One of the ways M2 reparative macrophages promote tissue remodeling, resolution of inflammation and healing is via elevated secretion of VEGF-A which is linked to stimulation of cardiac angiogenesis in the post-MI heart23–25. Our data showing an increase in vessel density in the border zone with HDAC inhibitor treatment is supportive of the early and robust recruitment of M2 macrophages contributing to angiogenesis.
Our in vivo data do not distinguish whether HDACs mediate macrophage polarization and regulation only by directly regulating gene expression in the macrophage or if it also includes the HDAC regulation of inflammatory cytokines secreted in the ischemic tissue. Our in vitro data do show that inhibition of HDACs is all that is required to polarize macrophages to a M2 phenotype. In addition, macrophages treated with SAHA and TSA activated a M2 gene expression pattern, with robust expression of Arginase 1, Il-10, moderately increased expression of CD206 (mannose receptor) and suppressed expression of the M1 marker, MMP-9, even in the presence of inflammatory LPS-stimulation. Our data are consistent with previous work showing that sodium valproate (a weak class I HDAC inhibitor) altered LPS-stimulated macrophage polarization18. Macrophages deficient in HDAC3 display a phenotype similar to the alternatively activated M2 macrophages and HDAC3 expression limits cytokine-stimulated alternative activation in vitro41. In addition, Carpio et al have shown that HDAC3 regulates the temporal and spatial expression of inflammatory response genes in chondrocytes which supports endochondral bone formation42. Although it is clear from this study and that of others, at least in an in vitro setting, class I HDACs are master phenotype regulators which can override a barrage of inflammatory signals, it remains to be determined if only inhibition of inflammatory macrophage class I HDACs is sufficient to increase M2 macrophage recruitment to the post-MI heart.
HDAC inhibition has been proven beneficial in prevention of adverse remodeling in MI, IR and cardiac hypertrophy, but this is the first account of this benefit being due in part to its ability to polarize macrophages, a cellular component underappreciated until recently in cardiac pathologies.43–46 It is clear that inflammation plays a critical role in LV remodeling in cardiovascular pathologies and therapies that promote the resolution of inflammation along with other consequences of the disease are significantly more beneficial. Finding new therapies which result in the preservation of ventricular function and prevent adverse remodeling are critical to improve clinical outcomes following MI. Here, we have demonstrated that HDAC inhibition promotes the early and robust recruitment of CD45+/CD11b+/CD206+ (M2) reparative macrophages in the very early inflammatory phase of healing following MI. The resulting robust expression of anti-inflammatory IL-4 and IL-10 cytokines may contribute to early inflammatory resolution and improve the environmental milieu surrounding nearby surviving cardiomyocytes, fibroblasts and endothelial cells. The early recruitment of reparative CD45+/CD11b+/CD206+ macrophages directly and indirectly appear to exert a cardioprotective effect which correlates with improved ventricular function and remodeling.
Highlights.
HDAC inhibition promotes the early and robust appearance of reparative M2 macrophages following MI.
HDAC inhibition promotes the significant upregulation of M2 markers and noninflammatory cytokines at one and five days post-MI.
HDAC inhibition following MI does not affect the recruitment of M1 inflammatory macrophages out to day 3 post-MI
HDAC inhibition promotes angiogenesis, limits LV dilation and preserves function in the post-MI ventricle
Acknowledgments
We want to thank Joy Buie for technical assistance.
Sources of Funding
This work was supported in part by the United States Department of Veterans Affairs Merit Review BX002327 (DRM), a pilot project from NIH/NCATS UL1 TR001450 (DRM), and by a Postdoctoral Fellowship (T32HL07260 to SHW and LGH).
Abbreviations
- HDAC
histone deacetylase enzyme
- MI
myocardial infarction
- MMPs
matrix metalloproteinases
- ECM
extracellular matrix
- HF
heart failure
- MCP-1
monocyte chemoattractant protein-1
- TLR
toll-like receptor
- IFN-γ
interferon gamma
- Trib1
Tribbles homolog 1
- SAHA
Suberoylanilide Hydroxamic Acid
- LPS
lipid polysaccharide
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Disclosures: None
References
- 1.Rizzoni D, Muiesan ML, Porteri E, De Ciuceis C, Boari GE, Salvetti M, et al. Vascular remodeling, macro- and microvessels: therapeutic implications. Blood Press. 2009;18:242–6. doi: 10.3109/08037050903254923. [DOI] [PubMed] [Google Scholar]
- 2.Sutton MG, Sharpe N. Left ventricular remodeling after myocardial infarction: pathophysiology and therapy. Circulation. 2000;101:2981–8. doi: 10.1161/01.cir.101.25.2981. [DOI] [PubMed] [Google Scholar]
- 3.French BA, Kramer CM. Mechanisms of Post-Infarct Left Ventricular Remodeling. Drug Discov Today Dis Mech. 2007;4:185–196. doi: 10.1016/j.ddmec.2007.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Jugdutt BI. Remodeling of the myocardium and potential targets in the collagen degradation and synthesis pathways. Curr Drug Targets Cardiovasc Haematol Disord. 2003;3:1–30. doi: 10.2174/1568006033337276. [DOI] [PubMed] [Google Scholar]
- 5.Lindsey ML, Zamilpa R. Temporal and spatial expression of matrix metalloproteinases and tissue inhibitors of metalloproteinases following myocardial infarction. Cardiovasc Ther. 2012;30:31–41. doi: 10.1111/j.1755-5922.2010.00207.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Vanhoutte D, Schellings M, Pinto Y, Heymans S. Relevance of matrix metalloproteinases and their inhibitors after myocardial infarction: a temporal and spatial window. Cardiovasc Res. 2006;69:604–13. doi: 10.1016/j.cardiores.2005.10.002. [DOI] [PubMed] [Google Scholar]
- 7.Lee TM, Lin MS, Chang NC. Inhibition of histone deacetylase on ventricular remodeling in infarcted rats. Am J Physiol Heart Circ Physiol. 2007;293:H968–77. doi: 10.1152/ajpheart.00891.2006. [DOI] [PubMed] [Google Scholar]
- 8.Xie M, Kong Y, Tan W, May H, Battiprolu PK, Pedrozo Z, et al. Histone deacetylase inhibition blunts ischemia/reperfusion injury by inducing cardiomyocyte autophagy. Circulation. 2014;129:1139–51. doi: 10.1161/CIRCULATIONAHA.113.002416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mani SK, Kern CB, Kimbrough D, Addy B, Kasiganesan H, Rivers WT, et al. Inhibition of class I histone deacetylase activity represses matrix metalloproteinase-2 and -9 expression and preserves LV function postmyocardial infarction. Am J Physiol Heart Circ Physiol. 2015;308:H1391–401. doi: 10.1152/ajpheart.00390.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Christia P, Frangogiannis NG. Targeting inflammatory pathways in myocardial infarction. Eur J Clin Invest. 2013;43:986–95. doi: 10.1111/eci.12118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Frantz S, Nahrendorf M. Cardiac macrophages and their role in ischemic heart disease. Cardiovasc Res. 2014 doi: 10.1093/cvr/cvu025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hilgendorf I, Gerhardt LM, Tan TC, Winter C, Holderried TA, Chousterman BG, et al. Ly-6Chigh monocytes depend on Nr4a1 to balance both inflammatory and reparative phases in the infarcted myocardium. Circ Res. 2014;114:1611–22. doi: 10.1161/CIRCRESAHA.114.303204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.van Amerongen MJ, Harmsen MC, van Rooijen N, Petersen AH, van Luyn MJ. Macrophage depletion impairs wound healing and increases left ventricular remodeling after myocardial injury in mice. Am J Pathol. 2007;170:818–29. doi: 10.2353/ajpath.2007.060547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nahrendorf M, Swirski FK. Abandoning M1/M2 for a Network Model of Macrophage Function. Circ Res. 2016;119:414–7. doi: 10.1161/CIRCRESAHA.116.309194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Nahrendorf M, Swirski FK. Monocyte and macrophage heterogeneity in the heart. Circulation research. 2013;112:1624–33. doi: 10.1161/CIRCRESAHA.113.300890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Shiraishi M, Shintani Y, Shintani Y, Ishida H, Saba R, Yamaguchi A, Adachi H, et al. Alternatively activated macrophages determine repair of the infarcted adult murine heart. J Clin Invest. 2016;126:2151–66. doi: 10.1172/JCI85782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Courties G, Heidt T, Sebas M, Iwamoto Y, Jeon D, Truelove J, Tricot B, et al. In Vivo Silencing of the Transcription Factor IRF5 Reprograms the Macrophage Phenotype and Improves Infarct Healing. J Am Coll Cardiol. 2014;63:1556–66. doi: 10.1016/j.jacc.2013.11.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wu C, Li A, Leng Y, Li Y, Kang J. Histone deacetylase inhibition by sodium valproate regulates polarization of macrophage subsets. DNA Cell Biol. 2012;31:592–9. doi: 10.1089/dna.2011.1401. [DOI] [PubMed] [Google Scholar]
- 19.Harel-Adar T, Ben Mordechai T, Amsalem Y, Feinberg MS, Leor J, Cohen S. Modulation of cardiac macrophages by phosphatidylserine-presenting liposomes improves infarct repair. Proc Natl Acad Sci U S A. 2011;108:1827–32. doi: 10.1073/pnas.1015623108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hockly E, Richon VM, Woodman B, Smith DL, Zhou X, Rosa E, et al. Suberoylanilide hydroxamic acid, a histone deacetylase inhibitor, ameliorates motor deficits in a mouse model of Huntington’s disease. Proc Natl Acad Sci U S A. 2003;100:2041–6. doi: 10.1073/pnas.0437870100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Prabhu SD, Frangogiannis NG. The Biological Basis for Cardiac Repair After Myocardial Infarction: From Inflammation to Fibrosis. Circ Res. 2016;119:91–112. doi: 10.1161/CIRCRESAHA.116.303577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hulsmans M, Sam F, Nahrendorf M. Monocyte and macrophage contributions to cardiac remodeling. J Mol Cell Cardiol. 2016;93:149–55. doi: 10.1016/j.yjmcc.2015.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Besnier M, Galaup A, Nicol L, Henry JP, Coquerel D, Gueret A, et al. Enhanced angiogenesis and increased cardiac perfusion after myocardial infarction in protein tyrosine phosphatase 1B-deficient mice. Faseb J. 2014;28:3351–61. doi: 10.1096/fj.13-245753. [DOI] [PubMed] [Google Scholar]
- 24.Barbay V, Houssari M, Mekki M, Banquet S, Edwards-Levy F, Henry JP, et al. Role of M2-like macrophage recruitment during angiogenic growth factor therapy. Angiogenesis. 2015;18:191–200. doi: 10.1007/s10456-014-9456-z. [DOI] [PubMed] [Google Scholar]
- 25.Chambers SE, O’Neill CL, O’Doherty TM, Medina RJ, Stitt AW. The role of immune-related myeloid cells in angiogenesis. Immunobiology. 2013;218:1370–5. doi: 10.1016/j.imbio.2013.06.010. [DOI] [PubMed] [Google Scholar]
- 26.Mani S, Kern CB, Addy B, Kasiganesan H, Rivers WT, Plyler RA, et al. Inhibition of Histone Deacetylase Activity Represses Matrix Metalloproteinase-9 Induction and Preserves Cardiac Function Post Myocardial Infarction. Circulation. 2008;118:498. [Google Scholar]
- 27.McWhorter FY, Wang T, Nguyen P, Chung T, Liu WF. Modulation of macrophage phenotype by cell shape. Proc Natl Acad Sci U S A. 2013;110:17253–8. doi: 10.1073/pnas.1308887110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Frangogiannis NG. Targeting the inflammatory response in healing myocardial infarcts. Curr Med Chem. 2006;13:1877–93. doi: 10.2174/092986706777585086. [DOI] [PubMed] [Google Scholar]
- 29.Troidl C, Mollmann H, Nef H, Masseli F, Voss S, Szardien S, et al. Classically and alternatively activated macrophages contribute to tissue remodelling after myocardial infarction. J Cell Mol Med. 2009;13:3485–96. doi: 10.1111/j.1582-4934.2009.00707.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zajac E, Schweighofer B, Kupriyanova TA, Juncker-Jensen A, Minder P, Quigley JP, Deryugina EI. Angiogenic capacity of M1- and M2-polarized macrophages is determined by the levels of TIMP-1 complexed with their secreted proMMP-9. Blood. 122:4054–67. doi: 10.1182/blood-2013-05-501494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Dreymueller D, Denecke B, Ludwig A, Jahnen-Dechent W. Embryonic stem cell-derived M2-like macrophages delay cutaneous wound healing. Wound Repair Regen. 21:44–54. doi: 10.1111/j.1524-475X.2012.00858.x. [DOI] [PubMed] [Google Scholar]
- 32.Zhang QZ, Su WR, Shi SH, Wilder-Smith P, Xiang AP, Wong A, et al. Human gingiva-derived mesenchymal stem cells elicit polarization of M2 macrophages and enhance cutaneous wound healing. Stem Cells. 28:1856–68. doi: 10.1002/stem.503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kigerl KA, Gensel JC, Ankeny DP, Alexander JK, Donnelly DJ, Popovich PG. Identification of two distinct macrophage subsets with divergent effects causing either neurotoxicity or regeneration in the injured mouse spinal cord. J Neurosci. 2009;29:13435–44. doi: 10.1523/JNEUROSCI.3257-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ben-Mordechai T, Holbova R, Landa-Rouben N, Harel-Adar T, Feinberg MS, Abd Elrahman I, et al. Macrophage subpopulations are essential for infarct repair with and without stem cell therapy. J Am Coll Cardiol. 62:1890–901. doi: 10.1016/j.jacc.2013.07.057. [DOI] [PubMed] [Google Scholar]
- 35.Fraccarollo D, Galuppo P, Bauersachs J. Novel therapeutic approaches to post-infarction remodelling. Cardiovasc Res. 94:293–303. doi: 10.1093/cvr/cvs109. [DOI] [PubMed] [Google Scholar]
- 36.Panizzi P, Swirski FK, Figueiredo JL, Waterman P, Sosnovik DE, Aikawa E, et al. Impaired infarct healing in atherosclerotic mice with Ly-6C(hi) monocytosis. J Am Coll Cardiol. 2010;55:1629–38. doi: 10.1016/j.jacc.2009.08.089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Courties G, Heidt T, Sebas M, Iwamoto Y, Jeon D, Truelove J, et al. In vivo silencing of the transcription factor IRF5 reprograms the macrophage phenotype and improves infarct healing. J Am Coll Cardiol. 2013 doi: 10.1016/j.jacc.2013.11.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Jung M, Ma Y, Iyer RP, DeLeon-Pennell KY, Yabluchanskiy A, Garrett MR, Lindsey ML. IL-10 improves cardiac remodeling after myocardial infarction by stimulating M2 macrophage polarization and fibroblast activation. Basic Res Cardiol. 2017;112:33. doi: 10.1007/s00395-017-0622-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Meschiari CA, Jung M, Iyer RP, Yabluchanskiy A, Toba H, Garrett MR, Lindsey ML. Macrophage overexpression of matrix metalloproteinase-9 in aged mice improves diastolic physiology and cardiac wound healing following myocardial infarction. Am J Physiol Heart Circ Physiol. 2017 doi: 10.1152/ajpheart.00453.2017. ajpheart 00453 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Majmudar MD, Keliher EJ, Heidt T, Leuschner F, Truelove J, Sena BF, et al. Monocyte-directed RNAi targeting CCR2 improves infarct healing in atherosclerosis-prone mice. Circulation. 2013;127:2038–46. doi: 10.1161/CIRCULATIONAHA.112.000116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mullican SE, Gaddis CA, Alenghat T, Nair MG, Giacomin PR, Everett LJ, et al. Histone deacetylase 3 is an epigenomic brake in macrophage alternative activation. Genes Dev. 2011;25:2480–8. doi: 10.1101/gad.175950.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Carpio LR, Bradley EW, McGee-Lawrence ME, Weivoda MM, Poston DD, Dudakovic A, et al. Histone deacetylase 3 supports endochondral bone formation by controlling cytokine signaling and matrix remodeling. Sci Signal. 2016;9:ra79. doi: 10.1126/scisignal.aaf3273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.McKinsey TA. Targeting inflammation in heart failure with histone deacetylase inhibitors. Mol Med. 2011;17:434–41. doi: 10.2119/molmed.2011.00022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bolego C, Cignarella A, Staels B, Chinetti-Gbaguidi G. Macrophage function and polarization in cardiovascular disease: a role of estrogen signaling? Arterioscler Thromb Vasc Biol. 33:1127–34. doi: 10.1161/ATVBAHA.113.301328. [DOI] [PubMed] [Google Scholar]
- 45.Cao X, Shen D, Patel MM, Tuo J, Johnson TM, Olsen TW, Chan CC. Macrophage polarization in the maculae of age-related macular degeneration: a pilot study. Pathol Int. 61:528–35. doi: 10.1111/j.1440-1827.2011.02695.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Gui T, Shimokado A, Sun Y, Akasaka T, Muragaki Y. Diverse roles of macrophages in atherosclerosis: from inflammatory biology to biomarker discovery. Mediators Inflamm. 2012:693083. doi: 10.1155/2012/693083. [DOI] [PMC free article] [PubMed] [Google Scholar]