

Abstract
Leishmania is a parasitic protozoan with more than two-dozen species causing the disease leishmaniasis. It is transmitted to humans through the bite of an infected female phlebotomine sand-fly vector. In the past two years the incidence of leishmaniasis has been drastically increasing in Lebanon. This was in parallel with the deterioration of the security in Syria forcing thousands to flee and seek shelter in poorly maintained refugee camps and collective shelters. Cutaneous leishmaniasis (CL) is now considered a public health problem, but its epidemiology has not been fully elucidated. To our knowledge, this is the first study comparing two different molecular methods for the detection and identification of Leishmania tropica in Lebanon.
Two molecular typing methods of 39 FFPE Leishmania isolates were used: the ITS1-PCR RFLP and the nested ITS1-5.8S rDNA gene amplification followed by sequencing and phylogenetic analysis. The efficiency of these two techniques in Leishmania identification was compared and the phylogenetic relationships among these isolates were illustrated based on the neighbor-joining (NJ) method. The results were statistically correlated with the parasitic index (PI). The DNA storage in formalin-fixed paraffin embedded (FFPE) tissues was assessed as well. The parasites identified were all L. tropica as determined by both techniques. ITS1-5.8S rDNA gene based typing proved to be more sensitive in the detection of parasites (positive in 69.2% of the isolates) as opposed to the ITS1-PCR RFLP method that was successful in identifying L. tropica in only 43.6% of the isolates. Sequencing and phylogenetic analysis revealed high levels of heterogeneity. A statistically significant correlation was observed between PI and the results of the nested ITS1-5.8S rDNA gene PCR. Genotyping at the species level is essential for monitoring the relative frequency of CL in the Mediterranean area that is correlated to three different Leishmania species (Leishmania infantum, Leishmania major and L. tropica), each characterized by distinct epidemiological features. The obtained results highlight the need to find a universally accepted diagnostic tool for Leishmania typing.
Abbreviations: Bp, base pair; CL, cutaneous leishmaniasis; FFPE, formalin-fixed paraffin embedded; ITS, internal transcribed spacer; L. tropica, Leishmania tropica; MLST, multilocus sequence typing; NJ, neighbor-joining; PCR, polymerase chain reaction; PI, parasitic index; RFLP, restriction fragment length polymorphism
Keywords: Leishmania, ITS1, 5.8S rDNA gene, RFLP
1. Background
Leishmania is a digenetic parasitic protozoan of the Leishmania genus, family Trypanosomatidae and Kinetoplastida order (Ramos et al., 2013). Leishmania parasites are transmitted to humans by phlebotomine female sand fly vectors (Teixeira et al., 2013) and are the causative agents of leishmaniasis. At least 21 species have showed to cause disease in humans (World Health Organization, 1990). The disease ranges from localized cutaneous leishmaniasis (CL), mucocutaneous leishmaniasis (MCL) to widespread visceral leishmaniasis (VL), also known as kala-azar which is fatal if left untreated (Tsukamaya et al., 2008, Zhang et al., 2013). CL is the most common with more than 95% of the cases occurring in the Americas, the Mediterranean basin, the Near East and Central Asia especially in Afghanistan, Algeria, Brazil, Colombia, Iran and Syria. In fact, leishmaniasis is sometimes referred to as “Aleppo boil” in the medical literature (Hayani et al., 2014). Although, an estimated of 0.7 to 1.3 million new cases of CL occur worldwide each year (World Health Organization, 2014), leishmaniasis is still considered one of the world's most neglected diseases (Ramos et al., 2013).
The crisis in the Syrian Arab Republic that started in March 2011 has resulted in outbreaks of several previously overlooked diseases (World Health Organization, 2013). Since then, cases of CL in Lebanon have been drastically increasing relative to the previous years (2001–2012) (Alawieh et al., 2014, Alvar et al., 2012). From 2004 to 2008 no CL cases were reported in Lebanon compared to 22,882 cases in Syria during the same period (World Health Organization, 2013). Recently, Saroufim et al. (2014) identified Leishmania major in 15% and L. tropica in 85% out of 948 Syrian refugee patients living in Lebanon, the latter species being endemic to the Aleppo region in Syria.
Since different Leishmania species have been shown to cause CL and due to population travel and migration, unexpected Leishmania species can appear in unexpected regions (Dujardin, 2006). Also, different species show different susceptibility to drugs (Blum et al., 2004) and primary and secondary resistance (Desjeux, 2004). The occurrence of natural interspecies hybrids and sexual recombination hinders species discrimination (Tojal da Silva et al., 2006, Nolder et al., 2007, Odiwuor et al., 2011a, Ravel et al., 2006).
Several PCR-based methods have been employed for typing Leishmania parasites. MLEE (Multilocus Enzyme Electrophoresis) is sometimes considered the gold standard for Leishmania identification (Bañuls et al., 2007, Schönian et al., 2001). DNA sequencing, PCR-RFLP (Restriction Fragment Length polymorphism) (Yehia et al., 2012) and MLST (Multilocus Sequence Typing) have all been employed for this purpose (da Silva et al., 2010, Montalvo et al., 2010, Maiden et al., 1998). In terms of sensitivity and validation, the ITS1 region offers the best resolution for Leishmania discrimination in the Old World (Odiwuor et al., 2011b). ITS1 is the sequence in between the 18S rRNA and 5.8S rRNA genes. It contains enough conservation to serve as a PCR target but sufficient polymorphisms to facilitate species typing and identification (Roelfsema et al., 2011). However, these PCR based methods are often hindered by low sensitivity results that fail to detect amastigotes in samples that proved to be positive by microscopy. This caveat in molecular typing is often due to a low parasite load in the original sample (Shahbazi et al., 2008).
In this study, the identification and typing of Leishmania isolates obtained from Syrian refugees in Lebanon using two molecular typing techniques: the ITS1-PCR-RFLP and the nested ITS1-5.8S rDNA gene PCR both followed by sequencing and phylogenetic tree analysis were conducted. The obtained results were used in comparing the efficiency of these two techniques in Leishmania identification and assessing the evolutionary and phylogenetic relationships among these isolates. The results were correlated with the parasitic index (PI). The DNA storage in formalin-fixed paraffin embedded (FFPE) tissues was assessed as well.
2. Methods
2.1. Ethical approval
This study was approved by the American University of Beirut Institutional Review Board and the patient data used in this study was anonymized.
2.2. Sample collection and description
A total of 39 FFPE blocks were obtained from the American University of Beirut Medical Center (AUBMC) in Lebanon. FFPE blocks contained skin biopsies from patients having histologically confirmed CL (PI ≥ 2) during the years of 2013–2014. Each patient had one biopsy performed, and one FFPE block per biopsy was available. Cases included in the study were restricted to cutaneous lesions of patients who did not receive treatment prior to the biopsy. Clinical information pertaining to the lesion was also collected including: number, duration, location and dermatologic appearance. In addition, the patient's age, gender and country of residency were tabulated. Organisms collected from cultures were used as positive controls to validate the analysis. IRB approval was granted prior to the initiation of this study (PALM I.K.01).
2.3. Histopathology
Sections from each of 39 FFPE tissue blocks were stained for hematoxylin and eosin, Giemsa, Acid Fast bacilli, Gomori Methylamine Silver and Periodic Acid-Schiff. All cases were reviewed by four pathologists (JS, RK, FF and IK) and classified according to the modified Ridley's parasitic index (PI) (Table 1). The correlation between the PI, the duration of the lesion, the age of the patients and the results of the ITS1-PCR-RFLP and the nested ITS1-5.8S rDNA gene PCR was performed using the Pearson's correlation test.
Table 1.
Modified Ridley's parasitic index (Ridley and Ridley, 1983).
| 1 + | 1 or more amastigotes per standard section |
| 2 + | 10 or more amastigotes per standard section |
| 3 + | 100 or more amastigotes per standard section |
| 4 + | 1000 or more amastigotes per standard section |
| 5 + | 10,000 or more amastigotes per standard section |
| 6 + | 100,000 or more amastigotes per standard section |
2.4. DNA extraction
DNA extraction was performed as described in Yehia et al. (2012). The resultant DNA was quantified with NanoDrop ND-100 spectrophotometer and Qubit fluorometer (Thermo Fisher Scientific, USA) and stored at 4 °C.
2.5. ITS1 PCR of Leishmania isolates
The extracted DNA was examined for the Leishmania-specific ribosomal ITS1 region by polymerase chain reaction (PCR) amplification using L5.8S and LITSR primers (Table 2) followed by RFLP analysis. The volume of DNA to be amplified was calculated to normalize the quantity among all isolates. 5 μL of DNA was used for ITS1 gene amplification in a 50 μL total reaction volume. A master mix was prepared consisting of 0.2 mM dNTPs, 1 X Taq buffer, 1.5 mM MgCl2, 500 nM of both the forward and the reverse primers and 2 U of Hot-Start Taq Gold DNA polymerase. The cycling conditions were 95 °C for 12 min followed by 32 amplification cycles, each consisting of three steps: 94 °C for 20 s, 53 °C for 30 s and 72 °C for 1 min, followed by a final extension at 72 °C for 6 min. All PCR assays were performed on a PerkinElmer GeneAmp 9700 thermal cycler (PerkinElmer, Wellesly, Massachusetts).
Table 2.
The primers used in this study, their corresponding sequences and target amplicon size.
| Targeted gene | Primer sequences | Amplicon size |
|---|---|---|
| LITSR | 5′-CTGGATCATTTTCCGATG-3′ | 300–350 bp |
| L5.8S | 5′-TGATACCACTTATCGCACTT-3′ | |
| IR1 | 5′-GCTGTAGGTGAACCTGCAGCAGCTGGATCATT-3′ | 320 bp |
| IR2 | 5′-GCGGGTAGTCCTGCCAAACACTCAGGTCTG-3′ | |
| ITS1F | 5′-GCAGCTGGATCATTTTC-C-3′ | 400 bp |
| ITS2R4 | 5′-ATATGCAGAAGAGAGGAGGC-3′ | |
| Actin | 5′-CGC TGC GCT GGT CGT CGA CA-3′ | 600 bp |
| 5′-GTC ACG CAC GAT TTC CCG CT-3′ | ||
| GAPDH | 5′-TGGTGCTCAGTGTAGCCCAG-3′ | 110 bp |
| 5′-GGACCTGACCTGCCGTCTAG-3′ |
Nested PCR was performed on all isolates that were negative by conventional PCR. 10 μL of the ITS1 PCR amplicon were amplified in a 50 μL total reaction volume using the same primers and the same cycling conditions.
PCR amplicons were analyzed by 1.5% agarose gel electrophoresis (1.5% TAE gels stained with ethidium bromide). 25 μL of PCR products were electrophoresed at 90 V in 1X TAE buffer (400 mM Tris-acetate, 10 mM EDTA, pH: 8.2–8.4) and compared to a standard 100 bp DNA ladder. A DNA band of 300–350 bp was considered as a positive indicator for the presence of Leishmania.
2.6. RFLP
10 μL of the PCR products were digested with 2 μL of the restriction endonuclease BsuRI (HaeIII) in 2 μL of 10 X Buffer R (Thermo Fisher Scientific, USA) (1 X of buffer R consists of 10 mM Tris HCl (pH 8.5 at 37 °C), 10 mM MgCl2, 100 mM KCl, 0.1 mg/mL BSA) and 18 μL H2O. Digestion was performed in a thermal cycler, in a total reaction volume of 32 μL, with the following conditions: 6 h at 37 °C followed by 20 min at 80 °C. 25 μL of the PCR product were run on 2.5% agarose gel in 1 X TAE (0.04 M Tris-acetate, 1 mM EDTA, pH = 8) for 1 h. The obtained bands were compared to 100 bp and 20 bp ladders.
2.7. Nested PCR of ITS1-5.8S rDNA genes of Leishmania isolates
The previously extracted DNA was further analyzed by targeting the ITS1-5.8S rDNA gene region. This step consisted of two stages of amplification: The first was performed using the forward IR1 and the reverse IR2 primers, while the second was performed with the nested-forward ITS1F and the nested-reverse ITS2R4 primers (Table 2). In the first stage of amplification: A total reaction volume of 20 μL was prepared of 1 X PCR reaction buffer, 1.5 mM of MgCl2, 60 μM of each dNTP, 1 μM of IR1 and 1 μM of IR2 primers, 1 U of Taq polymerase and 2 μL of the template DNA. PCR was performed under the following conditions: 95 °C for 12 min followed by 39 cycles consisting of 94 °C for 30 s, 58 °C for 30 s and 72 °C for 90 s. The extension step was further continued for 10 min after the last cycle.
In the second stage: The products obtained from the first PCR were further amplified by the nested-PCR technique. A total reaction volume of 20 μL was prepared consisting of 1 X PCR reaction buffer, 1.5 mM of MgCl2, 60 μM of each dNTP, 1 μM of ITS1F and 1 μM of ITS2R4 primers, 1 U of Taq polymerase and 2 μL of the previously obtained PCR product. Nested PCR was performed under the same conditions as the 1st stage of amplification. Bands were electrophoresed on a 1.5% agarose gel in 1 X TAE buffer and compared to a 100 bp ladder.
2.8. DNA sequencing reaction
The obtained PCR products of both PCR reactions, targeting the entire ITS1 and the ITS1-5.8S rDNA gene regions, were purified using ExoSAP-IT (Thermo Fisher Scientific, USA).
The amplicons were sequenced using the ABI Prism BigDye Terminator v3.1 Cycle Sequencing Kit (Applied Biosystem, USA). Two sequencing reactions were performed for each isolate. The sequencing reaction consisted of the BigDye premix, 0.2 pmol of either forward or reverse primer, and the cleaned PCR product in a total volume of 10 μL. The same primers used in the PCR were used for sequencing (Table 2). All sequencing reactions were performed with 25 cycles of 96 °C for 10 s, 50 °C for 5 s, and 60 °C for 4 min. PCR products were sequenced by Genetic Analyzer 3500 (Life Technologies, USA) using the BigDye XTerminator purification kit (Applied Biosystems, USA).
2.9. DNA sequencing and phylogenetic analysis
Sequences obtained were analyzed on CLC Main Workbench v5.5 and deposited to GenBank under the accession numbers indicated below. Sequences were aligned using the multiple alignment program MEGA6 (available at http://www.megasoftware.net/) (Tamura et al., 2013) and Clustal Omega multiple sequence alignment program (available at http://www.clustal.org/omega/) (Sievers et al., 2011) with default parameters. The evolutionary history was inferred using the Neighbor-Joining method (Saitou and Nei, 1987). The evolutionary distances were computed using the Maximum Composite Likelihood method (Tamura et al., 2004) and were in the units of the number of base substitutions per site. All positions containing gaps and missing data were eliminated. Bootstrap replicates were performed to estimate the node reliability, and values were obtained from 1000 randomly selected samples of the aligned sequence data. Sequences obtained from both procedures were compared to seven entries retrieved from Genbank (Table 3).
Table 3.
The sizes and G + C content of the reference sequences included in the phylogenetic trees. References were extracted from NCBI BLAST. References used cover the ITS 1, partial sequence; 5.8S rRNA gene, complete sequence and ITS 2, partial sequence.
2.10. DNA quality assessment
In order to assess the DNA quality of the extracted DNA, Actin and GAPDH amplification were performed under the following conditions: A total reaction volume of 50 μL was prepared consisting of 0.2 mM dNTPS, 1 X Taq buffer, 1.5 mM MgCl2, 500 nM of both forward and reverse primers and 2 U of Taq DNA polymerase. For actin, 10 μL of DNA were amplified under the following cycling conditions: 95 °C for 12 min followed by 35 cycles consisting of 94 °C for 20 s, 56 °C for 30 s and 72 °C for 1 min. Followed by a final extension step of 72 °C for 10 min. For actin, 10 μL of DNA were amplified under the following cycling conditions: Initial denaturation at 95 °C for 12 min followed by 40 cycles consisting of 94 °C for 20 s, 60 °C for 30 s and 72 °C for 1 min. Followed by a final extension step of 72 °C for 10 min. 25 μL of the obtained PCR product were loaded on a 1.5% agarose gel and compared to a 100 bp and a 20 bp ladders.
3. Results
3.1. DNA extraction
Skin biopsies from 39 patients from Lebanon and Syria with clinical diagnosis of CL were assessed. The specimen was obtained by punch biopsy, and the duration of the lesions ranged from 1 to 12 months (mean: 4.51 ± 2.36; median: 4). Ages of patients varied from 1 to 83 years (mean: 25.08 ± 23.22; median: 16). 53.9% of patients were females and 46.1% were males. Most of the samples (38.5%) were from the 0–10 year's old age group followed by the above 60 years old age group (15.4%). The obtained DNA concentrations ranged from 8.5 ng/μL to 462.3 ng/μL (mean: 66.16 ± 99.62 ng/μL; median: 37.6 ng/μL).
3.2. ITS1 PCR of Leishmania isolates
None of the isolates were positive for the first PCR step targeting the ITS1 region. After nested PCR using the same primers, 17 out of 39 isolates (43.6%) produced a band at around 350 bp, indicating the presence of Leishmania parasites (Fig. 1).
Fig 1.
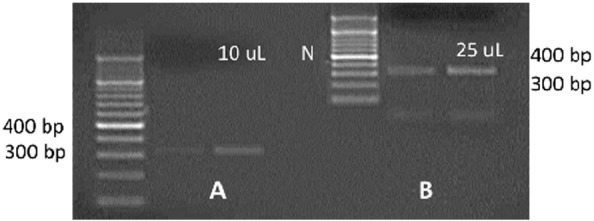
Agarose gel electrophoresis of the amplicons of ITS1-PCR for two representative isolates obtained after nested PCR. 10 μL (A) and 25 μL (B) of the PCR amplicon were loaded on the gel. Thicker bands at 350 bp were observed for loading 25 μL indicating the presence of Leishmania. A 100 bp ladder was used as a molecular marker. N: negative control.
3.3. RFLP
The digestion of the PCR product with endonuclease HaeIII revealed the profiles of L. tropica consisting of two bands at 200 bp and 60 bp, respectively.
3.4. Sequencing and phylogenetic tree analysis
Analysis of the ITS1 rDNA gene sequences further confirmed that they all contain L. tropica. Sequences showed a size range of 90 to 332 bp (mean: 236.06 ± 76.83 bp; median: 277 bp) and a G + C content ranging from 41.22% to 53.43% (mean: 45.74 ± 4.14%; median: 42.51%). GenBank accession numbers were obtained for 10 of the sequences [KT376735-KT376744] as the remaining sequences were too ambiguous even after re-sequencing. The complete alignment of the 10 sequences revealed a percent conservation of 6% to 94% (mean: 50.85 ± 19%; median: 56%) per nucleotide. The aligned sequences showed several blocks of conserved nucleotides in addition to a high occurrence of single nucleotide polymorphisms (SNPs) (Fig. 2). Variation was reflected by the low bootstrap values obtained in the NJ phylogenetic tree (Fig. 3).
Fig 2.

ITS1-PCR gene sequence alignment of L. tropica isolates using the Clustal Omega multiple sequence alignment program. Matching residues are highlighted with the same color. Gaps are denoted as dashes. “*” indicates positions which have a single, fully conserved residue. “:” indicates that a ‘strong’ group is fully conserved. “.” indicates that a ‘weaker’ group is fully conserved. These are all the positively scoring groups that occur in the Gonnet Pam250 matrix. The strong and weak groups are defined as strong score > 0.5 and weak score = < 0.5 respectively.
Fig 3.

Neighbor-joining (NJ) tree showing the relationships of the 17 L. tropica isolates based on the ITS1 sequences. Bootstrap values are based on 1000 replicates.
3.5. Nested PCR for amplifying the ITS1-5.8S rDNA gene
The first PCR reaction using the ITS1 and ITS2 primers showed no bands on gel electrophoresis for all 39 isolates. Nested PCR reaction produced bands at around 400 bp for 27 isolates (69.2%) confirming that they belong to L. tropica (Fig. 4).
Fig 4.
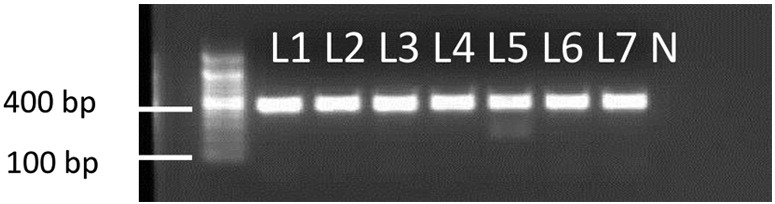
Nested-PCR of ITS1-5.8S rDNA gene of Leishmania DNA. Isolates positive for Leishmania gave a band around 400 bp characteristic of L. tropica. A 100 bp ladder was used as a molecular marker. N: negative control.
3.6. Sequencing and phylogenetic tree analysis
Analysis of the ITS1-5.8S rDNA gene sequences further confirmed that they all contain L. tropica. Alignment revealed a higher level of conservation along with the presence of numerous single point mutations (SNPs) (Fig. 5). Sizes ranged from 254 bp to 512 bp (mean: 426.79 ± 38.81 bp; median: 432 bp) and the G + C content from 38.17% to 50.58% (mean: 43.70 ± 2.84%; median: 39.9%). Percent conservation ranged from 4% to 100% (as per every nucleotide) (mean: 62.5% ± 29.75; median: 75%). Genbank accession numbers were obtained for the 27 sequences [KT363773-KT363799] (Table S2) (Fig 6.)
Fig 5.

ITS1-5.8S rDNA gene sequence alignment of L. tropica isolates using the Clustal Omega multiple sequence alignment program. Matching residues are highlighted with the same color. Gaps are denoted as dashes. “*” indicates positions which have a single, fully conserved residue.
Fig 6.

Neighbor-joining (NJ) tree showing the relationships of the 27 L. tropica isolates based on the ITS1-5.8S rDNA gene sequences. Bootstrap values are based on 1000 replicates.
3.7. Assessing DNA quality through actin and GAPDH amplification
In order to rule out possible DNA quality reasons affecting the results, two housekeeping genes, Actin and GAPDH, were amplified. The first PCR amplification yielded no bands. Nested PCR resulted in bands at around 600 bp and 200 bp for Actin and GAPDH, respectively. All the isolates used in this study were positive for Actin and GAPDH after performing nested PCR.
3.8. Statistical analysis
A statistically significant correlation was observed between the PI and the nested ITS1-5.8S rDNA gene PCR (p = 0.04 ≤ 0.05 level of significance). PIs of 2 and 3 were always correlated with positive molecular results. Increasing PI was correlated with an increase in the number of false negatives. These were microscopically determined to contain parasites but proved negative for molecular typing. False negatives appeared with ratios of 1:2, 1:1.6 and 2:1 for PI 4, 5 and 6, respectively. For PI = 6, a two fold increase in the number of false negatives might be due in part to a low frequency number (Fig. 7). No significant correlation was obtained between the PI and the results of the ITS-1-PCR RFLP, between the duration of the lesions and the results of the molecular tests and between PI and the duration of the lesion.
Fig 7.

Correlation between the parasitic index (PI) of the lesion and the results of the ITS1-5.8 S rDNA gene PCR.
4. Discussion
4.1. DNA extraction from FFPE tissue
In this study, commercial reagents (Qiagen GmbH) were used to extract purified DNA from FFPE skin biopsies (Yehia et al., 2012). Leishmania parasites contain only 10− 13 g of DNA, meaning that 10 million parasites would be required to generate 1 μg of workable DNA (Barker, 1989). This makes gene detection in Leishmania isolates extremely difficult. In order to obtain sufficient amount of DNA for further analysis, the process of DNA extraction has to be critically efficient (Momeni et al., 1996). In this study, all the conducted PCR reactions, including the isolates having a DNA concentration as high as 400 ng/μL, required a second PCR amplification to obtain a visible band, making the analysis process more costly, tedious and time consuming. Previously, it has been reported that DNA often gets degraded during the process of formalin fixation as well as during the extraction process itself (Momeni et al., 1996, Huijsmans et al., 2010). Also, negative results can be obtained despite a high yield due to the presence of crosslinks in the extracted DNA (Ludyga et al., 2012).
4.2. Leishmania typing using ITS1-PCR-RFLP and ITS1-5.8S rDNA gene
PCR-based methods have been proven to be highly sensitive and specific for the detection of Leishmania parasites in cultures or clinical samples (Ghatee et al., 2013). Previously, several studies exploited the ITS1 region amplification technique for the discrimination of Leishmania parasites (Roelfsema et al., 2011, Saitou and Nei, 1987, Schonian et al., 2003, Amro et al., 2009). Nested ITS1-5.8S rDNA gene PCR enhanced the sensitivity of this diagnostic tool by targeting two fragments in the ITS-rDNA region, one region consisting of ITS1 with the 5.8S rDNA gene (Parvizi and Ready, 2008). In this study, nested PCR of ITS1-5.8S rDNA gene proved to be more sensitive for Leishmania species identification. This proved to be consistent with a number of previously obtained results (Ajouad et al., 2013, El Tai et al., 2000, El Tai et al., 2001, Spanakos et al., 2008, Schallig and Oskam, 2002, Parvizi et al., 2008, Es-Sette et al., 2014). All of the cases studied were caused by L. tropica, a species endemic to the Aleppo region in Syria (Saroufim et al., 2014) thus supporting the import of this species into Lebanon from across the Syrian borders.
Diagnosis of CL relying solely on the microscopical examination of the lesions lacks enough sensitivity (Al-Jawabreh et al., 2004). And thus, it needs to be complemented by an efficient molecular typing method (Zhang et al., 2013) or preferably a combination of different techniques to achieve maximal sensitivity in parasite detection and eliminate false negative results. Several factors can affect the number of amastigotes in a lesion including the strain type, the stage of the disease and the immune defense of the host (Croft et al., 2006). Although histopathology-based diagnosis is the first step towards appropriate targeted therapy (Salman et al., 1999), it can be inadequate as all Leishmania species are morphologically similar and have variable numbers of amastigotes (Schonian et al., 2003, Singh et al., 2005). Furthermore, histopathological examination fails to detect amastigotes in up to 47% of cases (PI < 2) (Ameen, 2010). A PI of 2 or more is confirmatory of the presence of Leishmania parasites in the lesion. At this level the patient can start treatment (Ridley and Ridley, 1983). However, the species still needs to be determined because L. major and L. tropica infections are administered different treatment protocols (González et al., 2010). In this study, false negative results in ITS1-5.8S rDNA gene PCR were observed with Ridley's PI = 5 and 6 which could be attributed to the high parasite load (Yehia et al., 2012, Bensoussan et al., 2006). It can be inferred from the correlation analysis that samples with PI = 2 and 3 can be further subjected to molecular sub-species identification as opposed to the samples having a PI = 4 and more that are more likely to fail in ITS1-5.8S rDNA gene detection.
Moreover, ITS1 PCR-RFLP is the most widely used assay for direct detection and identification of Leishmania species in the Old World (Schonian et al., 2011), which identifies all clinically significant Leishmania species using only one enzyme (Yehia et al., 2012, Al-Nahhas and Kaldas, 2013). The comparison between three PCR-based methods involving two kinetoplast DNA (kDNA) PCRs and ITS1-PCR (LITSR/L5.8S primers) suggested that the ITS1 PCR-RFLP method was the most sensitive (Mouttaki et al., 2014). Based on the RFLP pattern of the ITS1 PCR product, isolates were identified as L. tropica (Schönian et al., 2001, Schonian et al., 2003, Al-Nahhas and Kaldas, 2013, Doudi et al., 2010, Rotureau et al., 2006). Previous attempts of extending the RFLP to include the 5.8S rDNA gene region did not show any advantage (Parvizi and Ready, 2008).
4.3. Sequencing and phylogenetic tree analysis
Although the ITS region serves as a marker for the differentiation of Leishmania at both the species and the strain levels, only few studies employed ITS sequence analysis to compare L. tropica isolates (Schönian et al., 2001, Khanra et al., 2011, Talmi-Frank et al., 2010, Mahdy et al., 2012, Ghatee et al., 2014). L. tropica is a diploid organism and such heterozygosity renders sequencing analysis more complicated (Yeo et al., 2011). Heterozygosity from electropherograms can be inferred by a double peak with two bases at the same variable bi-allelic site (Tavanti et al., 2005). A sequence of multiple bi-allelic sites is largely ambiguous. However, diploid sequence data can be modified, concatenated across multiple loci and applied in distance based phylogenetic methods for lineage assignment (Yeo et al., 2011, Tavanti et al., 2005). L. tropica is known to be a very heterogenous species (Schönian et al., 2001, Azmi et al., 2012, Le Blancq and Peters, 1986) and this was clearly observed in the alignment of the PCR amplification products and the low bootstrap values obtained on the phylogenetic tree. The reason behind this is the occurrence of at least two alleles for ITS in ribosomal DNA in Leishmania spp. (Ghatee et al., 2013, Mauricio et al., 2004).
Alignment based on the ITS1-5.8S rDNA gene revealed a significantly higher level of conservation than the ITS1 based alignment. ITS1 alone was very divers yielding several unreadable sequences. It was commonly reported that samples positive for leishmaniasis often show unreadable sequences upon ITS-rDNA fragment examination (Mirzaei et al., 2013). This can be explained by extremely high heterogeneity levels or sometimes may be due to mixed infections of two or more Leishmania species (Parvizi and Ready, 2008, Parvizi et al., 2008, Mirzaei et al., 2013, Strelkova et al., 2001). The obtained data also infers evolutionary relatedness and helps in detecting gene mosaics within or between homozygous gene loci (Odds and Jacobsen, 2005).
The total G + C content in Leishmania species was determined to be 57% (Alonso et al., 1992) whereas the G + C content in ITS1 and ITS1-5.8S rDNA gene was on average 45.74% and 43.70%, respectively. Although Leishmania species show a high preference for C- and G-ending codons (Langford et al., 1992), the low G + C content in the ITS region confirms that the selective pressure is not identical in coding and non-coding regions (Syvanen, 1994, Goncalves and Rosato, 2002).
In this study, two techniques for the molecular detection of Leishmania isolates were used: ITS1-PCR RFLP and ITS1-5.8S rDNA gene PCR amplification. ITS1-PCR RFLP profiles proved that the isolates were all L. tropica. This was further validated by sequencing the ITS1-PCR product as well as the ITS1-5.8S rDNA gene product. ITS1-5.8S rDNA PCR proved to be a more sensitive identification method for Leishmania isolates. Genotyping at the species level contributes to monitor the relative frequency of CL (Al-Jawabreh et al., 2004), especially that in the Mediterranean area CL can be caused by three different Leishmania species (Leishmania infantum, L. major and L. tropica), each characterized by distinct epidemiological features (Schonian et al., 2011). The obtained results highlight the need to find a universally accepted diagnostic tool for Leishmania typing, that is sensitive and capable of identifying all clinically significant Leishmania species. The risk of transmitting this “flesh-eating” parasite into the Lebanese community is high. Thus, effective prevention methods and appropriate therapy is critical. Prevention can be as simple as using nets treated with insecticide or spraying insecticides to kill sand-fly vectors (Barrett and Croft, 2012). For the differentiation of Leishmania at the sub-species level, it is recommended to perform multi-locus sequence typing (MLST) method. Moreover, whole-genome sequencing (WGS) provides a greater potential to identify genetic component of health problems and infectious diseases (Schonian et al., 2011, van El et al., 2013). Accessible international databases for cases of leishmaniasis in the MENA region (Middle East and North Africa) should be created for a better epidemiological assessment of these infectious agents and for tracing their patterns of migration between countries and continents.
The following are the supplementary data related to this article.
Clinical information of the patients having CL. The PI, the location from where the sample was collected, the age of the patient, the gender (F: female; M: male) and the duration of the lesion are depicted.
Sequences of Leishmania species based on ITS1. The biotype number, the size of the sequence (bp), the G + C content (%) and the GenBanK accession numbers are depicted. Some of the sequences were too ambiguous, and accession numbers could not be obtained.
Sequences of Leishmania species based on ITS1-5.8S rDNA gene. The biotype number, the size of the sequence (bp), the G + C content (%) and the GenBanK accession numbers are depicted.
Acknowledgments
The authors would like to thank the Lebanese American University, Byblos, Lebanon where the primary study was conducted and Miss Maya Farah for technical assistance in the lab. We also wish to thank the American University of Beirut Medical Center, AUBMC, Beirut, Lebanon, for providing the tested isolates.
Contributor Information
Tamara Salloum, Email: tamara.salloum@lau.edu.
Ibrahim Khalifeh, Email: ik08@aub.edu.lb.
Sima Tokajian, Email: stokajian@gmail.com.
References
- Ajouad M., Es-sette N., Hamdi S., El-Idrissi A., Riyad M., Lemrani M. Detection and molecular typing of Leishmania tropica from Phlebotomus sergenti and lesions of cutaneous leishmaniasis in an emerging focus of Morocco. Parasit. Vectors. 2013;6(1):217. doi: 10.1186/1756-3305-6-217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alawieh A., Musharrafieh U., Jaber A., Berry A., Ghosn N., Bizri A.R. Revisiting leishmaniasis in the time of war: the Syrian conflict and the Lebanese outbreak. Int. J. Infect. Dis. 2014;29:115–119. doi: 10.1016/j.ijid.2014.04.023. [DOI] [PubMed] [Google Scholar]
- Al-Jawabreh A., Schnur L.F., Nasereddin A., Schwenkenbecher J.M., Abdeen Z., Barghuthy F. The recent emergence of Leishmania tropica in Jericho (A'riha) and its environs, a classical focus of L. major. Tropical Med. Int. Health. 2004;9(7):812–816. doi: 10.1111/j.1365-3156.2004.01268.x. [DOI] [PubMed] [Google Scholar]
- Al-Nahhas S.A., Kaldas R.M. Characterization of Leishmania species isolated from cutaneous human samples from central region of Syria by RFLP analysis. ISRN Parasitol. 2013:5. doi: 10.5402/2013/308726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alonso G., Guevara P., Ramirez J.L. Trypanosomatidae codon usage and GC distribution. Mem. Inst. Oswaldo Cruz. 1992;87(4):517–523. doi: 10.1590/s0074-02761992000400009. [DOI] [PubMed] [Google Scholar]
- Alvar J., Velez I.D., Bern C., Herrero M., Desjeux P., Cano J. Leishmaniasis worldwide and global estimates of its incidence. PLoS One. 2012;7 doi: 10.1371/journal.pone.0035671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ameen M. Cutaneous leishmaniasis: advances in disease pathogenesis, diagnostics and therapeutics. Clin. Exp. Dermatol. 2010;35:699. doi: 10.1111/j.1365-2230.2010.03851.x. [DOI] [PubMed] [Google Scholar]
- Amro A., Azmi K., Schonian G., Nasereddin A., Alsharabati M.B., Sawalha S. Epidemiology of paediatric visceral leishmaniasis in Hebron district, Palestine. Trans. R. Soc. Trop. Med. Hyg. 2009;103(7):731–736. doi: 10.1016/j.trstmh.2008.10.008. [DOI] [PubMed] [Google Scholar]
- Azmi K., Schnur L., Schonian G., Nasereddin A., Pratlong F., El Baidouri F. Genetic, serological and biochemical characterization of Leishmania tropica from foci in northern Palestine and discovery of zymodeme MON-307. Parasit. Vectors. 2012;5(1):121. doi: 10.1186/1756-3305-5-121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bañuls A.L., Hide M., Prugnolle F. Leishmania and the leishmaniases: a parasite genetic update and advances in taxonomy, epidemiology and pathogenicity in humans. Adv. Parasitol. 2007;64:1–458. doi: 10.1016/S0065-308X(06)64001-3. [DOI] [PubMed] [Google Scholar]
- Barker D.C. Molecular approaches to DNA diagnosis. Parasitology. 1989;99(S1):S125–S146. doi: 10.1017/s0031182000083463. [DOI] [PubMed] [Google Scholar]
- Barrett M.P., Croft S.L. Management of trypanosomiasis and leishmaniasis. Br. Med. Bull. 2012;104:175–196. doi: 10.1093/bmb/lds031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bensoussan E., Nasereddin A., Jonas F., Schnur L.F., Jaffe C.L. Comparison of PCR assays for diagnosis of cutaneous leishmaniasis. J. Clin. Microbiol. 2006;44(4):1435–1439. doi: 10.1128/JCM.44.4.1435-1439.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blum J., Desjeux P., Schwartz E., Beck B., Hatz C. Treatment of cutaneous leishmaniasis among travelers. J. Antimicrob. Chemother. 2004;53(2):158–166. doi: 10.1093/jac/dkh058. [DOI] [PubMed] [Google Scholar]
- Croft S.L., Sundar S., Fairlamb A.H. Drug resistance in leishmaniasis. Clin. Microbiol. Rev. 2006;19(1):111–126. doi: 10.1128/CMR.19.1.111-126.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- da Silva L.A., de Sousa C.S., da Graca G.C., Porrozzi R., Cupolillo E. Sequence analysis and PCR-RFLP profiling of the hsp70 gene as a valuable tool for identifying Leishmania species associated with human leishmaniasis in Brazil. Infect. Genet. Evol. 2010;10(1):77–83. doi: 10.1016/j.meegid.2009.11.001. [DOI] [PubMed] [Google Scholar]
- Desjeux P. Leishmaniasis: current situation and new perspectives. Comp. Immunol. Microbiol. Infect. Dis. 2004;27(5):305–318. doi: 10.1016/j.cimid.2004.03.004. [DOI] [PubMed] [Google Scholar]
- Doudi M., Hejasi S.A., Razavi M.R., Eslami G. Comparative molecular epidemiology of Leishmania major and Leishmania tropica by PCR-RFLP technique in hyperendemic cities of Isfahan and Bam, Iran. Med. Sci. Monit. 2010;16(11):530–535. [PubMed] [Google Scholar]
- Dujardin J.C. Risk factors in the spread of leishmaniases: towards integrated monitoring? Trends Parasitol. 2006;22(1):4–6. doi: 10.1016/j.pt.2005.11.004. [DOI] [PubMed] [Google Scholar]
- El Tai N.O., El Fari M., Mauricio I., Miles M.A., Oskam L., El Safi S.H. Leishmania donovani: intra-specific polymorphisms of Sudanese isolates revealed by PCR-based analyses and DNA sequencing. Exp. Parasitol. 2000;97(1):35–44. doi: 10.1006/expr.2001.4592. [DOI] [PubMed] [Google Scholar]
- El Tai N.O., Osman O.F., El Fari M., Presber W., Schönian G. Genetic heterogeneity of ribosomal internal transcribed spacer in clinical samples of Leishmania donovani. Trans. R. Soc. Trop. Med. Hyg. 2001;94(5):575–579. doi: 10.1016/s0035-9203(00)90093-2. [DOI] [PubMed] [Google Scholar]
- Es-Sette N., Ajaoud M., Bichaud L., Hamdi S., Mellouki F., Charrel R.N. Phlebotomus sergenti a common vector of Leishmania tropica and Toscana virus in Morocco. J. Vector Borne Dis. 2014;51:86–90. [PubMed] [Google Scholar]
- Ghatee M., Sharifi I., Mirhendi H., Kanannejad Z., Hatam G. Investigation of double-band electrophoretic pattern of ITS-rDNA region in Iranian isolates of Leishmania tropica. Iran. J. Parasitol. 2013;8(2):264–272. [PMC free article] [PubMed] [Google Scholar]
- Ghatee M.A., Sharifi I., Kuhls K., Kanannejad Z., Harandi M.F., de Almeida M.E. Heterogeneity of the internal transcribed spacer region in Leishmania tropica isolates from southern Iran. Exp. Parasitol. 2014;144:44–51. doi: 10.1016/j.exppara.2014.06.003. [DOI] [PubMed] [Google Scholar]
- Goncalves E., Rosato Y. Phylogenetic analysis of Xanthomonas species based upon 16S-23S rDNA intergenic spacer sequences. Int. J. Syst. Microbiol. 2002;52:355–361. doi: 10.1099/00207713-52-2-355. [DOI] [PubMed] [Google Scholar]
- González U., Pinart M., Reveiz L., Rengifo-Pardo M., Tweed J., Macaya A. Designing and reporting clinical trials on treatments for cutaneous leishmaniasis. Clin. Infect. Dis. 2010;51(4):409–419. doi: 10.1086/655134. [DOI] [PubMed] [Google Scholar]
- Hayani K., Dandashli A., Weisshaar E. Cutaneous leishmaniasis in Syria: clinical features, current status and the effects of war. Acta Derm. Venereol. 2014;95:62–66. doi: 10.2340/00015555-1988. [DOI] [PubMed] [Google Scholar]
- Huijsmans C.J., Damen J., van der Linden J.C., Savelkoul P.H., Hermans M.H. Comparative analysis of four methods to extract DNA from paraffin-embedded tissues: effect on downstream molecular applications. BMC Res. Notes. 2010;3(1):239. doi: 10.1186/1756-0500-3-239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khanra S., Bandopadhyay S., Chakraborty P., Datta S., Mondal D., Chatterjee M. Characterization of the recent clinical isolates of Indian Kala-azar patients by RAPD-PCR method. J. Parasit. Dis. 2011;35(2):116–122. doi: 10.1007/s12639-011-0048-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langford I., Ullman B., Landfear M. Leishmania: codon utilization of nuclear genes. Exp. Parasitol. 1992;74:360–361. doi: 10.1016/0014-4894(92)90161-3. [DOI] [PubMed] [Google Scholar]
- Le Blancq S.M., Peters W. Leishmania in the old world: heterogeneity among L. tropica zymodemes. Trans. R. Soc. Trop. Med. Hyg. 1986;80(1):113–119. doi: 10.1016/0035-9203(86)90208-7. [DOI] [PubMed] [Google Scholar]
- Ludyga N., Grünwald B., Azimzadeh O., Engler S., Höfler H., Tapio S. Nucleic acids from long-term preserved FFPE tissues are suitable for downstream analyses. Virchows Arch. 2012;460(2):131–140. doi: 10.1007/s00428-011-1184-9. [DOI] [PubMed] [Google Scholar]
- Mahdy M.A., Al-Mekhlafi H.M., Al-Mekhlafi A.M., Lim Y.A., Shuaib N.O.B., Azazy A.A. Molecular characterization of Leishmania species isolated from cutaneous leishmaniasis in Yemen. PLoS One. 2012;5(9) doi: 10.1371/journal.pone.0012879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maiden M.C., Bygraves J.A., Feil E., Morelli G., Russell J.E., Urwin R. Multilocus sequence typing: a portable approach to the identification of clones within populations of pathogenic microorganisms. Proc. Natl. Acad. Sci. 1998;95(6):3140–3145. doi: 10.1073/pnas.95.6.3140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mauricio I.L., Stothard J.R., Miles M.A. Leishmania donovani complex: genotyping with ribosomal internal transcribed spacer and mini-exon. Parasitology. 2004;128(03):263–267. doi: 10.1017/s0031182003004578. [DOI] [PubMed] [Google Scholar]
- Mirzaei A., Rouhani A., Kazerooni P.A., Farahmand M., Parvizi P. Molecular detection and conventional identification of Leishmania species in reservoir hosts of zoonotic cutaneous leishmaniasis in Fars Province, South of Iran. Iran. J. Parasitol. 2013;8(2):280–288. [PMC free article] [PubMed] [Google Scholar]
- Momeni A.Z., Yotsumoto S., Mehregan D.R., Mehregan A.H., Mehregan D.A., Aminjavaheri M. Chronic lupoid leishmaniasis: evaluation by polymerase chain reaction. Arch. Dermatol. 1996;132(2):198–202. doi: 10.1001/archderm.132.2.198. [DOI] [PubMed] [Google Scholar]
- Montalvo A.M., Fraga J., Monzote L., Montano I., De Doncker S., Dujardin J.C. Heat-shock protein 70 PCR-RFLP: a universal simple tool for Leishmania species discrimination in the new and old world. Parasitology. 2010;137(08):1159–1168. doi: 10.1017/S0031182010000089. [DOI] [PubMed] [Google Scholar]
- Mouttaki T., Morales-Yuste M., Merino-Espinosa G., Chiheb S., Fellah H., Martin-Sanchez J. Molecular diagnosis of cutaneous leishmaniasis and identification of the causative Leishmania species in Morocco by using three PCR-based assays. Parasit. Vectors. 2014;7(1):420. doi: 10.1186/1756-3305-7-420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nolder D., Roncal N., Davies C.R., Llanos-Cuentas A., Miles M.A. Multiple hybrid genotypes of Leishmania (Viannia) in a focus of mucocutaneous leishmaniasis. Am.J.Trop. Med. Hyg. 2007;76(3):573–578. [PubMed] [Google Scholar]
- Odds F.C., Jacobsen M.D. Multilocus sequence typing of pathogenic Candida species. Eukaryot. Cell. 2005;7(7):1075–1084. doi: 10.1128/EC.00062-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Odiwuor S., De Doncker S., Maes I., Dujardin J.C., Van der Auwera G. Natural Leishmania donovani/Leishmania aethiopica hybrids identified from Ethiopia. Infect. Genet. Evol. 2011;11(8):2113–2118. doi: 10.1016/j.meegid.2011.04.026. [DOI] [PubMed] [Google Scholar]
- Odiwuor S.O., Saad A.A., De Doncker S., Maes I., Laurent T., El Safi S. Universal PCR assays for the differential detection of all old world Leishmania species. Eur. J. Clin. Microbiol. Infect. dis. 2011;30(2):209–218. doi: 10.1007/s10096-010-1071-3. [DOI] [PubMed] [Google Scholar]
- Parvizi P., Ready P.D. Nested PCRs and sequencing of nuclear ITS-rDNA fragments detect three Leishmania species of gerbils in sandflies from Iranian foci of zoonotic cutaneous leishmaniasis. Tropical Med. Int. Health. 2008;13(9):1159–1171. doi: 10.1111/j.1365-3156.2008.02121.x. [DOI] [PubMed] [Google Scholar]
- Parvizi P., Moradi G., Akbari G., Farahmand M., Ready P.D., Piazak N. PCR detection and sequencing of parasite ITS-rDNA gene from reservoir host of zoonotic cutaneous leishmaniasis in central Iran. Parasitol. Res. 2008;136(6):1289–1295. doi: 10.1007/s00436-008-1124-z. [DOI] [PubMed] [Google Scholar]
- Ramos J., González-Alcaide G., Bolaños-Pizarro M. Bibliometric analysis of leishmaniasis research in Medline (1945-2010) Parasit. Vectors. 2013;6:55. doi: 10.1186/1756-3305-6-55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ravel C., Cortes S., Pratlong F., Morio F., Dedet J.P., Campino L. First report of genetic hybrids between two very divergent Leishmania species: Leishmania infantum and Leishmania major. Int. J. Parasitol. 2006;36(13):1383–1388. doi: 10.1016/j.ijpara.2006.06.019. [DOI] [PubMed] [Google Scholar]
- Ridley D.S., Ridley M.J. The evolution of the lesion in cutaneous leishmaniasis. J. Pathol. 1983;141:83. doi: 10.1002/path.1711410109. [DOI] [PubMed] [Google Scholar]
- Roelfsema J.H., Nozari N., Herremans T., Kortbeek L.M., Pinelli E. Evaluation and improvement of two PCR targets in molecular typing of clinical samples of Leishmania patients. Exp. Parasitol. 2011;127(1):36–41. doi: 10.1016/j.exppara.2010.06.024. [DOI] [PubMed] [Google Scholar]
- Rotureau B., Ravel C., Couppie P., Pratlong F., Nacher M., Dedet J.P. Use of PCR-restriction fragment length polymorphism analysis to identify the main new world Leishmania species and analyze their taxonomic properties and polymorphism by application of the assay to clinical samples. J. Clin. Microbiol. 2006;44(2):459–467. doi: 10.1128/JCM.44.2.459-467.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saitou N., Nei M. The neighbor-joining method: a new method for reconstructing phylogenetic trees. Mol. Biol. Evol. 1987;4:406–425. doi: 10.1093/oxfordjournals.molbev.a040454. [DOI] [PubMed] [Google Scholar]
- Salman S.M., Rubeiz N.G., Kibbi A.G. Cutaneous leishmaniasis: clinical features and diagnosis. Clin. Dermatol. 1999;17:291. doi: 10.1016/s0738-081x(99)00047-4. [DOI] [PubMed] [Google Scholar]
- Saroufim M., Charafeddine K., Issa G., Khalifeh H., Habib R., Berry A. Ongoing epidemic of cutaneous leishmaniasis among Syrian refugees. Emerg. Infect. dis. 2014;20(10):1712. doi: 10.3201/eid2010.140288. (Retrieved from www.cdc.gov/eid) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schallig H.D., Oskam L. Molecular biological applications in the diagnosis and control of leishmaniasis and parasite identification. Tropical Med. Int. Health. 2002;7(8):641. doi: 10.1046/j.1365-3156.2002.00911.x. [DOI] [PubMed] [Google Scholar]
- Schönian G., Schnur L.F., El Fari M., Oskam L., Kolesnikov A.A., Sokolowska Köhler W. Genetic heterogeneity in the species Leishmania tropica revealed by different PCR-based methods. Trans. R. Soc. Trop. Med. Hyg. 2001;95(2):217–224. doi: 10.1016/s0035-9203(01)90173-7. [DOI] [PubMed] [Google Scholar]
- Schonian G., Nasereddin A., Dinse N., Schweynoch C., Schallig H.D., Presber W. PCR diagnosis and characterization of Leishmania in local and imported clinical samples. Diagn. Microbiol. Infect. Dis. 2003;41(1):349–358. doi: 10.1016/s0732-8893(03)00093-2. [DOI] [PubMed] [Google Scholar]
- Schonian G., Kuhls K., Mauricio L. Molecular approaches for a better understanding of the epidemiology and population genetics of Leishmania. Parasitology. 2011;138(04):405–425. doi: 10.1017/S0031182010001538. [DOI] [PubMed] [Google Scholar]
- Shahbazi F., Shahabi S., Kazemi B., Mohebali M., Abadi A., Zare Z. Evaluation of PCR assay in diagnosis and identification of cutaneous leishmaniasis: a comparison with the parasitological methods. Parasitol. Res. 2008;103(5):1159–1162. doi: 10.1007/s00436-008-1111-4. [DOI] [PubMed] [Google Scholar]
- Sievers F., Wilm A., Dineen D., Gibson T.J., Karplus K., Li W. Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol. Syst. Biol. 2011;7:539. doi: 10.1038/msb.2011.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh S., Dey A., Sivakumar R. Applications of molecular methods for Leishmania control. Expert. Rev. Mol. Diagn. 2005;5:251. doi: 10.1586/14737159.5.2.251. [DOI] [PubMed] [Google Scholar]
- Spanakos G., Piperaki E.T., Menounos P.G., Tegos N., Flemetakis A., Vakalis N.C. Detection and species identification of old world Leishmania in clinical samples using a PCR based method. Trans. R. Soc. Trop. Med. Hyg. 2008;102(1):46. doi: 10.1016/j.trstmh.2007.05.019. [DOI] [PubMed] [Google Scholar]
- Strelkova M.V., Eliseev L.N., Ponirovsky E.N., Dergacheva T.I., Annacharyeva D.K., Erokhin P.I. Mixed leishmanial infections in Rhombomys opimus: a key to the persistence of Leishmania major from one transmission season to the next. Ann. Trop. Med. Parasitol. 2001;95(8):811–819. doi: 10.1080/00034980120111154. [DOI] [PubMed] [Google Scholar]
- Syvanen M. Horizontal gene transfer: evidence and possible consequences. Annu. Rev. Genet. 1994;28:237–261. doi: 10.1146/annurev.ge.28.120194.001321. [DOI] [PubMed] [Google Scholar]
- Talmi-Frank D., Nasereddin A., Schnur L.F., Schönian G., Ozensoy Töz S., Jaffe C.L. Detection and identification of old world Leishmania by high resolution melt analysis. PLoS Negl. Trop. Dis. 2010;4(1):e581. doi: 10.1371/journal.pntd.0000581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamura K., Nei M., Kumar S. Prospects for inferring very large phylogenies by using the neighbor-joining method. Proc. Natl. Acad. Sci. U. S. A. 2004;101:11030–11035. doi: 10.1073/pnas.0404206101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamura K., Stecher G., Peterson D., Filipski A., Kumar S. MEGA6: molecular evolutionary genetics analysis version 6.0. Mol. Biol. Evol. 2013;30:2725–2729. doi: 10.1093/molbev/mst197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tavanti A., Davidson A.D., Johnson E.M., Maiden M.C., Shaw D.J., Gow N.A. Multilocus sequence typing for differentiation of strains of Candida tropicalis. J. Clin. Microbiol. 2005;43(11):5593–5600. doi: 10.1128/JCM.43.11.5593-5600.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teixeira D., Benchimol M., Rodrigues J., Crepaldi P., Pimenta P., de Souza W. The cell biology of Leishmania: how to teach using animations. PLoS Pathog. 2013;9(10) doi: 10.1371/journal.ppat.1003594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tojal da Silva A.C., Cupolillo E., Volpini A.C., Almeida R., Romero G.A. Species diversity causing human cutaneous leishmaniasis in Rio Branco, State of Acre, Brazil. Tropical Med. Int. Health. 2006;11(9):1388–1398. doi: 10.1111/j.1365-3156.2006.01695.x. [DOI] [PubMed] [Google Scholar]
- Tsukamaya P., Lucas C., Bacon J. Typing of four genetic loci discriminates among closely related species of new world Leishmania. Int. J. Parasit. 2008;39(3):355–362. doi: 10.1016/j.ijpara.2008.08.004. [DOI] [PubMed] [Google Scholar]
- van El C.G., Cornel M., Borry P., Hastings R., Fellmann F., Hodgson S. Whole-genome sequencing in health care: recommendations of the European society of human genetics. Eur. J. Hum. Genet. 2013;21:580–584. doi: 10.1038/ejhg.2013.46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- World Health Organization . 1990. Control of the Leishmaniasis, Report of a WHO Expert Committee. [Google Scholar]
- World Health Organization The WHO Leishmaniasis Control Team News. 2013. (Available: http://www.who.int/en/)
- World Health Organization . Regional Office for the Eastern Mediterranean; 2014. Framework for Action on Cutaneous Leishmaniasis in the Eastern Mediterranean Region 2014–2018. [Google Scholar]
- Yehia L., Adib-Houreih M., Raslan W.F., Kibbi A.G., Loya A., Firooz A. Molecular diagnosis of cutaneous leishmaniasis and species identification: analysis of 122 biopsies with varied parasite index. J. Cutan. Pathol. 2012;39(3):347–355. doi: 10.1111/j.1600-0560.2011.01861.x. [DOI] [PubMed] [Google Scholar]
- Yeo M., Mauricio I.L., Messenger L.A., Lewis M.D., Llewellyn M.S., Acosta N. Multilocus sequence typing (MLST) for lineage assignment and high resolution diversity studies in Trypanosoma cruzi. PLoS Negl. Trop. Dis. 2011;5(6) doi: 10.1371/journal.pntd.0001049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang C., Lu X., Du X., Jian J., Shu L., Ma Y. Phylogenetic and evolutionary analysis of Chinese Leishmania isolates based on multilocus sequence typing. PLoS One. 2013;8(4) doi: 10.1371/journal.pone.0063124. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Clinical information of the patients having CL. The PI, the location from where the sample was collected, the age of the patient, the gender (F: female; M: male) and the duration of the lesion are depicted.
Sequences of Leishmania species based on ITS1. The biotype number, the size of the sequence (bp), the G + C content (%) and the GenBanK accession numbers are depicted. Some of the sequences were too ambiguous, and accession numbers could not be obtained.
Sequences of Leishmania species based on ITS1-5.8S rDNA gene. The biotype number, the size of the sequence (bp), the G + C content (%) and the GenBanK accession numbers are depicted.