

Abstract
Through the action of two virus-encoded decapping enzymes (D9 and D10) that remove protective caps from mRNA 5′-termini, Vaccinia virus (VACV) accelerates mRNA decay and limits activation of host defenses. D9- or D10-deficient VACV are markedly attenuated in mice and fail to counter cellular double-stranded RNA-responsive innate immune effectors, including PKR. Here, we capitalize upon this phenotype and demonstrate that VACV deficient in either decapping enzyme are effective oncolytic viruses. Significantly, D9- or D10-deficient VACV displayed anti-tumor activity against syngeneic mouse tumors of different genetic backgrounds and human hepatocellular carcinoma xenografts. Furthermore, D9- and D10-deficient VACV hyperactivated the host anti-viral enzyme PKR in non-tumorigenic cells compared to wild-type virus. This establishes a new genetic platform for oncolytic VACV development that is deficient for a major pathogenesis determinant while retaining viral genes that support robust productive replication like those required for nucleotide metabolism. It further demonstrates how VACV mutants unable to execute a fundamental step in virus-induced mRNA decay can be unexpectedly translated into a powerful anti-tumor therapy.
Keywords: oncolytic virus, mRNA decay, decapping
Introduction
A key challenge in developing viral platforms for oncolytic virus (OV) immunotherapy lies in identifying an attenuated variant that retains sufficient replicative capacity in cancer cells.1, 2, 3 By manipulating the genetic backbone of what was to become the first clinically approved oncolytic HSV1 (oHSV1), a powerful solution to this problem was exposed and effectively validated.4, 5, 6, 7, 8 The oHSV1 is attenuated because it lacks a critical viral virulence gene required to antagonize innate immune defenses in normal cells.5, 9 This cell intrinsic immune response is triggered by double-stranded (ds) RNA, which accumulates in virus-infected cells and activates host anti-viral defense molecules, including PKR and RNase L.10, 11 Although host defenses restrict virus replication in normal cells, the oHSV1 can overcome the comparatively modest host dsRNA-induced, anti-viral responses in cancer cells,4, 12, 13, 14, 15, 16, 17 which often have altered innate immune responses.18 By taking advantage of this vulnerability, attenuated viruses can be engineered to preferentially replicate and spread within cancer cells, resulting in oncolysis and immunotherapeutic anti-tumor responses.6, 7, 8, 19, 20 Although targeting host dsRNA-induced defenses like PKR and RNase L has proven to be a successful strategy for developing an oHSV1, whether this tactic can be applied to other large DNA virus platforms, like vaccinia virus (VACV), remains unknown.
Like many viruses, VACV encodes effector molecules to antagonize dsRNA-induced host defenses.11, 21 Most notably, the viral E3L and K3L genes prevent PKR activation and limit inactivation of its substrate, the critical translation initiation factor eIF2.22 However, viruses deficient for E3L are profoundly attenuated and replicate poorly, likely the result of their inability to counter host dsRNA-induced defenses.23 In contrast, K3L-deficient viruses remain insufficiently attenuated for consideration as an OV candidate.24 Instead, much of the work on oncolytic VACV development has focused on strains deficient for the viral thymidine kinase (tk) gene, many of which are further attenuated by inactivation of additional virus genes, including the VACV growth factor VGF.25, 26, 27, 28 Indeed, several of these mutant VACV derivatives have entered into clinical trials,29, 30, 31, 32, 33 although one failed to meet its primary end point,34, 35 illustrating the need to investigate alternatives to multi-mutated tk-deficient VACV strains.
Unexpectedly, VACV mRNA decapping enzymes encoded by the D9 and D10 genes were recently found to limit dsRNA accumulation in infected cells.36, 37 By removing the m7GTP cap on the mRNA 5′ end, D9 and D10 accelerate mRNA decay by generating substrates for the cellular 5′-3′ mRNA exoribonuclease Xrn1.38, 39 Relative to WT VACV, D9- and D10-deficient strains accumulate higher levels of dsRNA and more effectively activate dsRNA-responsive host innate immune sensing pathways, including activation of PKR and RNase L.36, 37 Moreover, D9- and D10-deficient viruses were both attenuated in mice with D10-deficient recombinants displaying the greatest attenuation following intranasal administration.37, 40 Because D9 and D10 represent newly identified VACV-encoded virulence determinants controlling host dsRNA-activated defenses, the potential of D9- and D10-deficient VACV as a new genetic backbone for OV therapy was investigated.
Here, we establish that D9- and D10-deficient VACV replicates to near wild-type (WT) levels in several established murine cancer cell lines and have OV activity in syngeneic murine cancer in vivo models. D9- and D10-deficient VACV also reduced growth of an established human hepatocellular carcinoma (HCC) xenograft in athymic mice. Moreover, greater levels of VACV antigen accumulated in HCC tumors treated with D9- or D10-deficient VACV compared to the surrounding normal tissue. Whereas PKR was activated equivalently in a panel of HCC cells infected in vitro with either decapping-deficient or WT VACV, PKR was selectively hyperactivated in normal, non-tumorigenic cells. This shows that decapping-deficient VACV has anti-tumor activity against several murine syngeneic tumors and a human HCC model. Because D9- and D10-deficient VACV hyperactivates dsRNA innate immune defenses in non-tumorigenic cells, it further suggests a mechanism for its preferential replication in HCC tumors.
Results
Productive Replication of Decapping-Deficient VACV in Established Murine Cancer Cell Lines
To evaluate the capacity of the decapping-deficient VACV mutants to replicate in murine tumor cell lines, their ability to direct viral protein production was first tested. MBT2 murine bladder carcinoma and 4T1 murine breast carcinoma cells were infected with either WT VACV, D9-deficient (ΔD9) VACV, or D10-deficient (ΔD10) VACV. After 18 hr, cultures were metabolically radiolabeled with 35S amino acids. Total protein was subsequently harvested, fractionated by SDS-PAGE, and analyzed by autoradiography (Figure 1A) or immunoblotting (Figure 1B). Compared to control primary human fibroblasts (NHDFs), less virus-induced suppression of ongoing host cell protein synthesis (host cell shutoff) was observed in murine cancer cell lines infected with WT, D9-deficient, or D10-deficient VACV (Figure 1A). Despite the apparent absence of host cell shut-off, VACV proteins accumulated to similar levels in 4T1 or MBT2 cells infected with either WT, D9-deficient, or D10-deficient VACV (Figure 1B). Thus, viral proteins accumulate similarly in murine cancer cell lines infected with decapping-deficient VACVs lacking either the D9 or D10 genes compared to WT VACV.
Figure 1.

Protein Synthesis and Accumulation in Murine Cancer Cells Infected with D9- or D10-Deficient VACV
(A) Murine MBT2 bladder carcinoma, murine 4T1 breast carcinoma, or NHDFs were mock-infected (mock) or infected (MOI = 3) with WT VACV, D9-deficient VACV (ΔD9), or D10-deficient VACV (ΔD10). At 18 hours post-infection (hpi), cells were metabolically pulse labeled with [35S]Met-Cys for 30 min. Total protein was collected and separated by SDS-PAGE, and [35S]-labeled proteins were visualized by exposing the fixed, dried gel to X-ray film. Molecular mass standards (in kDa) are shown on the left. Representative radiolabeled proteins in mock-infected NHDFs that decrease in infected cells (consistent with host shut-off) are indicated (⋅). Representative radiolabeled proteins in mock-infected MBT2 or 4T1 cells that persist in infected cells are indicated (o). (B) Samples in (A) were analyzed by immunoblotting using anti-VACV polyclonal antisera as described.59
To compare the capacity of decapping-deficient VACV to productively replicate and spread in murine cancer cell lines, MBT2 (bladder carcinoma, H-2K) or 4T1 cells (breast carcinoma, H-2D) were infected with either WT, D9-deficient, or D10-deficient virus at low MOI (Figures 2A and 2B). Quantifying infectious virus production after 48 hr revealed decapping-deficient VACV mutants grow to similar levels as WT VACV, with only a minor reduction in yield (no more than 4-fold) detected in cells infected with either D9- or D10-deficient viruses. In addition, replication of decapping-deficient VACV mutants in MCA38 cells (colon adenocarcinoma, H-2B) was also comparable to WT virus (no more than 8-fold less) (Figure 2C). Thus, decapping-deficient VACV productively replicated and spread to near WT levels in representative murine cancer cell lines derived from different mouse genetic backgrounds.
Figure 2.
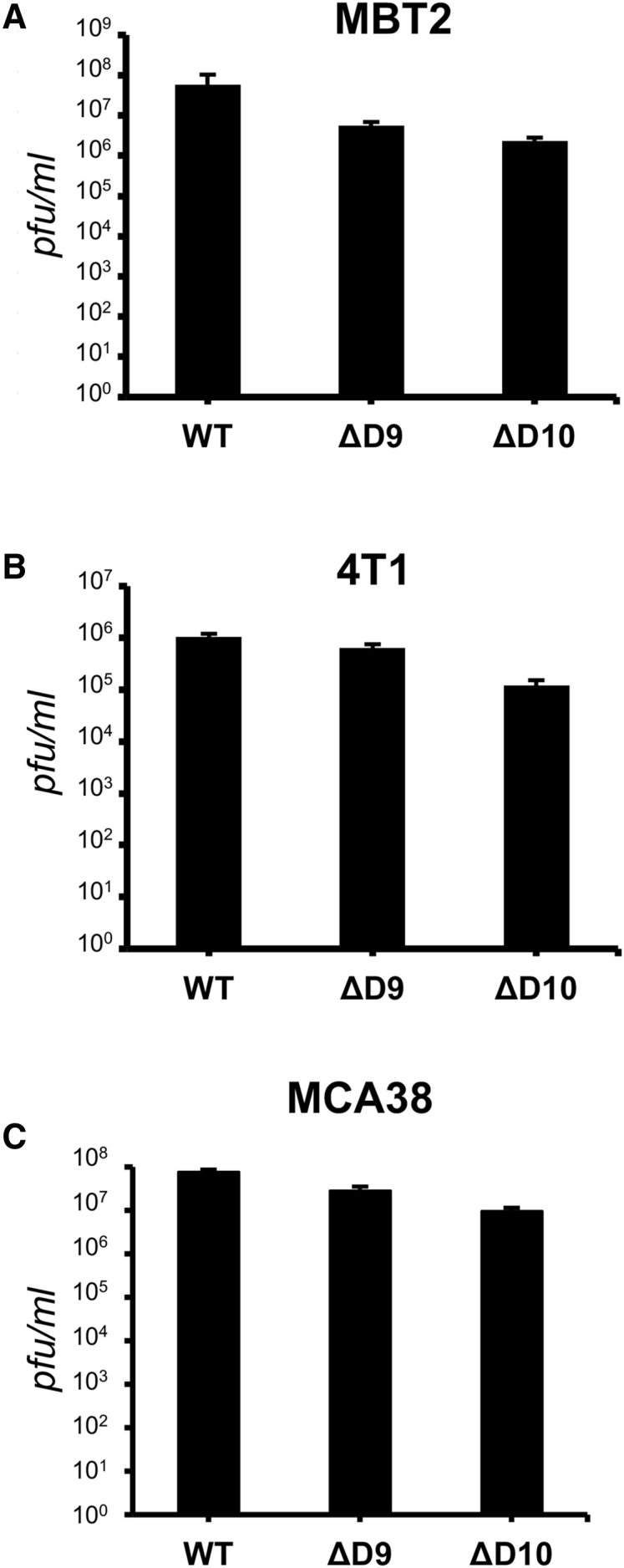
Replication of VACV D9- and D10-Deficient Mutants in Murine Cancer Cells
(A and B) MBT2 bladder carcinoma cells (A) or 4T1 breast carcinoma cells (B) seeded in 12-well dishes (approximately 5 × 105 cells/well) were infected (300 pfu/well) with either WT VACV, D9-deficient VACV (ΔD9), or D10-deficient VACV (ΔD10). After 48 hr, cultures were lysed by freeze thawing and the amount of infectious virus quantified by plaque assay in BSV40 cells. (C) As in (A) and (B), except murine MCA38, adenocarcinoma cells were infected (MOI = 1). Error bars represent SEM. N = 3 from three independent experiments.
Anti-tumor Activity of VACV Decapping-Deficient Mutants in Syngeneic Murine Cancer Models
To determine if decapping-deficient VACV lacking D10 could induce therapeutic anti-tumor responses, subcutaneous 4T1 tumors were established in syngeneic mice and once tumors reached approximately 50 mm3, they were directly injected with a virus-free control preparation from uninfected cells (mock) or D10-deficient VACV. Additional intra-tumoral injections were performed 3 and 6 days after the first treatment and tumor growth was measured over time. Between 6 and 9 days post-treatment, a statistically significant difference between mock and ΔD10-treated tumors was readily observed (Figure 3A). The volume of mock-treated tumors increased to a greater extent and more rapidly than D10-treated tumors. This difference increased and persisted through the entire course of the experiment (Figure 3A). This establishes that ΔD10 VACV treatment has anti-tumor activity against 4T1 tumors in syngeneic BALB/c mice and demonstrates that a decapping-deficient VACV, such as ΔD10, can be utilized as an oncolytic virus.
Figure 3.

Anti-tumor Activity of VACV D9- and D10-Deficient Mutants in Murine Cancer Cells
(A) 4T1 breast carcinoma cells (1 × 104) in DMEM without additives were injected s.c. into the right flank of 8-week-old, female BALB/c mice. When tumors reached approximately 50 mm3 (8 to 9 days after 4T1 inoculation), they were directly injected on days 0, 3, and 6 (indicated by downward pointing arrows) with 5.4 × 106 PFU of D10-deficient (ΔD10) VACV (N = 10 mice) or an equivalent volume of virus-free control preparation (mock) from uninfected cells (N = 10 mice). Tumor size was monitored and animals were euthanized when control-treated tumors reached approximately 1,200 mm3. Tumors were measured on the indicated days, and the average normalized values reflecting relative tumor size on each day were plotted. Initial tumor volume immediately before treatment was normalized to a relative size of 1.0. Error bars indicate SEM. p values were obtained by multiple t test. ***p < 0.001; **p < 0.01. (B) As in (A) except murine MCA38 colon adenocarcinoma cells (1 × 105) were injected s.c. into the flank of 6-week-old, female C57/Bl6 mice. When tumors reached approximately 50 mm3 (approximately 7 days after MCA38 inoculation), they were directly injected on days 0, 3, and 6 with 1.0 × 106 PFU of D10-deficient (ΔD10) VACV (N = 10 mice), 1.0 × 106 PFU of D9-deficient (ΔD9) VACV (N = 10 mice), or an equivalent virus-free control preparation from uninfected cells (N = 10 mice). Tumor size was monitored and animals were euthanized when control-treated tumors reached approximately 1,200 mm3. Between day 9 and 12, three mice died in the mock-treated group, two mice died in the ΔD10-treated group, and one mouse died in the ΔD9-treated group. Error bars indicate SEM. p values were obtained by multiple t test. Gray * above the mock-treated line indicates comparisons of mock versus ΔD10. Black * below the mock-treated line indicates comparisons of mock versus ΔD9. **p < 0.05; ***p ≤ 0.005.
To compare the anti-tumor properties of D9- and D10-deficient VACVs and determine if their OV activity was restricted to syngeneic BALB/c models, MCA38 tumors were established in C57BL/6 mice and treated as described above. By 6 days post-treatment, a statistically significant difference between mock and ΔD9- or ΔD10-treated tumors was readily observed (Figure 3B). The volume of mock-treated tumors increased to a greater extent and more rapidly than did ΔD9- or ΔD10-treated tumors and persisted through the entire time course of the experiment (Figure 3B). This establishes that D9- or D10-deficient VACV treatment has anti-tumor activity against MCA38 tumors in syngeneic C57BL/6 mice and demonstrates that mutant VACVs lacking either D9 or D10 decapping enzymes are effective oncolytic viruses. In addition, this activity is not limited to a particular murine genetic background. The MCA38 tumors were particularly aggressive, progressing more rapidly than 4T1, as evidenced by the death of 3 mice treated with the virus-free control preparation. The rapid growth of the tumors necessitated that the animals be euthanized on day 12, effectively ending the experiment. Although one mouse died in the ΔD9-treated group and two mice died in the ΔD10-treated group, the fatalities in each experimental group were less than the control group treated with a virus-free preparation.
Anti-tumor Activity of Decapping-Deficient VACV in a Human HCC Xenograft Cancer Model
To address the anti-tumor capacity of decapping-deficient VACV against human tumors, HepG2 HCC xenografts were established in athymic mice. When tumors reached approximately 50 mm3, they were injected with D10-deficient (ΔD10) VACV, D9-deficient (ΔD9) VACV, or an equivalent virus-free control preparation (mock) from uninfected cells and tumor volume was monitored. A statistically significant difference between tumors treated with a virus-free, mock preparation and ΔD9- or ΔD10-treated tumors was readily observed by day 14 (Figure 4). The volume of mock-treated tumors increased to a greater extent and more rapidly than tumors treated with VACV ΔD9 or ΔD10 (Figure 4). This difference increased and persisted through the entire 20-day course of the experiment, establishing that D9- or D10-deficient VACV treatment has anti-tumor activity against human HepG2 tumors in athymic, nude mice. Compared to the very large tumors in mice treated with a control, virus-free preparation, mice treated with either D9- or D10-deficient viruses had at most small tumors remaining and one animal had no palpable mass.
Figure 4.

Anti-tumor Activity of VACV D9- and D10-Deficient Mutants in Human Tumor Xenografts in Immunocompromised Mice
HepG2 human HCC cells were injected (1 × 107) s.c. into the flank of 8-week-old, female athymic, BALB/c nude mice. When tumors reached approximately 50 mm3 (approximately 7 days after HepG2 inoculation), they were directly injected on days 0, 3, 6, and 9 (indicated by downward black arrows) with 1.0 × 106 PFU of D10-deficient (ΔD10) VACV (N = 10 mice), 1.0 × 106 PFU of D9-deficient (ΔD10) VACV (N = 10 mice), or an equivalent volume of virus-free control preparation (mock) from uninfected cells (N = 10 mice). Tumors were measured on the indicated days, and the average normalized values reflecting relative tumor size on each day were plotted. Initial tumor volume immediately before treatment was normalized to a relative size of 1.0. Days on which the individual, moribund animals in the ΔD9 or ΔD10 treatment groups were sacrificed are indicated by symbols below (black + for ΔD9) or above (gray o for ΔD10) the respective data points. Error bars indicate SEM. p values were obtained by multiple t test. Gray * above the mock-treated line indicates comparisons of mock versus ΔD10. Black * below the mock-treated line indicates comparisons of mock versus ΔD9. **p < 0.05; ***p < 0.005.
All mice treated with a control, virus-free preparation survived until being euthanized on day 20, but 40% of the treated mice died (between day 17 and 18 for ΔD10 treated and between day 6 and 17 for ΔD9 treated) before the remaining animals were euthanized (day 20 for ΔD10 and mock treated and day 17 for ΔD9 treated). Thus, in athymic nude mice, the absence of the capacity to mount an acquired immune response likely accounts for the virulence of VACV deficient in only D9 or D10. Other VACV mutants that are attenuated in immunocompetent mouse models are likewise more virulent in athymic, immunocompromised mice, including strains containing multiple, engineered mutations being investigated in human trials as therapeutic OV candidates.41, 42
To analyze the anti-tumor response to ΔD9 and ΔD10 OV treatment, HCC tumors were fixed in formalin and embedded in paraffin. Sequential sections from representative tumors were stained with H&E and examined by light microscopy. All sections of the three mock-treated mice showed 45%–50% subcutaneous, viable HCC (Table 1). These tumors have a large main tumor and some smaller satellite tumor nodules, which represent tumor expansion outside the main tumor bed through infiltration or vascular invasion (Figure 5A). The viable HCC has a trabecular and solid growth pattern and is composed of cells with a scant to moderate amount of amphophilic cytoplasm and moderately pleomorphic nuclei with prominent nucleoli. Mitotic figures were easily identified, and approximately 50%–65% tumor necrosis was noted. A thin pseudo-capsule composed of reactive spindle/stromal cell fibroblasts with patchy, mild, polymorphonuclear cell infiltrates was seen around the tumor nodules (Figure 5D). Compared to mock-treated tumors, ΔD9 and ΔD10 OV-treated HCC xenografts showed very small foci of residual viable tumor and satellite tumor nodules were not observed (Figure 5, compare B and C to A). Significantly, one ΔD9 OV-treated mouse (#67) showed no residual viable tumor in three different sections through the tumor and a second ΔD9-treated mouse (#69) had only three small foci representing about 4% of the xenograft (Figure 5B; Table 1). Both ΔD10-treated mice examined (#51 and #52) showed small foci of residual tumor in two out of three examined sections, representing about 3% of the xenograft (Figure 5C; Table 1). Collectively, the pathological analysis summarized in Figure 5 and Table 1 clearly demonstrate that ΔD9 and ΔD10 OV treatment resulted in substantial tumor necrosis and reduction of viable HepG2 human HCC xenografts that is compatible with their potent antitumor effect.
Table 1.
Reduction in Viable Tumor by Treatment with Decapping-Deficient VACV
| Treatment Group | Mouse # | Tumor Necrosis | Residual Viable Tumor (HCC) (%) |
|---|---|---|---|
| Mock | 61 | yes | 50 |
| Mock | 64 | yes | 50 |
| Mock | 65 | yes | 45 |
| ΔD9 | 67 | yes | 0 |
| ΔD9 | 69 | yes | 4 |
| ΔD10 | 51 | yes | 3 |
| ΔD10 | 52 | yes | 3 |
On day 17 (mouse #67 and #69) or day 20 (mouse #51, #52, #61, #64, and #65), animals treated as in Figure 4 were sacrificed and explanted tumors were fixed in formalin. Paraffin-embedded sections were prepared and stained with H&E and evaluated by light microscopy.
Figure 5.

Histopathology of HCC Tumors Treated with Decapping-Deficient VACV OV
(A–F) On day 20 (A, C, D, and F) or day 17 (B and E) post-treatment, animals treated as in Figure 4 were sacrificed and explanted tumors were fixed in formalin. Paraffin-embedded sections were prepared and stained with H&E and evaluated by light microscopy. At low magnification (20X; A–C), the amount of residual viable tumor (HCC) in mock-treated mice (A) was substantially greater than that observed in ΔD9- (B) or ΔD10-treated mice (C). Viable HCC is highlighted between the blue lines; the red line shows a satellite tumor nodule present only in mock-treated mice (A). At high magnification (400X; D–F), viable HCC appears surrounded by a capsule composed of fibroblasts and few inflammatory cells in mock-treated mice (D). In contrast, ΔD9- (E) and ΔD10-treated (F) mice show a marked fibro-inflammatory response to the viable and necrotic HCC (left of the blue line: viable HCC; right of the blue line: fibro-inflammatory response). Tumor sections from individual mice are identified by the number at the top of the panels.
Although residual HCC in the OV-treated mice was similar in morphologic appearance to the control, mock-treated mice, the pseudo-capsule in the OV-treated mice focally shows a thick fibroblastic reaction, with abundant polymorphonuclear cell inflammatory infiltrates predominantly composed of neutrophils (Figures 5B, 5C, 5E, and 5F). To characterize the nature of these cellular infiltrates, immunohistochemistry was performed. Compared to mock-treated tumors, treatment with either ΔD9 or ΔD10 OVs resulted in an influx of slightly more F4/80+ myeloid cells (Figure 6) and a greater number of Ly6C+ cells (Figure 6), which are likely natural killer (NK) cells. These cell types reflect enhanced inflammation resultant from OV infection and are potentially related to the reduced rate of tumor growth in OV-treated tumors. The robust inflammatory infiltrate also appears activated by virtue of increased staining for Granzyme B (Figure 6), a marker of lytic function characteristic of NK cells.
Figure 6.

Characterization of Inflammatory Infiltrates in OV-Treated Tumors by Immunohistochemistry
On day 20 (mock, ΔD10) or day 17 (ΔD9) post-treatment, tumor xenographs from animals treated as in Figure 4 were harvested, fixed, and embedded. Samples were processed for immunohistochemistry (IHC) as described in Materials and Methods and stained with either control or Ab reactive with macrophage (F4/80) neutrophils (Ly6C) or granzyme B (GrzB) as indicated. Samples from two independent tumors are shown to demonstrate typical staining achieved.
To examine sites of VACV replication, sections from mock, ΔD9-treated, or ΔD10-treated tumor tissue sections were stained with antisera raised against VACV. In addition, human HCC cells were distinguished from normal mouse tissue by co-staining with anti-sera specific for a human nuclear mitotic apparatus component (NuMa). The HCC tumor is clearly demarcated from normal surrounding mouse tissue by NuMA staining in a representative mock-treated tumor and tumors from four independent ΔD9- or ΔD10-treated mice (Figure 7). At the greatest magnification shown, NuMA antigen accumulation is evident within nuclei. In ΔD9- and ΔD10-treated tumors isolated from multiple animals, VACV antigen is enriched and appears contained within NuMA-positive cells over a range of magnifications (Figure 7). VACV antigen is observed to be confined to the cytoplasm within cells with NuMA-staining nuclei, providing evidence that NuMA-positive cells are infected with VACV (Figure 7). Moreover, cytoplasmic VACV antigen was not detected in the neighboring normal, NuMA-negative mouse tissue. This establishes that VACV gene expression occurs in the HCC xenograft. Moreover, the enrichment of VACV antigen within cells with NuMA-positive nuclei is consistent with preferential replication of decapping-deficient VACV within the HCC tumor compared with the surrounding normal tissue.
Figure 7.

Preferential Enrichment of OV Antigens in Tumor Tissue
Tumors and surrounding tissue from animals treated as in Figure 4 were harvested and fixed on day 17 (ΔD9-treated mice #67 and #69) or day 20 (mock-treated mouse #64; ΔD10-treated mice #51 and #52). Samples were processed for IHC as described in Materials and Methods, stained with antibody reactive with human NuMA (purple nuclear staining) or VACV (brown cytoplasmic staining), and counterstained with hematoxylin. From left to right, 10X and 40X magnifications are shown. 40X magnifications highlight representative fields showing the intersection of NuMA-positive cells (magenta arrowhead) with normal mouse tissue (blue arrowhead) and show that VACV antigens preferentially accumulate in and appear restricted to NuMA-positive tumor cells (white arrowhead).
Altered Activation of Host dsRNA Responses by D9 or D10 Decapping-Deficient VACV in Human Tumor Cells Compared to Non-tumorigenic Cells
To determine if replication of D9 or D10 decapping-deficient VACVs might be preferentially restricted in non-tumorigenic human cells compared to tumor cells, the activation state of the interferon-induced, dsRNA-dependent protein kinase PKR was investigated. PKR is an interferon-induced host gene that is activated by dsRNA, a pathogen associated molecular pattern (PAMP) that accumulates in virus-infected cells and is a signature of virus infection. Upon activation, PKR phosphorylates the host translation initiation factor eIF2 on its alpha subunit, inactivating this critical translation initiation factor and restricting virus protein synthesis and replication (reviewed by Mohr et al.11). PKR activation is routinely measured by immunoblotting for the autophosphorylated form (on residue T446) using a phospho-specific anti-PKR antibody.36 Following mock-infection or infection (ΔD9, ΔD10, or WT VACV) of several tumorigenic human HCC cell lines (HepG2, Hep3B, and Huh7), the overall abundance of total PKR and phosphorylated (activated) PKR was assessed by immunoblotting. In HepG2 and Hep3B cells, PKR was similarly activated after infection with WT, ΔD9, or ΔD10 VACV beyond levels detected in mock-infected cells as evidenced by phosphorylated PKR abundance (Figure 8). Although the background level of activated PKR was detected in mock-infected Huh7 cells, activated PKR abundance was reduced similarly upon infection with WT, ΔD9, or ΔD10 VACV (Figure 8). Thus, in tumorigenic human cell lines, PKR was not detectably hyperactivated following infection with ΔD9 or ΔD10 VACV compared to WT VACV.
Figure 8.
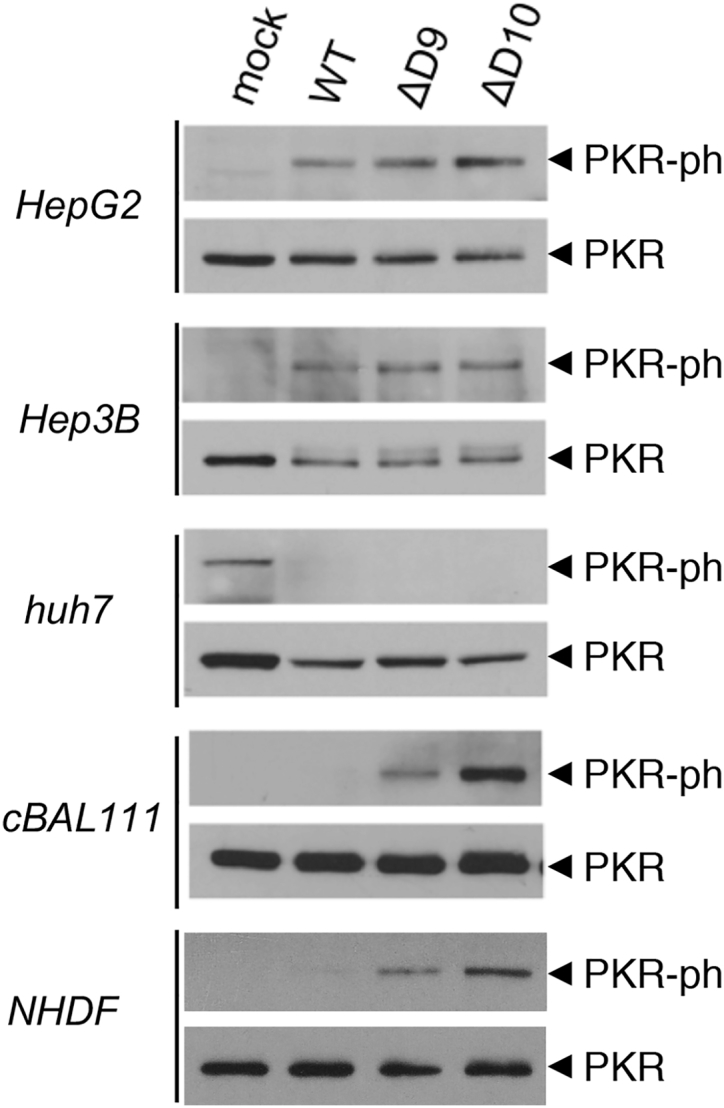
Hyperactivation of PKR in Non-tumorigenic Human Cells Following Infection with D9- or D10-Deficient VACV
Human HCC (HepG2, Hep3B, and Huh7) or untransformed, non-tumorigenic cBAL111 human liver cells were either mock-infected or infected (MOI = 5) with WT VACV, D9-deficient VACV (ΔD9), or D10-deficient VACV (ΔD10). After 18.5 hr, total protein was isolated and analyzed by immunoblotting with either total PKR or a PKR phospho-specific antibody as described.36
To compare PKR activation upon infection of non-tumorigenic cells with WT, ΔD9, or ΔD10 VACV, NHDFs and cBAL111 cells were either mock-infected or infected. Although NHDFs are primary cells, the cBAL111 cell line was derived from human fetal liver cells immortalized by overexpressing the telomerase reverse transcriptase.43 These cells display hepatic differentiated functionality similar to the parental cells prior to immortalization and express immature hepatocyte markers.43 Significantly, cBAL111 cells do not grow in soft agar and are not tumorigenic in nude mice.43 Remarkably, although phosphorylated PKR was undetectable in mock-infected or WT VACV-infected cBAL111 cells, activated, phosphorylated PKR was readily detected in cells infected by either ΔD9 or ΔD10 VACV (Figure 8). This demonstrates that PKR is selectively hyperactivated upon infection with ΔD9 or ΔD10 decapping-deficient VACV compared to WT VACV in non-tumorigenic cBAL111 cells and normal, primary human cells. Moreover, it establishes that D9 or D10 decapping-deficient VACV hyperactivates cell intrinsic antiviral responses selectively in non-tumorigenic cells compared to tumorigenic cells.
Discussion
Nearly all oncolytic VACV strains have been attenuated by removing viral tk or ribonucleotide reductase genes.25, 28, 41, 44, 45 Although presumed to restrict virus growth to dividing cells, these mutations over-attenuate the virus, restricting its capacity to directly destroy cancer cells and induce systemic, anti-tumor immune responses. Indeed, although largely safe, one first-generation VACV OV did not meet expectations in a phase 2b clinical trial.34, 35 In lieu of removing genes required for metabolism like tk that enable robust virus replication, a different attenuation strategy involves deleting genes required for pathogenesis. In this regard, VacV encodes two mRNA decapping enzymes (D9 and D10) that hydrolyze the m7GTP mRNA cap from 5′ termini that is critical for mRNA translation and stability.38, 39 Besides controlling virus and host gene expression, D9 and D10 antagonize host anti-viral responses by limiting dsRNA accumulation.36, 37 Although decapping enzymes are not essential for VACV productive growth, viruses deficient for D9 or D10 are attenuated.37, 40 Here, we demonstrate that VACV-deficient for either D9 or D10 decapping enzyme replicate to a similar extent as WT virus in murine cancer cell lines and are effective OVs with anti-tumor activity in different mouse genetic backgrounds. Decapping-deficient VACV also potently reduced tumor growth in a human HCC xenograft model, and viral antigen accumulation was preferentially enriched within the HCC tumor. Although host defenses are hyperactivated by D9- or D10-deficient VACV in a normal, non-tumorigenic hepatocyte model and primary human fibroblasts, this was not observed in HCC cells, in which the host anti-viral enzyme PKR was activated equivalently by WT, D9-deficient, or D10-deficient VACV. This establishes decapping-deficient VACV as a new, tk-positive platform for OV therapy. It further suggests that replication of decapping-deficient VACV in normal cells is restricted by cell intrinsic, anti-viral host defenses like PKR, which are often disabled in tumors, thereby enhancing the tumor specificity of VACV OVs.
Selective activation of dsRNA-dependent host defenses in normal, non-tumor tissue likely helps restrict virus replication and spread to cancer cells, a desirable feature for an OV. Replication of D9- and D10-deficient VacV D9 in normal cells and their virulence in animals is impaired because these viruses, although attenuated, potently activate cellular intrinsic anti-viral defenses.36, 37 In the absence of VACV decapping enzymes or the host endoribonuclease Xrn1, which degrades decapped mRNA, dsRNA accumulates in infected cells and activates host antiviral defense molecules, including the interferon-inducible, dsRNA-dependent eIF2α kinase PKR and RNase L.36, 37 Stimulation of PKR and RNase L arrests infected cell protein synthesis and virus replication.10, 11 The restriction of decapping-deficient VACV by host defenses, including PKR and RNase L, coupled with their attenuation, strongly resembled the phenotype of an HSV1 OV approved by US and European regulatory agencies.46, 47 Previously, we and others have established that HSV-1 ICP34.5-deficient derivatives, which are defective in controlling PKR and RNase L, are effective OVs.6, 7, 8 Although HSV1 ICP34.5-deficient viruses and derivative strains are strongly attenuated, they remain capable of destroying cancer cells because tumor cells have deficiencies in many cell intrinsic host defenses. Even though there are no homologs of ICP34.5 in VacV or any other poxvirus, it is remarkable that the PKR-RNase L host defense axis can be harnessed to attenuate VACV by deleting D9 or D10 to create effective OVs. This not only highlights the importance of dsRNA-responsive host defenses in controlling virulence, but hints at a powerful general strategy for OV design that may well have utility across virus families.
Having established decapping-deficient, attenuated VACV mutants as effective OVs with anti-tumor activity in murine and human models, how this platform might be developed and modified going forward can be rationally considered. Our data show that both innate and adaptive anti-tumor immune responses are elicited by VACV OV treatment. Inclusion of additional transgene armaments, such as GM-CSF or perhaps even agents that target the transporter associated with antigen processing (TAP) within tumor cells, could further increase anti-tumor immune responses.20, 26 Similarly, therapeutic synergy with an immune checkpoint blockade can be investigated.48, 49 As decapping-deficient VACV retains a functional tk gene, its more robust replication within cancer cells may engender more effective antitumor responses compared to tk-negative viruses. Should further attenuation be indicated once formal safety and toxicology studies are performed, for example, in treating immune compromised patients, the F4L gene encoding a ribonucleotide reductase subunit or the VACV growth factor gene (VGF), which does not significantly impact virus reproduction in a primate cell line, might also be removed.44, 50 Alternatively, a doubly deficient variant lacking both D9 and D10 might be evaluated for OV therapy, although it might be overly compromised for productive replication.37 Finally, the VACV tk gene could be replaced with the HSV1 tk gene. Prior studies have shown that VACV infection of cells expressing HSV tk is sensitive to acyclovir, an effective antiviral with a substantial safety record.51 In addition, VACV recombinants expressing HSV1 tk have been isolated.52 Significantly, replication of an attenuated VACV lacking one or more decapping enzymes and expressing HSV tk could be controlled by acyclovir. This could contribute greatly to the safety of VACV OVs in the clinic by providing access to a well-tested, widely used antiviral drug should any adverse clinical events occur.
Materials and Methods
Cells and Viruses
The D9- and D10-deficient recombinant VACVs in the western reserve strain genetic background38, 53 were kindly provided by Dr. B. Moss (NIAID). 4T1 and MCA38 cells were kindly provided by Dr. S. Demaria (Weil Cornell). NHDFs were purchased from Lonza (Walkersville, MD). Huh-7 cells, originally obtained from JCRB Genebank (Shinjuku, Japan), HepG2 and Hep3B cells, originally obtained from ATCC (ATCC HB-8064, CRL-2522), and cBAL111 cells were generously provided by Dr. A. Epstein (Lyon, France).
Mouse In Vivo Tumor Models
All animal procedures were performed in accordance with protocols approved by the institutional animal care & use committee at New York University (NYU) School of Medicine. 4T1 cells (1 × 104) in DMEM without additives were injected subcutaneously (s.c.) into the right flank of 8-week-old, female BALB/c mice anesthetized by intraperitoneal (i.p.) injection of ketamine (100 mg/kg) and xylazine (10 mg/kg). Tumor growth was monitored every day using an electronic digital caliper, and tumor volume was calculated as described.54 When tumors reached approximately 50 mm3 (8 to 9 days after 4T1 inoculation), they were directly injected on days 0, 3, and 6 with 5.4 × 106 PFU of D10-deficient VACV (N = 10 mice) or an equivalent volume of virus-free control preparation from uninfected cells (N = 10 mice). Tumor size was monitored over time, and animals were euthanized when control-treated tumors reached approximately 1,200 mm3.
MCA38 murine colon adenocarcinoma cells in media were injected (1 × 105) s.c. into the flank of 4- to 6-week-old, female C57/Bl6 mice. When tumors reached approximately 50 mm3 (approximately 7 days after MCA38 inoculation), they were directly injected on days 0, 3, and 6 with 1.0 × 106 PFU of D10-deficient (ΔD10) VACV (N = 10 mice), 1.0 × 106 PFU of D9-deficient (ΔD9) VACV (N = 10 mice), or an equivalent volume of virus-free control preparation from uninfected cells (N = 10 mice). Tumor size was monitored over time and animals were euthanized when control-treated tumors reached approximately 1,200 mm3.
HepG2 human HCC xenografts were established in the flanks of 8-week-old, female, athymic (nude) mice. HepG2 human HCC cells in media were injected (1 × 107) s.c. and when tumors reached approximately 50 mm3 (approximately 7 days after HepG2 inoculation), they were directly injected on days 0, 3, 6, and 9 with 1.0 × 106 PFU of D10-deficient (ΔD10) VACV (N-10 mice), 1.0 × 106 PFU of D9-deficient (ΔD10) VACV (N = 10 mice), or an equivalent virus-free control preparation from uninfected cells (N = 10 mice).
Histopathology and Immunohistochemistry
Sequentially cut sections (4 μm thick) from formalin-fixed, paraffin-embedded HepG2 xenograft tumors were stained with H&E, examined on an Olympus BX53 light microscope, and photographed. The first, fourth, and seventh sections were evaluated from each representative tumor. For immunohistochemistry, sections (4 μm thick) cut from formalin-fixed, paraffin-embedded HepG2 xenograft tumors were floated onto Plus slides (Fisher Scientific), air dried, and stored at room temperature. Immediately prior to staining, slides were incubated for 1 hr at 60°C in a convection oven. The following antibodies were used for chromogenic immunohistochemistry: rabbit anti-mouse Ly6G/6C, clone NIMP-R14 IgG2b (Abcam catalog # ab2557, Lot # GR135037-1, AB_303154), rat anti-mouse F480, clone BM8 IgG2a (eBioscience catalog # 14-4801, Lot # E016242, RRID: AB_2314387), and polyclonal rabbit anti-mouse Granzyme-B (Abcam catalog # ab4059, Lot # GR276728-4, RRID: AB_304251), polyclonal rabbit anti-Vaccinia virus (VAC, ViroStat catalog # 8101), and unconjugated, polyclonal rabbit anti-Human Nuclear Mitotic Apparatus Protein (NuMA, Abcam catalog # ab97585, Lot# GR268490-12, RRID: AB_10680001).55, 56, 57, 58 Immunohistochemistry was performed on a Ventana Medical Systems Discovery XT platform, with online deparaffinization, using Ventana’s reagents and detection kits unless otherwise noted. Slides stained for Ly6G/Ly6c were deparaffinized online and treated with protease-3 (Ventana Medical Systems) for 8 min. Samples for staining with F480 were heat retrieved using Cell Conditioner 2 (citrate pH 6.0) for 20 min, and samples for staining with Granzyme-B were heat retrieved using Cell Conditioner 1 (Tris-Borate-EDTA, pH 8.5) for 36 min. Endogenous peroxidase activity was blocked for all samples. Ly6G/Ly6c, F480, and Granzyme-B antibodies were diluted in Tris-buffered saline (Thermo Scientific) to 1:800, 1:100, and 1:100, respectively. Ly6G/Ly6c and Granzyme-B antibodies were incubated for 12 hr and F480 was incubated for 6 hr, each at room temperature. Ly6G/Ly6c and F480 antibodies were detected with goat anti-rat horseradish peroxidase-conjugated multimer incubated for 16 min, and anti-Granzyme-B was detected with goat anti-rabbit horseradish-peroxidase-conjugated multimer incubated for 8 min. The complex was visualized with 3,3 diaminobenzidene and enhanced with copper sulfate. Slides were washed in distilled water, counterstained with hematoxylin, dehydrated, and mounted with permanent media.
Anti-NuMa antibody was optimized and validated on tissue microarrays composed of both human and murine tissues. Anti-VACV was optimized on HepG2 HCC xenografts infected with VACV. Sequential chromogenic immunohistochemistry was performed on a Ventana Medical Systems Discovery XT platform as described in the preceding paragraph. Sections were deparaffinized online and endogenous peroxidase activity was blocked. Anti-VACV was diluted 1:1,500 in Tris-BSA (25 mM Tris, 0.15 M NaCL, and 1% BSA), incubated for 1 hr at 37°C, and detected with goat anti-rabbit horseradish-peroxidase-conjugated multimer for 8 min. The complex was visualized with 3,3 diaminobenzidene and enhanced with copper sulfate. Slides were washed in distilled water, and epitope retrieval was performed in a 1,200-W microwave oven at 100% power in 10 mM sodium citrate buffer, pH 6.0, for 10 min. Slides were allowed to cool for 30 min, rinsed in distilled water, and reloaded onto the instrument. Anti-NUMA was diluted 1:5,000 in Tris-BSA, incubated for 12 hr at room temperature, and detected with goat anti-rabbit horseradish-peroxidase-conjugated multimer followed by alpha-naphthol pyronin (8 min each). Slides were counterstained with hematoxylin, dehydrated, and mounted with permanent media. Double-negative controls substituted PBS for primary antibody in the sequential staining. Because both primary antibodies are made in a rabbit, we included an additional control test to eliminate cross-over detection. The test slide consisted of application of all reagents for anti-VACV antibody detection, except for the application of goat anti-rabbit secondary. The test slide was epitope retrieved (as described above) to strip off previous reagents and then re-probed with goat anti-rabbit horseradish-peroxidase-conjugated multimer as described above. Double-negative and cross-over controls did not have demonstrable labeling.
Antibodies
The following antibodies for immunoblotting were purchased from the indicated commercial sources: total PKR (Cell Signaling Technology # 12297) and phospho(T446)-PKR (Abcam # 32036).
Author Contributions
I.M. conceived the project. H.M.B., A.P., A.B.F., and I.M. designed the experiments. H.M.B., A.P., C.H.H., and L.C. performed the experiments. H.M.B., A.B.F., C.H.H., L.C., A.P., and I.M. wrote and edited the manuscript.
Conflicts of Interest
The authors declare no conflict of interest.
Acknowledgments
The authors thank B. Moss for generously providing the D9- and D10-deficient recombinant viruses. This work was supported by NIH grants AI126102, AI073898, and GM056927 (to I.M.) and awards from the NYU Office of Therapeutic Alliance (to I.M.) and from the Perlmutter Cancer Center at NYU Langone Medical Center (to I.M. and A.B.F.). The NYU Experimental Pathology Immunohistochemistry Core Laboratory is supported in part by the Laura and Isaac Perlmutter Cancer Center Support Grant (NIH/NCI P30CA016087) and NIH S10 grants (NIH/ORIP S10OD01058 and S10OD018338).
References
- 1.Russell S.J., Peng K.W. Oncolytic virotherapy: a contest between apples and oranges. Mol. Ther. 2017;25:1107–1116. doi: 10.1016/j.ymthe.2017.03.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Saha D., Wakimoto H., Rabkin S.D. Oncolytic herpes simplex virus interactions with the host immune system. Curr. Opin. Virol. 2016;21:26–34. doi: 10.1016/j.coviro.2016.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lichty B.D., Breitbach C.J., Stojdl D.F., Bell J.C. Going viral with cancer immunotherapy. Nat. Rev. Cancer. 2014;14:559–567. doi: 10.1038/nrc3770. [DOI] [PubMed] [Google Scholar]
- 4.Mohr I., Gluzman Y. A herpesvirus genetic element which affects translation in the absence of the viral GADD34 function. EMBO J. 1996;15:4759–4766. [PMC free article] [PubMed] [Google Scholar]
- 5.Mohr I., Sternberg D., Ward S., Leib D., Mulvey M., Gluzman Y. A herpes simplex virus type 1 gamma34.5 second-site suppressor mutant that exhibits enhanced growth in cultured glioblastoma cells is severely attenuated in animals. J. Virol. 2001;75:5189–5196. doi: 10.1128/JVI.75.11.5189-5196.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Taneja S., MacGregor J., Markus S., Ha S., Mohr I. Enhanced antitumor efficacy of a herpes simplex virus mutant isolated by genetic selection in cancer cells. Proc. Natl. Acad. Sci. USA. 2001;98:8804–8808. doi: 10.1073/pnas.161011798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Todo T., Martuza R.L., Rabkin S.D., Johnson P.A. Oncolytic herpes simplex virus vector with enhanced MHC class I presentation and tumor cell killing. Proc. Natl. Acad. Sci. USA. 2001;98:6396–6401. doi: 10.1073/pnas.101136398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Liu B.L., Robinson M., Han Z.Q., Branston R.H., English C., Reay P., McGrath Y., Thomas S.K., Thornton M., Bullock P. ICP34.5 deleted herpes simplex virus with enhanced oncolytic, immune stimulating, and anti-tumour properties. Gene Ther. 2003;10:292–303. doi: 10.1038/sj.gt.3301885. [DOI] [PubMed] [Google Scholar]
- 9.Chou J., Kern E.R., Whitley R.J., Roizman B. Mapping of herpes simplex virus-1 neurovirulence to gamma 134.5, a gene nonessential for growth in culture. Science. 1990;250:1262–1266. doi: 10.1126/science.2173860. [DOI] [PubMed] [Google Scholar]
- 10.Sadler A.J., Williams B.R. Interferon-inducible antiviral effectors. Nat. Rev. Immunol. 2008;8:559–568. doi: 10.1038/nri2314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mohr I., Sonenberg N. Host translation at the nexus of infection and immunity. Cell Host Microbe. 2012;12:470–483. doi: 10.1016/j.chom.2012.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mulvey M., Poppers J., Ladd A., Mohr I. A herpesvirus ribosome-associated, RNA-binding protein confers a growth advantage upon mutants deficient in a GADD34-related function. J. Virol. 1999;73:3375–3385. doi: 10.1128/jvi.73.4.3375-3385.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sànchez R., Mohr I. Inhibition of cellular 2′-5′ oligoadenylate synthetase by the herpes simplex virus type 1 Us11 protein. J. Virol. 2007;81:3455–3464. doi: 10.1128/JVI.02520-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mulvey M., Camarena V., Mohr I. Full resistance of herpes simplex virus type 1-infected primary human cells to alpha interferon requires both the Us11 and gamma(1)34.5 gene products. J. Virol. 2004;78:10193–10196. doi: 10.1128/JVI.78.18.10193-10196.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kew C., Lui P.Y., Chan C.P., Liu X., Au S.W., Mohr I., Jin D.Y., Kok K.H. Suppression of PACT-induced type I interferon production by herpes simplex virus 1 Us11 protein. J. Virol. 2013;87:13141–13149. doi: 10.1128/JVI.02564-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Herdy B., Jaramillo M., Svitkin Y.V., Rosenfeld A.B., Kobayashi M., Walsh D., Alain T., Sean P., Robichaud N., Topisirovic I. Translational control of the activation of transcription factor NF-κB and production of type I interferon by phosphorylation of the translation factor eIF4E. Nat. Immunol. 2012;13:543–550. doi: 10.1038/ni.2291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mulvey M., Poppers J., Sternberg D., Mohr I. Regulation of eIF2alpha phosphorylation by different functions that act during discrete phases in the herpes simplex virus type 1 life cycle. J. Virol. 2003;77:10917–10928. doi: 10.1128/JVI.77.20.10917-10928.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Critchley-Thorne R.J., Simons D.L., Yan N., Miyahira A.K., Dirbas F.M., Johnson D.L., Swetter S.M., Carlson R.W., Fisher G.A., Koong A. Impaired interferon signaling is a common immune defect in human cancer. Proc. Natl. Acad. Sci. USA. 2009;106:9010–9015. doi: 10.1073/pnas.0901329106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mohr I. To replicate or not to replicate: achieving selective oncolytic virus replication in cancer cells through translational control. Oncogene. 2005;24:7697–7709. doi: 10.1038/sj.onc.1209053. [DOI] [PubMed] [Google Scholar]
- 20.Pourchet A., Fuhrmann S.R., Pilones K.A., Demaria S., Frey A.B., Mulvey M., Mohr I. CD8(+) T-cell immune evasion enables oncolytic virus immunotherapy. EBioMedicine. 2016;5:59–67. doi: 10.1016/j.ebiom.2016.01.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jan E., Mohr I., Walsh D. A cap-to-tail guide to mRNA translation strategies in virus-infected cells. Annu. Rev. Virol. 2016;3:283–307. doi: 10.1146/annurev-virology-100114-055014. [DOI] [PubMed] [Google Scholar]
- 22.Langland J.O., Jacobs B.L. The role of the PKR-inhibitory genes, E3L and K3L, in determining vaccinia virus host range. Virology. 2002;299:133–141. doi: 10.1006/viro.2002.1479. [DOI] [PubMed] [Google Scholar]
- 23.Rice A.D., Turner P.C., Embury J.E., Moldawer L.L., Baker H.V., Moyer R.W. Roles of vaccinia virus genes E3L and K3L and host genes PKR and RNase L during intratracheal infection of C57BL/6 mice. J. Virol. 2011;85:550–567. doi: 10.1128/JVI.00254-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Brandt T.A., Jacobs B.L. Both carboxy- and amino-terminal domains of the vaccinia virus interferon resistance gene, E3L, are required for pathogenesis in a mouse model. J. Virol. 2001;75:850–856. doi: 10.1128/JVI.75.2.850-856.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.McCart J.A., Ward J.M., Lee J., Hu Y., Alexander H.R., Libutti S.K., Moss B., Bartlett D.L. Systemic cancer therapy with a tumor-selective vaccinia virus mutant lacking thymidine kinase and vaccinia growth factor genes. Cancer Res. 2001;61:8751–8757. [PubMed] [Google Scholar]
- 26.Kim J.H., Oh J.Y., Park B.H., Lee D.E., Kim J.S., Park H.E., Roh M.S., Je J.E., Yoon J.H., Thorne S.H. Systemic armed oncolytic and immunologic therapy for cancer with JX-594, a targeted poxvirus expressing GM-CSF. Mol. Ther. 2006;14:361–370. doi: 10.1016/j.ymthe.2006.05.008. [DOI] [PubMed] [Google Scholar]
- 27.Thorne S.H., Hwang T.H., O’Gorman W.E., Bartlett D.L., Sei S., Kanji F., Brown C., Werier J., Cho J.H., Lee D.E. Rational strain selection and engineering creates a broad-spectrum, systemically effective oncolytic poxvirus, JX-963. J. Clin. Invest. 2007;117:3350–3358. doi: 10.1172/JCI32727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Breitbach C.J., Parato K., Burke J., Hwang T.H., Bell J.C., Kirn D.H. Pexa-Vec double agent engineered vaccinia: oncolytic and active immunotherapeutic. Curr. Opin. Virol. 2015;13:49–54. doi: 10.1016/j.coviro.2015.03.016. [DOI] [PubMed] [Google Scholar]
- 29.Downs-Canner S., Guo Z.S., Ravindranathan R., Breitbach C.J., O’Malley M.E., Jones H.L., Moon A., McCart J.A., Shuai Y., Zeh H.J. Phase 1 study of intravenous oncolytic poxvirus (vvDD) in patients with advanced solid cancers. Mol. Ther. 2016;24:1492–1501. doi: 10.1038/mt.2016.101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Breitbach C.J., Burke J., Jonker D., Stephenson J., Haas A.R., Chow L.Q., Nieva J., Hwang T.H., Moon A., Patt R. Intravenous delivery of a multi-mechanistic cancer-targeted oncolytic poxvirus in humans. Nature. 2011;477:99–102. doi: 10.1038/nature10358. [DOI] [PubMed] [Google Scholar]
- 31.Heo J., Reid T., Ruo L., Breitbach C.J., Rose S., Bloomston M., Cho M., Lim H.Y., Chung H.C., Kim C.W. Randomized dose-finding clinical trial of oncolytic immunotherapeutic vaccinia JX-594 in liver cancer. Nat. Med. 2013;19:329–336. doi: 10.1038/nm.3089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Park S.H., Breitbach C.J., Lee J., Park J.O., Lim H.Y., Kang W.K., Moon A., Mun J.H., Sommermann E.M., Maruri Avidal L. Phase 1b trial of biweekly intravenous Pexa-Vec (JX-594), an oncolytic and immunotherapeutic vaccinia virus in colorectal cancer. Mol. Ther. 2015;23:1532–1540. doi: 10.1038/mt.2015.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zeh H.J., Downs-Canner S., McCart J.A., Guo Z.S., Rao U.N., Ramalingam L., Thorne S.H., Jones H.L., Kalinski P., Wieckowski E. First-in-man study of western reserve strain oncolytic vaccinia virus: safety, systemic spread, and antitumor activity. Mol. Ther. 2015;23:202–214. doi: 10.1038/mt.2014.194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Carroll, J. (2013). Korean CRO grabs failed cancer vaccine in $150M-max Jennerex buyout. https://www.fiercebiotech.com/financials/korean-cro-grabs-failed-cancer-vaccine-150m-max-jennerex-buyout.
- 35.ClinicalTrials.gov (2016). A study of recombinant vaccinia virus prior to sorafenib to treat unresectable primary hepatocellular carcinoma. https://clinicaltrials.gov/ct2/show/study/NCT01171651.
- 36.Burgess H.M., Mohr I. Cellular 5′-3′ mRNA exonuclease Xrn1 controls double-stranded RNA accumulation and anti-viral responses. Cell Host Microbe. 2015;17:332–344. doi: 10.1016/j.chom.2015.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Liu S.W., Katsafanas G.C., Liu R., Wyatt L.S., Moss B. Poxvirus decapping enzymes enhance virulence by preventing the accumulation of dsRNA and the induction of innate antiviral responses. Cell Host Microbe. 2015;17:320–331. doi: 10.1016/j.chom.2015.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Parrish S., Moss B. Characterization of a second vaccinia virus mRNA-decapping enzyme conserved in poxviruses. J. Virol. 2007;81:12973–12978. doi: 10.1128/JVI.01668-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Parrish S., Resch W., Moss B. Vaccinia virus D10 protein has mRNA decapping activity, providing a mechanism for control of host and viral gene expression. Proc. Natl. Acad. Sci. USA. 2007;104:2139–2144. doi: 10.1073/pnas.0611685104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Liu S.W., Wyatt L.S., Orandle M.S., Minai M., Moss B. The D10 decapping enzyme of vaccinia virus contributes to decay of cellular and viral mRNAs and to virulence in mice. J. Virol. 2014;88:202–211. doi: 10.1128/JVI.02426-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Buller R.M., Smith G.L., Cremer K., Notkins A.L., Moss B. Decreased virulence of recombinant vaccinia virus expression vectors is associated with a thymidine kinase-negative phenotype. Nature. 1985;317:813–815. doi: 10.1038/317813a0. [DOI] [PubMed] [Google Scholar]
- 42.McCart, J.A., Bartlett, D.L., and Moss, B. (2013). Vaccinia virus expression vector for selective replication in a tumor cell and introduction of exogenous nucleotide sequence into a tumor cell. U.S. patent 8506947.
- 43.Deurholt T., van Til N.P., Chhatta A.A., ten Bloemendaal L., Schwartlander R., Payne C., Plevris J.N., Sauer I.M., Chamuleau R.A., Elferink R.P. Novel immortalized human fetal liver cell line, cBAL111, has the potential to differentiate into functional hepatocytes. BMC Biotechnol. 2009;9:89. doi: 10.1186/1472-6750-9-89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Gammon D.B., Gowrishankar B., Duraffour S., Andrei G., Upton C., Evans D.H. Vaccinia virus-encoded ribonucleotide reductase subunits are differentially required for replication and pathogenesis. PLoS Pathog. 2010;6:e1000984. doi: 10.1371/journal.ppat.1000984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Potts K.G., Irwin C.R., Favis N.A., Pink D.B., Vincent K.M., Lewis J.D., Moore R.B., Hitt M.M., Evans D.H. Deletion of F4L (ribonucleotide reductase) in vaccinia virus produces a selective oncolytic virus and promotes anti-tumor immunity with superior safety in bladder cancer models. EMBO Mol. Med. 2017;9:638–654. doi: 10.15252/emmm.201607296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.U.S. Food and Drug Administration (2015). IMLYGIC (talimogene laherparepvec). https://www.fda.gov/BiologicsBloodVaccines/CellularGeneTherapyProducts/ApprovedProducts/ucm469411.htm.
- 47.European Medicines Agency (2017). Imlygic. http://www.ema.europa.eu/ema/index.jsp?curl=pages/medicines/human/medicines/002771/human_med_001941.jsp&mid=WC0b01ac058001d124.
- 48.Liu Z., Ravindranathan R., Kalinski P., Guo Z.S., Bartlett D.L. Rational combination of oncolytic vaccinia virus and PD-L1 blockade works synergistically to enhance therapeutic efficacy. Nat. Commun. 2017;8:14754. doi: 10.1038/ncomms14754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Rojas J.J., Sampath P., Hou W., Thorne S.H. Defining effective combinations of immune checkpoint blockade and oncolytic virotherapy. Clin. Cancer Res. 2015;21:5543–5551. doi: 10.1158/1078-0432.CCR-14-2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Buller R.M., Chakrabarti S., Cooper J.A., Twardzik D.R., Moss B. Deletion of the vaccinia virus growth factor gene reduces virus virulence. J. Virol. 1988;62:866–874. doi: 10.1128/jvi.62.3.866-874.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Darby G., Larder B.A., Bastow K.F., Field H.J. Sensitivity of viruses to phosphorylated 9-(2-hydroxyethoxymethyl)guanine revealed in TK-transformed cells. J. Gen. Virol. 1980;48:451–454. doi: 10.1099/0022-1317-48-2-451. [DOI] [PubMed] [Google Scholar]
- 52.Panicali D., Paoletti E. Construction of poxviruses as cloning vectors: insertion of the thymidine kinase gene from herpes simplex virus into the DNA of infectious vaccinia virus. Proc. Natl. Acad. Sci. USA. 1982;79:4927–4931. doi: 10.1073/pnas.79.16.4927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Parrish S., Moss B. Characterization of a vaccinia virus mutant with a deletion of the D10R gene encoding a putative negative regulator of gene expression. J. Virol. 2006;80:553–561. doi: 10.1128/JVI.80.2.553-561.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Demaria S., Kawashima N., Yang A.M., Devitt M.L., Babb J.S., Allison J.P., Formenti S.C. Immune-mediated inhibition of metastases after treatment with local radiation and CTLA-4 blockade in a mouse model of breast cancer. Clin. Cancer Res. 2005;11:728–734. [PubMed] [Google Scholar]
- 55.Hiebert P.R., Boivin W.A., Zhao H., McManus B.M., Granville D.J. Perforin and granzyme B have separate and distinct roles during atherosclerotic plaque development in apolipoprotein E knockout mice. PLoS One. 2013;8:e78939. doi: 10.1371/journal.pone.0078939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Mackler A.M., Green L.M., McMillan P.J., Yellon S.M. Distribution and activation of uterine mononuclear phagocytes in peripartum endometrium and myometrium of the mouse. Biol. Reprod. 2000;62:1193–1200. doi: 10.1095/biolreprod62.5.1193. [DOI] [PubMed] [Google Scholar]
- 57.Perez-Aso M., Montesinos M.C., Mediero A., Wilder T., Schafer P.H., Cronstein B. Apremilast, a novel phosphodiesterase 4 (PDE4) inhibitor, regulates inflammation through multiple cAMP downstream effectors. Arthritis Res. Ther. 2015;17:249. doi: 10.1186/s13075-015-0771-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Puentes F., van der Star B.J., Victor M., Kipp M., Beyer C., Peferoen-Baert R., Ummenthum K., Pryce G., Gerritsen W., Huizinga R. Characterization of immune response to neurofilament light in experimental autoimmune encephalomyelitis. J. Neuroinflammation. 2013;10:118. doi: 10.1186/1742-2094-10-118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Walsh D., Arias C., Perez C., Halladin D., Escandon M., Ueda T., Watanabe-Fukunaga R., Fukunaga R., Mohr I. Eukaryotic translation initiation factor 4F architectural alterations accompany translation initiation factor redistribution in poxvirus-infected cells. Mol. Cell. Biol. 2008;28:2648–2658. doi: 10.1128/MCB.01631-07. [DOI] [PMC free article] [PubMed] [Google Scholar]