

INTRODUCTION
Despite Dr. Naughton's hypotheses, we remain convinced that central sleep apnea with Hunter-Cheyne-Stokes breathing (CSA-HCSB) is “a bad bedfellow” for patients suffering from heart failure with reduced ejection fraction (HFrEF).1 His fundamental argument that CSA-HCSB may be a potentially beneficial compensatory mechanism is not supported by experimental or clinical evidence and stands in contradiction to findings from decades of empirical research, some of which was provided by Dr. Naughton himself. Dr Naughton has postulated2 a variety of mechanisms to support his opinion. These proposed mechanisms can be summarized as:
Hyperventilation-related increases in end-expiratory lung volume (EELV) and positive pressure augment cardiac stroke volume;
Attenuation of excessive sympathetic nervous activity;
Maintaining a state of respiratory alkalosis is cardioprotective; and
Provision of periodic rest to fatigue-prone respiratory pump muscles.
The negative results of a recent trial brought Naughton's point of view to the forefront. His writings were used by the authors as a possible explanation for the negative findings of their trial.3 However, the trial was widely criticized for numerous deficiencies that are detailed elsewhere.4 To date, the investigators have not provided a credible response to this and other critiques.
Below, we address each of Naughton's proposed mechanisms separately.
PROPOSED MECHANISMS
Hyperventilation-Related Increases in EELV and Positive Pressure Augment Stroke Volume in Patients With HFrEF
Naughton refers to the study of Brack et al.5 in which the investigators measured lung and tidal volumes by direct current-coupled respiratory inductance plethysmography in 12 patients with HFrEF. They observed that following central apneas, during the crescendo phase of hyperventilation, tidal volume progressively increased and averaged 867 mL at the peak compared to 567 mL during baseline stable breathing. Because of inadequate time to fully exhale this increased magnitude of tidal volume, EELV increased (214 mL). It follows that, as a result of the rise in EELV, the elastic recoil of the lung increases, and during the next exhalation, positive intrathoracic pressure is generated.
The development of positive pressure and the increase in lung volume have at least three known effects, depending on whether right or left ventricular function is being considered. Venous return to the right ventricle will decrease, possibly resulting in lower cardiac output, whereas a fall in transmural pressure across the left ventricular myocardium would reduce afterload and tend to augment stroke volume. The latter is useful, but the former effect could be disadvantageous depending on preexisting right ventricular function. A third adverse cardiac consequence of the rise in EELV relates to increased pulmonary vascular resistance, which is dependent on lung volume. This increases right ventricular afterload, adversely affecting right ventricular function, and raises oxygen consumption (see Javaheri et al.4). Therefore, the direction in which the rise in EELV in each patient with HFrEF affects cardiac hemodynamics is unclear. Further, in order to achieve an increase in lung volume and intrinsic positive pressure during the hyperventilatory phase of HCSB, equally large negative swings in pleural (Figure 1) and alveolar pressure are required to increase inspired volume, a difficult task in the face of decreased respiratory system compliance because of pulmonary vascular congestion and edema and an edematous chest wall. These negative swings in intrathoracic pressure will necessarily oppose the hemodynamics of the positive swings, and also increase the work of breathing. Even if we accept the argument that an increase in EELV and positive pressure have beneficial effect in every single patient, then devices that provide extrinsic positive airway pressure (ie, continuous or bilevel positive airway pressure/adaptive servoventilation) should be even more beneficial. Positive airway pressure counters the negative pressure swings of hyperventilation, and by all accounts should improve cardiac hemodynamics rather than act as a detriment.
Figure 1. Work of breathing with central apnea.

A 30-second epoch of central sleep apnea (CSA). From top to bottom: 2 channels of electro-oculogram (EOG), chin electromyogram (CHIN EMG), 2 channels of electroencephalogram (EEG), electrocardiogram (EKG), airflow (CO2), rib cage (RC) and abdominal (ABD) sum as well as in separate channels, esophageal pressure (PES), and oxyhemoglobin saturation (SaO2) measured by pulse oximetry. Note absence of airflow, and excursions in pleural pressure, thorax, and abdomen with CSA when work of breathing should be zero. However, following CSA there is hyperventilation with relatively large negative swings in esophageal pressure with hyperventilation which should increase WOB. Reprinted with permission of the American Thoracic Society. Copyright © 2018 American Thoracic Society. Modified from Dowdell WT, Javaheri S, McGinnis W. Cheyne-Stokes respiration presenting as sleep apnea syndrome: clinical and polysomnographic features. Am Rev Respir Dis. 1990;141:871–879. The American Review of Respiratory Disease is an official journal of the American Thoracic Society.
Attenuation of Excessive Sympathetic Nerve Activity by Lung Inflation During Post-Apnea Hyperventilation
Excessive sympathetic nerve activity (SNA) is increased in HFrEF (Figure 2) and known to represent a maladaptive mechanism in heart failure with adverse consequences.
Figure 2. Heart failure with reduced ejection fraction is a hyperadrenergic state.

There are multiple mechanisms contributing to increased sympathetic activity, and presence of CSA further contributes. Locus coeruleus is the brainstem arousal network, and norepinephrine is the neurotransmitter. Modified and reprinted from Journal of the American College of Cardiology, Vol. 69, 2017, Javaheri S, Barbe F, Campos-Rodriguez F, et al. Sleep apnea types, mechanisms, and clinical cardiovascular consequences, pages 841–858, © 2018, with permission from Elsevier.
In patients with HF, breathing spontaneously, Naughton and colleagues6 showed an inverse correlation between muscle sympathetic nerve activity (MSNA) and tidal volume. The mechanism underlying this effect is attributed to the stimulation of pulmonary vagal afferents by the lung inflation required to achieve the increase in tidal volume, which inhibit central sympathetic outflow to peripheral vascular beds. Although this is a well-known physiologic effect of lung inflation, the opposite occurs in the absence of lung inflation (ie, central apneas) and this along with hypoxia and arousals should increase sympathetic activity (Figure 2) as is the case in relation to obstructive apneas. Indeed, in a group of patients with HFrEF with either predominantly obstructive or central sleep apnea and matched for left ventricle ejection fraction, apnea-hypopnea index (AHI), and desaturation, Spaak and colleagues showed similar MSNA augmentation.7 Therefore, these increases and decreases in MSNA from the aforementioned opposing phenomena are not equal; the overall balance favors an increase in SNA (Figure 2). Given the existence of this effect, there has developed overwhelming evidence that in patients with HFrEF, CSA-HCSB induces a hyperadrenergic state that is reversed when sleep-disordered breathing is suppressed, regardless of the therapeutic modality. First, peroneal microneurography, a measurement known to reflect central nervous system sympathetic outflow, has shown a close association between HCSB and increased sympathetic activity.8 Second, studies comparing overnight urinary norepinephrine and morning catecholamines in patients with HFrEF with and without CSA-HCSB have shown significant differences, with higher levels in patients with CSA-HCSB. For instance, Naughton et al.9 matched two groups of patients with HFrEF: one group that demonstrated CSA-HCSB and one group with normal breathing during sleep. Overnight urinary and morning plasma catecholamine values were significantly higher in those with CSAHCSB compared to those without sleep-disordered breathing. Moreover, they went on to randomize the patients with CSAHCSB to continuous positive airway pressure (CPAP) versus control for 1 month. Urinary and morning plasma catecholamine values significantly decreased with CPAP in the treated group, almost to a level similar to those without CSA.
We should note, however, that a study from Naughton's laboratory using right heart catheterization in 55 patients with HF reported total body and cardiac norepinephrine (NE) spillover via radioisotope dilution.10 Patients were separated into groups depending on whether nocturnal polysomnography (PSG) revealed normal breathing during sleep, obstructive sleep apnea (OSA), or CSA-HCSB. Interestingly, the highest levels of NE spillover were found in the patients with CSA-HCSB; their NE levels were even higher than the levels in patients with OSA. However, stepwise regression demonstrated that only mean pulmonary artery pressure independently correlated with total body and cardiac NE spillover. Naughton concluded that CSA severity bore no relationship to markers of SNA. The issue here is that measurements were performed during daytime, rather than during sleep. In contrast, the studies that measured overnight urinary norepinephrine consistently found increased SNA, during the time period when sleep-disordered breathing is present.
The most important question to be answered is whether therapy would attenuate these high levels of increased overnight SNA. In addition to the aforementioned CPAP trial by Naughton et al.,8 there are three randomized controlled trials (RCTs) using different treatment paradigms that have demonstrated decreases in overall SNA when CSA-HCSB is treated. The Canadian Continuous Positive Airway Pressure for Patients with Central Sleep Apnea and Heart Failure (CANPAP) trial randomized patients with severe CSA-HCSB (mean AHI of 40 ± 16 events/h) to a CPAP plus guideline-based treatment arm and a guideline-based only treatment arm.11 After 3 months, there was a significant reduction in plasma NE in the treated patients compared with the controls. These values approached the values of patients with HFrEF without CSA-HCSB. The remaining two studies were both randomized double-blind placebo-controlled trials of either adaptive servoventilation (ASV) or supplemental nasal oxygen, with respective shams. In the ASV trial, Pepperell and associates treated two groups of patients with HF and CSA-HCSB with either therapeutic ASV or sham ASV. After 1 month of randomization, catecholamines decreased in the active treatment arm compared to the control arm.12 In a double-arm, crossover placebo controlled trial of 1-month duration, Staniforth and colleagues randomized patients with HFrEF at baseline to nocturnal supplemental oxygen from a concentrator versus a similar amount of airflow from a concentrator set to provide only room air. The patients underwent PSG under three conditions, one at baseline, one on oxygen, and one on sham oxygen. Oxygen therapy significantly reduced the CSA index (18 versus 4 events/h; P = .05) and reduced urinary norepinephrine excretion.13
In summary, in patients with HFrEF, CSA-HCSB is associated with both increased MSNA burst activity and increased overnight urinary and morning plasma catecholamines, and importantly, three RCTs with CPAP, ASV, and oxygen therapy have shown that attenuation of CSA-HCSB significantly decreases indices of sympathetic activity. Collectively, these data demonstrate convincingly that, in patients with HFrEF, CSA-HCSB contributes to an already hyperadrenergic state (Figure 2) and that attenuation of central sleep-disordered breathing reduces this sympathetic overactivity. The evidence therefore overwhelmingly supports a conclusion that treatment of CSA-HCSB reduces sympathetic tone. This is extremely important, in view of the well-known association of increased sympathetic activity with poor prognosis in patients with HFrEF, and the therapeutic triumph of beta-blocker therapy that has unequivocally led to better outcomes.
Maintaining a State of Respiratory Alkalosis
The implications of this hypothesis are twofold: first, that treatment of CSA-HCSB results in hypercapnic acidosis (or at least abolishes the respiratory alkalosis associated with CSA-HCSB in HF) and second, that hypocapnic alkalosis is cardioprotective. There is evidence to refute the first assertion and little to support the second. In a study by Teschler et al.,14 patients with HFrEF underwent PSG under five conditions: no treatment; oxygen via nasal cannula; CPAP; bilevel positive airway pressure with backup respiratory rate, and ASV. Treatments were performed in random order.14 Transcutaneous PCO2 (PtcCO2) was measured every 5 minutes across every night. Mean ± standard error PtcCO2 values (mmHg) were 32.1 ± 0.7, 37.2 ± 0.5, 34.6 ± 1.0, 34.5 ± 0.8, and 34.2 ± 0.7, respectively. Consequently, when comparing each treatment modality to the night without treatment, none of the modalities induced respiratory acidosis and all except oxygen maintained some degree of respiratory alkalosis. Teschler and colleagues also measured evening and morning PaCO2. The respective morning values were 30.8 ± 0.8, 39.8 ± 0.9, 34.0 ± 1.5, 33.6 ± 1.2, and 35.2 ± 0.8 mmHg consistent with the overnight data. We can conclude that morning PaCO2 values also tend toward maintaining a mild degree of respiratory alkalosis (other than with oxygen treatment) and do not induce hypercapnic acidosis. In another study, Naughton and colleagues monitored PtcCO2 during stage N2 sleep in 12 consecutive patients with HFrEF and CSA-HCSB, before and after one month of treatment with CPAP, as well as in 6 untreated controls. In the treated patients, AHI decreased while mean PtcCO2 increased from 34.6 ± 1.4 to 40.8 ± 1.1 mmHg, P < .001 with CPAP compared to controls.15 These findings do support the shift of nocturnal hypocapnia associated with CSA-HCSB to more normal values when CPAP is applied, but do not demonstrate the development of hypercapnic acidosis. As will be noted in the following paragraphs, with treatment, the hyperventilatory phase of HCSB is attenuated tending to normalize ventilation and PCO2 and at the same time diminishing the work of breathing.
With respect to the second aspect of Naughton's theory, he posits that the nocturnal hypocapnia associated with CSAHCSB in HF may be cardioprotective.2 He cites an in vitro study by Bing et al.16 of rat isolated left ventricular papillary muscles under various pH and hypoxic conditions. In these experiments, the solution containing the papillary muscles was adjusted to a pH of 6.8 or 7.8, which resulted in decreases of muscle tension to 93.7% and 90% of control values, respectively. These differences are small and such extreme pH values are not even reached during HCSB. The same experiments were repeated under anoxia (95% N2/5% CO2) with a duration of 1 hour. Compared to pH 7.8, muscle tension declined more rapidly at pH 6.8, with no significant difference as anoxia continued. Interestingly, after 15 minutes of reoxygenation at pH 7.4, preparations previously hypoxic at pH 7.8 redeveloped approximately 50% of prehypoxia tension, whereas those previously hypoxic at pH 6.8 developed tension that returned to almost 100% of prehypoxia values. The title of this paper could not be more directly in opposition to Naughton's hypothesis: “Heart muscle viability following hypoxia: protective effect of acidosis”! As Bing et al. point out, their findings relate to a more “static” cardiac ischemia/myocardial infarction scenario, and in our opinion, are not even relevant to the dynamics of HCSB, where the perturbations in blood gas chemistry (PO2 and PCO2) are rapid and repetitive but of much lower amplitude. Naughton also refers to the fact that a small proportion of patients presenting with acute pulmonary edema exhibit respiratory acidosis as evidence that maintaining patients with HF in an alkalotic state might be beneficial. We could find virtually no data in human studies of HF to support this speculation.
Although Dr. Naughton writes about debatable beneficial effects of hypocapnia on the myocardium, he does not mention that hypocapnia is a profound vasoconstrictor of both cerebral and coronary vessels. It has been shown that hyperventilation can be a specific test for diagnosing coronary artery spasm.17 In the context of HCSB, concomitant hypocapnia and hypoxemia could decrease myocardial oxygen delivery, adversely affecting the failing heart. Furthermore, hypocapnia and alkalemia are known to be arrhythmogenic and arrhythmias are common in patients with HFrEF, especially in those with CSA-HCSB.18–20 In two studies of patients with HFrEF and CSA-HCSB, overnight Holter monitoring revealed that low arterial PCO220 and alkalemia19 were associated with nocturnal ventricular tachycardia. These data strongly support the possibility that alkalemia is involved in the demise of patients from what is aptly described as sudden death in the cardiology literature.
Provision of Periodic Rest to Fatigue-Prone Respiratory Pump Muscles
In contrast to the increase that occurs during an obstructive apnea (Figure 3), during a central apnea, esophageal pressure is maintained at a resting level, and the diaphragm is indeed not performing any work (Figure 1). However, CSA-related hypoxia and the rise in PaCO2 profoundly stimulate ventila-tory drive, and the resumption of breathing entails significant hyperventilation (Figure 1). Achieving this degree of hyper-ventilation in a patient with the reduced respiratory system compliance described earlier requires relatively large negative swings in intrathoracic pressure (Figure 1), and this must be generated by intense diaphragmatic and accessory respiratory muscle contractions. In this regard, Kee et al. (with Naughton)21 measured work of breathing (WOB) in a group of 25 patients with HFrEF during normal breathing, while the patient exhibited obstructive apneas, and while the patient exhibited central apneas. At baseline, WOB averaged 10.2 ± 0.8 J/min, increased to 12.6 ± 0.9 J/min during episodes of obstructive apneas, and averaged 12.0 ± 0.8 J/min during episodes of central apneas (Figure 4). From these data, it would appear that both phenotypes of sleep apnea are associated with increased WOB. Therefore, although Dr. Naughton is correct that WOB is reduced with CSA, the predominant effect of hyperventilation results in an overall increase in WOB, not a decrease. In fact, Naughton and colleagues further confirmed this impression in a study in which they measured nocturnal minute ventilation in a group of patients with HFrEF during episodes of periodic breathing without treatment and while breathing were stabilized on CPAP.22 During periodic breathing measured minute ventilation was 9.8 L/min and decreased to 4.8 L/min when CPAP induced stable breathing. Overall, these studies indicate that in chronic heart failure, HCSB increases the WOB and WOB improves with therapy. As part of the same study,21 Naughton and colleagues showed that the pressure time product is reduced in CSA, suggesting greater efficiency and reduced fatigability. Yet, the same group performed another study that suggested that treatment of CSA-HCSB actually improves inspiratory muscle strength.22 In this randomized trial, patients with HF and CSA-HCSB were treated with CPAP for a period of 3 months, achieving a decrease in average AHI from 49 ± 7 to 17 ± 7 events/h (P < .001). In the treated group, maximum inspiratory pressure increased from a mean of 79 ± 8 to 91 ± 10 cmH2O (P < .02). At the same time, symptoms of dyspnea and fatigue were alleviated. No signifi-cant changes were observed in the control group. The authors concluded that nightly application of CPAP in patients with CHF and CSA-HCSB improves inspiratory muscle strength and relieves dyspnea and fatigue.
Figure 3. Work of breathing with obstructive apnea.

An epoch of polysomnogram showing obstructive sleep apnea (OSA). Channels as in Figure 1 except absence of electro-oculogram. During OSA, there is progressive decrease in esophageal pressure against a closed upper airway, increasing work of breathing, which should diminish as the upper airway opens and pressure swings diminish.
Figure 4. Work of breathing with periodic breathing.
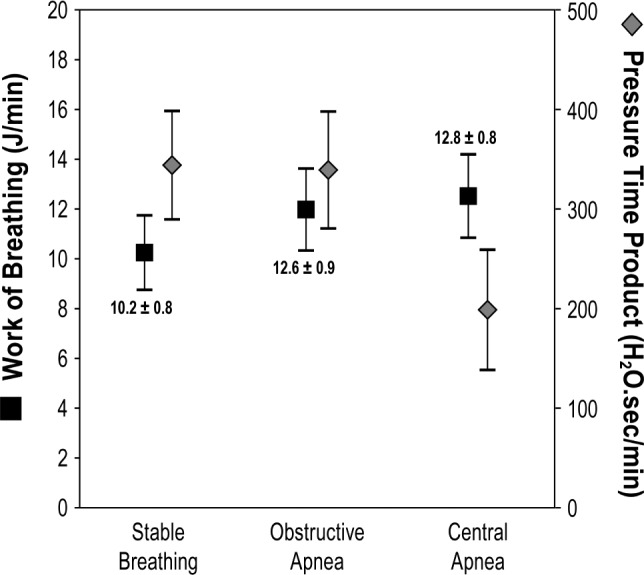
Work of breathing (WOB, Y axis on theft) measured in 25 patients with HFrEF while they exhibited normal breathing or periods of obstructive or central sleep apnea, showing that WOB is equally increased in sleep apnea compared to normal breathing. However, pressure time product was lower with central sleep apnea (Y axis on the right). Reprinted with permission of the American Thoracic Society. Copyright © 2018 American Thoracic Society. Modified from Kee K, Sands SA, Stuart-Andrews, et al. Effect of apnea type on work of breathing and respiratory fatigability during sleep in humans with heart failure [abstract]. Am J Respir Crit Care Med. 2014;189:A3892. The American Journal of Respiratory and Critical Care Medicine is an official journal of the American Thoracic Society.
SUMMARY AND CONCLUSIONS WITH RESPECT TO NAUGHTON'S HYPOTHESES
Experimental evidence from multiple physiologic studies indicate that CSA-HCSB is associated with a number of pathological consequences including creation of a hyperadrenergic state, which in HFrEF is known to carry a grim prognosis.
Multiple RCTs show that this pathological state is reversible when CSA-HCSB is treated with CPAP, ASV, or supplemental oxygen. The use of these treatments to suppress CSA-HCSB consistently result in a diminution of sympathetic activity. These findings are not dissimilar to those reported in the treatment of OSA with CPAP.1
In patients with HFrEF, in the face of reduced respiratory system compliance hyperventilation is associated with increased WOB, largely due to a substantial increase in negative swings in intrathoracic pressure.
Attenuation of periodic breathing results in an overall decrease in ventilation and increased diaphragmatic strength, as reflected by increased MIP.
Based on the aforementioned data we firmly conclude that CSA-HCSB in HFrEF is maladaptive, and should be considered a “bad bedfellow” that in most cases leads to untoward consequences and over time, excess mortality.23 Moreover, because the SERVE-HF results have been misused to add credibility to Naughton's hypotheses, it is important to carefully consider the validity of the SERVE-HF study itself. We believe that the failure of the SERVE-HF trial to demonstrate a positive outcome in patients with HFrEF treated with ASV relates more to its multiple deficiencies, highlighted previously, and therefore should not be used to justify a theory that CSA is cardioprotective.
In summary, our message is twofold: Naughton's hypotheses are riddled with contradictions and lack of a firm basis from known physiology; and the SERVE-HF study, imperfect as it is, does not supply added credibility to Naughton's message.
DISCLOSURE STATEMENT
The authors report no conflicts of interest.
CITATION
Javaheri S, Brown LK, Khayat R. Con: persistent central sleep apnea/Hunter-Cheyne-Stokes breathing, despite best guideline-based therapy of heart failure with reduced ejection fraction, is not a compensatory mechanism and should be suppressed. J Clin Sleep Med. 2018;14(6):915–921.
REFERENCES
- 1.Javaheri S, Barbe F, Campos-Rodriguez F, et al. Sleep apnea types, mechanisms, and clinical cardiovascular consequences. J Am Coll Cardiol. 2017;69(7):841–858. doi: 10.1016/j.jacc.2016.11.069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Naughton MT. Cheyne-Stokes respiration: friend or foe? Thorax. 2012;67(4):357–360. doi: 10.1136/thoraxjnl-2011-200927. [DOI] [PubMed] [Google Scholar]
- 3.Cowie MR, Woehrle H, Wegscheider K, et al. Adaptive servo-ventilation for central sleep apnea in systolic heart failure. N Engl J Med. 2015;373(12):1095–1105. doi: 10.1056/NEJMoa1506459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Javaheri S, Brown LK, Randerath W, Khayat R. SERVE-HF: more questions than answers [commentary] Chest. 2016;149(4):900–904. doi: 10.1016/j.chest.2015.12.021. [DOI] [PubMed] [Google Scholar]
- 5.Brack T, Jubran A, Laghi F, et al. Fluctuations in end-expiratory lung volume during Cheyne-Stokes respiration. Am J Respir Crit Care Med. 2005;171(12):1408–1413. doi: 10.1164/rccm.200503-409OC. [DOI] [PubMed] [Google Scholar]
- 6.Naughton MT, Floras JS, Rahman MA, et al. Respiratory correlates of muscle sympathetic nerve activity in heart failure. Clin Sci. 1998;95(3):277–285. [PubMed] [Google Scholar]
- 7.Spaak J, Egri ZT, Kubo T, Yu E, et al. Muscle sympathetic nerve activity during wakefulness in heart failure patients with and without sleep apnea. Hypertension. 2005;46(6):1327–1332. doi: 10.1161/01.HYP.0000193497.45200.66. [DOI] [PubMed] [Google Scholar]
- 8.van de Borne P, Oren R, Abouassaly C, Anderson E, Somers VK. Effect of Cheyne-Stokes respiration on muscle sympathetic nerve activity in severe congestive heart failure secondary to ischemic or idiopathic dilated cardiomyopathy. Am J Cardiol. 1998;81(4):432–436. doi: 10.1016/s0002-9149(97)00936-3. [DOI] [PubMed] [Google Scholar]
- 9.Naughton MT, Benard DC, Liu PP, Rutherford R, Rankin F, Bradley TD. Effects of nasal CPAP on sympathetic activity in patients with heart failure and central sleep apnea. Am J Respir Crit Care Med. 1995;152(2):473–479. doi: 10.1164/ajrccm.152.2.7633695. [DOI] [PubMed] [Google Scholar]
- 10.Mansfield D, Kaye DM, Brunner La Rocca H, Solin P, Esler MD, Naughton MT. Raised sympathetic nerve activity in heart failure and central sleep apnea is due to heart failure severity. Circulation. 2003;107(10):1396–1400. doi: 10.1161/01.cir.0000056520.17353.4f. [DOI] [PubMed] [Google Scholar]
- 11.Bradley TD, Logan AG, Kimoff RJ, et al. Continuous positive airway pressure for central sleep apnea and heart failure. N Engl J Med. 2005;353(19):2025–2033. doi: 10.1056/NEJMoa051001. [DOI] [PubMed] [Google Scholar]
- 12.Pepperell JC, Maskell NA, Jones DR, et al. A randomized controlled trial of adaptive ventilation for Cheyne-Stokes breathing in heart failure. Am J Respir Crit Care Med. 2003;168(9):1109–1114. doi: 10.1164/rccm.200212-1476OC. [DOI] [PubMed] [Google Scholar]
- 13.Staniforth AD, Kinnear WJ, Starling R, Hetmanski DJ, Cowley AJ. Effect of oxygen on sleep quality, cognitive function and sympathetic activity in patients with chronic heart failure and Cheyne-Stokes respiration. Eur Heart J. 1998;19(6):922–928. doi: 10.1053/euhj.1997.0861. [DOI] [PubMed] [Google Scholar]
- 14.Teschler H, Döhring J, Wang YM, Berthon-Jones M. Adaptive pressure support servo-ventilation: a novel treatment for Cheyne-Stokes respiration in heart failure. Am J Respir Crit Care Med. 2001;164(4):614–619. doi: 10.1164/ajrccm.164.4.9908114. [DOI] [PubMed] [Google Scholar]
- 15.Naughton MT, Benard DC, Rutherford R, Bradley TD. Effect of continuous positive airway pressure on central sleep apnea and nocturnal PCO2 in heart failure. Am J Respir Crit Care Med. 1994;150(6 Pt 1):1598–1604. doi: 10.1164/ajrccm.150.6.7952621. [DOI] [PubMed] [Google Scholar]
- 16.Bing OH, Brooks WW, Messer JV. Heart muscle viability following hypoxia: protective effect of acidosis. Science. 1973;180(4092):1297e8. doi: 10.1126/science.180.4092.1297. [DOI] [PubMed] [Google Scholar]
- 17.Nakao K, Ohgushi M, Yoshimura M, et al. Hyperventilation as a specific test for diagnosis of coronary artery spasm. Am J Cardiol. 1997;80(5):545–549. doi: 10.1016/s0002-9149(97)00419-0. [DOI] [PubMed] [Google Scholar]
- 18.Lanfranchi PA, Somers VK, Braghiroli A, Corra U, Eleuteri E, Giannuzzi P. Central sleep apnea in left ventricular dysfunction: prevalence and implications for arrhythmic risk. Circulation. 2003;107(5):727–732. doi: 10.1161/01.cir.0000049641.11675.ee. [DOI] [PubMed] [Google Scholar]
- 19.Javaheri S, Shukla R, Wexler L. Association of smoking, sleep apnea, and plasma alkalosis with nocturnal ventricular arrhythmias in men with systolic heart failure. Chest. 2012;141(6):1449–1456. doi: 10.1378/chest.11-1724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Javaheri S, Corbett WS. Association of low arterial PCO2 with central sleep apnea and ventricular arrhythmias in ambulatory patients with stable heart failure. Ann Intern Med. 1998;128(3):204–207. doi: 10.7326/0003-4819-128-3-199802010-00006. [DOI] [PubMed] [Google Scholar]
- 21.Kee K, Sands SA, Stuart-Andrews, et al. Effect of apnea type on work of breathing and respiratory fatigability during sleep in humans with heart failure [abstract] Am J Respir Crit Care Med. 2014;189:A3892. [Google Scholar]
- 22.Granton JT, Naughton MT, Benard DC, Liu PP, Goldstein RS, Bradley TD. CPAP improves inspiratory muscle strength in patients with heart failure and central sleep apnea. Am J Respir Crit Care Med. 1996;153(1):277–282. doi: 10.1164/ajrccm.153.1.8542129. [DOI] [PubMed] [Google Scholar]
- 23.Oldenburg O, Wellmann B, Buchholz A, et al. Nocturnal hypoxaemia is associated with increased mortality in stable heart failure patients. Eur Heart J. 2016;37(21):1695–1703. doi: 10.1093/eurheartj/ehv624. [DOI] [PubMed] [Google Scholar]