

Abstract
A novel strategy is developed to expand the scope of chemoenzymatic synthetic products by designing a chemoenzymatic synthon. The synthon is enzymatically converted to carbohydrate analogs which are readily derivatized chemically to produce desired targets. The strategy is demonstrated for synthesizing glycosides containing 7,9-di-N-acetyllegionaminic acid (Leg5,7Ac2), a bacterial nonulosonic acid (NulO) analog of sialic acid. A versatile library of α2–3/6-linked Leg5,7Ac2- glycosides is built using chemically synthesized 2,4-diazido-2,4,6-trideoxy mannose as a chemoenzymatic synthon for highly efficient one-pot multienzyme (OPME) sialylation followed by a downstream chemical conversion of the azido groups to acetamido groups. The overall yields of the syntheses are 34–52% in 10 steps from commercially available D-fucose, representing significant improvements over previous methods. Free Leg5,7Ac2 monosaccharide is also synthesized using a sialic acid aldolase-catalyzed reaction.
Keywords: carbohydrates, chemoenzymatic synthesis, glycosylation, legionaminic acid, sialic acid
Graphical abstract
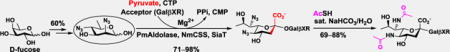
Chemoenzymatic synthesis: A versatile library of α2–3/6-linked Leg5,7NAc2-containing glycosides has been built by highly efficient one-pot multienzyme (OPME) sialylation systems using chemically synthesized 6deoxyMan2,4diN3 as a chemoenzymatic synthon followed by facile chemical converting the azido groups to acetamido groups.
The high efficiency of enzymatic and chemoenzymatic synthetic approaches in carbohydrate synthesis has been increasingly recognized. The methods, however, rely heavily on the access to related enzymes and can be limited by the substrate specificity of the enzymes that are available.
Here we develop a novel strategy to expand the scope of chemoenzymatic synthetic products using chemoenzymatic synthons, which are designed to be used by enzymatic reactions to produce carbohydrate analogs that can be readily converted to desired targets by chemical derivatization. The strategy is demonstrated for the synthesis of a versatile library of glycosides containing a terminal α2–3 or α2–6-linked di-N-acetyllegionaminic acid (Leg5,7Ac2, 1) (Figure 1). Leg5,7Ac2, or 5,7-diacetamido-3,5,7,9-tetradeoxy-D-glycero-D-galacto-non-2-ulosonic acid, is a bacterial nine-carbon α-keto acid (nonulosonic acid, NulO)[1] that has been found as a component of the serological-specificity-defining structures of several pathogenic bacteria.[2] It is the di-N-acetyl derivative of legionaminic acid (Leg, 2) (5,7-diamino-3,5,7,9-tetradeoxy-D-glycero-D-galacto-non-2-ulosonic acid) and has the exact the same stereochemistry of N-acetylneuraminic acid (Neu5Ac, 3), the most common sialic acid form in nature.[1b] It differs from Neu5Ac at two sites where the C-7 and C-9 hydroxyl groups of Neu5Ac are substituted by C-7 acetamido group and C-9 hydrogen, respectively.[1b, 3]
Figure 1.
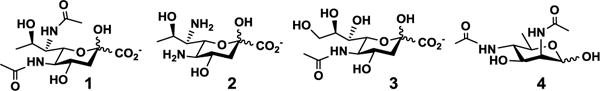
Structures of 5,7-di-N-acetyllegionaminic acid (Leg5,7Ac2, 1), legionaminic acid (Leg, 2), N-acetylneuraminic acid (Neu5Ac, 3), and 2,4-diacetamido-2,4,6-trideoxy-D-mannose (6deoxyManNAc4NAc, 4) which is the six-carbon biosynthetic precursor of Leg5,7Ac2 (1).
Leg5,7Ac2 (1) and its N-acyl derivatives and related 4- or 8-epimers have been found in lipopolysaccharides (LPSs) or extracellular polysaccharides of many pathogenic Gram-negative bacteria.[2, 4] Pathogenic bacterial LPSs containing Leg5,7Ac2 and derivatives are attractive targets for developing bacterial polysaccharide vaccines and diagnostic tools (e.g. anti-carbohydrate antibodies).
Several examples of chemical synthesis of Leg5,7Ac2 and its simple glycosides have been reported. Condensation of oxalacetic acid with chemically synthesized 2,4-diacetamido-2,4,6-trideoxy-D-mannose (6deoxyManNAc4NAc, 4, Figure 1) produced both Leg5,7Ac2 and its C4-epimer with 7% and 10% yields, respectively.[5] De novo synthesis of a β-glycoside (not the desired α-glycoside) of legionaminic acid from D-threonine was achieved with a yield of 7% in 17 steps.[6] From Neu5Ac (3), a protected thio-adamentyl diazido-legionaminic acid donor was obtained with a yield of 17% in 15 steps which was used for chemical glycosylation followed by additional chemical transformation to produce an α2–3-Leg5,7Ac2-terminated methyl β-galactoside with an overall yield of 7% in 19 steps.[7] All these efforts resulted in low yields and demonstrated challenges for synthesizing Leg5,7Ac2-glycosides including low stereo- and regio- selectivities.[6–7]
Biosynthetically, Leg5,7Ac2 is produced from 6deoxyManNAc4NAc (4) and phosphoenolpyruvate (PEP) catalyzed by a Leg5,7Ac2 synthase.[8] 6deoxyManNAc4NAc itself is formed from uridine 5’-diphosphate-N-acetylglucosamine (UDP-GlcNAc, e.g. in Legionella penumophila) or guanosine 5’-diphosphate-N-acetylglucosamine (GDP-GlcNAc, e.g. in Campylobacter jejuni) by a process involving four reactions catalyzed by different enzymes.[8] Leg5,7Ac2 is activated by a cytidine 5’-monophosphate-Leg5,7Ac2 (CMP-Leg5,7Ac2) synthetase[3] to provide CMP-Leg5,7Ac2 as the glycosyltransferase donor for the synthesis of desired structures.
Although a native Leg5,7Ac2-glycosylltransferase has yet to be identified, several mammalian and bacterial sialyltransferases have been tested for catalyzing the transfer of Leg5,7Ac2 from CMP-Leg5,7Ac2 to galactosides. Porcine ST3Gal I, human ST6Gal I,[9] Pasteurella multocida sialyltransferase 1 (PmST1),[10] and Neisseria meningitides MC58 α2–3-sialyltransferase[11] showed reasonable activity in forming Leg5,7Ac2-glycosides. Nevertheless, these enzymatic syntheses relied on a complex process of producing CMP-Leg5,7Ac2 either from UDP-GlcNAc by multiple enzymes[12] in vitro with chemical acetylation of the 4-amino group[10] or from Leg5,7Ac2 produced de novo using Escherichia coli engineered with combined biosynthetic pathways from Saccharomyces cerevisiae, Campylobacter jejuni,[13] and Legionella pneumophila.[14]
Here we show that a versatile library of α2–3 and α2–6-linked Leg5,7Ac2-glycosides can be produced readily from chemically synthesized 2,4-diazido-2,4,6-trideoxy mannose (6deoxyMan2,4diN3) as a chemoenzymatic synthon in highly efficient one-pot multienzyme (OPME) sialylation systems[15] using commercially available enzymes with downstream chemical derivatization.
We started by developing an efficient method for the production of 6deoxyManNAc4NAc (4), the six-carbon precursor of Leg5,7Ac2 (1). Inspired by a method reported recently by the Kulkarni group,[16] commercially available D-fucose (5) was chosen as the starting material to allow simultaneous inversion of its stereochemistry at C-2 and C-4 to form the desired mannose derivative. As shown in Scheme 1, per-O-acetylation of D-fucose (5) followed by BF3·Et2O-catalyzed nucleophilic displacement of the anomeric acetate with p-methoxyphenol in CH2Cl2 produced intermediate 6 in 84% yield. De-O-acetylation with sodium methoxide in methanol produced triol. Dimethyltin chloride (Me2SnCl2)-catalyzed regio-selective benzoyl protection[16] of 3-OH formed D-fucosyl-2,4-diol (7) in two steps in 94% yield. Compound 7 was then treated with trifluoromethanesulfonic anhydride (Tf2O) and pyridine to form the corresponding 2,4-bistriflate[16] which upon treating with 2.5 equivalent of tetrabutylammonium azide (TBAN3) in anhydrous toluene at 70 °C to reflux,[17] 6-deoxy-D-mannose derivative 8 was formed in 2 hours in 93% yield. Debenzoylation and ceric ammonium nitrate-catalyzed removal of the p-methoxyphenyl group[18] produced 2,4-diazido-2,4,6-trideoxy mannose (6deoxyMan2,4diN3, 9) in 81% yield. Overall, the production of 6deoxyMan2,4diN3 (9) from D-fucose (5) was achieved in eight steps with an overall yield of 60%. ManNAc derivative 6deoxyManNAc4NAc (4) was obtained readily from 9 in 73% yield by treating with thioacetic acid in pyridine[19] at room temperature for 20 hours.
Scheme 1.
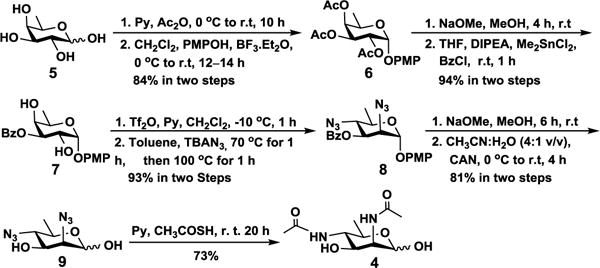
Chemical synthesis of 6deoxyManNAc4NAc (4) from commercially available D-fucose (5) via a diazido intermediate 2,4-diazido-2,4,6-trideoxy mannose (6deoxyMan2,4diN3, 9).
To our delight, 6deoxyManNAc4NAc (4) was a suitable substrate for both recombinant Escherichia coli (EcAldolase)[20] and Pasteurella multocida (PmAldolase)[21] sialic acid aldolases. PmAldolase was found to be more efficient and was used for preparative-scale synthesis of Leg5,7Ac2 (1) with a 71% yield (Scheme 2).
Scheme 2.
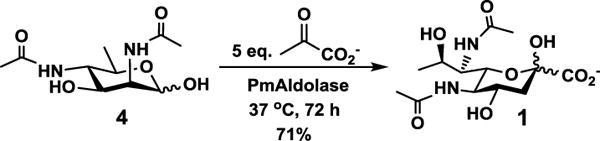
PmAldolase-catalyzed synthesis of Leg5,7Ac2 (1) from 6deoxyManNAc4NAc (4) and sodium pyruvate.
Nevertheless, the resulting Leg5,7Ac2 (1) was not a suitable substrate for Neisseria meningitidis CMP-sialic acid synthetase (NmCSS)[20] for the synthesis of the corresponding CMP- Leg5,7Ac2.
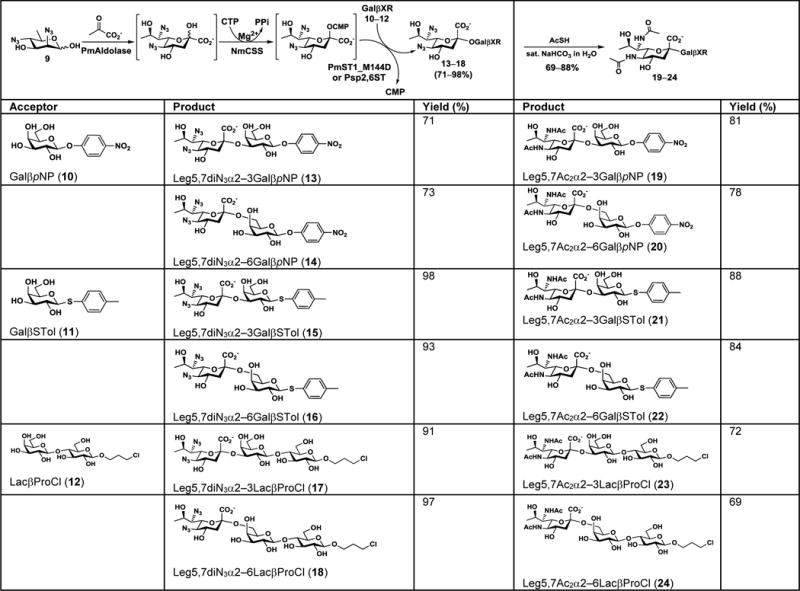
As azido derivatives of N-acetylmannosamine (ManNAc) and mannose have been shown to be suitable starting materials for OPME sialylation systems[15] for the synthesis of various α2–3/6-linked sialosides[22] including those containing 7-azido-[23] or 9-deoxy-derivative[24] of Neu5Ac, the diazido compound 9 was tested for the synthesis of glycosides. To our delight, 9 was well tolerated by the OPME α2–3/6-sialylation systems as demonstrated using three different acceptors including para-nitrophenyl β-galactoside (GalβpNP,10), thiotolyl β-galactoside (GalβSTol, 11), and lactosyl β-propylchloride (LacβProCl, 12). In these systems, 9 was coupled with pyruvate to form the diazido-derivative of Leg (Leg5,7diN3) by a PmAldolase-catalyzed reaction. Leg5,7diN3 reacted with cytidine 5'-triphosphate (CTP) using an NmCSS-catalyzed reaction to form CMP-Leg5,7diN3 which was used by a sialyltransferase (e.g. PmST1_M144D[25] or Psp2,6ST[26]) to produce α2–3/6-linked Leg5,7diN3-containing glycosides (13–18) (Table 1).
Table 1.
Production of Leg5,7Ac2-containing glycosides (19–24) by one-pot multienzyme (OPME) synthesis of Leg5,7diN3-containing glycosides (13–18) followed by chemical conversion of the azido group to N-acetyl group.
|
Using PmST1_M144D[25] as the sialyltransferase in the OPME sialylation system containing PmAldolase and NmCSS (Table 1), α2–3-linked Leg5,7diN3-containing glycosides (13, 15, and 17) were obtained in good (71%) to excellent (98% and 91%) yields with GalβpNP (10), GalβSTol (11), and LacβProCl (12) as the sialyltransferase acceptors, respectively. Similarly, using Psp2,6ST[26] as the sialyltransferase, α2–6-linked Leg5,7diN3-containing glycosides (14, 16, and 18) were obtained in good (73%) to excellent (93% and 97%) yields. Compared to GalβSTol (11) and LacβProCl (12), GalβpNP (10) was a less effective sialyltransferase acceptor, resulting in lower sialylation yields in OPME sialylation systems.
Different strategies were tested to convert the azido groups in the glycosides synthesized (13–18) to N-acetyl groups to form desired Leg5,7Ac2-containing glycosides (19–24). A conventional Perlman catalyst-mediated reduction of azide to amine[27] followed by selective acetylation of the amine worked for 13, 15, 16, and 18 with ~50% yields in two steps but hydrogenation also converted the aromatic nitro group of 13 and 14 to the corresponding amine which was undesirable. PMe3-mediated Staudinger reaction[28] worked well for all compounds and quantitatively produced the corresponding diamine derivatives. However, selective acetylation of amine by a combination of acetylchloride and triethyl amine in tetrahydrofuran and water (4:1 v/v) produced the di-N-acetyl derivative in poor yields (~40%). An alternative N-acetylation strategy using thioacetic acid and catalytic copper sulfate in methanol[29] produced Leg5,7Ac2-glycosides with very poor yields (<20%). Finally, thioacetic acid-mediated one-pot conversion of azido to acetamido group[19] using saturated sodium bicarbonate in water was found to be the optimal condition and the Leg5,7Ac2-containing glycosides (19–24) were produced in 69–88% yields (Table 1). In comparison, poorer yields (45–70%) were obtained when pyridine was used as the solvent in the same method (data not shown).
It is worth to note that Leg5,7Ac2α2–3Gal- and Leg5,7Ac2α2–6Gal-containing structures have been found in O-antigens[4b, 4c, 30] and an extracellular polysaccharide fraction,[4e] respectively, of various opportunistic pathogens. Therefore, the obtained thiotolyl β-glycosides (compounds 15–16 and 21–22) can be used to form building blocks for efficient chemical synthesis of more complex glycosides similar to those described before for Neu5Ac-containing sialosides.[31]
The propyl chloride aglycon in compounds 23 and 24 was readily converted to propyl azide by treating them with sodium azide (NaN3) and a catalytic amount of sodium iodide (NaI) in dimethylformamide (DMF) at 60 °C for 12 hours[32] to produce Leg5,7Ac2α2–3LacβProN3 (25) and Leg5,7Ac2α2–6LacβProN3 (26) in 85% and 92% yields, respectively (Figure 2). The azido group in the final products can be easily reduced to an amine group in future for microarray study and for synthesizing glycoconjugates.
Figure 2.
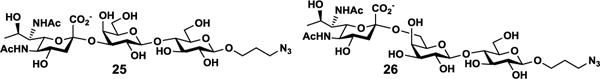
Structures of Leg5,7Ac2α2–3LacβProN3 (25) and Leg5,7Ac2α2–6LacβProN3 (26).
Leg5,7diN3α2–3/6GalβpNP (13–14) and Leg5,7Ac2α2–3/6GalβpNP (19–20) were tested as potential substrates for recombinant human cytosolic sialidase hNEU2[33] and several bacterial sialidases including three commercially available sialidases from Arthrobacter ureafaciens, Vibrio cholerae, and Clostridium perfringens (CpNanH), as well as five recombinant bacterial sialidases such as the α2–3-sialidase activity of multifunctional PmST1,[22a] Bifidobacterium infantis sialidase BiNanH2,[34] Streptococcus pneumoniae sialidases SpNanA,[35] SpNanB,[35] and SpNanC).[36] It was interesting to note that Leg5,7Ac2α2–3GalβpNP (19), but not Leg5,7diN3α2–3GalβpNP (13), was a substrate for the α2–3-sialidase activity of PmST1.[22a] Compared to Neu5Acα2–3GalβpNP (kcat/KM = 117 min−1 mM−1), the α2–3-sialidase activity of PmST1 (in the presence of 0.4 mM CMP) for Leg5,7Ac2α2–3GalβpNP (19) (kcat/KM = 4 min−1 mM−1) was about 30-fold less efficient (Table S1, ESI). Other sialidases tested did not show any activity for the compounds used (13, 14, 19, and 20) indicating the sialidase activity-blocking effect of 7-NAc substitution in Leg5,7Ac2-glycosides because 9-deoxy substitution of Neu5Ac-glycosides was tolerated by several sialidases.[24] The lack of PmST1 α2–3-sialidase activity for Leg5,7diN3α2–3GalβpNP (13) is advantageous for synthesizing α2–3-linked Leg5,7diN3-glycosides. Indeed, commercially available PmST1 was found to be as effective as its M144D mutant in synthesizing α2–3-linked Leg5,7Ac2-glycosides 13, 15, and 17. Gram-scale (1.85 g) synthesis of Leg5,7diN3α2–3GalβSTol (15) was readily achieved in an excellent 93% yield using the OPME sialylation system containing PmST1. Psp2,6ST A336G mutant[37] with a higher expression level and commercially available Photobacterium damselae α2–6-sialyltransferase (Pd2,6ST)[22b] were also suitable sialyltransferases for OPME synthesis of α2–6-linked Leg5,7diN3-glycosides 14, 16, and 18 (data not shown).
In conclusion, 2,4-diazido-2,4,6-trideoxy mannose (6deoxyMan2,4diN3) has been designed as an easy-to-obtained and highly effective chemoenzymatic synthon. It was readily synthesized from commercially available D-fucose by chemical methods in eight steps with an overall yield of 60% and was successfully used for highly efficient chemoenzymatic synthesis of a library of α2–3- and α2–6-linked di-N-acetyllegionaminic acid (Leg5,7Ac2)-containing glycosides in 57–86% yields. The chemoenzymatic method described here allows high-yield synthesis of a diverse array of biologically important Leg5,7Ac2-containing glycosides using commercially available enzymes. The method of designing chemoenzymatic synthons for enzymatic formation of glycosides followed by chemical derivatization can be a general strategy for producing complex N-acetyl-containing glycosides.
Supplementary Material
Acknowledgments
This work is supported by National Institutes of Health (NIH) grants R01AI130684 and U01GM125288. Bruker Avance-800 NMR spectrometer was funded by NSF grant DBIO-722538.
Footnotes
Supporting information for this article is given via a link at the end of the document.
Experimental Section
Detailed synthetic procedures, nuclear magnetic resonance (NMR) spectroscopy and high-resolution mass spectrometry (HRMS) data, and NMR spectra for products are available in the supporting information.
References
- 1.a) Lewis AL, Desa N, Hansen EE, Knirel YA, Gordon JI, Gagneux P, Nizet V, Varki A. Proc Natl Acad Sci U S A. 2009;106:13552–13557. doi: 10.1073/pnas.0902431106. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Chen X, Varki A. ACS Chem Biol. 2010;5:163–176. doi: 10.1021/cb900266r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.a) Hashii N, Isshiki Y, Iguchi T, Hisatsune K, Kondo S. Carbohydr Res. 2003;338:1055–1062. doi: 10.1016/s0008-6215(03)00077-6. [DOI] [PubMed] [Google Scholar]; b) Knirel YA, Shashkov AS, Tsvetkov YE, Jansson PE, Zahringer U. Adv Carbohydr Chem Biochem. 2003;58:371–417. doi: 10.1016/s0065-2318(03)58007-6. [DOI] [PubMed] [Google Scholar]
- 3.Schoenhofen IC, Young NM, Gilbert M. Methods Enzymol. 2017;597:187–207. doi: 10.1016/bs.mie.2017.06.042. [DOI] [PubMed] [Google Scholar]
- 4.a) Hashii N, Isshiki Y, Iguchi T, Kondo S. Carbohydr Res. 2003;338:1063–1071. doi: 10.1016/s0008-6215(03)00078-8. [DOI] [PubMed] [Google Scholar]; b) MacLean LL, Vinogradov E, Pagotto F, Perry MB. Carbohydr Res. 2011;346:2589–2594. doi: 10.1016/j.carres.2011.09.003. [DOI] [PubMed] [Google Scholar]; c) Sun Y, Arbatsky NP, Wang M, Shashkov AS, Liu B, Wang L, Knirel YA. FEMS Immunol Med Microbiol. 2012;66:323–333. doi: 10.1111/j.1574-695X.2012.01013.x. [DOI] [PubMed] [Google Scholar]; d) Filatov AV, Wang M, Wang W, Perepelov AV, Shashkov AS, Wang L, Knirel YA. Carbohydr Res. 2014;392:21–24. doi: 10.1016/j.carres.2014.01.012. [DOI] [PubMed] [Google Scholar]; e) Kodali S, Vinogradov E, Lin F, Khoury N, Hao L, Pavliak V, Jones CH, Laverde D, Huebner J, Jansen KU, Anderson AS, Donald RG. J Biol Chem. 2015;290:19512–19526. doi: 10.1074/jbc.M115.655852. [DOI] [PMC free article] [PubMed] [Google Scholar]; f) Vinogradov E, Maclean L, Xu HH, Chen W. Carbohydr Res. 2014;390:42–45. doi: 10.1016/j.carres.2014.03.001. [DOI] [PubMed] [Google Scholar]; g) Shashkov AS, Kenyon JJ, Senchenkova SN, Shneider MM, Popova AV, Arbatsky NP, Miroshnikov KA, Volozhantsev NV, Hall RM, Knirel YA. Glycobiology. 2016;26:501–508. doi: 10.1093/glycob/cwv168. [DOI] [PubMed] [Google Scholar]; h) Shashkov AS, Senchenkova SN, Popova AV, Mei Z, Shneider MM, Liu B, Miroshnikov KA, Volozhantsev NV, Knirel YA. Russian Chem Bull Int Ed. 2015;64:1196–1199. [Google Scholar]
- 5.Tsvetkov YE, Shashkov AS, Knirel YA, Zahringer U. Carbohydr Res. 2001;331:233–237. doi: 10.1016/s0008-6215(01)00041-6. [DOI] [PubMed] [Google Scholar]
- 6.Matthies S, Stallforth P, Seeberger PH. J Am Chem Soc. 2015;137:2848–2851. doi: 10.1021/jacs.5b00455. [DOI] [PubMed] [Google Scholar]
- 7.Popik O, Dhakal B, Crich D. J Org Chem. 2017;82:6142–6152. doi: 10.1021/acs.joc.7b00746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Morrison MJ, Imperiali B. Biochemistry. 2014;53:624–638. doi: 10.1021/bi401546r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Watson DC, Wakarchuk WW, Gervais C, Durocher Y, Robotham A, Fernandes SM, Schnaar RL, Young NM, Gilbert M. Glycoconj J. 2015;32:729–734. doi: 10.1007/s10719-015-9624-4. [DOI] [PubMed] [Google Scholar]
- 10.Watson DC, Leclerc S, Wakarchuk WW, Young NM. Glycobiology. 2011;21:99–108. doi: 10.1093/glycob/cwq135. [DOI] [PubMed] [Google Scholar]
- 11.Watson DC, Wakarchuk WW, Leclerc S, Schur MJ, Schoenhofen IC, Young NM, Gilbert M. Glycobiology. 2015;25:767–773. doi: 10.1093/glycob/cwv017. [DOI] [PubMed] [Google Scholar]
- 12.Glaze PA, Watson DC, Young NM, Tanner ME. Biochemistry. 2008;47:3272–3282. doi: 10.1021/bi702364s. [DOI] [PubMed] [Google Scholar]
- 13.Schoenhofen IC, Vinogradov E, Whitfield DM, Brisson JR, Logan SM. Glycobiology. 2009;19:715–725. doi: 10.1093/glycob/cwp039. [DOI] [PubMed] [Google Scholar]
- 14.Hassan MI, Lundgren BR, Chaumun M, Whitfield DM, Clark B, Schoenhofen IC, Boddy CN. Angew Chem Int Ed Engl. 2016;55:12018–12021. doi: 10.1002/anie.201606006. [DOI] [PubMed] [Google Scholar]
- 15.Yu H, Chokhawala HA, Huang S, Chen X. Nat Protoc. 2006;1:2485–2492. doi: 10.1038/nprot.2006.401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sanapala SR, Kulkarni SS. J Am Chem Soc. 2016;138:4938–4947. doi: 10.1021/jacs.6b01823. [DOI] [PubMed] [Google Scholar]
- 17.Tsvetkov YE, Shashkov AS, Knirel YA, Zähringer U. Carbohydr Res. 2001;335:221–243. doi: 10.1016/s0008-6215(01)00235-x. [DOI] [PubMed] [Google Scholar]
- 18.Ghosh S, Nishat S, Andreana PR. J Org Chem. 2016;81:4475–4484. doi: 10.1021/acs.joc.6b00195. [DOI] [PubMed] [Google Scholar]
- 19.Shangguan N, Katukojvala S, Greenberg R, Williams LJ. J Am Chem Soc. 2003;125:7754–7755. doi: 10.1021/ja0294919. [DOI] [PubMed] [Google Scholar]
- 20.Yu H, Yu H, Karpel R, Chen X. Bioorg Med Chem. 2004;12:6427–6435. doi: 10.1016/j.bmc.2004.09.030. [DOI] [PubMed] [Google Scholar]
- 21.Li Y, Yu H, Cao H, Lau K, Muthana S, Tiwari VK, Son B, Chen X. Appl Microbiol Biotechnol. 2008;79:963–970. doi: 10.1007/s00253-008-1506-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.a Yu H, Chokhawala H, Karpel R, Yu H, Wu B, Zhang J, Zhang Y, Jia Q, Chen X. J Am Chem Soc. 2005;127:17618–17619. doi: 10.1021/ja0561690. [DOI] [PubMed] [Google Scholar]; b Yu H, Huang S, Chokhawala H, Sun M, Zheng H, Chen X. Angew Chem Int Ed Engl. 2006;45:3938–3944. doi: 10.1002/anie.200600572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Khedri Z, Li Y, Muthana S, Muthana MM, Hsiao CW, Yu H, Chen X. Carbohydr Res. 2014;389:100–111. doi: 10.1016/j.carres.2014.02.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Khedri Z, Muthana MM, Li Y, Muthana SM, Yu H, Cao H, Chen X. Chem Commun. 2012;48:3357–3359. doi: 10.1039/c2cc17393j. [DOI] [PubMed] [Google Scholar]
- 25.Sugiarto G, Lau K, Qu J, Li Y, Lim S, Mu S, Ames JB, Fisher AJ, Chen X. ACS Chem Biol. 2012;7:1232–1240. doi: 10.1021/cb300125k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ding L, Yu H, Lau K, Li Y, Muthana S, Wang J, Chen X. Chem Commun. 2011;47:8691–8693. doi: 10.1039/c1cc12732b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Corey EJ, Link JO. J Am Chem So. 1992;114:1906–1908. [Google Scholar]
- 28.Leffler JE, Temple RD. J Am Chem Soc. 1967;89:5235–5246. [Google Scholar]
- 29.Mali SM, Bhaisare RD, Gopi HN. J Org Chem. 2013;78:5550–5555. doi: 10.1021/jo400701v. [DOI] [PubMed] [Google Scholar]
- 30.Filatov AV, Wang M, Wang W, Perepelov AV, Shashkov AS, Wang L, Knirel YA. Carbohydr Res. 2014;392:21–24. doi: 10.1016/j.carres.2014.01.012. [DOI] [PubMed] [Google Scholar]
- 31.a) Mehta S, Gilbert M, Wakarchuk WW, Whitfield DM. Org Lett. 2000;2:751–753. doi: 10.1021/ol990406k. [DOI] [PubMed] [Google Scholar]; b) Cao H, Huang S, Cheng J, Li Y, Muthana S, Son B, Chen X. Carbohydr Res. 2008;343:2863–2869. doi: 10.1016/j.carres.2008.06.020. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Yan F, Mehta S, Eichler E, Wakarchuk WW, Gilbert M, Schur MJ, Whitfield DM. J Org Chem. 2003;68:2426–2431. doi: 10.1021/jo026569v. [DOI] [PubMed] [Google Scholar]
- 32.Lau K, Thon V, Yu H, Ding L, Chen Y, Muthana MM, Wong D, Huang R, Chen X. Chem Commun. 2010;46:6066–6068. doi: 10.1039/c0cc01381a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Li Y, Cao H, Yu H, Chen Y, Lau K, Qu J, Thon V, Sugiarto G, Chen X. Mol BioSyst. 2011;7:1060–1072. doi: 10.1039/c0mb00244e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sela DA, Li Y, Lerno L, Wu S, Marcobal AM, German JB, Chen X, Lebrilla CB, Mills DA. J Biol Chem. 2011;286:11909–11918. doi: 10.1074/jbc.M110.193359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tasnima N, Yu H, Li Y, Santra A, Chen X. Org Biomol Chem. 2016;15:160–167. doi: 10.1039/c6ob02240e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Li W, Xiao A, Li Y, Yu H, Chen X. Carbohydr Res. 2017;451:51–58. doi: 10.1016/j.carres.2017.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ding L, Zhao C, Qu J, Li Y, Sugiarto G, Yu H, Wang J, Chen X. Carbohydr Res. 2015;408:127–133. doi: 10.1016/j.carres.2014.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.