

Abstract
Genetic variation at rs4240624 on chromosome 8 is associated with an attenuated signal on hepatic computerized tomography (CT), which has been attributed to changes in hepatic fat. The closest coding gene to rs4240624, PPP1R3B, encodes a protein that promotes hepatic glycogen synthesis. Here we performed studies to determine if the X-ray attenuation associated with rs4240624 is due to changes in hepatic glycogen or hepatic triglyceride content (HTGC). A sequence variant in complete linkage disequilibrium (LD) with rs4240624, rs4841132, was genotyped in the Dallas Heart Study (DHS), the Dallas Liver Study, and the Copenhagen Cohort (n=112,428) of whom 1,539 had non-viral liver disease. The minor A-allele of rs4841132 was associated with increased hepatic X-ray attenuation (n=1,572, P=4×10−5), but not with HTGC (n=2,674, P=0.58). Rs4841132-A was associated with modest, but significant elevations in serum ALT in the Copenhagen Cohort (P=3×10−4) and the DHS (P=0.004), and with odds ratios for liver disease of 1.13 (95% CI, 0.97-1.31) and 1.23 (1.01-1.51), respectively. Mice lacking PPP1R3B were deficient in hepatic glycogen, whereas HTGC was unchanged. Hepatic overexpression of PPP1R3B caused accumulation of hepatic glycogen and elevated plasma levels of ALT, but did not change HTGC. Conclusion: These observations are consistent with the notion that the minor allele of rs4841132 promotes a mild form of hepatic glycogenosis that is associated with hepatic injury.
Keywords: hepatic steatosis, hepatic glycogen, liver injury, metabolism
Fatty liver disease (FLD) has become the most common liver disease in the Western world, affecting approximately 30% of adults in the U.S.(1,2) The disease is characterized by accumulation of hepatic fat, which over time can cause inflammation (steatohepatitis), and ultimately end-stage liver disease (cirrhosis, liver cancer).(1) Obesity is the major risk factor for FLD, and the increased prevalence of the disorder mirrors the increase in adiposity.(1) Other risk factors for FLD include a high intake of alcohol, a sedentary lifestyle, and a diet rich in refined sugars.(1, 3, 4)
Apart from these risk factors, genetics plays an important role in FLD development. Several studies report clustering of the disease in families.(5, 6) The first genome-wide association study (GWAS) for FLD found a common variant in PNPLA3 (I148M) that is associated with a 2-3 fold increase in HTGC, as measured by proton magnetic resonance spectroscopy (1H-MRS).(7) A subsequent GWAS, which used computerized tomography (CT) to assess HTGC, replicated the association with PNPLA3, and identified four additional loci (NCAN, LYPLAL1, GCKR, and PPP1R3B) that were associated with hepatic steatosis.(8) Three of these loci (NCAN, LYPLAL1, and GCKR) were also associated with histologic evidence of NAFLD. The fourth locus, PPP1R3B (defined by the SNP rs4240624, which is located 175 kb upstream of PPP1R3B) was strongly associated with HTGC (P=10−18), but not with histologically determined FLD. Thus, PPP1R3B may be associated with a benign form of hepatic steatosis that does not progress to steatohepatitis. (8)
Speliotes et. al. (8) noted that the discrepant effects of variation in PPP1R3B on liver fat and histologic FLD may have other explanations. The apparent discrepancy may represent a statistical artifact: either a false positive association with liver fat or a false negative association with histologic indices of disease. Alternatively, genetic variation in PPP1R3B locus may be associated with other factors, such as iron or glycogen, that affect hepatic X-ray attenuation independently of HTGC.(9)
The nature of the association between PPP1R3B and hepatic steatosis has important implications for FLD. If rs4240624 confers a benign form of hepatic steatosis, then liver TG per se is not pathogenic, and other factors must explain the progression of FLD that occurs in a subset of individuals. Subsequent studies replicated the association between rs4240624 and liver fat measured by hepatic X-ray signal attenuation (10) and by ultrasound.(11) These findings make false positive association unlikely. They also support the contention that variation in PPP1R3B is associated with HTGC, rather than glycogen, although neither CT nor ultrasound directly measures liver fat content. Gorden et al.(12) failed to find an association between rs4240624 and FLD using histologically determined liver fat content. Therefore, the relationship between the PPP1R3B and liver fat content remains equivocal. The notion that rs4240624 is not associated with histologic FLD also requires confirmation. In the study reported by Speliotes et al.(8), the sample that was available for association with NAFLD (n=592) was much smaller than the sample used for analysis of HTGC (n=7176); thus, the possibility of a false negative association cannot be excluded.
The close proximity of rs4240624 to PPP1R3B raises the possibility that the association between the SNP and hepatic X-ray attenuation is due to differences in hepatic glycogen rather than TG. PPP1R3B encodes a 285-residue protein that plays a key role in hepatic glycogen synthesis(13–15)(Fig. 1). PPP1R3B binds both protein phosphatase 1 (PP1) and glycogen (Fig. 1A). PP1 then dephosphorylates and activates glycogen synthase, which catalyzes the addition of UDP-glucose to glycogen (Fig. 1B). PP1 also dephosphorylates glycogen phosphorylase, inactivating the enzyme that catalyzes release of glucose from glycogen.(16) Thus, the overall effect of PPP1R3B is to promote formation of glycogen.(16) Here we tested the hypothesis that the association between rs4841132 (a proxy variant in complete linkage with rs4240624) and hepatic X-ray attenuation is due to changes in hepatic glycogen rather than TG content. Given that excessive glycogen accumulation is known to cause liver damage in patients with glycogen storage diseases (17), we also tested if rs4841132 confers susceptibility to liver disease. Finally, to confirm that the changes observed in hepatic X-ray attenuation are consistent with effects of alteration in hepatic PPP1R3B activity, we altered expression of the protein in mice and determined the effects on hepatic glycogen and TG content.
FIG 1.
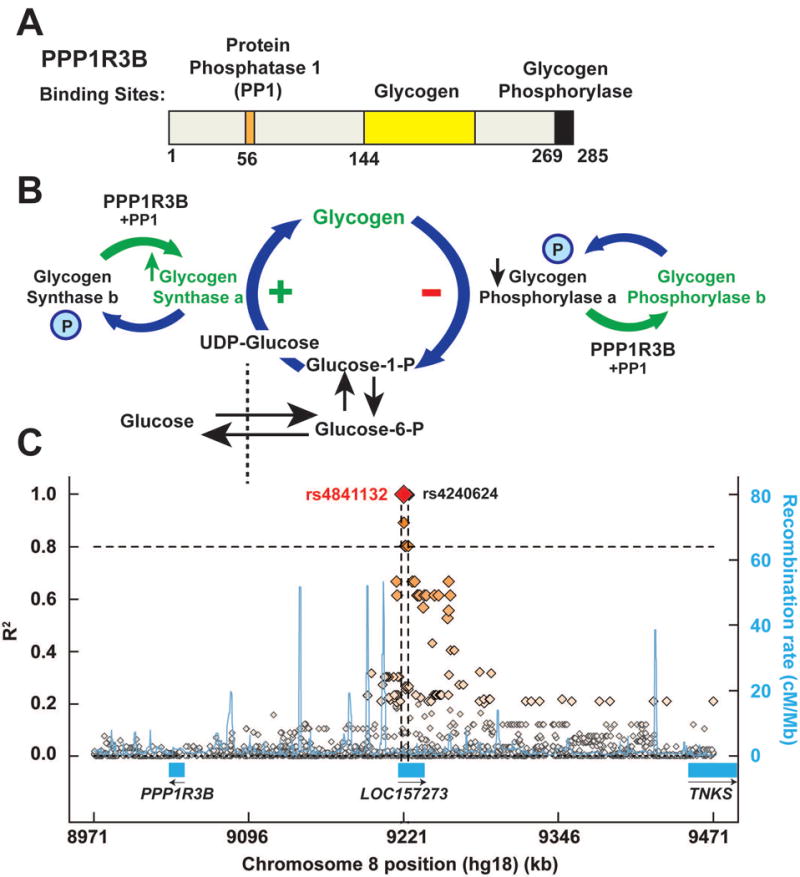
Structure (A), activity (B) and genomic location (C) of PPP1R3B. (A) Functional domains of the PPP1R3B protein showing the regions of the protein that bind protein phosphatase 1 (PP1), glycogen and glycogen phosphorylase. Figure adapted from Doherty et al.(15) (B) Schematic of hepatic glycogen metabolism and the function of PPP1R3B. Glucose taken up by the liver is phosphorylated and then converted to UDP-glucose prior to being incorporated into glycogen by glycogen synthase. Glycogen phosphorylase breaks down glycogen, releasing glucose-1-phosphate. PPP1R3B targets protein phosphatase 1 (PP1) to glycogen synthase and glycogen phosphorylase. PP1 then dephosphorylates these enzymes, leading to activation of glycogen synthase, and de-activation of glycogen phosphorylase. The overall effect of PPP1R3B is to promote glycogen formation. (C) Genomic region flanking rs4841132 (500 kb). The diamonds indicate individual genetic variants. Linkage disequilibrium of the variants with rs4841132 (R2) is given on Y axis (left) and the recombination hotspots are shown in blue with the recombination rates provided on Y axis (right). Note that both rs4841132 and rs4240624 are located in the liver-specific lncRNA LOC157273 (exon 2 and intron 2, respectively).
Experimental Procedures
Studies were approved by IRB and ethics committees of the University of Texas Southwestern Medical Center and Copenhagen and were conducted according to the Declaration of Helsinki. Written informed consent was obtained from all participants.
Human Subjects
We included participants from the DHS, the Dallas Liver Study, the Copenhagen City Heart Study (CCHS), and the Copenhagen General Population Study (CGPS).
The DHS is a multiethnic, probability-based sample of Dallas County residents that was collected between 2000 and 2002,(18) and 2007 and 2009. Ethnicity was self-reported in accordance with U.S. census categories. For this study we included 2,674 DHS participants in whom HTGC was measured using 1H-MRS, and 1,275 additional individuals for the analysis of serum ALT levels, and for use as controls for the Dallas Liver Study.
The Dallas Liver Study is a multiethnic sample of patients with liver disease of non-viral etiology. Participants were recruited from liver clinics at UT Southwestern and Parkland Health and Hospital System in Dallas, TX. Participants completed a questionnaire on ethnic/racial background, medical history, lifestyle factors, and family history of liver disease and other diseases. Additional clinical information was extracted from medical records by a trained technician. We included all African-Americans, European-Americans, and Hispanic-Americans (n=514). As healthy controls we used 514 age-matched and ethnicity-matched participants from the DHS. All individuals with a known etiology to their liver disease were not included.
The CCHS and CGPS are prospective studies of the Danish general population initiated in 1976 and 2003, respectively.(19) All participants from the CCHS and CGPS were white and of Danish descent, as determined by the National Danish Civil Registration System. We combined the CCHS and CGPS into one cohort, totaling 107,447 indivdiuals (Copenhagen Cohort). Individuals with the following ICD codes were included: ICD8: 57109 (alcoholic cirrhosis), 57111 (nonalcoholic steatohepatitis), 57119 (fatty liver), 57192 (unspecified cirrhosis), and 57199 (nonalcoholic cirrhosis), and ICD10: K70.3 (alcoholic cirrhosis), K74.0 (fibrosis or cirrhosis), K74.6 (unspecified cirrhosis), K76.0 (fatty liver), and K76.9 (liver disease, unspecified). These ICD-codes were selected since they are the ones most likely to be used in patients with liver disease of unknown etiology. Data were collected from the National Danish Patient Registry, and the National Danish Causes of Death Registry from January 1, 1977 to November 10, 2014. The National Danish Patient Registry has information on all patient contacts with clinical hospital departments in Denmark, including emergency wards and outpatient clinics (from 1994). The National Danish Causes of Death Registry contains data on the causes of all deaths in Denmark, as reported by hospitals and general practitioners. The diagnoses were combined into a dichotomous liver disease endpoint. In total, 1,025 participants in the Copenhagen Cohort had a diagnosis of liver disease, defined by ICD-codes as described above.
Measurement of HTGC and CT X-ray Attenuation
HTGC was measured in the DHS using 1H-MRS as previously described.(20) Hepatic CT attenuation was measured in 1,572 participants from the CGPS. A random sample of participants from the CGPS was invited to undergo a CT scan of the thorax and upper abdomen (Aquilion One; Toshiba Medical Systems Corporation, Tokyo, Japan). For liver fat assessment, a single, non-contrast, 16 cm volume scan of the upper abdomen was acquired and reconstructed using AIDR3D (Bodyfilter FC12).(21) CT-X-ray attenuation in liver segments 5-6 was measured using circular ~1.5 cm2 regions of interest.
Supplemental Methods
See the Supplemental Methods for details on i) other measurements and genotyping, ii) generation of Ppp1r3b KO mice, iii) generation of mice lacking a 10.6 kb region corresponding to LOC157273, iv) animal experimental procedures, v) generation of Adeno-PPP1R3B and overexpression of PPP1R3B in mice, vi) transfection of cells with LOC157273 and PPP1R3B, vii) RT-PCR and RNA-sequencing, and viii) statistical analysis.
Results
Baseline characteristics of the cohorts used in this study are shown (Table 1). The DHS and Dallas Liver Study are multiethnic, and the Copenhagen Cohort is 100% White Europeans. The frequency of rs4841132-A was 9% in Whites, 12% in African-Americans, and 24% in Hispanic-Americans.
Table 1.
Baseline characteristics of study participants.
| Study | Dallas Heart Study | Copenhagen Cohort | Dallas Liver Study |
|---|---|---|---|
| N | 4,467 | 107,447 | 514 cases |
| Age, y | 44 (36-53) | 58 (48-67) | 55 (46-61) |
| Female, n (%) | 2,571 (58) | 53,227 (55) | 298 (58) |
| BMI, kg/m2 | 29 (26-35) | 26 (23-28) | 31 (28-35) |
| European American, % | 30 | 100 | 26 |
| African American, % | 53 | 0 | 5 |
| Hispanic American,% | 17 | 0 | 69 |
Values are median and interquartile ranges, N or %. Data on age, gender, and BMI were available in ~97,000 from the Copenhagen Cohort.
ASSOCIATION BETWEEN RS4841132 AND HEPATIC X-RAY ATTENUATION
Previously, rs4240624 was found to be associated with HTGC.(8) Analysis of SNPs within the region revealed another variant, rs4841132 that is 635 bp downstream of rs4240624. The two SNPs are in complete LD (Fig. 1C). rs4841132 is located in exon 2 of the major transcript of an intergenic long noncoding RNA (lncRNA), LOC157273, which is located 175 kb upstream of PPP1R3B (Fig. 1C). rs4841132-A (minor allele) was associated with increased hepatic X-ray attenuation among 1572 participants from the Copenhagen Cohort (Fig. 2A, left). Mean Hounsfield units (HU) were 58.8, 60.6, and 63.2 for GG-homozygotes, GA-heterozygotes and AA-homozygotes, respectively (P-trend=4×10−5). To assess changes in attenuation due to differences in HTGC, we examined the same population after stratifying by the steatogenic PNPLA3(I148M) genotype (Fig. 2A). As expected, 148M was associated with decreased X-ray attenuation (P-trend=3×10−4). The absolute per-allele effect size on X-ray attenuation of rs4841132-A was approximately twice that of PNPLA3(148M) (+2.4 vs −1.4 HU per allele). Thus, the A-allele would be predicted to be associated with a substantial reduction in HTGC.
FIG. 2.
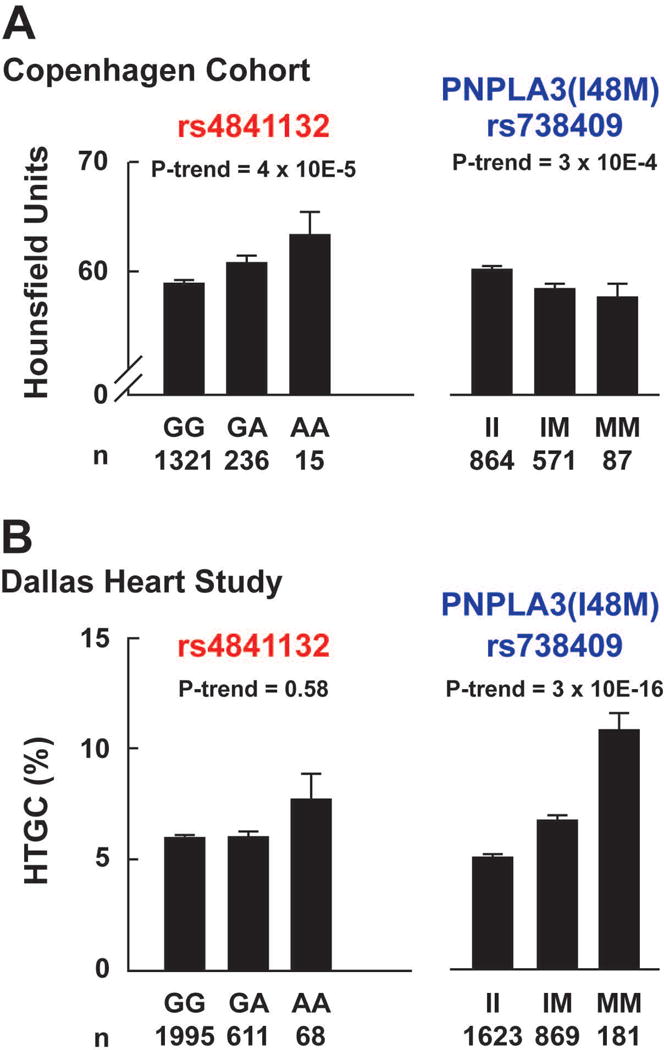
X-ray attenuation and HTGC as a function of rs4841132 and PNPLA3(I148M) (rs738409) genotype. (A) Hepatic CT was measured in 1,572 participants from the Copenhagen Cohort. (B) Magnetic resonance spectroscopy (MRS) was available in 2,674 participants from the Dallas Heart Study. Rs4841132 and rs738409 were genotyped as described in the Methods. P-values are from linear regression, adjusted for age, sex, body mass index and ethnicity. Bars and error bars indicate means and standard errors of the mean. Note: the Y-axes for hepatic CT measurements are cut at 50.
ASSOCIATION BETWEEN RS4841132 AND HTGC
Next we examined the relationship between rs4841132-A and HTGC in the DHS where HTGC was measured using 1H-MRS.(20) No association was found between HTGC and rs4841132-A in the entire cohort (Fig. 2B), or after stratifying participants by ethnicity (P>0.40, Fig. S1). Using a recessive model (AA vs GG+GA) did not change the null association (P=0.37). As shown previously, PNPLA3(148M) was strongly associated with HTGC in the DHS (P=3×10−16) (Fig. 2B, right).(7) Individuals homozygous for the risk variant (148M) had a 2-fold increase in HTGC compared to individuals homozygous for the non-risk allele (148I). This finding does not support the hypothesis that rs4841132 is associated with HTGC.
ASSOCIATION BETWEEN RS4841132 AND SERUM LIVER ENZYMES
To assess the effect of rs4841132 on hepatic inflammation, we examined the association between the variant and circulating liver enzyme levels. In both the Copenhagen Cohort and the DHS, the A-allele was associated with increased alanine aminotransaminase (ALT) levels (Fig. 3A, P-trend=3×10−4 and 0.004, respectively). A similar but weaker association were seen for AST in the larger cohort (P=0.04, Table S1), but not in the DHS (P= 0.27, Table S2). The A-allele was also associated with increased levels of alkaline phosphatase (ALP) in both the DHS (P=0.001) and Copenhagen Cohort (P= 9×10−25) (Fig. S2). Consistent with these findings, the minor allele of rs6984305, a sequence variant in LD with rs4841132 (r2=0.61), was associated with increased ALT (P=0.043) in a large GWAS of serum liver enzymes.(22) No association was found between rs4841132 and serum levels of bilirubin or gamma-glutamyltransferase in either the Copenhagen Cohort (Table S1) or DHS (Table S2). The association of rs4841132-A with increased plasma ALT levels is not consistent with the hypothesis that the allele is associated with protection from FLD.
FIG 3.
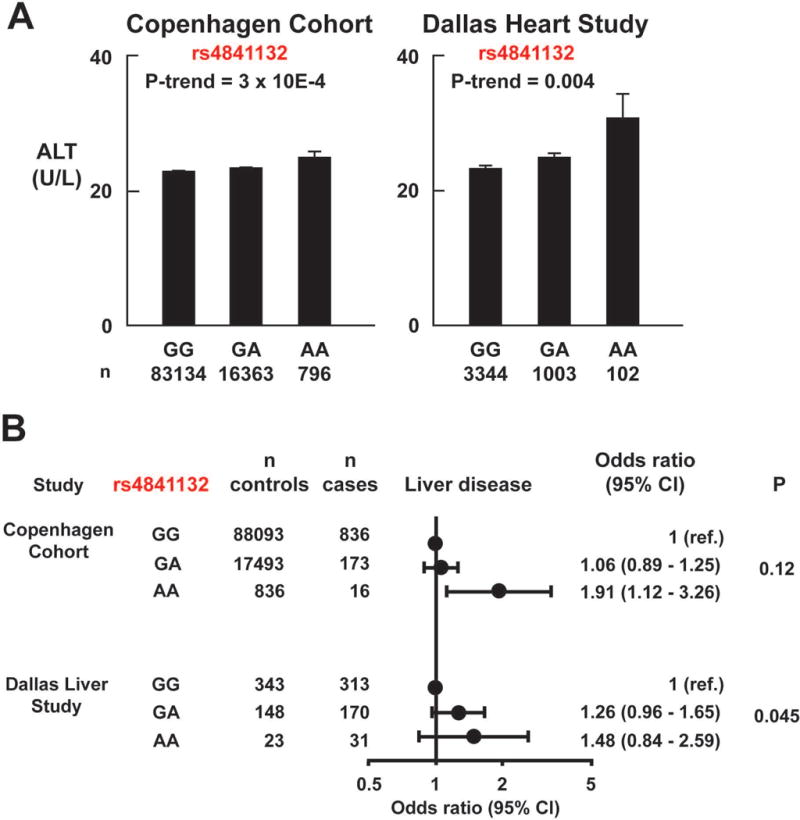
Risk of liver disease as a function of rs4841132 genotype. (A) Association of serum ALT levels with rs4841132 in the Copenhagen Cohort and Dallas Heart Study. Bars and error bars indicate means and standard errors of the mean. P-values are by linear regression, adjusted for age, sex, body mass index and ethnicity. (B) Risk of liver disease in the Copenhagen Cohort and the Dallas Liver Study. Subjects without an ICD-code for liver diseases were used as controls in the Copenhagen Cohort. Healthy subjects from the Dallas Heart Study were used as ethnicity and age-matched controls for the Dallas Liver Study cases. Homozygotes for the major G-allele acted as the reference group in each study. P-values are tests for trend, adjusted for age, gender and body mass index in the Copenhagen Cohort, and for gender in the Dallas Liver Study.
ASSOCIATION BETWEEN RS4841132 AND NONVIRAL LIVER DISEASE
Given that the rs4841132-A was associated with elevated liver enzymes, we tested if the variant was associated with increased risk of liver disease. For this analysis we used two cohorts of patients who had liver disease due to obesity (NAFLD) or to alcohol: the Copenhagen Cohort, which included 1,025 patients with liver disease, and 106,422 controls and the Dallas Liver Study, which included 514 patients with liver disease (65% Hispanic-American, 30% European-American, 5% African-American), and 514 age- and ethnicity-matched controls from the DHS (Fig. 3B). Patients with viral hepatitis were excluded. In the Copenhagen Cohort, the odds ratio for liver disease for rs4841132-AA versus –GG homozygotes was 1.91 (95%CI: 1.12-3.26). In both cohorts the rs4841132-A tended towards an association with liver disease, but the association did not reach significance (P=0.12) in the Copenhagen Cohort. Excluding individuals with ICD-codes specific for NAFLD (n=151) and/or alcoholic cirrhosis (n=168) did not change the results (data not shown). The trend was significant in the Dallas Liver Study (P=0.045), but the confidence intervals for all genotypes included 1. Meta-analysis across the two studies yielded a summary per-allele odds ratio of 1.16 (1.03-1.31, P=0.01). These data are consistent with an association between the rs4841132-A allele and liver disease.
As a positive control for the cohort, we tested the association of PNPLA3 (148M) with liver disease and plasma ALT levels in the Copenhagen Cohort, the DHS, and the Dallas Liver Study (Table S3). As expected, the M-allele was robustly associated with elevated ALT(19), and with increased risk of liver disease.
Rs4841132 MAPS TO A LIVER-SPECIFIC NONCODING RNA, LOC157273, THAT IS ASSOCIATED WITH INCREASED EXPRESSION OF PPP1R3B
Fig. 1C shows a LD map of the chromosomal region surrounding rs4841132. No significant LD was observed between rs4841132 and any of the SNPs in either flanking gene: PPP1R3B or TNKS, which encodes tankyrase, a telomere-associated poly-ADP ribose polymerase. Thus, the association between rs4841132 and hepatic glycogen cannot be attributed to LD with a sequence variation in either of the adjacent coding genes.
As noted above, rs4841132 is located in exon 2 of LOC157273, a liver-specific lncRNA expressed in humans and great apes. LOC157273 resides 175 kb upstream of PPP1R3B and 230 kb upstream of TNKS (Fig. 1C and 4A). To determine if the variant is associated with differences in levels of PPP1R3B or TNKS mRNA, we examined an eQTL database (n=707 liver samples) collected at Massachusetts General Hospital (Table S4).(23, 24) In that database, the rs4841132-A was associated with a significantly higher level of PPP1R3B mRNA in human livers (P=9×10−17). This finding supports the hypothesis that the minor allele of rs4841132 increases expression of hepatic PPP1R3B, which would be predicted to increase hepatic glycogen synthesis (Fig. 1B). No association was found between rs4841132-A and TNKS mRNA levels. In the same database, the A-allele was associated with reduced RNA levels of LOC157273 in human liver (P=3×10−8), raising the possibility that effects of rs4841132-A on PPP1R3B are mediated by LOC157273.
FIG 4.
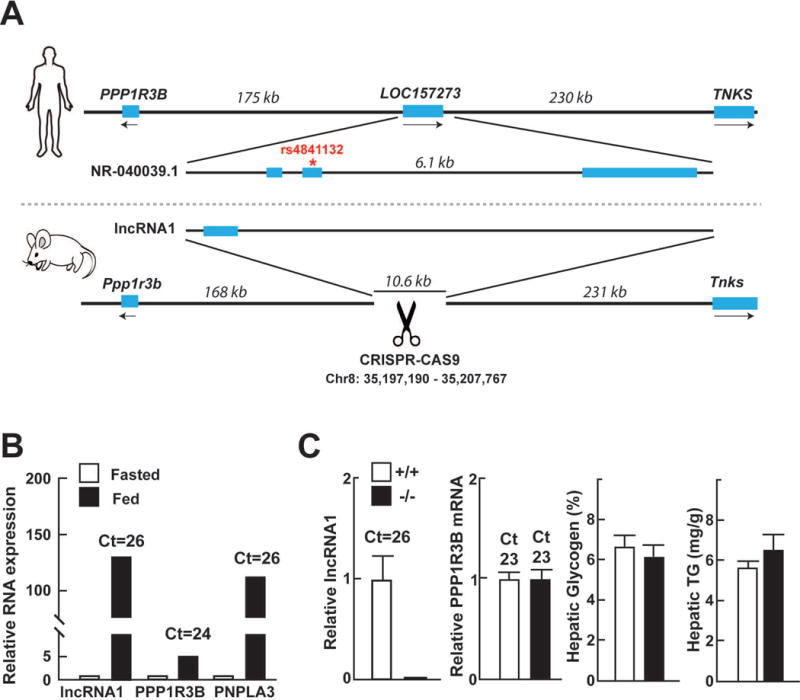
Genomic region of rs4841132 and knock-out of the corresponding region in mice. (A) LOC157273 is not conserved in mice, but RNA-sequencing of mouse liver identified a lncRNA in the same region. CRISPR/Cas9 was used to generate mice lacking a 10.6 kb region corresponding to LOC157273, including the mouse lncRNA. (B) Relative levels of hepatic lnc-RNA1, PPP1R3B, and PNPLA3 mRNA assayed by RT-qPCR in fasted mice and 4 h after refeeding with a chow diet. PNPLA3, which is induced by feeding,(40) served as a positive control in the experiment. (C) Levels of hepatic PPP1R3B mRNA, glycogen, and TG were not significantly different in mice homozygous for the genomic deletion (−/−) compared to WT controls (+/+).
To determine if differences in LOC157273 expression alter PPP1R3B mRNA levels or the glycogen content of cells, we expressed LOC157273 in three human cell lines that synthesize glycogen: embryonic kidney cells (293 cells),(25) and two immortalized human hepatocellular carcinoma cell lines, HepG2 and HuH7-cells. As a positive control, we expressed PPP1R3B in 293 cells, which was associated with a 20-fold increase in cellular glycogen content when compared to cells expressing vector alone (Fig. S3A).(13) In contrast, robust expression of LOC157273 (Ct-values 14-17 by RT-PCR) in the three cell lines failed to alter cellular levels of PPP1R3B mRNA (Ct-values 27-31) or glycogen (Fig. S3B). Thus, LOC157273 expression appears not to alter expression levels of PPP1R3B, or cause changes in glycogen content, at least in these cultured cell lines.
LOC157273 is not conserved in mice (Fig. 4A). To identify transcripts that might serve a function similar to LOC157273 in mice, we performed whole transcriptome analysis of mouse liver and identified transcripts that mapped to the interval between Ppp1r3b and Tnks. An unannotated transcript from the locus, which we have called lncRNA1 (mm10, Chr8: 35206516-35207482) was identified (GenBank Accession ID: MF573324). The transcript is predicted to have a single 967-bp exon and is robustly regulated by food intake (Fig. 4B).
To determine if the mouse lncRNA transcribed from the region, or the genomic region per se, plays a functional role in regulating Ppp1r3b, we used CRISPR to generate mice with a 10.6 kb deletion (mm10, Chr8: 35197190-35207767) that included both the lncRNA1 and the genomic region corresponding to human LOC157273. Mice homozygous for the deletion were viable and exhibited no overt phenotype. As anticipated, no lncRNA1 transcript was present in mice homozygous for the deletion (Fig. 4C). Levels of PPP1R3B mRNA, glycogen and Tg did not differ in the KO mice (Fig. 4C). Serum glucose levels did not differ between the strains (data not shown). Thus, lncRNA1 and/or the genomic region corresponding to LOC157273, appear not to influence expression of Ppp1r3b or alter hepatic glycogen or fat content in mice.
HEPATIC GLYCOGEN CONTENT WAS REDUCED AND HTGC WAS UNCHANGED IN Ppp1r3b−/− MICE
To test the hypothesis that hepatic X-ray attenuation associated with rs4841132-A is due to an increase in hepatic glycogen content caused by increased expression of PPP1R3B, we examined the hepatic glycogen and TG levels in the livers of mice that either lack (KO) or overexpress PPP1R3B. As we were performing these studies, Rader and his colleagues developed liver-specific Ppp1r3b KO mice.(26) These mice had reduced hepatic glycogen due to a reduction in incorporation of glucose into glycogen. The mice also had lower blood glucose levels on both an ad lib diet and after a 4 h fast. The effect of Ppp1r3b inactivation on liver TG content was not reported.
We used CRISPR to generate mice with a frameshift mutation in Ppp1r3b that is predicted to truncate the protein at residue 22 (p.Pro14Leufs*22). Breeding heterozygous carriers of the deletion yielded 14% homozygotes (Ppp1r3b−/−) among the offspring (56% Ppp1r3b+/− and 30% Ppp1r3b+/+). Despite being born in less than the expected Mendelian ratio, Ppp1r3b−/− mice were viable, fertile, and had a similar body weight at 8 weeks. In chow-fed male mice (8 weeks), fed for 4 h and then killed, hepatic glycogen content was markedly reduced in Ppp1r3b−/− mice compared to WT littermates (0.5% vs 4.4% glycogen/liver weight, P<0.001). HTGC was similar in KO and WT mice (Fig. 5A). Plasma levels of cholesterol, TG, glucose and insulin (both fed and fasted for 18 h) were unchanged (Fig. 5A and S4). These findings indicate that variation in Ppp1r3b expression has a major impact on liver glycogen levels but is not associated with significant changes in HTGC.
FIG 5.
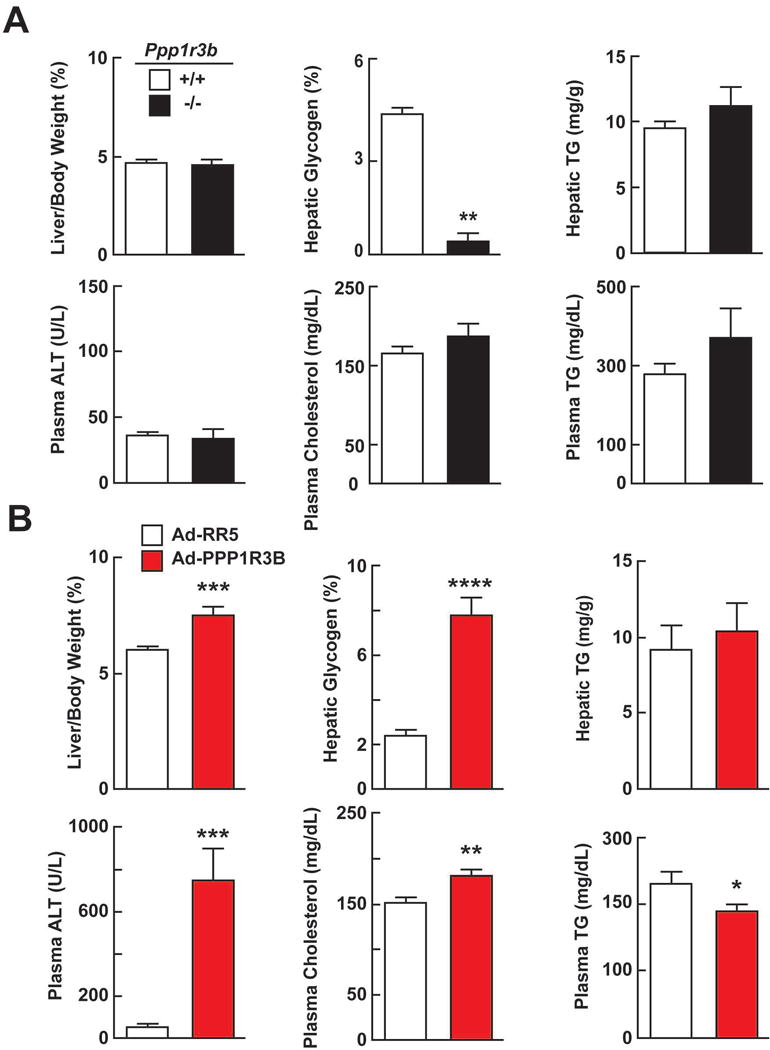
Phenotype of Ppp1r3b-KO mice (A) and mice infected with Ad-mouse PPPR1R3B. (A) Food intake was synchronized for 3 days (18 h fasting, 6 h feeding) in male mice (8 wk old, n=3-5/group). At the end of the fasting cycle on the 4th day, the mice were killed 4 h after they were fed. The experiment was repeated and the results were similar. P-values by t-test. * = P<0.05; ** = P<0.001 (B) Phenotype of mice overexpressing mouse PPP1R3B. The tail veins of 8-week old male mice (10/group) were injected with adenovirus expressing PPP1R3B (Ad-PPP1R3B) or an empty control virus (Ad-RR5) (1.25×1011 viral particles per mouse). The chow-fed mice were sacrificed 48 h after the injection. P-values by t-test. * = P<0.05; ** = P<0.01; ***P<0.001’ ****P<0.0001.
ADENOVIRUS- MEDIATED OVEREXPRESSION OF PPP1R3B IN MOUSE LIVER INCREASES GLYCOGEN CONTENT
Overexpression of PPP1R3B in livers of mice and rats is associated with increased hepatic glycogen content.(26, 27) Similarly, we found that adenovirus-mediated expression of PPP1R3B at supra-physiological levels in the livers of mice caused a dramatic increase in liver weight and hepatic glycogen content (Fig. 5B). Forty eight hours after injection of recombinant adenoviruses, livers of the mice expressing the mouse PPP1R3B transgene were visibly larger than those injected with the control virus (7.5% vs 6.0% liver/body weight, P<0.001). The hepatic glycogen content was increased in these mice (7.8% vs. 2.4% glycogen/liver weight, P<0.001) (Fig. 5B). The levels of hepatic PPP1R3B mRNA were 4-fold higher in mice expressing the PPP1R3B transgene (Ct=18 vs 20, Fig. S5). No changes in HTGC were seen with PPP1R3B overexpression (Fig. 5B). Plasma levels of ALT, ALP, and cholesterol were increased (Fig. 5B and Fig. S5) whereas TG levels were reduced in the PPP1R3B-treated mice (Fig. 5B); plasma levels of glucose and insulin were unchanged in these animals (Fig. S5).
Discussion
The main finding of this study is that the minor A-allele of rs4841132 is associated with increased hepatic X-ray attenuation on CT scan, but not with hepatic fat content. Our data support a model in which rs4841132-A is associated with increased activity of PPP1R3B, an activator of glycogen synthase and inhibitor of glycogen phosphorylase (Fig. 1). The net effect of the variant is to promote glycogen accumulation in the liver, which increases the attenuation of X-rays by this organ.(9) The accumulation of glycogen in carriers of rs4841132-A is associated with elevated circulating levels of ALT and also appears to confer increased susceptibility to liver disease. Observations in mice were consistent with this model: genetic manipulation of Ppp1r3b activity in mice resulted in large changes in liver glycogen content with no change in HTGC.
In agreement with prior studies, we found that variation at the PPP1R3B locus was strongly associated with hepatic X-ray attenuation.(8) However in our population, the genotype (GG) that was associated with decreased attenuation (consistent with increased fat) was associated with a reduce plasma ALT levels and reduced risk of liver disease. In contrast, the PNPLA3 genotype associated with decreased attenuation and increased HTGC was associated with higher ALT levels and with liver disease. These data are consistent with our observation that rs4841132 was not associated with HTGC (Fig. 2) and with previous studies where histology was used to assay hepatic TG.(8, 12) A study in the NHANES III cohort reported an association between the rs4240624 variant and HTGC measured using ultrasonography (OR: 1.28; 95% CI, 1.03-1.59; P=0.03).(11) However, the lack of association between liver fat content and PNPLA3(148M) [OR 1.14 (95% CI, 0.97-1.35)] in that study raises concerns about other reported associations. Taken together, the available data support the hypothesis that hepatic glycogen is the factor underlying the robust X-ray attenuation associated with rs4841132-A.
PPP1R3B increases hepatic glycogen accumulation by promoting dephosphorylation and activation of glycogen synthase and dephosphorylation and inactivation of glycogen phosphorylase, the enzymes governing synthesis and breakdown of hepatic glycogen (Fig. 1B).(31) The finding that rs4841132-A is associated with higher levels of PPP1R3B mRNA in human livers supports the hypothesis that the allele increases hepatic glycogen content by augmenting PPP1R3B activity.(23, 24) The mechanism by which rs4841132 influences the transcription of PPP1R3B is unknown. Intriguingly, rs4841132 maps to exon 2 of the main transcript of LOC157273, a liver-specific lncRNA that is encoded by 3 exons (NR-040039.1) (Fig. 4A). The mature LOC157273 transcript contains several open reading frames (longest is 119 residues) but these do not appear to be evolutionarily conserved. Could rs4841132 mediate its effect on PPP1R3B expression via LOC157273, as has been documented for regulation of other protein-coding genes by neighboring lncRNAs? Unfortunately, this hypothesis cannot be tested in a small animal model such as mice because LOC157273, like 30% of other lncRNAs, is not present outside of primates.(32)
We used whole transcriptome sequencing of mouse liver RNA to identify transcripts from the locus that might perform the same function as LOC157273. A single exon-containing lncRNA (lncRNA1 in this paper) sharing no sequence identity with LOC157273 was regulated in a coordinate fashion with PPP1R3B in response to fasting and refeeding (Fig. 4B). Noncoding RNAs can be expressed from the same location across species, but share little sequence conservation.(33) We hypothesized that the genomic region per se, or transcriptional activity at the region, might play a functional role in regulating Ppp1r3b. However, genetic deletion of a 10.6 kb genomic region that corresponds to LOC157273 and includes lncRNA1 did not affect Ppp1r3b expression or hepatic glycogen content (Fig. 4C). Thus, we found no evidence that this transcript, or any other sequences syntenic to LOC157273, affected expression levels of Ppp1r3b.
Rs4841132 is an eQTL for PPP1R3B (minor allele is associated with increased expression) and LOC157273 (decreased expression) in human liver. Thus, the SNP may affect PPP1R3B by altering expression of LOC157273, (ie. in trans). We examined the effects of expressing LOC157273 in immortalized human hepatocyte cell lines (HuH7, HepG2 cells) where endogenous LOC157273 is expressed at low levels (Ct value>30). Overexpression of the transcript did not influence mRNA levels of Ppp1r3b or glycogen content (Fig. S3B). Thus, the mechanism by which sequence variation at the locus affects PPP1R3B expression, and the role of the lncRNA LOC157273 in this interaction, remains unknown. We suspect that replacing the entire ~200 kb genomic region in mice with the corresponding human region might be required to get an experimental handle on this question.(34)
To determine if the associations observed in human carriers of rs4841132 are consistent with what would be expected from perturbing PPP1R3B expression, we generated mice that either lacked or overexpressed the gene and compared their phenotype with the human associations. While we were performing these studies, Mehta et al.(26) reported that liver-specific Ppp1r3b-KO mice had almost no liver glycogen, a slightly more rapid reduction in plasma glucose levels upon acute fasting, and increased plasma ketone levels after prolonged fasting.(26) The phenotypes of these mice are broadly consistent with those of the whole-body Ppp1r3b-KO mice studied by us. In addition, we found that HTGC was unchanged in Ppp1r3b-KO mice when compared to WT littermates. These data support the hypothesis that genetic variation in PPP1R3B primarily affects hepatic glycogen levels and has little effect on HTGC. Overexpression of PPP1R3B in the livers of rats (27) and mice (26) robustly increased hepatic glycogen content. In agreement with these prior studies, we found that hepatic overexpression of PPP1R3B in mice caused a marked increase in hepatic glycogen content, and increased plasma markers of liver damage, likely reflecting a glycogen-mediated hepatopathy. HTGC was not affected by PPP1R3B overexpression. Thus, genetic perturbation of PPP1R3B expression in mice changes hepatic glycogen content without affecting HTGC. These observations are consistent with the hypothesis that the increased CT attenuation in human carriers of rs4841132-A is due to elevations in hepatic glycogen content and not to changes in HTGC.
Excessive hepatic glycogen accumulation causes liver damage in patients with glycogen storage diseases,(17) but it is not known whether more modest elevations in hepatic glycogen content increase risk of liver disease. There is a clear dose-response relationship between HTGC and liver damage, and even modest increases in HTGC increase risk of liver damage.(19) We found that rs4841132-A was associated with a modest, but statistically significant and reproducible increase in plasma ALT, a marker of liver injury. Consistent with this observation, the minor allele of rs4240624 (in complete LD with rs4841132) was associated with higher mean ALT among 740 obese Mexicans.(35) We hypothesize that the increase in plasma ALT reflects a mild glycogen-induced hepatopathy.
There are limitations to our study that merit consideration. Definite proof that rs4841132 is glycogenic requires measurement of hepatic glycogen in humans.(36) To our knowledge, hepatic glycogen has not been measured in a large enough sample to detect modest effects. Potential methods to quantitate hepatic glycogen include histology (PAS staining), biochemical measurement in liver biopsies, or 13C-MRS of the liver in vivo. (36) Assessing the sample size required to detect an association between rs4841132 and hepatic glycogen content is complicated because the effect size of the variant and the natural variation of hepatic glycogen content in human populations are not known. Previous studies found that a 1% increase in biochemically measured hepatic glycogen content increases hepatic X-ray attenuation by ~3 HU.(9,37) Given that the per-allele effect of rs4841132 on X-ray attenuation was 2.4 HU in our study, we estimate that the per-allele effect of rs4841132 on hepatic glycogen content is ~0.8% (2.4 HU × 1%/3.0 HU). The hepatic glycogen content varies ~2-fold within a 24 h period.(38) In this context, an 0.8% increase (eg. from 4.0% to 4.8%) appears to be modest. It is therefore likely that a large sample size where feeding status is rigorously controlled will be required to detect association of rs4841132 with hepatic glycogen content.
Another limitation of this study is that the association between rs4841132 and liver disease only reached statistical significance in one of the two liver patient cohorts examined. The association with liver disease will need to be validated in large patient cohorts. Unfortunately, most liver patient cohorts are of European ancestry, and the frequency of rs4841132-A (9% in Whites vs 12% in Blacks and 24% in Hispanics), limits the power to detect associations. In a German/UK GWAS(39) of alcoholic liver disease (n=712 alcoholic cirrhotics and 1,426 alcoholic controls), rs4841132-A was not associated with alcoholic cirrhosis (OR: 0.97 [95% CI, 0.76-1.25]). Larger patient cohorts, preferably of Hispanic ancestry, will be required to definitively refute or confirm an association with liver disease.
There were only 1,025 cases with liver disease in the Copenhagen Cohort. Potential explanations for the low prevalence of disease in this population include: 1) the registry-based method of ascertainment selects for symptomatic patients; 2) the median BMI in the Danish cohort was 26 kg/m2, lower than in Americans; 3) until recently, NAFLD was under-diagnosed. Nonetheless, the validity of the endpoints used in this paper was supported by the finding that PNPLA3(I148M) was strongly associated with FLD in this study.
Finally, the causal SNP, and the mechanism by which it influences PPP1R3B transcription, remains unknown. We studied rs4841132, but there are several other SNPs at the locus that are in high LD, any of which could contribute to the association. We speculated that the lncRNA LOC157273 could play a role in mediating the effect of rs4841132 on PPP1R3B transcription. An alternative possibility is that the causal SNP at the locus acts directly on PPP1R3B transcription, and that LOC157273 is irrelevant.
In conclusion, the minor allele of r4841132 is associated with increased hepatic X-ray attenuation, and with modestly elevated plasma markers of liver damage. These observations are consistent with the notion that rs4841132 promotes a mild form of hepatic glycogenosis.
Supplementary Material
Acknowledgments
We thank Fang Xu, Liangcai Nie, and Vanessa Schmid for excellent technical assistance, Mohammed Kanchwala for analyzing the RNA-sequencing data, and Dermot Reilly (Merck, Inc.) for providing us data from the MGH-liver eQTL database. This work was supported by National Institutes of Health grants to R24HL123879, R01HG003988, and UM1HG009421, and research was conducted at the E.O. Lawrence Berkeley National Laboratory and performed under Department of Energy Contract DE-AC02-05CH11231, University of California.
Financial Support: SS was supported by a Sapere Aude grant from the Danish Medical Research Council (4004-00398). JCC and HHH were supported by grants from the National Institute of Health: RO1 DK090066 and PO1 HL20948.
Abbreviations
- ALP
alkaline phosphatase
- ALT
alanine aminotransferase
- AST
aspartate aminotransferase
- FLD
fatty liver disease
- HTGC
hepatic triglyceride content
- 1H-MRS
proton magnetic resonance spectroscopy
- KO
knockout
- NAFLD
nonalcoholic liver disease
- lncRNA
long noncoding RNA
- PNPLA3
patatin-like phospholipase-domain containing protein 3
- PP1
protein phosphatase 1
- PPP1R3B
Protein phosphatase 1 regulatory subunit 3B
- TG
triglycerides
- WT
wildtype
- HU
Hounsfield units
Footnotes
Potential conflict of interest: none
References
- 1.Cohen JC, Horton JD, Hobbs HH. Human fatty liver disease: old questions and new insights. Science. 2011;332:1519–1523. doi: 10.1126/science.1204265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ludwig J, Viggiano TR, McGill DB, Oh BJ. Nonalcoholic steatohepatitis: Mayo Clinic experiences with a hitherto unnamed disease. Mayo Clin Proc. 1980;55:434–438. [PubMed] [Google Scholar]
- 3.York LW, Puthalapattu S, Wu GY. Nonalcoholic fatty liver disease and low-carbohydrate diets. Annu Rev Nutr. 2009;29:365–379. doi: 10.1146/annurev-nutr-070208-114232. [DOI] [PubMed] [Google Scholar]
- 4.Dietrich P, Hellerbrand C. Non-alcoholic fatty liver disease, obesity and the metabolic syndrome. Best Pract Res Clin Gastroenterol. 2014;28:637–653. doi: 10.1016/j.bpg.2014.07.008. [DOI] [PubMed] [Google Scholar]
- 5.Schwimmer JB, Celedon MA, Lavine JE, Salem R, Campbell N, Schork NJ, et al. Heritability of nonalcoholic fatty liver disease. Gastroenterology. 2009;136:1585–1592. doi: 10.1053/j.gastro.2009.01.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Brouwers MC, Cantor RM, Kono N, Yoon JL, van der Kallen CJ, Bilderbeek-Beckers MA, et al. Heritability and genetic loci of fatty liver in familial combined hyperlipidemia. J Lipid Res. 2006;47:2799–2807. doi: 10.1194/jlr.M600312-JLR200. [DOI] [PubMed] [Google Scholar]
- 7.Romeo S, Kozlitina J, Xing C, Pertsemlidis A, Cox D, Pennacchio LA, et al. Genetic variation in PNPLA3 confers susceptibility to nonalcoholic fatty liver disease. Nat Genet. 2008;40:1461–1465. doi: 10.1038/ng.257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Speliotes EK, Yerges-Armstrong LM, Wu J, Hernaez R, Kim LJ, Palmer CD, et al. Genome-wide association analysis identifies variants associated with nonalcoholic fatty liver disease that have distinct effects on metabolic traits. PLoS Genet. 2011;7:e1001324. doi: 10.1371/journal.pgen.1001324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dwyer A, Doppman JL, Adams AJ, Girton ME, Chernick SS, Cornblath M. Influence of glycogen on liver density: computed tomography from a metabolic perspective. J Comput Assist Tomogr. 1983;7:70–73. doi: 10.1097/00004728-198302000-00012. [DOI] [PubMed] [Google Scholar]
- 10.Palmer ND, Musani SK, Yerges-Armstrong LM, Feitosa MF, Bielak LF, Hernaez R, et al. Characterization of European ancestry nonalcoholic fatty liver disease-associated variants in individuals of African and Hispanic descent. Hepatology. 2013;58:966–975. doi: 10.1002/hep.26440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hernaez R, McLean J, Lazo M, Brancati FL, Hirschhorn JN, Borecki IB, Harris TB, et al. Association between variants in or near PNPLA3, GCKR, and PPP1R3B with ultrasound-defined steatosis based on data from the Third National Health and Nutrition Examination Survey. Clin Gastroenterol Hepatol. 2013;11:1183–1190. e1182. doi: 10.1016/j.cgh.2013.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gorden A, Yang R, Yerges-Armstrong LM, Ryan KA, Speliotes E, Borecki IB, Harris TB, et al. Genetic variation at NCAN locus is associated with inflammation and fibrosis in non-alcoholic fatty liver disease in morbid obesity. Hum Hered. 2013;75:34–43. doi: 10.1159/000346195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gasa R, Jensen PB, Berman HK, Brady MJ, DePaoli-Roach AA, Newgard CB. Distinctive regulatory and metabolic properties of glycogen-targeting subunits of protein phosphatase-1 (PTG, GL, GM/RGl) expressed in hepatocytes. J Biol Chem. 2000;275:26396–26403. doi: 10.1074/jbc.M002427200. [DOI] [PubMed] [Google Scholar]
- 14.Armstrong CG, Doherty MJ, Cohen PT. Identification of the separate domains in the hepatic glycogen-targeting subunit of protein phosphatase 1 that interact with phosphorylase a, glycogen and protein phosphatase 1. Biochem J. 1998;336(Pt 3):699–704. doi: 10.1042/bj3360699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Doherty MJ, Moorhead G, Morrice N, Cohen P, Cohen PT. Amino acid sequence and expression of the hepatic glycogen-binding (GL)-subunit of protein phosphatase-1. FEBS Lett. 1995;375:294–298. doi: 10.1016/0014-5793(95)01184-g. [DOI] [PubMed] [Google Scholar]
- 16.Newgard CB, Brady MJ, O’Doherty RM, Saltiel AR. Organizing glucose disposal: emerging roles of the glycogen targeting subunits of protein phosphatase-1. Diabetes. 2000;49:1967–1977. doi: 10.2337/diabetes.49.12.1967. [DOI] [PubMed] [Google Scholar]
- 17.Kilimann MW, Oldfors A. Glycogen pathways in disease: new developments in a classical field of medical genetics. J Inherit Metab Dis. 2015;38:483–487. doi: 10.1007/s10545-014-9785-5. [DOI] [PubMed] [Google Scholar]
- 18.Victor RG, Haley RW, Willett DL, Peshock RM, Vaeth PC, Leonard D, et al. The Dallas Heart Study: a population-based probability sample for the multidisciplinary study of ethnic differences in cardiovascular health. Am J Cardiol. 2004;93:1473–1480. doi: 10.1016/j.amjcard.2004.02.058. [DOI] [PubMed] [Google Scholar]
- 19.Stender S, Kozlitina J, Nordestgaard BG, Tybjaerg-Hansen A, Hobbs HH, Cohen JC. Adiposity amplifies the genetic risk of fatty liver disease conferred by multiple loci. Nat Genet. 2017;49:842–847. doi: 10.1038/ng.3855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Szczepaniak LS, Nurenberg P, Leonard D, Browning JD, Reingold JS, Grundy S, et al. Magnetic resonance spectroscopy to measure hepatic triglyceride content: prevalence of hepatic steatosis in the general population. Am J Physiol Endocrinol Metab. 2005;288:462–468. doi: 10.1152/ajpendo.00064.2004. [DOI] [PubMed] [Google Scholar]
- 21.Fuchs A, Mejdahl MR, Kühl JT, Stisen ZR, Nilsson EJ, Køber LV, et al. Normal values of left ventricular mass and cardiac chamber volumes assessed by 320-detector computed tomography angiography in the Copenhagen General Population Study. Eur Heart J Cardiovasc Imaging. 2016;17:1009–17. doi: 10.1093/ehjci/jev337. [DOI] [PubMed] [Google Scholar]
- 22.Chambers JC, Zhang W, Sehmi J, Li X, Wass MN, Van der Harst P, et al. Genome-wide association study identifies loci influencing concentrations of liver enzymes in plasma. Nat Genet. 2011;43:1131–1138. doi: 10.1038/ng.970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhong H, Beaulaurier J, Lum PY, Molony C, Yang X, Macneil DJ, et al. Liver and adipose expression associated SNPs are enriched for association to type 2 diabetes. PLoS Genet. 2010;6:e1000932. doi: 10.1371/journal.pgen.1000932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Teslovich TM, Musunuru K, Smith AV, Edmondson AC, Stylianou IM, Koseki M, et al. Biological, clinical and population relevance of 95 loci for blood lipids. Nature. 2010;466:707–713. doi: 10.1038/nature09270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Oligschlaeger Y, Miglianico M, Chanda D, Scholz R, Thali RF, Tuerk R, et al. The recruitment of AMP-activated protein kinase to glycogen is regulated by autophosphorylation. J Biol Chem. 2015;290:11715–11728. doi: 10.1074/jbc.M114.633271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mehta MB, Shewale SV, Sequeira RN, Millar JS, Hand NJ, Rader DJ. Hepatic protein phosphatase 1 regulatory subunit 3B (Ppp1r3b) promotes hepatic glycogen synthesis and thereby regulates fasting energy homeostasis. J Biol Chem. 2017;292:10444–10454. doi: 10.1074/jbc.M116.766329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gasa R, Clark C, Yang R, DePaoli-Roach AA, Newgard CB. Reversal of diet-induced glucose intolerance by hepatic expression of a variant glycogen-targeting subunit of protein phosphatase-1. J Biol Chem. 2002;277:1524–1530. doi: 10.1074/jbc.M107744200. [DOI] [PubMed] [Google Scholar]
- 28.Roudot-Thoraval F, Halphen M, Larde D, Galliot M, Rymer JC, Galacteros F. Evaluation of liver iron content by computed tomography: its value in the follow-up of treatment in patients with idiopathic hemochromatosis. Hepatology. 1983;3:974–979. doi: 10.1002/hep.1840030615. [DOI] [PubMed] [Google Scholar]
- 29.Dixon AK, Walshe JM. Computed tomography of the liver in Wilson disease. J Comput Assist Tomogr. 1984;8:46–49. doi: 10.1097/00004728-198402000-00010. [DOI] [PubMed] [Google Scholar]
- 30.Patrick D, White FE, Adams PC. Long-term amiodarone therapy: a cause of increased hepatic attenuation on CT. Br J Radiol. 1984;57:573–576. doi: 10.1259/0007-1285-57-679-573. [DOI] [PubMed] [Google Scholar]
- 31.Agius L. Role of glycogen phosphorylase in liver glycogen metabolism. Mol Aspects Med. 2015;46:34–45. doi: 10.1016/j.mam.2015.09.002. [DOI] [PubMed] [Google Scholar]
- 32.Derrien T, Johnson R, Bussotti G, Tanzer A, Djebali S, Tilgner H, et al. The GENCODE v7 catalog of human long noncoding RNAs: analysis of their gene structure, evolution, and expression. Genome Res. 2012;22:1775–1789. doi: 10.1101/gr.132159.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Johnsson P, Lipovich L, Grandér D, Morris KV. Evolutionary conservation of long non-coding RNAs; sequence, structure, function. Biochim Biophys Acta. 2014;1840:1063–71. doi: 10.1016/j.bbagen.2013.10.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bassett AR, Akhtar A, Barlow DP, Bird AP, Brockdorff N, Duboule D, et al. Considerations when investigating lncRNA function in vivo. Elife. 2014;3:e03058. doi: 10.7554/eLife.03058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Flores YN, Velazquez-Cruz R, Ramirez P, Banuelos M, Zhang ZF, Yee HF, Jr, et al. Association between PNPLA3 (rs738409), LYPLAL1 (rs12137855), PPP1R3B (rs4240624), GCKR (rs780094), and elevated transaminase levels in overweight/obese Mexican adults. Mol Biol Rep. 2016;43:1359–1369. doi: 10.1007/s11033-016-4058-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Buehler T, Bally L, Dokumaci AS, Stettler C, Boesch C. Methodological and physiological test-retest reliability of (13) C-MRS glycogen measurements in liver and in skeletal muscle of patients with type 1 diabetes and matched healthy controls. NMR Biomed. 2016;29:796–805. doi: 10.1002/nbm.3531. [DOI] [PubMed] [Google Scholar]
- 37.Leander P, Månsson S, Pettersson G. Glycogen content in rat liver. Importance for CT and MR imaging. Acta Radiol. 2000;41:92–6. [PubMed] [Google Scholar]
- 38.Wise S, Nielsen M, Rizza R. Effects of hepatic glycogen content on hepatic insulin action in humans: alteration in the relative contributions of glycogenolysis and gluconeogenesis to endogenous glucose production. J Clin Endocrinol Metab. 1997;82:1828–1833. doi: 10.1210/jcem.82.6.3971. [DOI] [PubMed] [Google Scholar]
- 39.Buch S, Stickel F, Trepo E, Way M, Herrmann A, Nischalke HD, et al. A genome-wide association study confirms PNPLA3 and identifies TM6SF2 and MBOAT7 as risk loci for alcohol-related cirrhosis. Nat Genet. 2015;47:1443–1448. doi: 10.1038/ng.3417. [DOI] [PubMed] [Google Scholar]
- 40.Huang Y, Cohen JC, Hobbs HH. Expression and characterization of a PNPLA3 protein isoform (I148M) associated with nonalcoholic fatty liver disease. J Biol Chem. 2011;286:37085–37093. doi: 10.1074/jbc.M111.290114. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.