

Abstract
Natural IgM binds to glomerular epitopes in several progressive kidney diseases. Previous work has shown that IgM also binds within the glomerulus after ischemia/reperfusion (I/R) but does not fully activate the complement system. Factor H is a circulating complement regulatory protein, and congenital or acquired deficiency of factor H is a strong risk factor for several types of kidney disease. We hypothesized that factor H controls complement activation by IgM in the kidney after I/R, and that heterozygous factor H deficiency would permit IgM-mediated complement activation and injury at this location. We found that mice with targeted heterozygous deletion of the gene for factor H developed more severe kidney injury after I/R than wild-type controls, as expected, but that complement activation within the glomeruli remained well controlled. Furthermore, mice that are unable to generate soluble IgM were not protected from renal I/R, even in the setting of heterozygous factor H deficiency. These results demonstrate that factor H is important for limiting injury in the kidney after I/R, but it is not critical for controlling complement activation by immunoglobulin within the glomerulus in this setting. IgM binds to glomerular epitopes after I/R, but it is not a significant source of injury.
Keywords: complement, kidney, IgM, factor H, ischemia
Graphical Abstract
The complement cascade is activated in the kidney by two distinct mechanisms after ischemia/reperfusion. In the glomerulus, IgM binds to neoepitopes expressed on damaged cells and activates the classical pathway, but activation remains controlled at the level of C4. In the tubulointerstitium, decreased complement regulation permits activation of the alternative pathway. Soluble factor H controls this process, and more severe injury is seen in mice that are deficient in factor H.
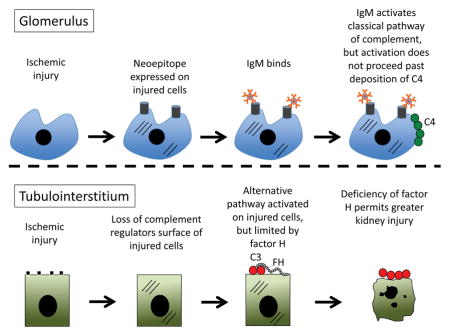
Introduction
The alternative pathway of complement mediates kidney injury in many different diseases, including ischemic acute kidney injury (AKI) [1], anti-neutrophil cytoplasmic autoantibody (ANCA)-associated vasculitis [2], atypical hemolytic uremic syndrome (aHUS) [3], and C3 glomerulopathy (C3G) [4]. Factor H is the main endogenous regulator of the alternative pathway [5], and pathologic alternative pathway activation in these diseases presumably involves impaired or insufficient regulation by factor H within the kidney. Indeed, factor H mutations are strongly associated with the development of atypical hemolytic uremic syndrome and C3G [6], and these diseases are limited to the kidneys even though factor H controls complement activation throughout the body. These observations suggest that the kidney is uniquely dependent on factor H to prevent alternative pathway-mediated injury.
Although genetic defects of factor H are associated with C3G and aHUS, the morphologic patterns of injury in these diseases differs. C3G is a rare and severe form of glomerulonephritis. Inflammatory changes are seen in the glomeruli, but the histologic pattern of injury varies among patients [7]. By definition, C3 deposits are always seen in the glomeruli of patients with C3G, and can be located within the mesangium and/or the capillary loops. In aHUS, thrombi are often seen within the glomerular capillaries but C3 is not prominently deposited [7]. Chronically, aHUS can cause a membranoproliferative pattern of injury by light microscopy, similar to what is seen in some patients with C3G. Nevertheless, the morphologic differences between these diseases possibly reflect complement activation on distinct surfaces within the glomeruli [8].
Although the alternative pathway is involved in the pathogenesis of glomerular and tubulointerstitial diseases, complement activation at various locations within the kidney is likely caused by distinct molecular events that depend on the local microenvironment [9]. Alternative pathway activation in ischemic AKI, for example, appears to involve specific responses of tubular epithelial cells (TECs) to injury. The cells produce complement proteins, such as C3, increasing local activation [10]. Basolateral expression of the cell surface complement regulator Crry on TECs decreases in response to hypoxia [11], further promoting complement activation on the cell surface [12]. We have also found that complement regulation by factor H is impaired on the surface of hypoxic TECs, [13] possibly due to the production of molecules – such as annexin A2 - that block factor H through protein-protein interactions [14]. Kidney ischemia, therefore, simultaneously increases the local concentration of C3 while reducing complement regulation on the TEC surface by factor H and Crry.
Factor H is also critical for controlling alternative pathway activation in the glomerulus. No complement regulatory proteins are intrinsically expressed as a component of the glomerular basement membrane (GBM), and factor H deficiency (either genetic or acquired) permits alternative pathway activation on this surface [15, 16]. Even though immune complexes activate the complement system through the classical pathway, amplification through the alternative pathway also contributes to tissue injury in immune-complex glomerulonephritis [17]. Factor H controls this process, and consequently deficiency of factor H increases the severity of immune-complex-mediated glomerular injury [18, 19].
Factor H also controls alternative pathway activation after I/R. Recurrence of aHUS is very common in the peri-transplant period, possibly because of ischemia-induced complement activation within the kidney [20]. Similarly, acute liver failure after transplantation has been reported in patients with factor H mutations [21, 22]. Extensive complement activation was observed within the livers of these patients, suggesting that deficiency of factor H permitted uncontrolled complement activation within the liver in the setting of ischemia similar to what occurs in the kidney. In ischemic AKI, alternative pathway activation primarily occurs in the tubulointerstium [1]. We have previously found that IgM deposits in the glomeruli of mice after I/R and activates the classical pathway of complement, as manifest by C4 deposition [23]. The alternative pathway remains very well controlled at this location after ischemia, however, and glomerular classical pathway activation does not result in subsequent C3 deposition and engagement of the amplification loop. Given the importance of factor H for controlling alternative pathway activation in the glomerulus, we hypothesized that glomerular IgM would cause complement activation and injury in factor H deficient mice after renal I/R. To test this hypothesis, we studied mice with a heterozygous targeted deletion of the gene for factor H (fH+/− mice [16]). To further explore the role of IgM in ischemic AKI, we crossed the fH+/− mice with another strain of mice that has a mutation in the μs exon of the IgM gene (sIgM−/− mice [24]). This mutation prevents secretion of soluble IgM by B cells. We then compared kidney injury and complement activation in wild-type, fH+/−, and fH+/−sIgM−/− mice after renal I/R.
Results
Ischemic AKI is more severe in mice with deficiency of factor H than wild-type control mice
We previously reported that mice with homozygous deletion of the gene for factor H are protected from ischemic AKI, likely because the lack of factor H-mediated control of spontaneous activation in the circulation causes almost complete consumption of C3 [25]. In the current study we compared the severity of ischemic AKI in mice with heterozygous deficiency of factor H, which in contrast maintain functional C3 levels, with wild-type controls. Serum urea nitrogen (SUN) and creatinine levels were significantly higher in fH+/− mice after I/R compared to wild-type controls (Figure 1A), indicating more severe kidney injury than in control mice. Tubular damage was seen in both fH+/− mice and wild-type controls, but the glomeruli were normal appearing in both strains of mice (Figure 1B).
Figure 1. Factor H protects mice from ischemic acute kidney injury.
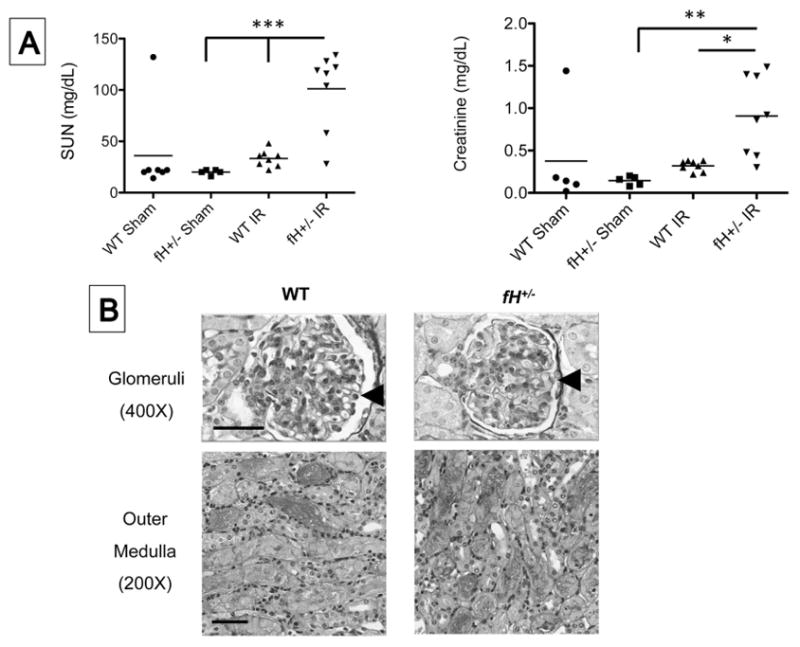
Wild-type (WT; C57BL/6) and mice with heterozygous deficiency of factor H (fH+/−) were subjected to ischemia/reperfusion of the kidneys. A) After 24 hours the serum urea nitrogen (SUN) and creatinine levels were measured using a NADH oxidation assay and a picric acid chromogenic assay, respectively. Values were compared by ANOVA with Tukey post test. **P < 0.01, ***P < 0.001. Each mouse is represented by a data-point, and the mean for each group is shown. Data shown is pooled from four independent experiments with 5–8 mice per experiment. Each mouse is represented by a data-point, and the mean for each group is shown. B) One representative image of two number of independent experiments (5–8 mice in each group) showing kidney sections stained with periodic acid Schiff. Scale bar = 100 μm for 200X images and 50 μm for 400X images.
Factor H deficiency is not associated with glomerular complement activation after I/R
Although plasma C3 levels are lower in unmanipulated fH+/− mice than in wild-type controls, heterozygous expression of the factor H gene is sufficient to control C3 consumption in the fluid phase and to support complement activation within tissues [16]. Western blot analysis confirmed that intact C3 is present in the plasma of fH+/− mice (Figure 2A). Although we expected heterozygous factor H deficiency to permit greater complement activation within the kidneys after I/R, Western blot analysis of kidney lysates suggested a greater abundance of C3b fragments in the kidneys of wild-type mice than in fH+/− mice (Figure 2B).
Figure 2. Complement activation in the kidneys of mice with ischemic acute kidney injury.
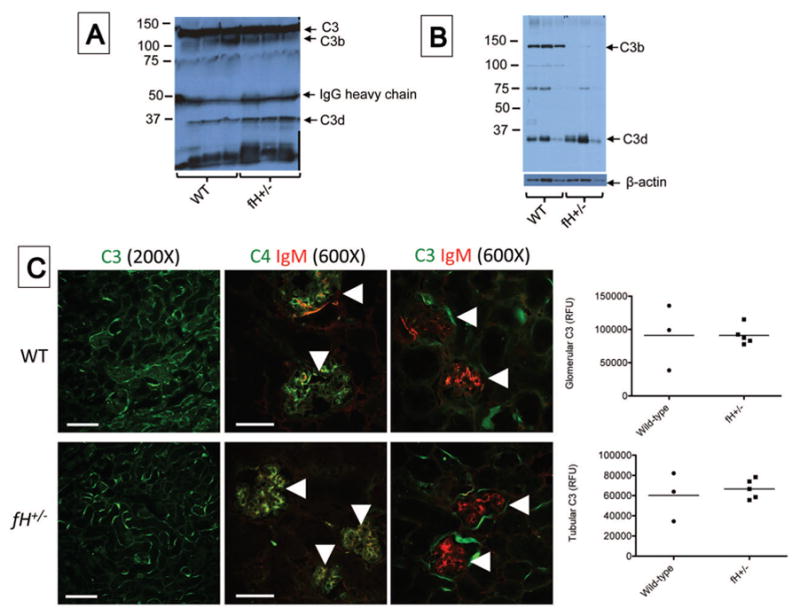
Wild-type (WT; C57BL/6) and mice with heterozygous deficiency of factor H (fH+/−) were subjected to ischemia/reperfusion of the kidneys. After 24 hours serum and tissues were collected. A) Western blot analysis of plasma from wild-type and factor H deficient mice showing levels of circulating intact C3. C3 fragments were detected using a monoclonal antibody to C3d (mAB 3d11) B) Western blot analysis of kidney lysates for C3b in factor H deficient mice with ischemic acute kidney injury compared to wild-type controls. The blot was re-probed with an antibody for β-actin as a loading control. C) Tissue sections were stained using antibodies to murine C3, C4, and IgM. Immunofluorescence microscopy for C3, C4, and IgM revealed C3 deposits throughout the tubulointerstitium of both strains of mice. High-powered views of the glomeruli (indicated with arrowheads) demonstrates the deposits of IgM and C4 co-localize within the glomeruli of both strains of mice. unpaired student t test showed no significant difference in both genotypes. Scale bar = 100 μm for 200X images and 50 μm for 600X images. Fluorescence data shown is representative of two experiments from which 3–5 mice from each group were analyzed. Each data point represents one mouse, and the bar represents the group mean.
Previously it was shown that alternative pathway activation causes C3 deposition on the renal tubules of wild-type ice after I/R, but C3 deposition is not seen in the glomeruli [1]. To determine whether heterozygous factor H deficiency would permit greater complement activation in either of these compartments after I/R we performed immunofluorescence staining for C3, which revealed C3 deposits throughout the tubulointerstitium of both wild-type and fH+/− mice (Figure 2C). We previously found that IgM (but not IgG) binds within the glomeruli of mice with homozygous deficiency of factor H and contributes to kidney injury [26]. In the current study we found that glomerular IgM was seen in both wild-type and fH+/− mice after I/R (Figure 2C). The IgM co-localized with C4, indicating classical pathway activation. C3 did not co-localize with IgM in the glomeruli of either strain of mice, however, indicating that complement activation by glomerular IgM remains well controlled even in mice with heterozygous deficiency of factor H. Staining of kidney sections for C6 demonstrated deposits in kidneys of wild-type and fH+/− mice (Figure 3). C6 appeared more abundant in the fH+/− mice, suggesting that factor H deficiency may be associated with worse kidney injury due to greater activation of the terminal complement pathway.
Figure 3. C6 deposition in the kidneys of mice with ischemic acute kidney injury.
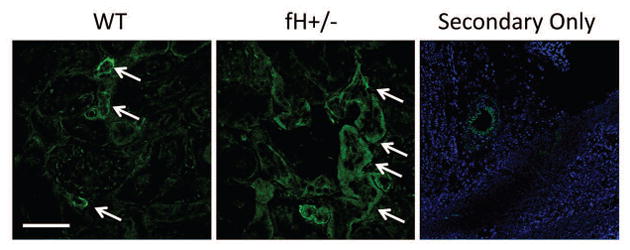
Wild-type (WT; C57BL/6) and mice with heterozygous deficiency of factor H (fH+/−) were subjected to ischemia/reperfusion of the kidneys. After 24 hours tissues were collected, and kidney sections from four mice in each group were stained for C6.. Original magnification x600 and scale bar = 50 μm for the WT and fH+/− panels. Original magnification x200 and scale bar = 100 μm for the secondary only panel. Image shown is representative of four mice that were analyzed from two independent experiments.
Injection of mice with recombinant factor H does not protect them from ischemic AKI
To test whether injection of factor H would protect factor H deficient mice from ischemia AKI, we produced recombinant factor H protein (Figure 4A). fH+/− mice were injected with 0.5 mg of factor H or with an equal volume of vehicle control, and ischemic AKI was then induced. Surprisingly, injection of the mice with purified factor H did not protect them from injury (Figure 4B). To test whether the injected factor H reached the kidney, we fluorescently tagged the protein in two mice. The labeled factor H was detectable in glomeruli and in the tubulointerstitium after I/R, but the distribution of factor H was less extensive than the deposited C3d (Figure 4C). In mice with complete deficiency of factor H (fH−/− mice), in contrast, the distribution of injected factor H in the glomerulus was similar to that of C3d deposits. It is possible that complement activation in the fH+/− mice occurs in the fluid phase or on other surfaces not easily examined by microscopy, such as on circulating platelets or leukocytes. It is also possible that factor H is produced within the kidney, and that injected factor H does fully access all of these sites. Indeed, it has long been noted that plasma exchange is usually ineffective in aHUS patients, even when deficiency of factor H is the main risk factor for the disease [3].
Figure 4. Reconstitution of fH+/− mice with recombinant factor H does not protect them from kidney injury after ischemia/reperfusion.
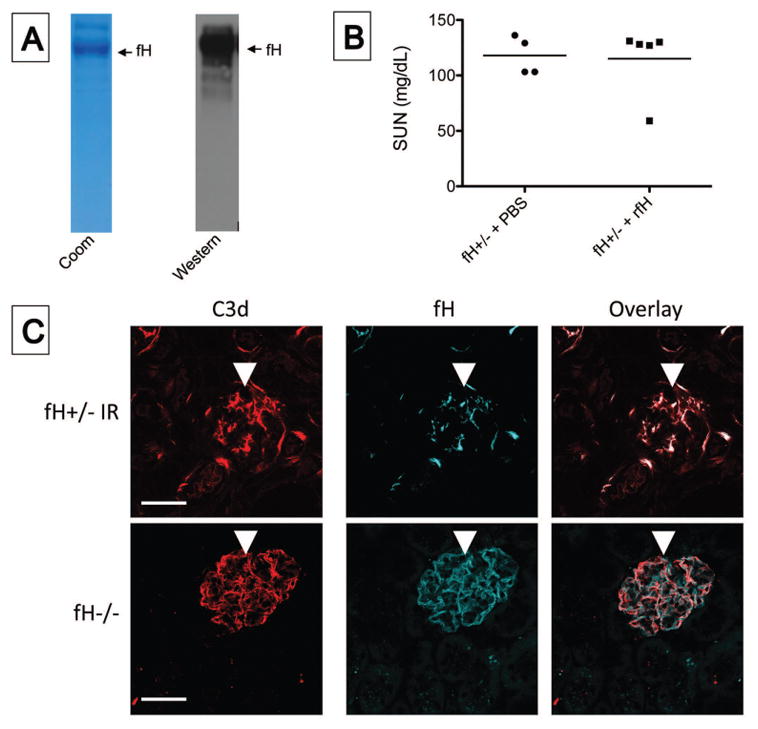
fH+/− mice were injected intra-peritoneally with recombinant factor H (fH) or with PBS as a vehicle control and subjected to ischemia/reperfusion of the kidneys. A) The purity and identify of the ~150 kD fH was confirmed by Coomassie staining (Coom) and Western blot analysis. B) After 24 hours the serum urea nitrogen (SUN) levels were measured using a NADH oxidation assay. Values in the two groups of mice were not significantly different by unpaired student t test. C) After 24 hours tissues were collected, and kidney sections from the two mice injected with fluorescently tagged factor H were stained for C3d and imaged by confocal microscopy. Original magnification x600 and scale bar = 50 μm. Data shown is representative of four mice that were analyzed from two independent experiments.
IgM deficiency does not protect mice from ischemic AKI
We previously found that glomerular IgM contributes to kidney injury in several chronic models of disease [26, 27]. To determine whether glomerular IgM deposits contribute to ischemic AKI, we compared renal injury in wild-type and sIgM−/− mice. There was a trend toward lower SUN and creatinine values in sIgM−/− mice 24 hours after I/R, but the differences were not statistically significant (Figure 5A). To determine whether factor H limits the inflammatory effects of glomerular IgM, we next compared ischemic injury in fH+/− and fH+/−sIgM−/− mice. There was again a trend towards lower SUN and creatinine values in fH+/−sIgM−/− mice compared to fH+/− mice 24 hours after I/R, but this did not reach statistical significance (Figure 5B). As one would expect, glomerular IgM was not seen in the sIgM−/− mouse strains (Figure 5C). Glomerular C3 deposition was not detected in any of the strains, indicating that complement regulation in the glomerulus remains effective even in mice with reduced expression of factor H.
Figure 5. IgM does not contribute to kidney injury after ischemia/reperfusion.
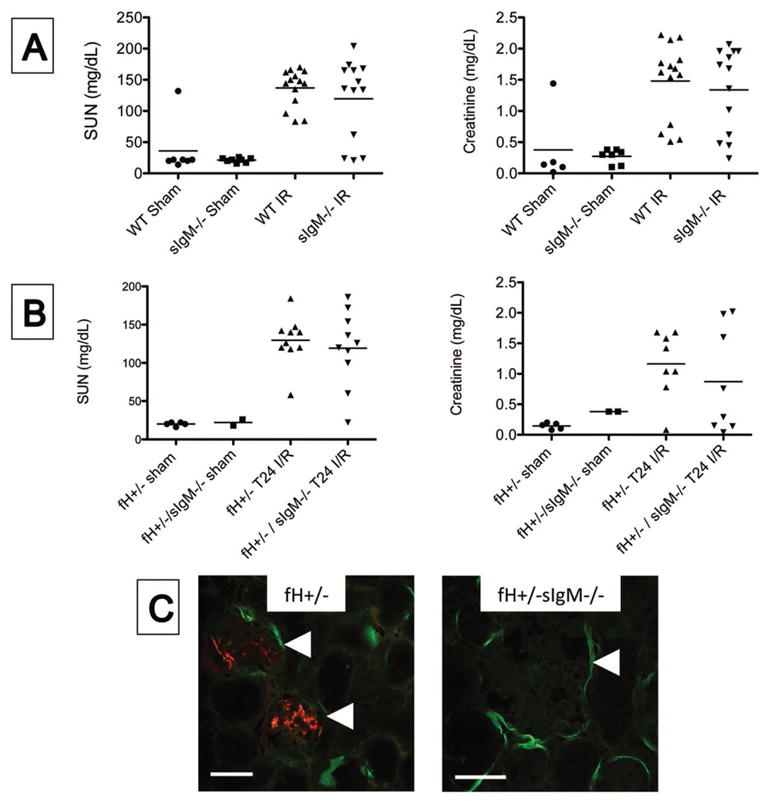
Wild-type (WT) and mice that cannot generate soluble IgM (sIgM−/−) were subjected to ischemia/reperfusion of the kidneys. A) After 24 hours the serum urea nitrogen (SUN) and serum creatinine levels were measured using a NADH oxidation assay and a picric acid chromogenic assay, respectively. There was not a significant difference between WT and sIgM−/− mice when compared by ANOVA with Tukey post test. B) Factor H deficient (fH+/− mice) and mice deficient in both factor H and soluble IgM (fH+/−sIgM−/− mice) were subjected to ischemia/reperfusion of the kidneys. After 24 hours the SUN and serum creatinine levels were using the same assays as in A. There was not a significant difference between the two strains when compared using ANOVA with Tukey post test. C) Tissue sections were stained with antibodies to murine C3 and IgM. Immunofluorescence microscopy for C3 (green) and IgM (red) revealed IgM deposits in the glomeruli of fH+/− mice but not of fH+/−sIgM−/− mice. Original magnification 200X for fH+/− and 600X for fH+/−sIgM−/−. Glomeruli are indicated with arrowheads. Scale bar = 100 μm for 200X and 50 μm for 600X. Data shown A is from three ischemia experiments with 6–14 mice per experiment, and data in B is from four experiments 5–9 mice per experiment. Each mouse is represented by a data-point, and the mean for each group is shown. Images in C are representative of three mice from two experiments.
Natural IgM clones bind to renal cells in vitro
To further explore control of complement activation by IgM in the glomerulus, we tested the binding of a panel of IgM hybridomas to murine microvascular endothelial cells, glomerular endothelial cells, and podocytes (Figure 6). As we have previously found with mesangial cells [26], a natural IgM clone (monoclonal antibody C2) binds to glomerular endothelial cells and podocytes. Binding of natural IgM (including C2) to the different glomerular cell types does not cause C3 deposition on the cells, however, demonstrating that the different glomerular cell types are effective at controlling IgM-mediated complement activation on the cell surface.
Figure 6. Natural IgM binds to glomerular cells in vitro.
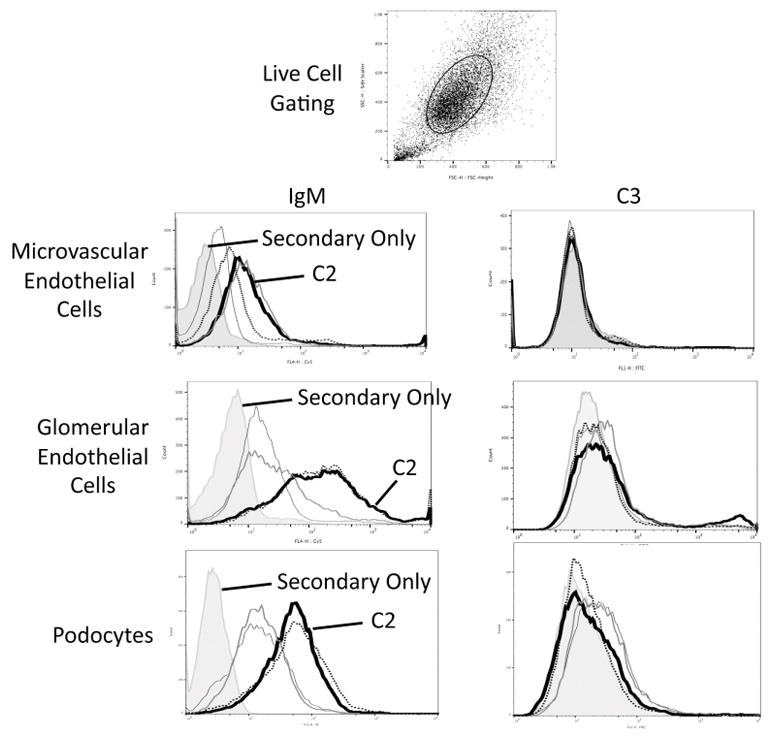
Murine microvascular endothelial cells, glomerular endothelial cells, and podocyte cell lines were grown in culture and were incubated with several different natural IgM clones. Binding of IgM to live cells was examined by flow cytometry using an allophycocyanin (APC)-conjugated antibody to mouse IgM (μ chain). The gating strategy to examine live cells is shown. Several of the IgM clones bound to cells. Secondary antibody only staining is shown with the shaded graphs. One of the monoclonal IgM antibodies, C2, bound to all three cell types and is represented with a dark black line. In a separate set of experiments the cells were incubated with purified IgM and serum from IgM deficient mice, and C3 deposition on the cells was measured by flow cytometry using a fluorescein (FITC)-conjugated antibody to C3. Each experiment was performed at least three times independently, and representative results are shown.
Factor H-deficiency is associated with decreased levels of arginine and citrulline after renal I/R
As natural IgM binds to microvascular and glomerular endothelial cells, we sought to further explore whether natural IgM may cause endothelial injury after renal I/R. To do this we measured the levels of metabolites involved in the nitric oxide (NO) pathway and vascular dysfunction in the serum of mice with AKI (Figure 7). Endothelial nitric oxide synthase (eNOS) converts arginine and citrulline into nitric oxide. Levels of both of these metabolites were lower in fH+/− mice compared to wild-type mice, suggesting that there is consumption of these metabolites in the kidney after I/R, possibly due to endothelial stress. Deficiency of soluble IgM prevented this decrease, indicating that IgM is a mediator of this effect. Homocysteine and asymmetric dimethylarginine (ADMA) inhibit eNOS. ADMA is a marker of vascular injury, and levels are increased in patients with chronic kidney disease [28]. Surprisingly, however, ADMA levels tended to be lower in fH+/− mice after renal I/R compared to wild-type controls, although this was not statistically significant.
Figure 7. Soluble IgM affects the levels of metabolites in serum after kidney ischemia/reperfusion.
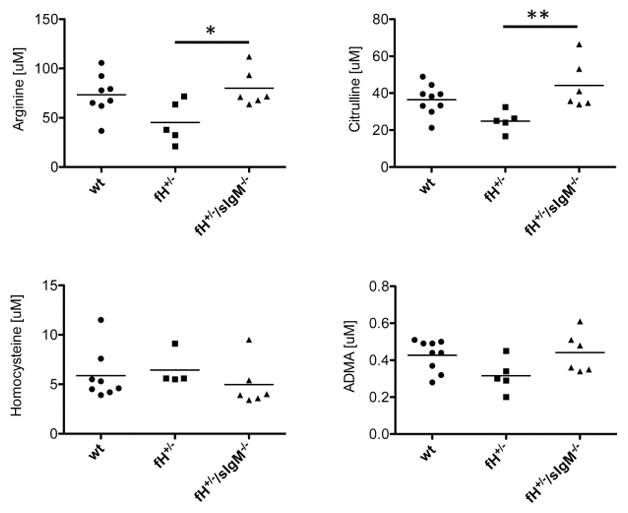
Several metabolites were measured in the plasma of mice with ischemic acute kidney injury by mass spectrometry and tested statistically using ANOVA with Tukey post test, *P < 0.05, **P < 0.01. Data shown is pooled from four experiments with 4–6 mice per experiment. Each mouse is represented by a data-point, and the mean for each group is shown.
Monoclonal IgM C2 targets the glomeruli after I/R but does not cause complement activation
Because mAb C2 binds to endothelial cells, glomerular endothelial cells, mesangial cells, and podocytes in vitro, we examined whether C2 would target glomerular structures in vivo. We induced ischemic AKI in sIgM−/− mice and reconstituted them with either vehicle control or 100 μg of C2. There was a trend towards higher levels of SUN and creatinine in the C2 injected mice, but this did not reach statistical significance (P = 0.25 and 0.34, respectively; Figure 8A). Immunofluorescence microscopy revealed that the C2 bound to the mesangium and glomerular capillaries, and the peritubular capillaries (Figure 8B). However, C2 deposition was not associated with C3 deposition on these structures. This experiment was repeated in fH+/−sIgM−/− mice to determine whether deficiency of factor H would permit complement activation by the glomerular IgM deposits. Again, there was a trend toward higher SUN and creatinine levels in the C2 injected mice (P = 0.14 and 0.15, respectively; Figure 9A), but binding of C2 within the glomeruli did not result in full complement activation and C3 deposition (Figure 9B).
Figure 8. Monoclonal IgM binds to glomerular epitopes after ischemia/reperfusion but does not cause C3 deposition.
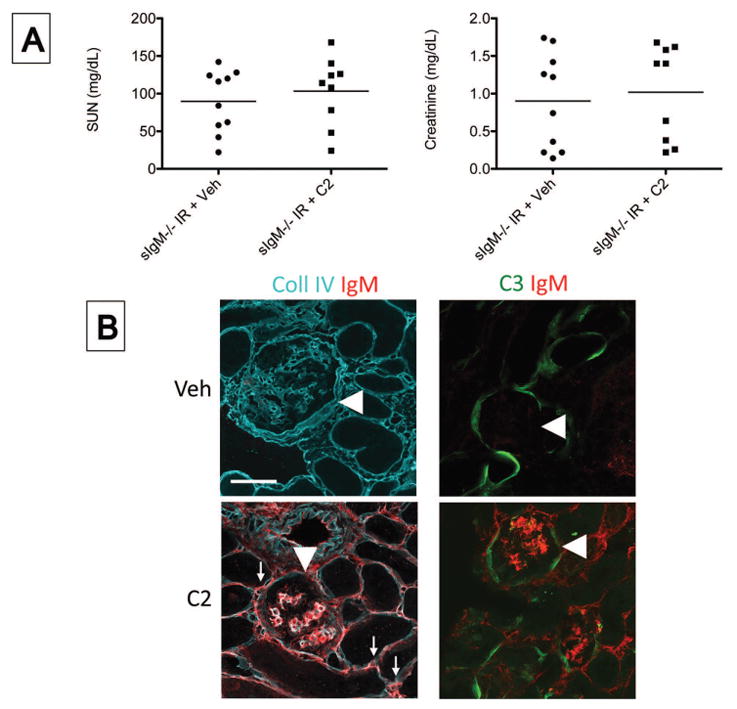
IgM deficient mice (sIgM−/− mice) were injected intravenously with purified C2 IgM or with an equal volume of a vehicle control (Veh), and the mice were then subjected to ischemia/reperfusion of the kidneys. After 24 hours the mice were sacrificed. A) The serum urea nitrogen (SUN) and serum creatinine levels in Veh and C2 injected mice were measured using a NADH oxidation assay and a picric acid chromogenic assay, respectively. There was not a significant difference by unpaired student t test. B) Tissue sections were stained using antibodies for murine collagen type IV (Coll IV), IgM, and C3. Immunofluorescence microcopy revealed that IgM bound within the glomeruli (arrowheads) and peritubular capillaries (arrows). Original magnification x600. Scale bar = 50 μm. Symbols in A represent single mice. Data shown in A is pooled from two independent experiments. Images in B are representative of eight mice for each group from two independent experiments.
Figure 9. Monoclonal IgM binds to glomerular epitopes in factor H deficient mice after ischemia/reperfusion but does not cause C3 deposition.
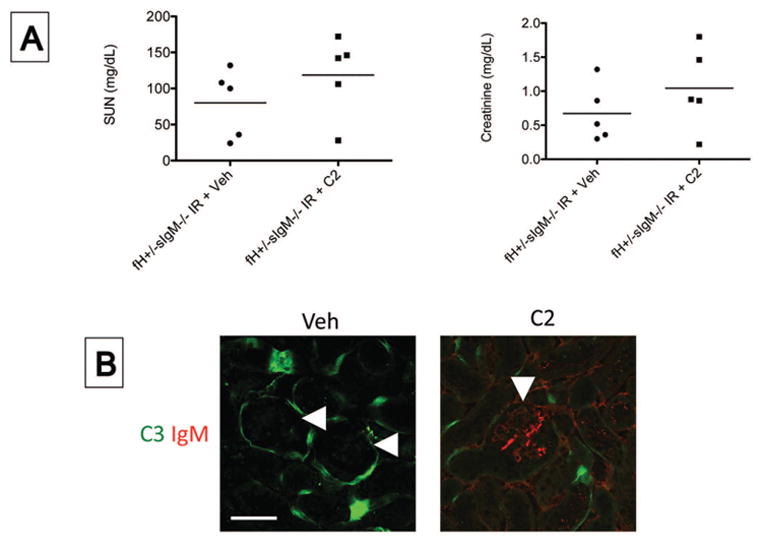
Mice deficient in factor H and soluble IgM deficient mice (fH+/−sIgM−/− mice) were injected intravenously with purified C2 IgM or with an equal volume of a vehicle control (Veh), and the mice were then subjected to ischemia/reperfusion of the kidneys. After 24 hours, the mice were sacrificed. A) The serum urea nitrogen (SUN) and serum creatinine levels in Veh and C2 injected mice were measured using a NADH oxidation assay and a picric acid chromogenic assay, respectively. No significant difference was observed (unpaired student t test). B) Tissue sections were stained with antibodies to murine C3 and IgM. Immunofluorescence microcopy for IgM and C3. Original magnification x600. Scale bar = 50 μm. Data shown in A is pooled from one experiment. Each data point represents one mouse, and the bar represents the group mean. Images in B are representative of six mice for each group from two independent experiments.
Discussion
The alternative pathway of complement is activated on tubular epithelial cells after kidney I/R and causes kidney injury [1, 29]. Factor H is the main regulator of the alternative pathway. In the current study we found that mice deficient in factor H develop more severe ischemic AKI injury than wild-type controls. Surprisingly, C3 deposition on the tubules of fH+/− mice was not increased relative to wild-type mice, indicating that pathologic complement activation in the fH+/− mice may occur in an anatomic location that is harder to detect by microscopy, such as the microvasculature. C6 deposits did appear more extensive in the tubulointerstitium of fH+/− mice, however. It is possible, therefore, that the greater injury seen in the fH+/− mice is due to greater activity of the C5 convertase and terminal complement activation.
We previously found that natural IgM binds to glomerular structures after I/R but that it does not activate complement to the level of C3 [23]. Factor H is critical for controlling complement activation in the glomerulus [15], and we hypothesized that factor H deficiency would be associated with increased glomerular complement activation by the deposited IgM after I/R. Although the glomerular IgM co-localized with C4 in both wild-type and fH+/− mice, indicating classical pathway activation by IgM, glomerular C3 was not detected in either strain. Thus, complement regulation in the glomerulus remains robust even in mice with reduced levels of factor H, and glomerular IgM does not activate the complement system to the level of C3. Substrates for eNOS generation were reduced in the fH+/− mice compared to wild-type mice. This was not seen in IgM deficient mice, suggesting that IgM causes vascular stress in these mice even though it does not cause detectable complement activation.
Specific natural IgM clones bind to glomerular structures and contribute to injury in other diseases [15]. We tested a panel of IgM hybridomas against endothelial cells, glomerular endothelial cells, and podocytes. We found good binding of one of these clones, mAb C2, to all of these cell types. Even though C2 bound to glomerular endothelial cells, however, it did not cause C3 deposition on the cell surface in vitro. Similarly, when injected into IgM deficient mice C2 bound within the glomeruli, but it did not cause detectable C3 deposition or a significant degree of injury.
Natural IgM contributes to ischemia of other organs, including intestine and skeletal muscle [30, 31]. We have also previously found that natural IgM contributes to glomerular injury in two models of chronic glomerular injury, and that C2 exacerbates glomerular injury when injected into mice with chronic glomerulonephritis [26, 27]. In the current study we did not find an important role for natural IgM or mAb C2 in kidney ischemia. It remains to be determined why natural IgM does not cause injury in ischemic AKI whereas it does cause tissue damage after ischemia of other organs and in chronic models of glomerular disease. Likely, this is due to persistent, effective complement regulation on the glomerular structures after I/R. Similarly, factor H deficiency exacerbates glomerular IC mediated injury in other models [18, 19]. Complement activation by glomerular IgM remained well controlled in the post-ischemic kidney, however, even in mice with partial factor H deficiency.
There was a trend towards mild protection in the sIgM−/− and fH+/−sIgM−/− mice compared to control mice, and mild exacerbation of injury in C2 injected mice. It is possible that complement activation by deposited IgM does make a minor contribution to kidney injury after I/R, even though C3 is not detected by immunofluorescence microscopy. Deposited IgM may also have pathologic effects that are not mediated through the complement system. There is a receptor for IgM, for example, and this receptor may mediate a mild pathologic effect, although the expression and function of this receptor is incompletely understood [32]. Nevertheless, the overall effect of IgM in this model seems to be modest.
In conclusion, activation of the alternative pathway is an important cause of kidney injury after I/R, and we found that mice with heterozygous deficiency of factor H develop more severe ischemic AKI than wild-type mice. Complement activation to the level of C3 is limited to the tubulointerstitium, however, and C3 deposits are not seen in the glomeruli. This is in spite of the fact that IgM deposits in the glomeruli and engages the classical pathway. Although factor H is important for controlling complement activation by glomerular immune-complexes in other diseases, full expression of factor H is not necessary to control glomerular complement activation after I/R. It is important to understand the endogenous mechanisms for controlling complement activation on host cells, and to identify the molecular events that lead to complement dysregulation in many glomerular diseases. Greater understanding of these processes will allow the development of new therapies that can block or reverse these pathogenic processes.
Materials and Methods
Animals
Mice with targeted deletion of the gene for factor H[16] or with a mutation in the μs exon of the IgM gene (sIgM−/− mice [24]) were generated as previously described. Both strains were back-crossed onto a C57BL/6 background for more than 9 generations, and fH−/− mice were crossed with C57BL/6 mice to generate fH+/− mice. The fH−/− and sIgM−/− mice were intercrossed to generate fH+/−sIgM−/− mice. Mice were housed and maintained in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals, and the University of Colorado Center for Laboratory Animal Care approved all experiments.
Ischemia-reperfusion protocol
Kidney I/R was induced as previously described [23]. Briefly, mice were anesthetized with ketamine and xylazine injected intraperitoneally and placed on a heating pad to maintain their body temperature during surgery. Laparotomies were performed and the renal pedicles were located and isolated by blunt dissection. The pedicles were clamped with surgical clips (Miltex Instrument Company, Inc., Dedham, MA), and occlusion of blood flow was confirmed by visual inspection of the kidneys. The clamps were left in place for 24 minutes and then released. The kidneys were observed for approximately one minute to ensure reperfusion, and then the fascia and skin were sutured with 4-0 silk (United States Surgical Corporation, Norwalk, CT). Sham surgery was performed in an identical fashion, except that the renal pedicles were not clamped. The mice were volume resuscitated with 0.5 ml of normal saline injected subcutaneously and kept in an incubator at 29°C for one hour to maintain body temperature while recovering from anesthesia.
Reagents
The following antibodies were used in these studies: FITC-conjugated goat IgG to mouse Complement C3 (MP Biomedicals, Santa Ana, CA), a monoclonal antibody (mAb) that only recognizes the iC3b and C3d fragments of C3 (mAb 3d29) [33], a mAb that recognizes all of the C3 fragments by Western blot analysis (mAb 3d11 [33]), a Cy3-conjugated goat antibody against mouse IgM, μ-chain (Jackson ImmunoResearch, West Grove, PA), an isotype control (PE-conjugated rat IgG isotype control; Novus Biologicals, Littleton, CO), antibodies to murine C4 and C6 (Hycult Biotech, Uden, the Netherlands), an antibody to β-actin (Sigma-Aldrich, St. Louis, MO), and an antibody to collagen IV (EMD Millipore, Billerica, MA). Monoclonal natural IgM hybridomas were generated as previously described [34]. Recombinant factor H was produced and purified as previously described [15]. The recombinant protein was fluorescently labeled using a DyLight 650 Labeling Kit (Thermo Scientific, Waltham, MA) according to the manufacturer’s instructions.
Confocal microscopy
Kidneys were snap frozen in OCT compound (Sakura Finetek USA Inc., Torrance, CA) at the time of harvest and stored at −80°C. Four μm sagittal sections were cut using a cryostat, warmed to room temperature, and fixed with acetone. Non-specific binding was blocked with 10% heat-inactivated goat serum. Primary antibodies were diluted in 2% heat-inactivated goat serum and incubated overnight at 4°C. Autofluorescence was blocked with 0.05% Sudan Black B in 70% ethanol for 20 minutes at room temperature, followed by two 10 minute washes in deionized water [35]. High-resolution images were obtained with an Olympus FV1000 FCS/RICS confocal microscope (Olympus Life Science, Waltham, MA). Olympus FV10-ASW software was used for intensity and co-localization analyses.
Murine mesangial, podocyte, and glomerular endothelial cell culture and flow cytometry
Immortalized murine glomerular mesangial cells were grown in culture (MES-13 cells, from ATCC, Manassas, VA). Murine glomerular endothelial cells and podocytes were grown in culture as previously described [26, 36]. Murine pancreatic microvascular endothelial cells were also used as previously described [37]. To detect binding of specific natural IgM clones to the cells, the cells were incubated with the antibodies at the indicated concentration for 35 minutes at 37° C. The cells were then washed with PBS and stained with an antibody to mouse IgM. To measure complement activation on the cells by specific IgM clones, the cells were incubated with 10% serum from sIgM−/− mice with 10 μg of purified IgM for 30 minutes at 37° C. The cells were washed with PBS and then stained for deposited C3. Flow cytometric analysis was performed on a FACS Calibur machine (BD Biosciences, San Jose, CA) and analyzed by CellQuest Pro software (BD Biosciences, San Jose, CA). Flow cytrometry was performed in accordance with the “Guidelines for the use of flow cytometry and cell sorting in immunological studies” (http://onlinelibrary.wiley.com/doi/10.1002/eji.201646632/pdf).
Metabolomics
Metabolic markers of vascular function were measured using a mass spectroscopy assay. Briefly, to 100μL aliquots of mouse plasma, 50μL of solution containing internal standards, and 40μL of 500mM DTT solution were combined. For protein precipitation, 400μL of 0.05% trifluoric acid in acetonitrile were added, the sample was vortexed for 5 minutes and centrifuged for 10 minutes at 13,000g.
Twenty μL of the supernatant were injected onto a 4.6x12.5 mm guard column (Eclipse XDB-C8, 5μm, Agilent Technologies, Palo Alto, CA) inline with a 3.0x150 mm analytical column (RP-Amide, 3.5μm, Supelco, CA). The gradient started at 3% methanol and 97% 10mM ammonium formate buffer and was maintained at a flow of 0.8mL/min throughout the assay. At minute 4.5, the solvent gradient reached 25% methanol; hereafter, this the methanol content was raised to 98% and held for additional 1.5 minutes, after which the columns were re-equilibrated to the starting conditions for the remaining 2 minutes of the assay. The HPLC system was interfaced using turbo electrospray with a triple quadrupole tandem mass spectrometry detector (LC-MS/MS) (API4000, Applied Biosystems, Foster City, CA), run in positive multiple reaction monitoring (MRM) mode. Peak area ratios of the mass transitions of the analytes and their corresponding isotope-labeled internal standards were used for the quantification of arginine, citrulline, homocysteine and asymmetric dimethylarginine (ADMA).
Western blot analysis
Plasma samples were diluted 1:10 in PBS and reduced by boiling with 0.15 M dithiothreitol for 10 min. at 100° C. Renal tissue was homogenized in RIPA lysis buffer containing 1% Triton X-100, 0.5% deoxycholic acid, 150 mM NaCl, 20 mM β-glycerophosphate, 20 mM Tris·HCl (pH 8.0), 5 mM EGTA, 3 mM MgCl2, 0.1% SDS, 1mM DTT, 50μM Na3VO4, and one tablet of complete, EDTA-free, protease inhibitor cocktail (Roche Applied Science, Indianapolis, IN). Homogenates were centrifuged at 14,000 rpm for 15 min. at 4° C and the supernatant was collected. Fifty μg of protein for each sample was used. The proteins were resolved by electrophoresis with a 10% Tris-HCl gel (Bio-Rad, Hercules, CA), and transferred to a polyvinylidene fluoride membrane.
Renal histology
Kidney tissue was fixed in formalin, embedded in paraffin, cut into 4 μm sections, and stained with periodic acid-Schiff. Sections were assessed for the extent of glomerular hypercellularity, glomerulosclerosis, and tubular injury.
Statistical analysis
Statistical analysis between two groups was assessed by unpaired t-test and analysis of multiple groups was performed using one-way analysis of variance (ANOVA) with a Tukey post test (GraphPad Prism, La Jolla, CA). A P value of less than 0.05 was considered statistically significant.
Acknowledgments
This work was supported by the KIDNEEDS Foundation (JMT) and National Institutes of Health Grants R01 DK076690 and DK113586 (JMT).
Abbreviations
- I/R
ischemia/reperfusion
- AKI
acute kidney injury
- TECs
tubular epithelial cells
- GBM
glomerular basement membrane
Footnotes
Conflict of interest.
JMT and VMH receive royalties from Alexion Pharmaceuticals, Inc. JMT and VMH are also consultants for AdMIRx, Inc., a company developing complement inhibitors. They also hold stock and will receive royalty income from AdMIRx. All other authors declare no commercial or financial conflict of interest.
References
- 1.Thurman JM, Ljubanovic D, Edelstein CL, Gilkeson GS, Holers VM. Lack of a functional alternative complement pathway ameliorates ischemic acute renal failure in mice. J Immunol. 2003;170:1517–1523. doi: 10.4049/jimmunol.170.3.1517. [DOI] [PubMed] [Google Scholar]
- 2.Xiao H, Schreiber A, Heeringa P, Falk RJ, Jennette JC. Alternative complement pathway in the pathogenesis of disease mediated by anti-neutrophil cytoplasmic autoantibodies. Am J Pathol. 2007;170:52–64. doi: 10.2353/ajpath.2007.060573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Noris M, Remuzzi G. Atypical hemolytic-uremic syndrome. N Engl J Med. 2009;361:1676–1687. doi: 10.1056/NEJMra0902814. [DOI] [PubMed] [Google Scholar]
- 4.Pickering MC, D’Agati VD, Nester CM, Smith RJ, Haas M, Appel GB, Alpers, et al. C3 glomerulopathy: consensus report. Kidney Int. 2013;84:1079–1089. doi: 10.1038/ki.2013.377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zipfel PF, Skerka C. Complement regulators and inhibitory proteins. Nat Rev Immunol. 2009;9:729–740. doi: 10.1038/nri2620. [DOI] [PubMed] [Google Scholar]
- 6.Pickering MC, Cook HT. Translational mini-review series on complement factor H: renal diseases associated with complement factor H: novel insights from humans and animals. Clin Exp Immunol. 2008;151:210–230. doi: 10.1111/j.1365-2249.2007.03574.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cook HT, Pickering MC. Histopathology of MPGN and C3 glomerulopathies. Nat Rev Nephrol. 2015;11:14–22. doi: 10.1038/nrneph.2014.217. [DOI] [PubMed] [Google Scholar]
- 8.Thurman JM, Nester CM. All Things Complement. Clin J Am Soc Nephrol. 2016 doi: 10.2215/CJN.01710216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Thurman JM. Complement in kidney disease: core curriculum 2015. Am J Kidney Dis. 2015;65:156–168. doi: 10.1053/j.ajkd.2014.06.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Farrar CA, Zhou W, Lin T, Sacks SH. Local extravascular pool of C3 is a determinant of postischemic acute renal failure. Faseb J. 2006;20:217–226. doi: 10.1096/fj.05-4747com. [DOI] [PubMed] [Google Scholar]
- 11.Thurman JM, Ljubanovic D, Royer PA, Kraus DM, Molina H, Barry NP, Proctor G, et al. Altered renal tubular expression of the complement inhibitor Crry permits complement activation after ischemia/reperfusion. J Clin Invest. 2006;116:357–368. doi: 10.1172/JCI24521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Miao J, Lesher AM, Miwa T, Sato S, Gullipalli D, Song WC. Tissue-specific deletion of Crry from mouse proximal tubular epithelial cells increases susceptibility to renal ischemia-reperfusion injury. Kidney Int. 2014;86:726–737. doi: 10.1038/ki.2014.103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Renner B, Coleman K, Goldberg R, Amura C, Holland-Neidermyer A, Pierce K, Orth HN, et al. The complement inhibitors Crry and factor H are critical for preventing autologous complement activation on renal tubular epithelial cells. J Immunol. 2010;185:3086–3094. doi: 10.4049/jimmunol.1000111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Renner B, Tong HH, Laskowski J, Jonscher K, Goetz L, Woolaver R, Hannan J, et al. Annexin A2 Enhances Complement Activation by Inhibiting Factor H. J Immunol. 2016;196:1355–1365. doi: 10.4049/jimmunol.1500793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Laskowski J, Renner B, Le Quintrec M, Panzer S, Hannan JP, Ljubanovic D, Ruseva MM, et al. Distinct roles for the complement regulators factor H and Crry in protection of the kidney from injury. Kidney Int. 2016;90:109–122. doi: 10.1016/j.kint.2016.02.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pickering MC, Cook HT, Warren J, Bygrave AE, Moss J, Walport MJ, Botto M. Uncontrolled C3 activation causes membranoproliferative glomerulonephritis in mice deficient in complement factor H. Nat Genet. 2002:424–428. doi: 10.1038/ng912. [DOI] [PubMed] [Google Scholar]
- 17.Watanabe H, Garnier G, Circolo A, Wetsel RA, Ruiz P, Holers VM, Boackle SA, et al. Modulation of renal disease in MRL/lpr mice genetically deficient in the alternative complement pathway factor B. J Immunol. 2000;164:786–794. doi: 10.4049/jimmunol.164.2.786. [DOI] [PubMed] [Google Scholar]
- 18.Bao L, Haas M, Quigg RJ. Complement factor H deficiency accelerates development of lupus nephritis. J Am Soc Nephrol. 2011;22:285–295. doi: 10.1681/ASN.2010060647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Alexander JJ, Pickering MC, Haas M, Osawe I, Quigg RJ. Complement factor h limits immune complex deposition and prevents inflammation and scarring in glomeruli of mice with chronic serum sickness. J Am Soc Nephrol. 2005;16:52–57. doi: 10.1681/ASN.2004090778. [DOI] [PubMed] [Google Scholar]
- 20.Le Quintrec M, Zuber J, Moulin B, Kamar N, Jablonski M, Lionet A, Chatelet V, et al. Complement Genes Strongly Predict Recurrence and Graft Outcome in Adult Renal Transplant Recipients with Atypical Hemolytic and Uremic Syndrome. Am J Transplant. 2013 doi: 10.1111/ajt.12077. [DOI] [PubMed] [Google Scholar]
- 21.Remuzzi G, Ruggenenti P, Codazzi D, Noris M, Caprioli J, Locatelli G, Gridelli B. Combined kidney and liver transplantation for familial haemolytic uraemic syndrome. Lancet. 2002;359:1671–1672. doi: 10.1016/S0140-6736(02)08560-4. [DOI] [PubMed] [Google Scholar]
- 22.Remuzzi G, Ruggenenti P, Colledan M, Gridelli B, Bertani A, Bettinaglio P, Bucchioni S, et al. Hemolytic uremic syndrome: a fatal outcome after kidney and liver transplantation performed to correct factor h gene mutation. Am J Transplant. 2005;5:1146–1150. doi: 10.1111/j.1600-6143.2005.00783.x. [DOI] [PubMed] [Google Scholar]
- 23.Renner B, Strassheim D, Amura CR, Kulik L, Ljubanovic D, Glogowska MJ, Takahashi K, et al. B cell subsets contribute to renal injury and renal protection after ischemia/reperfusion. J Immunol. 2010;185:4393–4400. doi: 10.4049/jimmunol.0903239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Boes M, Esau C, Fischer MB, Schmidt T, Carroll M, Chen J. Enhanced B-1 cell development, but impaired IgG antibody responses in mice deficient in secreted IgM. J Immunol. 1998;160:4776–4787. [PubMed] [Google Scholar]
- 25.Renner B, Ferreira VP, Cortes C, Goldberg R, Ljubanovic D, Pangburn MK, Pickering MC, et al. Binding of factor H to tubular epithelial cells limits interstitial complement activation in ischemic injury. Kidney Int. 2011;80:165–173. doi: 10.1038/ki.2011.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Panzer SE, Laskowski J, Renner B, Kulik L, Ljubanovic D, Huber KM, Zhong W, et al. IgM exacerbates glomerular disease progression in complement-induced glomerulopathy. Kidney Int. 2015;88:528–537. doi: 10.1038/ki.2015.120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Strassheim D, Renner B, Panzer S, Fuquay R, Kulik L, Ljubanovic D, Holers VM, Thurman JM. IgM Contributes to Glomerular Injury in FSGS. J Am Soc Nephrol. 2013;24:393–406. doi: 10.1681/ASN.2012020187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Martens CR, Edwards DG. Peripheral vascular dysfunction in chronic kidney disease. Cardiol Res Pract. 2011;2011:267257. doi: 10.4061/2011/267257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Thurman JM, Royer PA, Ljubanovic D, Dursun B, Lenderink AM, Edelstein CL, Holers VM. Treatment with an inhibitory monoclonal antibody to mouse factor B protects mice from induction of apoptosis and renal ischemia/reperfusion injury. J Am Soc Nephrol. 2006;17:707–715. doi: 10.1681/ASN.2005070698. [DOI] [PubMed] [Google Scholar]
- 30.Zhang M, Alicot EM, Chiu I, Li J, Verna N, Vorup-Jensen T, Kessler B, et al. Identification of the target self-antigens in reperfusion injury. J Exp Med. 2006;203:141–152. doi: 10.1084/jem.20050390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Fleming SD, Shea-Donohue T, Guthridge JM, Kulik L, Waldschmidt TJ, Gipson MG, Tsokos GC, Holers VM. Mice deficient in complement receptors 1 and 2 lack a tissue injury-inducing subset of the natural antibody repertoire. J Immunol. 2002;169:2126–2133. doi: 10.4049/jimmunol.169.4.2126. [DOI] [PubMed] [Google Scholar]
- 32.Kubagawa H, Kubagawa Y, Jones D, Nasti TH, Walter MR, Honjo K. The Old but New IgM Fc Receptor (FcmuR) Curr Top Microbiol Immunol. 2014;382:3–28. doi: 10.1007/978-3-319-07911-0_1. [DOI] [PubMed] [Google Scholar]
- 33.Thurman JM, Kulik L, Orth H, Wong M, Renner B, Sargsyan SA, Mitchell LM, et al. Detection of complement activation using monoclonal antibodies against C3d. J Clin Invest. 2013;123:2218–2230. doi: 10.1172/JCI65861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kulik L, Fleming SD, Moratz C, Reuter JW, Novikov A, Chen K, Andrews KA, et al. Pathogenic natural antibodies recognizing annexin IV are required to develop intestinal ischemia-reperfusion injury. J Immunol. 2009;182:5363–5373. doi: 10.4049/jimmunol.0803980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sun Y, Yu H, Zheng D, Cao Q, Wang Y, Harris D, Wang Y. Sudan black B reduces autofluorescence in murine renal tissue. Arch Pathol Lab Med. 2011;135:1335–1342. doi: 10.5858/arpa.2010-0549-OA. [DOI] [PubMed] [Google Scholar]
- 36.Rops AL, van der Vlag J, Jacobs CW, Dijkman HB, Lensen JF, Wijnhoven TJ, van den Heuvel LP, et al. Isolation and characterization of conditionally immortalized mouse glomerular endothelial cell lines. Kidney Int. 2004;66:2193–2201. doi: 10.1111/j.1523-1755.2004.66009.x. [DOI] [PubMed] [Google Scholar]
- 37.Renner B, Klawitter J, Goldberg R, McCullough JW, Ferreira VP, Cooper JE, Christians U, Thurman JM. Cyclosporine induces endothelial cell release of complement-activating microparticles. J Am Soc Nephrol. 2013;24:1849–1862. doi: 10.1681/ASN.2012111064. [DOI] [PMC free article] [PubMed] [Google Scholar]