

Abstract
Idiopathic pulmonary arterial hypertension (IPAH) is a devastating pulmonary vascular disease in which autoimmune and inflammatory phenomena are implicated. B cells and autoantibodies have been associated with IPAH and identified as potential therapeutic targets. However, the specific populations of B cells involved and their roles in disease pathogenesis are not clearly defined. We aimed to assess the levels of activated B cells (plasmablasts) in IPAH, and to characterize recombinant antibodies derived from these plasmablasts. Blood plasmablasts are elevated in IPAH, remain elevated over time, and produce IgA autoantibodies. Single-cell sequencing of plasmablasts in IPAH revealed repertoires of affinity-matured antibodies with small clonal expansions, consistent with an ongoing autoimmune response. Recombinant antibodies representative of these clonal lineages bound known autoantigen targets and displayed an unexpectedly high degree of polyreactivity. Representative IPAH plasmablast recombinant antibodies stimulated human umbilical vein endothelial cells to produce cytokines and overexpress the adhesion molecule ICAM-1. Together, our results demonstrate an ongoing adaptive autoimmune response involving IgA plasmablasts that produce anti-endothelial cell autoantibodies in IPAH. These antibodies stimulate endothelial cell production of cytokines and adhesion molecules, which may contribute to disease pathogenesis. These findings suggest a role for mucosally-driven autoimmunity and autoimmune injury in the pathogenesis of IPAH.
Keywords: Autoimmunity, B cells, Antibodies, Clinical Immunology, VDJ Recombination
Introduction
Pulmonary arterial hypertension (PAH) is a devastating pulmonary vascular disease frequently associated with concurrent autoimmune and inflammatory conditions [1, 2]. Endothelial cell apoptosis combined with myofibroblast and smooth muscle cell hyper proliferation causes narrowing of the small pulmonary arteries, which increases pulmonary arterial pressure and can lead to right heart failure and death [3, 4]. Cell proliferation pathways, such as bone morphogenic protein receptor type 2 (BMPR2), contribute to disease pathogenesis, but autoimmune and inflammatory effects have also been implicated [3, 5-7].
The reported prevalence of autoantibodies in PAH varies greatly between studies, as do their antigenic targets. In addition to autoantibodies that bind phospholipids [8], RNPs [9, 10], Sm [10], CENP-A [11], lamin A/C, tubulin-β, vinculin [12], and other individual proteins, PAH patients also produce anti-endothelial cell autoantibodies (AECAs) that bind endothelial cells via undefined antigen targets. AECAs purified from PAH patient serum induce a pro-inflammatory, pro-adhesive phenotype in endothelial cells in vitro [13, 14]. Furthermore, anti-endothelin receptor type A (ETAR) and angiotens in receptor type-1 (AT1R) autoantibodies from PAH patients with systemic sclerosis promote PAH in murine models [15], and a recent study of PAH patients with systemic sclerosis identified anomalies in B cell development, including altered V(D)J gene usage and increased somatic mutations in expanded B cell lineages [16]. They further showed increased IgD-producing cells with few mutations and an increase in antibody-secreting cells [16]. Interest in B cell immunity as a driving force in PAH resulted in a currently-enrolling NIH trial to test the efficacy of rituximab in a double-blinded, placebo-controlled, multi-center trial (NCT01086540).
To assess whether activated B cell populations are specifically increased in IPAH and if such cells encode disease-promoting autoantibodies, we utilized flow cytometry in combination with single-cell barcoding to sequence the antibody repertoire in IPAH [17, 18]. We focused our analysis on blood plasmablasts, a population of activated B cells that is generated and released from germinal centers into the blood during an adaptive immune response. Plasmablasts are transiently elevated in the blood in self-limited immune responses such as to Influenza vaccination [17], and chronically elevated in persistent immune responses such as in chronic autoimmune diseases [18, 19]. Characterization of IgA- and IgG-producing plasmablasts in IPAH enabled direct analysis of the antibodies encoded by the active B cell response, thereby separating this response from the >108 specificities in a human B cell repertoire [20]. Bioinformatic analyses of the plasmablast antibody repertoire were used to identify clonal lineages, and to select representative antibodies for recombinant expression. The expressed antibodies were characterized in binding and functional assays to gain insights into the underlying pathogenesis of the disease.
Results
Characteristics of the study subjects
We obtained blood from 22 healthy control and 25 IPAH subjects, with no significant differences in age, gender, or race between the groups (Table 1). IPAH subjects had a median age of 62, and 76% were female. The median mPAP and PVR were 40 mm Hg and 7.75 Wood Units, respectively. While 32% of patients were treatment-naïve, the remainder were on monotherapy (36%) or dual/triple therapy (16% for dual and 16% for triple). Longitudinal samples were available for five subjects with IPAH.
Table 1. Baseline cohort characteristics.
| Idiopathic PAH | Healthy controls | |||
|---|---|---|---|---|
| n = 25 | n = 22 | |||
| Demographics | ||||
| Age, years, median (IQR) | 62 | (23.5) | 60.5 | (17) |
| Female sex, n (%) | 19 | (76%) | 15 | (68.2%) |
| Race, n (%) | ||||
| White | 15 | (60%) | 15 | (68.2%) |
| Hispanic | 6 | (24%) | 1 | (4.5%) |
| Asian/Pacific Islander | 1 | (4%) | 5 | (22.7%) |
| African American | 0 | (0%) | 1 | (4.5%) |
| Other | 3 | (12%) | 0 | 0 |
|
| ||||
| Non-invasive clinical metrics | ||||
| NYHA functional class, n (%) | ||||
| Class I or II | 13 | (52%) | ||
| Class III | 10 | (40%) | ||
| Class IV | 2 | (8%) | ||
| Six minute walk distance, m, median (IQR) | 422 | (207) | ||
| NT-proBNP, pg/ml, median (IQR) | 129 | (398) | ||
| DLCO, % of predicted, median (IQR) | 72 | (30) | ||
|
| ||||
| RHC hemodynamic measurements | ||||
| Mean pulmonary arterial pressure, mm Hg, median (IQR) | 40 | (14) | ||
| Pulmonary vascular resistance, Wood units, median (IQR) | 7.75 | (6.2) | ||
| Mean right atrial pressure, mm Hg, median (IQR) | 9 | (6) | ||
| Cardiac index, ml/min/m2, median (IQR) | 2.19 | (0.49) | ||
|
| ||||
| Background PAH guided therapy | ||||
| Number of agents, n (%) | ||||
| Treatment naïve | 8 | (32%) | ||
| Monotherapy | 9 | (36%) | ||
| Dual therapy | 4 | (16%) | ||
| Triple therapy | 4 | (16%) | ||
| Class of agents, n (%) | ||||
| Phosphodiesterase-5 inhibitor | 12 | (48%) | ||
| Endothelin receptor antagonist | 6 | (24%) | ||
| Prostanoid | 11 | (44%) | ||
Elevated levels of IgA-producing plasmablasts in IPAH
While total B cells were not significantly elevated (Figure 1A), plasmablasts (as a percent of B cells) were significantly elevated in subjects with IPAH (Figure 1B). As the percent of plasmablasts may be skewed in subjects with low total B cell frequency, we calculated the plasmablasts per 106 live single cells, which was also significantly increased in IPAH (Figure 1C). Plasmablast numbers were stable over time for five IPAH subjects analyzed at serial time points including 6 and/or 12 months following the initial visit (Figure 1D). Most IPAH plasmablasts expressed surface IgA (Figure 1E), while IgM was the predominant isotype of total CD19+IgD- cells (not shown). Despite elevated circulating IgA-positive plasmablasts, no difference was detected in total serum IgA between IPAH and healthy controls (Figure 1F). Assessments of plasmablast numbers in relation to measurements of clinical disease (Supplemental Tables S1 and S3) and comorbid conditions (Supplemental Tables S2 and S3) did not reveal any statistically significant correlations. Thus, our findings suggest that elevated IgA plasmablasts represent a characteristic feature of evolving disease in IPAH.
Figure 1. Increased IgA-producing blood plasmablasts in IPAH.
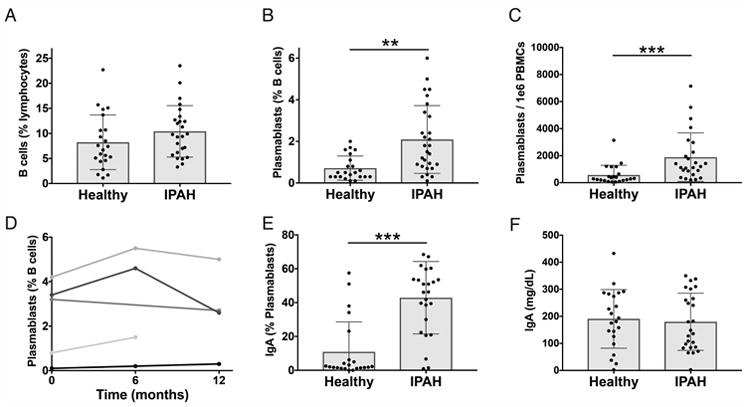
Peripheral blood mononuclear cells (PBMCs) and plasma were isolated from the blood of IPAH and healthy individuals. (A-C) Cells were analyzed by flow cytometry for CD19+ B cells (A), CD19+/INTCD3-CD14-CD20-CD27+CD38hi plasmablasts as a percent of CD19+ cells (B), and plasmablasts per 1×106 PBMCs (C). Each bar represents the mean ± SD of all subjects in a group and each dot represents the measured value for one subject with n = 25 (IPAH), n = 22 (Healthy).(D) Stability of plasmablast numbers over time in five individual IPAH patients for whom serial samples were available. Each line represents an individual subject. (E) Proportion of circulating plasmablasts with surface IgA expression. Each bar represents the mean ± SD of all subjects in a group and each dot represents the measured value for one subject with n = 25 (IPAH), n = 22 (Healthy). (F) Total serum IgA, measured by ELISA. Each bar represents the mean ± SD of all subjects in a group, and each dot represents the mean value of two replicate wells from an individual subject with n = 25 (IPAH), n = 22 (Healthy). IgA ELISA data was collected in a single independent experiment, while flow cytometry measurements were performed in batches of 1 to 5 patients, together with controls, at the time of the patient's medical visit. Cumulative data from all batches of flow cytometry is shown. ***P<0.001, **P<0.01 by Student's t-test.
Sequencing the plasmablast antibody repertoire in IPAH
Plasmablasts isolated from four representative IPAH patients were used for sequencing of the paired heavy chain (HC) and light chain (LC) variable regions. We previously pioneered this method to sequence plasmablast antibody repertoires in a diverse set of diseases [17, 18, 21-24]. With single-cell barcoding of the cDNA derived from each cell, we paired the HCs and LCs expressed by individual plasmablasts, determined error-corrected consensus sequences for the HC and LC variable regions, and generated phylogenetic trees to visualize the antibody repertoires (Figure 2). Phylogenetic trees representing paired HC and LC sequences from IgA and IgG plasmablasts were generated as previously described [17, 18, 21]. The phylogenetic trees are rooted based on HC V gene usage, and provide an overview of the plasmablast repertoire, including the size and frequency of plasmablast lineage expansions (Figure 2). As plasmablasts typically comprise only a small fraction of cells in healthy individuals, control subjects were not sequenced due to insufficient plasmablast numbers. Phylogenetic trees from all repertoires are presented in Supplemental Figure S1. Phylogenetic trees of IPAH plasmablast sequences revealed small, sparse clonal expansions in both the IgG and IgA repertoires.
Figure 2. Representative phylogenetic trees of plasmablast antibody repertoires in IPAH.
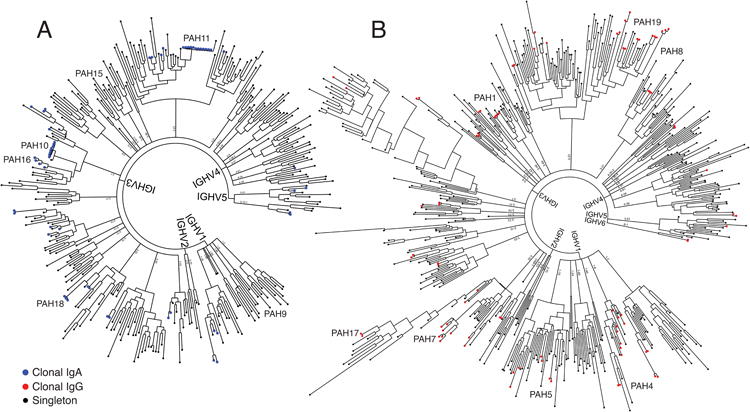
The paired HC and LC variable regions of individual plasmablasts isolated by flow cytometry were sequenced from four individuals with IPAH, and a representative phylogenetic tree from one subject is presented (see Supplemental Figure S1 for phylogenetic trees from the other subjects). The paired, consensus HC and LC antibody sequences from each plasmablast were binned and ordered by HC V-gene usage and clustered to create phylogenetic trees. Antibodies from clonally expanded lineages are represented by colored tips, with blue representing clonal lineage members expressing the IgA isotype (A) and red representing clonal lineage members expressing the IgG isotype (B). Tips representing non-clonal (singleton) sequences of either isotype are colored black. Representative sequences expressed as recombinant mAbs are labeled with text on each tree. The presented trees include antibodies derived from n=340 IgA expressing and n=519 IgG expressing plasmablasts.
Molecular features of IPAH plasmablast antibodies
Plasmablast lineage expansions were defined based on shared V and J gene usage and 60% CDR3 amino acid identity as previously described [21]. IPAH plasmablasts displayed a small number of lineage expansions, similar to the level of lineage expansions previously observed in RA [18, 21] (Genbank numbers KJ423107-KJ424184). These results are in contrast to the large lineage expansions observed in response to influenza vaccination [17, 22]. Both RA and IPAH exhibited clonality significantly lower than that observed in response to influenza vaccination (Figure 3A-C). IPAH plasmablast HC CDR3s exhibited a median length of 16 amino acids (Figure 3D), which is similar to the reported HC CDR3 length in healthy individuals [25, 26]. The rearranged V genes of IPAH plasmablasts possessed a median of 20 nucleotide mutations as compared to their IMGT-predicted germline V genes, indicating that these B cells had undergone affinity maturation. Despite comparable median mutation numbers between IgA and IgG plasmablasts, some IgA plasmablasts possessed un-mutated sequences. For all the sequenced subjects, 10% of the IgA sequences exhibited 0-10 mutations from germline, while 0% of the IgG sequences exhibited 1-10 mutations from germline (Figure 3E).
Figure 3. Cellular and molecular characteristics of IPAH plasmablast antibody repertoires.
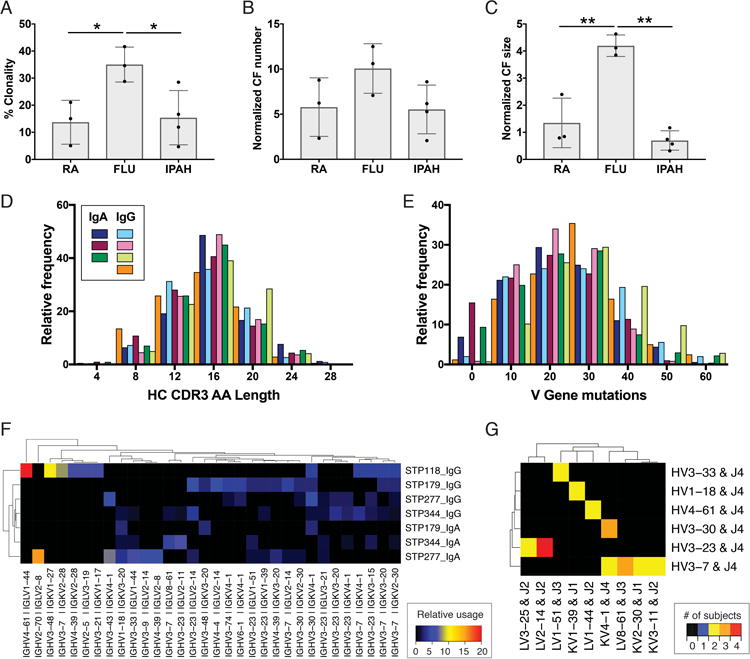
Paired HC and LC antibody sequences from individual plasmablasts underwent bioinformatic analyses of the clonal relationships and predicted germline gene segment usage of each cell. (A-C) Comparison of IPAH repertoire clonality with previously published rheumatoid arthritis (RA, GenBank Accession Numbers KJ423107-KJ424184) and Influenza vaccine (FLU, GenBank Accession Numbers KF994033-KF994563) repertoires. Each bar represents the mean ± SD of all subjects in a group, and each dot represents the value derived from an individual subject. (D) HC CDR3 amino acid length for the IgG and IgA IPAH repertoires. Each colored bar represents an individual patient's IgA or IgG plamablast repertoire. (E) HC V gene mutations from the predicted germline sequence. (F) Heatmap visualization of the most frequent HC and LC V gene usage combinations for each patient. Relative usage is defined as the frequency of a given gene combination normalized relative to sequencing depth. Gene combinations with 3% or greater usage in at least one repertoire are included on the plot. (G) Heatmap visualization of V and J gene usage across subjects. Color indicates the number of subjects who used a given HC V-J plus LC V-J gene combination at least two times. Genes with a combination used by at least two subjects are included on the plot. ** P<0.01, *P<0.05 by one-way ANOVA with Tukey's post-hoc test. For (A-G) n = 4 (IPAH); for (A-C) n = 3 (FLU), 3 (RA).
Gene segment usage
To assess if common B cell responses exist across IPAH subjects, we compared the frequency of HC and LC V and J gene usage across the IgA and IgG isotypes in these subjects (Figure 3F). The IGHV1-18 + IGKV3-20 combination had a significantly higher frequency in the IgA as compared to the IgG repertoires, while IGHV3-7 paired with several different J genes was significantly more frequent in IgG as compared to the IgA repertoires. The combination of IGHV3-30 + J4 with IGLV2-14 + J2 was present in multiple cells from all four subjects (Figure 3G). However, the CDR3 sequences from these pairs were highly divergent, indicating that they do not represent a shared amino acid sequence motif among subjects (Supplemental Figure S2). Clustering of CDR3 sequences with the cd-hit algorithm [27, 28], which analyzes the sequences independent of gene segment usage, did not reveal significant cross-patient patterns (Supplemental Figure S3). Ultimately, we observed preferred usage of certain antibody gene combinations but did not identify cross-patient shared CDR3 sequence motifs.
IPAH-derived monoclonal antibodies are autoreactive and polyreactive
To further investigate the relationships between the antibody repertoire features described above and the specificity of each plasmablast, we recombinantly expressed antibody proteins from representative single cells. Characteristics of the expressed antibody sequences are presentedin Table S5 (Online Supplement). To assess whether IPAH-sequence derived mAbs are autoreactive, they were analyzed using autoantigen micro arrays [29-31]. Unexpectedly, mAbs from IPAH plasmablasts displayed a high degree of polyreactivity, binding multiple antigens on the array (Figure 4A). To address the possibility that this polyreactivity was due to non-specific binding, the targets of two mAbs (mAb PAH9 and mAb PAH15) were validated by ELISA (Figure 4B). These mAbs were selected for target validation and in vitro functional characterization (described below), as they were derived from representative affinity-matured sequences from IgA clonal families, with representative levels of polyreactive binding. No binding was observed using ELISA plates coated with the negative control protein BSA (Figure 4B). Plasmablast sequence-derived mAbs from subjects with bacterial infection (B1) or those who received an Influenza vaccination (FLU57, FLU62) were not polyreactive in nature. Sequence-derived mAbs from IPAH also bound to the surface of live HUVECs in a cyto-ELISA (Figure 4C). These results demonstrate that blood plasmablasts in IPAH produce antibodies that function as AECAs.
Figure 4. PAH antigen microarray analysis and ELISA confirmation of IPAH patient recombinant mAbs.
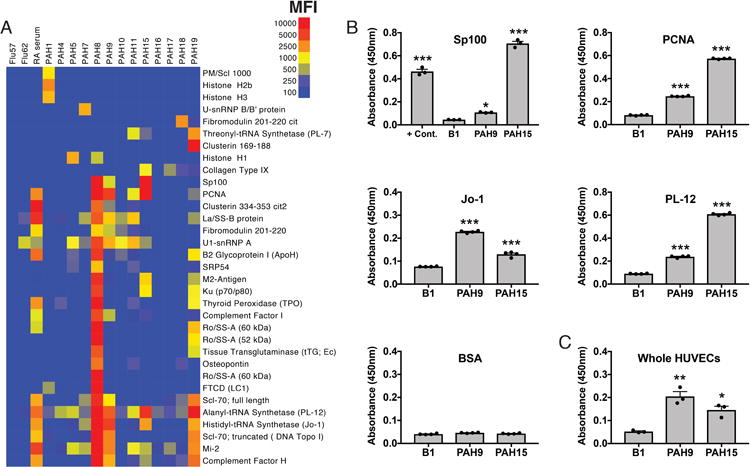
Representative IPAH plasmablast-derived clonal family sequences were selected for recombinant expression and analysis of recombinant mAb binding specificities. A summary of the sequence and binding characteristics of these mAbs is presented in supplemental table S5. Negative control antibodies were derived from sequencing the plasmablasts of subjects following Influenza vaccination (FLU57 and FLU62) or bacterial infection (B1). (A) A set of 108 IPAH-related and connective tissue disease candidate protein and peptide antigens were printed on epoxy microarray slides, and the IPAH antigen microarrays probed with IPAH clonal family mAbs, followed by a PE-conjugated anti-human IgG secondary antibody. Antigens with at least one positive hit of mean fluorescence intensity (MFI) >1000 are displayed, out of 108 total antigens included on the array. Blue represents no reactivity, yellow moderate reactivity, and orange-red high reactivity. (B) ELISA validation of microarray hits for mAb polyreactivity. (C) Cyto-ELISA of cell surface binding to live HUVECs, to assess anti-endothelial cell reactivity. ***P<0.001, **P<0.01, *P<0.05 versus the B1 isotype control antibody by one-way ANOVA with Tukey's post-hoc test. For ELISA results, each value represents the mean ± SD of three replicate wells, with similar results confirmed by at least two independent experiments.
Polyreactive IPAH mAbs stimulate endothelial cells in vitro
Total IgG purified from PAH serum can activate endothelial cells in vitro, inducing an activated pro-inflammatory phenotype characterized by increased surface expression of ICAM-1 (CD54), ICAM-2 (CD102), and E-Selectin (CD62L) as well as secretion of the vasoconstrictor Endothelin-1 [13, 14]. We assessed whether plasmablast-derived mAbs alone were sufficient to stimulate HUVECs to produce inflammatory mediators and markers. Stimulation of HUVECs with mAb PAH9 resulted in a significant increase in the surface expression of ICAM-1, an adhesion molecule that recruits leukocytes to a site of inflammation via interaction with LFA-1 (Figure 5A-B). Flow cytometry analysis demonstrated increased surface staining for E-selectin, another cell adhesion molecule expressed on activated endothelial cells (Figure 5B). HUVECs also secreted the inflammatory cytokines IL-8 and MCP-1 into culture supernatants following stimulation (Figure 5C). Unlike reported experiments with total serum IgG [13, 14], Endothelin-1 secretion was not induced by IPAH mAbs (Figure 5C). Corroborating these results, RT-PCR analysis showed increased expression of mRNA encoding il8, mcp1, and icam1 following stimulation with PAH9 (Figure 5D). Overall, mAb PAH9 induced secretion of inflammatory cytokines and increased surface ICAM-1 expression by HUVECs, as measured by ELISA, flow cytometry, and qPCR. Thus, IPAH plasmablast-derived sequences encode autoantibodies that can induce a pathogenic effect on endothelial cells in vitro, and may ultimately prove to do so in vivo.
Figure 5. Induction of pro-inflammatory and pro-adhesive mediators in HUVECs by recombinant IPAH plasmablast mAbs.
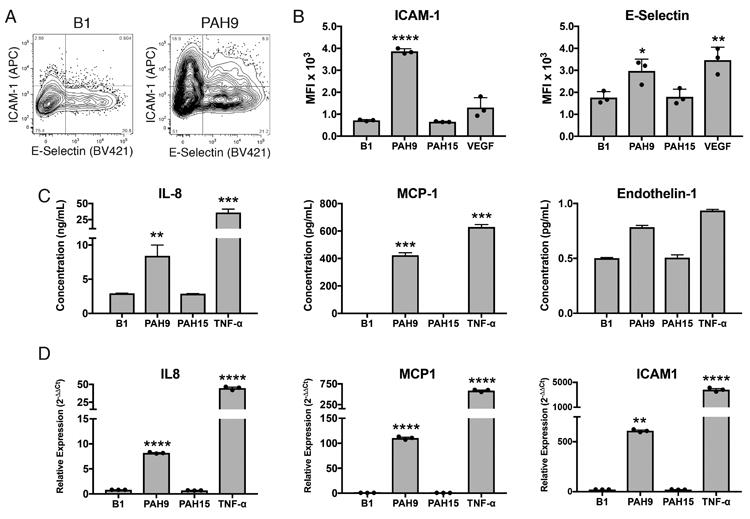
HUVEC cells were stimulated with IPAH mAbs, isotype control mAb, or positive control cytokines. Cells were harvested after 6 hours and stained for ICAM-1 and E-selectin. Additional cultures were harvested after 24 hours for qPCR and cell supernatant ELISAs. Negative control antibody B1 was derived from sequencing the plasmablasts of a patient with bacterial infection. (A) Representative flow cytometric staining of mAb-stimulated HUVECs. Full gating strategy is displayed in Supplemental Figure S5. (B) Mean fluorescence intensity of ICAM-1 and E-selectin staining on HUVECS 8 hours following addition of the mAb. Data shown is representative of two independent experiments with three samples per experiment. (C) ELISA measurement of MCP-1, IL-8, and endothelin-1 secreted by mAb-stimulated HUVECs. (D) qPCR gene expression analysis of HUVECS following overnight stimulation. For panels (B-D), Each value represents the mean ± SD of three replicate samples per experiment, with similar results confirmed by at least two independent experiments. ***P<0.001, **P<0.01, *P<0.05 by one-way ANOVA with Tukey's post-hoc test.
Discussion
Despite prior investigations of serum antibodies in IPAH, the B cell response is not well characterized. Herein we show that blood plasmablasts are significantly elevated in IPAH and are predominantly IgA-producing. In healthy individuals, plasmablasts are present in low numbers and rapidly mobilize in response to immune stimuli; they peak approximately one week following infection/vaccination, and return to baseline levels 10-14 days after the antigen stimulus is cleared [32, 33]. The persistent elevations in IgA plasmablasts in IPAH suggest the presence of a sustained mucosal antigen stimulus. Such a stimulus could arise from a persistent microbial stimulus (e.g., a mucosal microbe, mucosal microbial dysbiosis, or EBV) or from perpetually damaged host endothelium. Regulatory T cells (Tregs) normally limit pulmonary vascular inflammation, and the Treg abnormalities strongly associated with PAH could conceivably permit an anomalous expansion of autoreactive plasmablasts that give rise to IPAH [34, 35].
To characterize blood plasmablasts in IPAH, we performed single-cell sequencing of the paired HC and LC genes from IPAH patients, using a cell barcoding method that we developed [17, 18, 21]. Sequences from the HC constant regions confirmed the isotypes (IgA versus IgG) that had been identified by flow cytometry. IPAH plasmablasts displayed small clonal expansions, consistent with those observed in chronic autoimmune conditions and in contrast to the large expansions observed following acute microbial stimuli [17, 18, 21]. We observed usage of common antibody HC and LC gene segment combinations across multiple subjects. In addition to preferred antibody gene segment usage, future studies involving deeper sequencing of the antibody repertoire in a larger number of subjects could provide the opportunity to identify antibody paratopes shared across subjects with IPAH.
Diagnostic subgroups of PAH include those associated with drug use, connective tissue diseases, and congenital defects [36]. We limited the confounding effects of these factors by focusing our study on individuals with IPAH. Although previous reports have largely focused on IgG, lymphatic drainage occurs on a regional level, and cells from the pulmonary vasculature likely encounter mucosally-derived cells and molecules in their draining lymph nodes. Additionally, significant IgA autoantibody responses are present in autoimmune diseases in which mucosal surfaces are not the primary disease target, including RA [37, 38] and SLE [39, 40]. The increased prevalence of IgA plasmablasts, and the autoantigen targets identified by sequencing these IgA plasmablasts, point to the importance of including analysis of IgA and potential mucosal triggers in future studies of the autoantibody response in PAH. FcαRI (CD89) signaling on macrophages is a known checkpoint for tolerance versus activation – crosslinking monomeric IgA promotes tolerance, while crosslinking IgA immune complexes promotes an activated, inflammatory phenotype [41]. The role of macrophages and macrophage-derived LTB4 is well established in PAH animal models [7]. It remains possible that activation of macrophages by autoantigen-IgA immune complexes represents a key functional pathway by which autoimmunity mediates the pathogenesis of IPAH.
Unlike polyclonal serum, mAbs most often bind a single antigen target. Surprisingly, we observed polyreactive binding of mAbs to multiple antigens on planar microarrays. Antigen microarrays enable high-throughput screening of antibody binding, but with some potential for non-specific reactivity. Conversely, ELISA measurement of autoantibody-autoantigen interactions is more reproducible, but has a much lower throughput. Accordingly, we took a strategy of autoantigen array screening, followed by ELISA validation. Polyreactive mAbs derived from PAH patient plasmablasts bound to multiple autoantigens by ELISA (Sp100, PCNA, Jo-1, and PL-12) as well as to the surface of HUVECs, which classifies them as AECAs. These results suggest true polyclonal reactivity, rather than artifact. Polyreactive antibodies are characteristic of many autoimmune diseases. In some cases, this is due to specificity for a common post-translational modification (PTM) found on multiple antigens, such as citrullination or carbamylation [42]. A wide assortment of PTMs could potentially be shared across the autoantigen targets observed in this study, including the SUMO family of ubiquitin-related molecules [43, 44]. SUMOylation is present at relatively low levels in vascular endothelial cells, but increases in response to hypoxia, DNA damage, and inflammation [45]. Thus, a feedback loop of hypoxia, cellular damage, inflammation, and further SUMOylation could lead to continual autoimmune stimulation that promotes the pathogenesis of IPAH.
Polyreactive autoantibodies have been observed in healthy individuals, and it is possible that the IPAH-derived mAbs investigated in this study could be similarly derived. However, natural autoantibody (NAAb) clones in peripheral circulation are most frequently of the IgM isotype and not derived from affinity-matured sequences of highly antibody-secreting cells, such as plasmablasts. It was recently shown that microbiota-specific unmutated natural IgA antibodies are present in the intestines of mice [46], but such antibodies are not present at significant levels in human peripheral circulation [47]. Plasmablast sequence-derived mAbs used as negative controls in this study did not show autoreactivity/polyreactivity, and we have previously observed that >80% of mAbs derived from Influenza vaccine subject plasmablasts are highly specific for Influenza hemagglutinin [17]. Such results suggest that polyreactivity is not a broad characteristic of the plasmablast population. Polyreactive NAAbs from healthy subjects carry out a variety of beneficial functions, including tissue homeostasis and viral clearance, while they can also be upregulated in response to cellular damage to promote complement-mediated tissue destruction [47]. However, given the degree of somatic hypermutation and derivation from IgA-producing plasmablasts, it is likely that the IPAH mAbs described herein are induced as part of the PAH disease process and are distinct from NAAbs that have been observed in healthy individuals.
A representative IPAH patient-derived plasmablast mAb from an affinity-matured clonal family (PAH9) stimulated HUVEC endothelial cells to express inflammatory mediators. HUVECs were selected over human pulmonary artery endothelial cells (HPAECs) as they have been well characterized in a large number of studies, and the conditions for stimulation of cytokine production and adhesion molecule overexpression with positive control stimuli have been optimized and widely reported. The effects observed following HUVEC stimulation with mAb PAH9 in our system were consistent with those that have been reportedfor immunoglobulins purified from the serum of PAH patients [13, 14]. However, we did not observe significant functional effects of mAb PAH15, a different polyreactive mAb, on HUVECs. This difference suggests that polyreactive binding and/or cell surface binding to endothelial cells is not sufficient to induce endothelial inflammatory mediator production. Targeting a specific antigen or group of antigens by IPAH autoantibodies is likely necessary to promote inflammatory mediator production and the pathogenesis of IPAH. Future studies are needed to define the targets and pathways by which IPAH plasmablast sequence-derived mAbs activate endothelial cells, and whether they align with studies that have identified agonistic endothelin 1 and angiotensin II IgG autoantibodies as pathogenic pathways in PAH associated with systemic sclerosis [15]. Additionally, the variable regions of these IPAH mAbs can be expressed on mouse or rat immunoglobulin Fc regions to enable investigation of their pathogenicity in vivo in murine models of PAH.
Overall, we report that blood plasmablasts are significantly elevated in IPAH, remain elevated over time, and produce antibodies of the IgA isotype. Further, a representative recombinant IPAH plasmablast-derived mAb stimulated endothelial cells to produce inflammatory mediators implicated in the pathogenesis of IPAH. Our results suggest a role for mucosally-driven autoimmunity and autoimmune injury in the pathogenesis of IPAH, which provides new diagnostic and therapeutic opportunities.
Materials and Methods
Collection and processing of human samples
Samples were obtained under protocols approved by the Stanford University Institutional Review Board (IRB), and written informed consent was obtained from all patients. IPAH samples were acquired via the Stanford Pulmonary Hypertension Biobank (IRB #14083) from patients treated at Stanford between 2013 and 2017. Diagnosis of IPAH was based on clinical and hemodynamic criteria (mPAP≥25, PVR>3, PAWP≤15, and exclusion of non-WHO Group I factors). Healthy control samples were acquired from Stanford Blood Center. Blood was collected in sodium heparin tubes, and PBMCs processed with Ficoll using standard procedures.
Flow cytometry and cell sorting
Freshly isolated PBMCs were stained in Hank's Balanced Salt Solution (HBSS) with 2% fetal bovine serum using the following antibodies: CD20 (clone L27), CD38 (clone HB7), IgD (clone IA6-2), CD3 (clone UCHT1), and CD14 (clone MφP9) from BD Biosciences (San Jose, CA, USA); CD19 (clone HIB19), CD27 (clone O323), and IgM (clone MHM-88) from BioLegend (San Diego, CA, USA); and IgA (clone IS11-8E10) from Miltenyi Biotec (San Diego, CA, USA). Cells were stained for viability by the addition of Sytox blue dye (Thermo Fisher Scientific; Waltham, MA, USA) 10 minutes prior to analysis. Plasmablasts, defined as CD19+/INTCD3-CD14-CD20-CD27+CD38hi live single cells, were single-cell sorted into 96-well plates by a FACSAria II instrument (BD Biosciences). Plasmablast numbers were compared between IPAH subjects and healthy controls, and isotype-specific surface staining used to identify the isotype of the antibody expressed by each cell. Due to low IgG surface expression on plasmablasts, IgG-producing plasmablasts were defined by the absence of both IgA and IgM staining, and the antibody isotype expressed was further confirmed based on gene-specific PCR and the antibody constant region sequences recovered from each cell. Flow cytometry analyses were in accordance with the European Journal of Immunology's guidelines for the use of flow cytometry and cell sorting in immunological studies [48].
Single-cell antibody sequencing
IgA- and IgG-producing plasmablasts were single-cell sorted into 96-well plates, their cDNA labeled with oligonucleotide cell barcodes, and their paired antibody heavy and light chain variable regions sequenced as previously described [17, 18, 21]. Well-specific barcode sequences (TruGrade DNA oligos; IDT; Coralville, IA, USA) were then added to the cDNA of each cell by template switching (Maxima Reverse Transcriptase, Thermo Fisher Scientific, Waltham, MA, USA). Barcoded cDNA were pooled, gene-specific PCR used to amplify the immunoglobulin heavy and light chain variable regions, and MiSeq 2×330 paired-end sequencing performed (Illumina, Inc.; San Diego, CA, USA). Oligonucleotide sequences are provided in Supplemental Table S4.
Sequence data processing and analysis
Fastq generation and plate demultiplexing were performed using the onboard MiSeq Generate FASTQ workflow. After quality filtering, paired reads were stitched, separated by well ID, and consensus sequences determined by clustering well ID reads into operational taxonomic units (OTUs) [49]. Consensus sequences were analyzed with version 1.3.1 of IMGT HighV-QUEST [50]. Clonal families (CF) of plasmablasts were defined based on sharing IMGT-predicted HC (Heavy Chain) and LC (Light Chain) VJ genes and exhibiting>60% amino acid identity within the HC and LC CDR3s. Percent clonality was calculated as the percent of paired sequences that fall into any CF. Normalized CF number and size were calculated as the total number and average size of CFs in a repertoire, normalized to a sequencing depth of 100.
Recombinant expression
Antibodies were selected for recombinant expression based on previously identified criteria including clonal relationships, gene usage, and somatic mutations [17, 18, 22], and were synthesized and cloned as previously described [24]. Expression was performed by transient transfection of Expi293 cells (performed in the Robinson lab; cells from Thermo Fisher Scientific) or Tuna293 cells (performed at Lake Pharma, Inc., San Carlos, CA, USA), followed by purification with Protein A Plus resin (Thermo Fisher Scientific). To enable comparison of binding using the same secondary antibody, variable domains from both IgA and IgG plasmablasts were expressed on the human IgG1 Fc constant region. Negative control antibodies were derived from sequencing the plasmablasts of subjects following Influenza vaccination (FLU57 and FLU62) or bacterial infection (B1).
Antigen identification
The binding specificities of recombinant antibodies were identified using autoantigen microarrays. Protein and peptide autoantigens were printed onto glass slides (Arrayit; Sunnyvale, CA, USA), probed with recombinant antibodies, and analyzed as previously described [29-31]. The Sp100 autoantibody ELISA was performed according to the manufacturer's instructions (Inova Diagnostics; San Diego, CA, USA). PCNA, PL-12, and Jo-1 antigens were purchased from Surmodics (Eden Prairie, MN), with anti-human IgG-HRP (Bethyl Laboratories; Montgomery, TX, USA) as a secondary antibody.
In vitro stimulation assays
To determine if IPAH-derived monoclonal antibodies (mAbs) increased the production of inflammatory/adhesion molecules in vitro, human umbilical vein endothelial cells (HUVECs) were stimulated with 10μg/mL of the selected recombinant plasmablast antibodies, 20ng/mL VEGF (R&D Systems; Minneapolis, MN, USA), or 5ng/mL TNF-α (R&D Systems) between the third and fifth passages. Cells were harvested after 6 hours for surface staining of ICAM-1 and E-selectin, and supernatants from replicate cultures harvested after 24 hours for ELISA measurement of IL-8 (Abcam; Cambridge, UK), Endothelin-1 (Abcam), and MCP-1 (Lifespan Biosciences; Seattle, WA, USA).
qPCR
Cells from 24 hour stimulations were harvested in buffer RLT plus and purified using the RNEasy Plus kit (Qiagen; Hilden, Germany). Analyses were performed using the High Capacity cDNA Reverse Transcription Kit and Taqman gene expression assays for il8: Hs00174103_m1, mcp1: Hs00234140_m1, and icam1: Hs00164932_m1 (Thermo Fisher Scientific).
Statistical Analysis
Data are presented as the mean ± standard deviation (SD). Student's t tests (two-tailed, unpaired) were used to assess comparisons between two groups, and multiple comparisons were performed using one-way ANOVA with Tukey's post-hoc test. All tests were performed with Prism version 7.0c (Graphpad Software, Inc., La Jolla, CA, USA).
Supplementary Material
Acknowledgments
We thank Stanford Functional Genomics Center and Stanford Shared FACS Facility for advice and technical assistance. This work was supported by funding from NIH (R01HL138473-01 to M.R.N., R01 HL122887-01A1 to M.R.N., 2P01 HL014985-41A1 to M.R.N, K12 RFA-HL-07-004:CDP to L.K.B., T32 AR050942 to L.K.B.) and by the Vera Moulton Wall Center for Pulmonary Vascular Diseases.
Abbreviations
- IPAH
idiopathic pulmonary arterial hypertension
- RA
pulmonary arterial hypertension (PAH), rheumatoid arthritis
- SLE
systemic lupus erythematosus
- AECAs
anti-endothelial cell autoantibodies
- HC
immunoglobulin heavy chain
- LC
immunoglobulin light chain
- HUVECs
human umbilical vein endothelial cells
- PTM
post-translational modification
- SUMO
small ubiquitin-like modifier
- mAb
monoclonal antibody
- CF
peripheral blood mononuclear cells, clonal family
- NAAbs
natural autoantibodies
Footnotes
Conflict of interest: W.H.R. is a founder, member of the Board of Directors, consultant for, and owner of equity in Atreca, Inc. All other authors declare no financial or commercial conflicts.
Publisher's Disclaimer: This is the peer reviewed version of the following article: Blum LK et al. Eur J Immunol. 2018 Jan 25. doi: 10.1002/eji.201747460, which has been published in final form at http://dx.doi.org/10.1002/eji.201747460. This article may be used for non-commercial purposes in accordance with Wiley Terms and Conditions for Self-Archiving.
References
- 1.Galie N, Manes A, Farahani KV, Pelino F, Palazzini M, Negro L, Romanazzi S, et al. Pulmonary arterial hypertension associated to connective tissue diseases. Lupus. 2005;14:713–717. doi: 10.1191/0961203305lu2206oa. [DOI] [PubMed] [Google Scholar]
- 2.Bigna JJ, Nansseu JR, Um LN, Noumegni SR, Sime PS, Aminde LN, Koulla-Shiro S, et al. Prevalence and incidence of pulmonary hypertension among HIV-infected people in Africa: a systematic review and meta-analysis. BMJ Open. 2016;6:e011921. doi: 10.1136/bmjopen-2016-011921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Schermuly RT, Ghofrani HA, Wilkins MR, Grimminger F. Mechanisms of disease: pulmonary arterial hypertension. Nat Rev Cardiol. 2011;8:443–455. doi: 10.1038/nrcardio.2011.87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tonelli AR, Arelli V, Minai OA, Newman J, Bair N, Heresi GA, Dweik RA. Causes and circumstances of death in pulmonary arterial hypertension. Am J Respir Crit Care Med. 2013;188:365–369. doi: 10.1164/rccm.201209-1640OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rabinovitch M, Guignabert C, Humbert M, Nicolls MR. Inflammation and immunity in the pathogenesis of pulmonary arterial hypertension. Circ Res. 2014;115:165–175. doi: 10.1161/CIRCRESAHA.113.301141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Harper RL, Reynolds AM, Bonder CS, Reynolds PN. BMPR2 gene therapy for PAH acts via Smad and non-Smad signalling. Respirology. 2016;21:727–733. doi: 10.1111/resp.12729. [DOI] [PubMed] [Google Scholar]
- 7.Tian W, Jiang X, Tamosiuniene R, Sung YK, Qian J, Dhillon G, Gera L, et al. Blocking macrophage leukotriene b4 prevents endothelial injury and reverses pulmonary hypertension. Sci Transl Med. 2013:5. doi: 10.1126/scitranslmed.3006674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zuily S, Domingues V, Suty-Selton C, Eschwege V, Bertoletti L, Chaouat A, Chabot F, et al. Antiphospholipid antibodies can identify lupus patients at risk of pulmonary hypertension: A systematic review and meta-analysis. Autoimmun Rev. 2017;16:576–586. doi: 10.1016/j.autrev.2017.04.003. [DOI] [PubMed] [Google Scholar]
- 9.Sobanski V, Giovannelli J, Lynch BM, Schreiber BE, Nihtyanova SI, Harvey J, Handler CE, et al. Characteristics and Survival of Anti-U1 RNP Antibody-Positive Patients With Connective Tissue Disease-Associated Pulmonary Arterial Hypertension. Arthritis Rheumatol. 2016;68:484–493. doi: 10.1002/art.39432. [DOI] [PubMed] [Google Scholar]
- 10.Wang J, Qian J, Wang Y, Zhao J, Wang Q, Tian Z, Li M, et al. Serological biomarkers as risk factors of SLE-associated pulmonary arterial hypertension: a systematic review and meta-analysis. Lupus. 2017;26:1390–1400. doi: 10.1177/0961203317702255. [DOI] [PubMed] [Google Scholar]
- 11.Perosa F, Favoino E, Favia IE, Vettori S, Prete M, Corrado A, Cantatore FP, et al. Subspecificities of anticentromeric protein A antibodies identify systemic sclerosis patients at higher risk of pulmonary vascular disease. Medicine (Baltimore) 2016:95. doi: 10.1097/MD.0000000000003931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dib H, Tamby MC, Bussone G, Regent A, Berezne A, Lafine C, Broussard C, et al. Targets of anti-endothelial cell antibodies in pulmonary hypertension and scleroderma. Eur Respir J. 2012;39:1405–1414. doi: 10.1183/09031936.00181410. [DOI] [PubMed] [Google Scholar]
- 13.Arends SJ, Damoiseaux JG, Duijvestijn AM, Debrus-Palmans L, Vroomen M, Boomars KA, Brunner-La Rocca HP, et al. Immunoglobulin G anti-endothelial cell antibodies: inducers of endothelial cell apoptosis in pulmonary arterial hypertension? Clin Exp Immunol. 2013;174:433–440. doi: 10.1111/cei.12166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Arends SJ, Damoiseaux JG, Duijvestijn AM, Debrus-Palmans L, Boomars KA, Brunner-La Rocca HP, Cohen Tervaert JW, et al. Functional implications of IgG anti-endothelial cell antibodies in pulmonary arterial hypertension. Autoimmunity. 2013;46:463–470. doi: 10.3109/08916934.2013.812080. [DOI] [PubMed] [Google Scholar]
- 15.Becker MO, Kill A, Kutsche M, Guenther J, Rose A, Tabeling C, Witzenrath M, et al. Vascular receptor autoantibodies in pulmonary arterial hypertension associated with systemic sclerosis. Am J Respir Crit Care Med. 2014;190:808–817. doi: 10.1164/rccm.201403-0442OC. [DOI] [PubMed] [Google Scholar]
- 16.de Bourcy CFA, Dekker CL, Davis MM, Nicolls MR, Quake SR. Dynamics of the human antibody repertoire after B cell depletion in systemic sclerosis. Sci Immunol. 2017:2. doi: 10.1126/sciimmunol.aan8289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tan YC, Blum LK, Kongpachith S, Ju CH, Cai X, Lindstrom TM, Sokolove J, et al. High-throughput sequencing of natively paired antibody chains provides evidence for original antigenic sin shaping the antibody response to influenza vaccination. Clin Immunol. 2014;151:55–65. doi: 10.1016/j.clim.2013.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tan YC, Kongpachith S, Blum LK, Ju CH, Lahey LJ, Lu DR, Cai X, et al. Barcode-enabled sequencing of plasmablast antibody repertoires in rheumatoid arthritis. Arthritis Rheumatol. 2014;66:2706–2715. doi: 10.1002/art.38754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Szabo K, Papp G, Szanto A, Tarr T, Zeher M. A comprehensive investigation on the distribution of circulating follicular T helper cells and B cell subsets in primary Sjogren's syndrome and systemic lupus erythematosus. Clin Exp Immunol. 2016;183:76–89. doi: 10.1111/cei.12703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Robinson WH. Sequencing the functional antibody repertoire--diagnostic and therapeutic discovery. Nat Rev Rheumatol. 2015;11:171–182. doi: 10.1038/nrrheum.2014.220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kinslow JD, Blum LK, Deane KD, Demoruelle MK, Okamoto Y, Parish MC, Kongpachith S, et al. Elevated IgA Plasmablast Levels in Subjects at Risk of Developing Rheumatoid Arthritis. Arthritis Rheumatol. 2016;68:2372–2383. doi: 10.1002/art.39771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lu DR, Tan YC, Kongpachith S, Cai X, Stein EA, Lindstrom TM, Sokolove J, et al. Identifying functional anti-Staphylococcus aureus antibodies by sequencing antibody repertoires of patient plasmablasts. Clin Immunol. 2014;152:77–89. doi: 10.1016/j.clim.2014.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hansmann L, Blum L, Ju CH, Liedtke M, Robinson WH, Davis MM. Mass cytometry analysis shows that a novel memory phenotype B cell is expanded in multiple myeloma. Cancer Immunol Res. 2015;3:650–660. doi: 10.1158/2326-6066.CIR-14-0236-T. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nair N, Feng N, Blum LK, Sanyal M, Ding S, Jiang B, Sen A, et al. VP4- and VP7-specific antibodies mediate heterotypic immunity to rotavirus in humans. Sci Transl Med. 2017:9. doi: 10.1126/scitranslmed.aam5434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kitaura K, Yamashita H, Ayabe H, Shini T, Matsutani T, Suzuki R. Different Somatic Hypermutation Levels among Antibody Subclasses Disclosed by a New Next-Generation Sequencing-Based Antibody Repertoire Analysis. Front Immunol. 2017;8:389. doi: 10.3389/fimmu.2017.00389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rosner K, Winter DB, Tarone RE, Skovgaard GL, Bohr VA, Gearhart PJ. Third complementarity-determining region of mutated VH immunoglobulin genes contains shorter V, D, J, P, and N components than non-mutated genes. Immunology. 2001;103:179–187. doi: 10.1046/j.1365-2567.2001.01220.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Fu L, Niu B, Zhu Z, Wu S, Li W. CD-HIT: accelerated for clustering the next-generation sequencing data. Bioinformatics. 2012;28:3150–3152. doi: 10.1093/bioinformatics/bts565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Li W, Godzik A. Cd-hit: a fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics. 2006;22:1658–1659. doi: 10.1093/bioinformatics/btl158. [DOI] [PubMed] [Google Scholar]
- 29.Hueber W, Kidd BA, Tomooka BH, Lee BJ, Bruce B, Fries JF, Sonderstrup G, et al. Antigen microarray profiling of autoantibodies in rheumatoid arthritis. Arthritis Rheum. 2005;52:2645–2655. doi: 10.1002/art.21269. [DOI] [PubMed] [Google Scholar]
- 30.Robinson WH, DiGennaro C, Hueber W, Haab BB, Kamachi M, Dean EJ, Fournel S, et al. Autoantigen microarrays for multiplex characterization of autoantibody responses. Nat Med. 2002;8:295–301. doi: 10.1038/nm0302-295. [DOI] [PubMed] [Google Scholar]
- 31.Sokolove J, Bromberg R, Deane KD, Lahey LJ, Derber LA, Chandra PE, Edison JD, et al. Autoantibody epitope spreading in the pre-clinical phase predicts progression to rheumatoid arthritis. PLoS One. 2012;7:e35296. doi: 10.1371/journal.pone.0035296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Arumugakani G, Stephenson SJ, Newton DJ, Rawstron A, Emery P, Doody GM, McGonagle D, et al. Early Emergence of CD19-Negative Human Antibody-Secreting Cells at the Plasmablast to Plasma Cell Transition. J Immunol. 2017;198:4618–4628. doi: 10.4049/jimmunol.1501761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Fink K. Origin and Function of Circulating Plasmablasts during Acute Viral Infections. Front Immunol. 2012;3:78. doi: 10.3389/fimmu.2012.00078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Nicolls MR, Voelkel NF. The Roles of Immunity in the Prevention and Evolution of Pulmonary Arterial Hypertension. Am J Respir Crit Care Med. 2017;195:1292–1299. doi: 10.1164/rccm.201608-1630PP. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Jang E, Cho WS, Cho ML, Park HJ, Oh HJ, Kang SM, Paik DJ, et al. Foxp3+ regulatory T cells control humoral autoimmunity by suppressing the development of long-lived plasma cells. J Immunol. 2011;186:1546–1553. doi: 10.4049/jimmunol.1002942. [DOI] [PubMed] [Google Scholar]
- 36.Ryan JJ, Thenappan T, Luo N, Ha T, Patel AR, Rich S, Archer SL. The WHO classification of pulmonary hypertension: A case-based imaging compendium. Pulm Circ. 2012;2:107–121. doi: 10.4103/2045-8932.94843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kokkonen H, Mullazehi M, Berglin E, Hallmans G, Wadell G, Ronnelid J, Rantapaa-Dahlqvist S. Antibodies of IgG, IgA and IgM isotypes against cyclic citrullinated peptide precede the development of rheumatoid arthritis. Arthritis Res Ther. 2011;13:R13. doi: 10.1186/ar3237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Agrawal S, Misra R, Aggarwal A. Autoantibodies in rheumatoid arthritis: association with severity of disease in established RA. Clin Rheumatol. 2007;26:201–204. doi: 10.1007/s10067-006-0275-5. [DOI] [PubMed] [Google Scholar]
- 39.Villalta D, Bizzaro N, Bassi N, Zen M, Gatto M, Ghirardello A, Iaccarino L, et al. Anti-dsDNA antibody isotypes in systemic lupus erythematosus: IgA in addition to IgG anti-dsDNA help to identify glomerulonephritis and active disease. PLoS One. 2013;8:e71458. doi: 10.1371/journal.pone.0071458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Matsiota P, Druet P, Dosquet P, Guilbert B, Avrameas S. Natural autoantibodies in systemic lupus erythematosus. Clin Exp Immunol. 1987;69:79–88. [PMC free article] [PubMed] [Google Scholar]
- 41.Ben Mkaddem S, Rossato E, Heming N, Monteiro RC. Anti-inflammatory role of the IgA Fc receptor (CD89): from autoimmunity to therapeutic perspectives. Autoimmunity reviews. 2013;12:666–669. doi: 10.1016/j.autrev.2012.10.011. [DOI] [PubMed] [Google Scholar]
- 42.Burska AN, Hunt L, Boissinot M, Strollo R, Ryan BJ, Vital E, Nissim A, et al. Autoantibodies to posttranslational modifications in rheumatoid arthritis. Mediators Inflamm. 2014;2014:492873. doi: 10.1155/2014/492873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sternsdorf T, Jensen K, Will H. Evidence for covalent modification of the nuclear dot-associated proteins PML and Sp100 by PIC1/SUMO-1. J Cell Biol. 1997;139:1621–1634. doi: 10.1083/jcb.139.7.1621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Stelter P, Ulrich HD. Control of spontaneous and damage-induced mutagenesis by SUMO and ubiquitin conjugation. Nature. 2003;425:188–191. doi: 10.1038/nature01965. [DOI] [PubMed] [Google Scholar]
- 45.Jiang Y, Wang J, Tian H, Li G, Zhu H, Liu L, Hu R, et al. Increased SUMO-1 expression in response to hypoxia: Interaction with HIF-1alpha in hypoxic pulmonary hypertension. Int J Mol Med. 2015;36:271–281. doi: 10.3892/ijmm.2015.2209. [DOI] [PubMed] [Google Scholar]
- 46.Bunker JJ, Erickson SA, Flynn TM, Henry C, Koval JC, Meisel M, Jabri B, et al. Natural polyreactive IgA antibodies coat the intestinal microbiota. Science. 2017;358 doi: 10.1126/science.aan6619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lutz HU. Homeostatic roles of naturally occurring antibodies: an overview. J Autoimmun. 2007;29:287–294. doi: 10.1016/j.jaut.2007.07.007. [DOI] [PubMed] [Google Scholar]
- 48.Cossarizza A, Chang HD, Radbruch A, Akdis M, Andra I, Annunziato F, Bacher P, et al. Guidelines for the use of flow cytometry and cell sorting in immunological studies. Eur J Immunol. 2017;47:1584–1797. doi: 10.1002/eji.201646632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Edgar RC. UPARSE: highly accurate OTU sequences from microbial amplicon reads. Nat Methods. 2013;10:996–998. doi: 10.1038/nmeth.2604. [DOI] [PubMed] [Google Scholar]
- 50.Alamyar E, Duroux P, Lefranc MP, Giudicelli V. IMGT((R)) tools for the nucleotide analysis of immunoglobulin (IG) and T cell receptor (TR) V-(D)-J repertoires, polymorphisms, and IG mutations: IMGT/V-QUEST and IMGT/HighV-QUEST for NGS. Methods Mol Biol. 2012;882:569–604. doi: 10.1007/978-1-61779-842-9_32. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.