

Abstract
Background
Risk stratification of newly diagnosed patients with mantle cell lymphoma (MCL) is primarily based on the MCL international prognostic index (MIPI) and Ki67. Single-center studies have reported inferior outcomes in patients with a complex karyotype (CK), but this remains an area of controversy.
Methods
We retrospectively reviewed 483 patients from 5 academic centers in the United States and describe the effect of a CK on survival outcomes in MCL.
Results
CK was associated with inferior OS (4.5 vs 11.6 years, p<0.01) and PFS (1.9 vs 4.4 years, p<0.01). In patients who underwent high-intensity induction followed by ASCT in CR1, CK was associated with poor OS (5.1 vs 11.6 years, p=0.04) and PFS (3.6 vs 7.8 years, p<0.01). Among patients with a CK, high-intensity induction had no effect on OS (4.5 vs 3.8 years, p=0.77) nor PFS (2.3 vs 1.5 years, p=0.46). Similarly, ASCT in CR1 did not improve PFS (3.5 vs 1.2 years, p=0.12), nor OS (5.1 vs 4.0 years, p=0.27). In multivariable analyses with Ki67 and MIPI, only CK predicted for OS (HR 1.98, 95%CI 1.12 – 3.49, p=0.02), while both CK (HR 1.91, 95%CI 1.17 – 3.12, p=0.01) and Ki67>30% (HR 1.86, 95%CI 1.06 – 3.28, p=0.03) were associated with inferior PFS. Multivariable analysis did not identify any specific cytogenetic abnormalities associated with inferior survival.
Conclusions
CK is independently associated with inferior outcomes in MCL regardless of the intensity of induction therapy and receipt of ASCT. Cytogenetics should be incorporated into workup of a new diagnosis of MCL and novel therapeutic approaches should be investigated for patients with CK.
Keywords: Mantle Cell Lymphoma, Cytogenetics, Prognostic markers, Chemotherapy, Complex Karyotype
Introduction
Risk-stratification of newly diagnosed patients with mantle cell lymphoma (MCL) has not routinely informed clinical practice and has been inconsistently applied despite the fact that initial clinical presentation can range from indolent disease which can potentially be observed to very aggressive behavior commonly managed with intensive therapy1,2. The MCL International Prognostic Index (MIPI) uses baseline laboratory and clinical features to distinguish high-, intermediate-, and low-risk patients, and this distinction has been associated with differences in overall survival (OS) in several series3–5. However, in more recent analyses, the difference between the low- and intermediate-risk groups has become less apparent4. The European MCL Network recently described a modified combination of MIPI and Ki67 (MIPI-C) which defined 4 risk groups. In this series, the 5-year OS ranged from 17% for the highest-risk patients to 85% for low risk patients6. Other biologically based prognostic markers such as SOX-11, p53, and miR-18b have been investigated, but are not often utilized in routine clinical practice.7,8
Cytogenetics are utilized in several hematologic malignancies to predict outcome but have been evaluated on a more limited basis in MCL. In smaller series, both a complex karyotype (CK; ≥3 chromosomal abnormalities) and specific cytogenetic abnormalities such as del(17p) have been associated with inferior outcomes in the pre- and post-rituximab era.9–12 In the pre-rituximab era, increasing karyotypic complexity in a small cohort of patients was found to have adverse effect on survival in MCL,10 and this was later also seen in larger post-rituximab cohorts.13 In a single institution study of 80 patients treated between 2002 and 2011, a CK resulted in decreased 2-year PFS estimate of 48% as compared to 70% in patients with a non-CK (p=0.02), and a CK was also associated with inferior 2-year OS (58% vs 85%, p=0.02). However, this difference was not found to be significant when controlling for other variables such as MIPI in a multivariable analysis.11 Espinet et al also found that the degree of karyotypic complexity did not predict OS in a series of 145 patients with untreated and/or relapsed MCL, and that survival was more influenced by the specific cytogenetic abnormalities including del(17p) and add(3q).9 Given these findings, we conducted a multi-institutional retrospective study to evaluate the effect of a CK and individual abnormalities on prognosis and patient outcomes in MCL.
Methods
Patients ≥ 18 years old with previously untreated MCL diagnosed between 1993 and 2015 and evaluated at 5 academic centers were eligible. Patients without survival follow-up were excluded from the analysis. We collected data for each patient including demographic, laboratory, pathologic, and treatment-related variables of interest. Specific cytogenetic abnormalities were also collected from conventional metaphase karyotyping. This project was approved by the institutional review board at all participating sites with waiver of informed consent.
With the assistance of a clinical cytogeneticist, the full karyotypes were analyzed with aberrations such as derivative chromosomes and isochromosomes categorized into specific abnormalities for statistical analysis. We analyzed the 11 most common cytogenetic abnormalities (10 individual autosome abnormalities + sex chromosome loss). CK was defined as having ≥3 unrelated cytogenetic abnormalities not including t(11;14), based on a conventional karyotype exclusive of FISH, SNP array, or other genomic assessments. We assessed for differences between patients with CK and without CK with regards to clinical presentation, baseline prognostic factors, and therapy.
Categorical variables were compared across CK status using chi-squared tests or Fisher’s Exact tests, where appropriate, and numeric variables were compared using ANOVA. We also evaluated associations of CK with other prognostic markers in MCL including MIPI and Ki67 proliferative index (using a cutoff of 30% to define high Ki67). Therapy received was evaluated by individual regimen as well as grouped as intensive vs non-intensive based on whether or not an induction regimen incorporated high dose cytarabine. OS was calculated from the date of diagnosis to the date of death from any cause, with living patients censored at last follow-up. PFS was evaluated from the date of diagnosis to the date of progression or death from any cause, with patients alive and progression-free censored at last follow-up. Survival distributions were estimated using the Kaplan-Meier method, and were compared using the log-rank tests. Univariate and multivariable Cox proportional hazards models were fit for OS and PFS using available clinical, biologic, and treatment-related variables. Univariate analysis was performed that included CK and the 11-most common cytogenetic abnormalities. Variables that were significantly associated with survival in the univariate model were then fit in a multivariable model. Abnormalities were subjected to backwards elimination with a p-value for removal of 0.1. Remaining significant variables from the CK/specific abnormality model were then fit into a model with MIPI and Ki67. For both MIPI and Ki67, patients with missing data were set to an “unknown” category. Treatment site effect was fit as a random effect in multivariable models. The proportional hazards assumption was checked, and significance was assessed at the 0.05 level. Statistical analyses were performed using SAS 9.4 (SAS Institute Inc., Cary, NC).
Results
Patient characteristics
Of 483 MCL cases identified, 18 patients were excluded due to missing survival data, resulting in a sample of 465 patients. 188 of the 465 patients did not have karyotypic data and were excluded from analyses of karyotypic complexity and specific cytogentic abnormalities. Also excluded were an additional two patients who were known to have a CK but for whom no data existed regarding presence of specific abnormalitites. An additional patient only had florescence in-situ hybridization results, but no conventional cytogenetic analysis. Patient characteristics for 274 patients available for analysis with a known karyotype are described in Table 1. Of 274 patients, 189 (69%) were male, 20 (7.5%) had stage III disease, 236 (89%) had stage IV disease, and the median age at diagnosis was 63 years (range: 32-83). Fifty-three patients (19%) had a CK. Among patients with available pre-treatment MIPI risk score (n=168); 29%, 40%, and 31% were low-, intermediate-, and high–risk, respectively. In patients with available proliferative index (n=125), Ki67 was > 30% in 47% of patients. Of the 252 patients with known induction data, 48% underwent intensive cytarabine containing induction. Of 227 patients with known transplant status, 111(49%) underwent autologous stem cell transplant (ASCT) in first remission (CR1).
Table 1. Patient characteristics at diagnosis and treatments.
P-values are for comparisons of non-CK vs CK groups. Percentages only refer to patients for whom data for a given variable is available, missing data not included.
| Characteristic | All patients | Complex karyotype | Non-complex | p |
|---|---|---|---|---|
| (n=274) | (n=53) | (n=221) | ||
| Median age (range) | 63 (32-83) | 63 (43-81) | 63(32-83) | 0.57 |
| Male sex, n(%) | 189 (69) | 34 (64) | 155 (70) | 0.4 |
| MIPI Group, n(%) | <0.01 | |||
| Low | 49 (29) | 4 (12) | 45 (33) | |
| Intermediate | 67 (40) | 11 (33) | 56 (41) | |
| High | 52 (31) | 18 (55) | 34 (25) | |
| Ki67 expression, n(%) | 0.19 | |||
| Ki67 > 30% | 59 (47) | 11 (58) | 48 (45) | |
| Ki67 ≤ 30% | 66 (53) | 8 (42) | 58 (55) | |
| ECOG PS, n(%) | 0.05 | |||
| 0 or 1 | 211 (92) | 38 (84) | 173 (94) | |
| 2, 3 or 4 | 19 (8) | 7 (16) | 12 (6) | |
| Stage | 0.15 | |||
| I/II | 9 (3) | 0 (0) | 9 (4) | |
| III/IV | 256 (97) | 52 (100) | 204 (96) | |
| Bone marrow involvement | <0.01 | |||
| Yes | 220 (83) | 47 (96) | 173 (80) | |
| No | 45 (17) | 2(4) | 43 (20) | |
| GI involvement | 0.37 | |||
| Yes | 33 (18) | 2 (11) | 31 (20) | |
| No | 150 (82) | 17 (89) | 188 (81) | |
| Splenomegaly | <0.01 | |||
| Yes | 144 (57) | 41 (80) | 103 (52) | |
| No | 107 (42) | 10 (20) | 97 (49) | |
| Elevated LDH | <0.01 | |||
| Yes | 91 (41) | 28 (60) | 63 (36) | |
| No | 132 (59) | 19 (40) | 113 (64) | |
| WBC > 10,000 | <0.01 | |||
| Yes | 87(35) | 26 (53) | 61 (31) | |
| No | 159 (65) | 23 (47) | 136 (69) | |
| Nodal mass > 10cm | 0.78 | |||
| Yes | 12 (5) | 2 (4) | 10 (5) | |
| No | 236 (95) | 47 (96) | 189 (95) | |
| Constitutional symptoms | <0.01 | |||
| Yes | 86(33) | 24 (50) | 62 (29) | |
| No | 176 (67) | 24 (50) | 152 (71) | |
| High intensity induction regimen | 0.34 | |||
| Yes | 117 (48) | 26 (54) | 91 (46) | |
| No | 127 (52) | 22 (46) | 105 (54) | |
| ASCT in CR1 | 0.88 | |||
| Yes | 111 (49) | 20 (50) | 91 (49) | |
| No | 116 (51) | 20 (50) | 96 (51) | |
| Year of diagnosis | <0.01 | |||
| 2006 and earlier | 44 (16) | 10 (19) | 34 (15) | |
| 2007-2009 | 64 (23) | 8 (15) | 56 (25) | |
| 2010-2012 | 95 (35) | 28 (53) | 67 (30) | |
| 2013-2015 | 71 (26) | 7 (13) | 64 (29) |
Compared to those patients without CK, patients with CK were more likely to have bone marrow involvement (p<0.01), elevated LDH at diagnosis (p<0.01), and white blood cell count greater than 10,000 (p<0.01). In addition, patients with CK were more likely to have a high-risk MIPI score (p<0.01). There was no significant difference between CK and non-CK groups in the frequency of intensive induction (54% vs 46%, p=0.34) or ASCT in CR1 (50% vs 51%, p=0.88).
The patients without available cytogenetics (188 of 465 patients) were analyzed, and in general, the patients with unknown karyotype had a presentation similar to those with a non-complex karyotype (Supplemental Table 1). Specifically, the unknown and non-complex karyotype were similar compared to the patients with CK with respect to the known prognostic markers of high-risk MIPI (24% and 25% vs 55% respectively) and high Ki67 expression (47% and 45% vs 55%). While the reason for having an unknown karyotype was not known for all patients, most patients had an unknown karyotype due to the test not being ordered at the time of diagnosis, and there were no recorded instances of a failed test.
Predictors of Progression-free and Overall Survival
Patients with a CK had a median OS of 4.5 years compared to 11.6 years for those without CK (p<0.01). Median PFS was 1.9 years in the CK group and 4.4 years in the non-CK group (p<0.01). 2-year OS was 68% (95%CI: 52%-80%) in the CK group and 91% (95%CI: 85%-94%) in the non-CK group. 2-year PFS was 48% (95%CI: 33%-62%) in the CK group and 76% (95%CI: 69%-82%) in the non-CK group (Figure 1).
Figure 1. Survival in MCL patients stratified by karyotypic complexity.
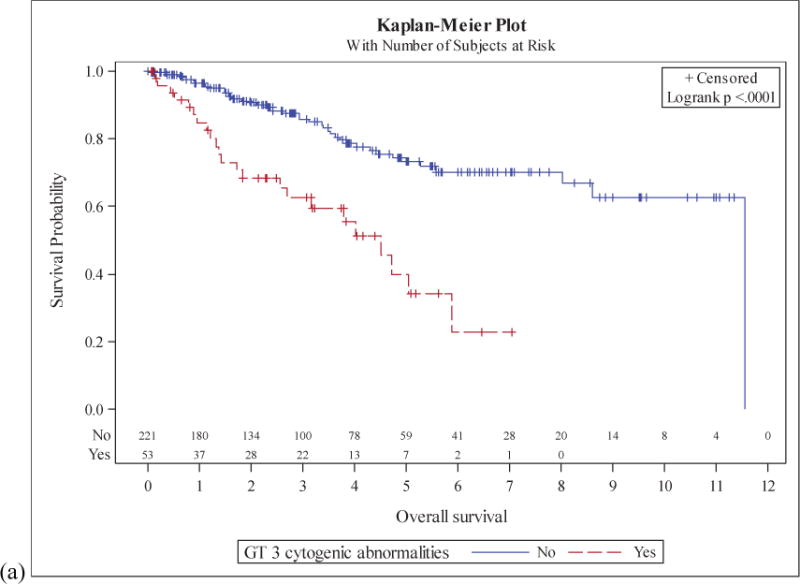
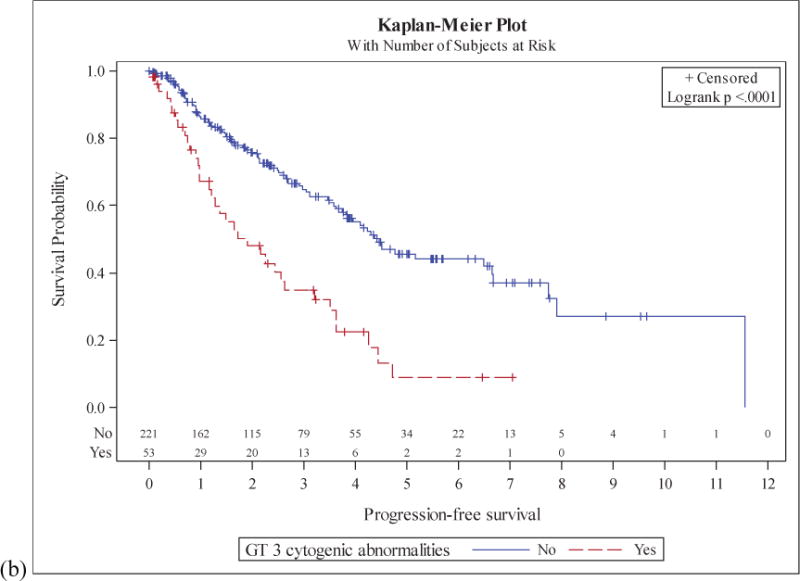
Overall survival (a) and progression free survival (b) in patients with and without a complex karyotype.
228 of the 465 patient had MIPI data available. Median OS of the high and intermediate MIPI risk groups was 5.9 and 8.0 years respectively, while the median OS of the low risk group was not reached (p=0.06). Median PFS of the high, intermediate, and low risk groups was 2.6, 3.6, and 7.9 years respectively (p=0.03).
125 of the 465 patients had Ki67 proliferative index information available. When we applied a cutoff of 30%, median OS was not reached in both the high and low Ki67 groups (p=0.21). 5-year OS was 56% (95% CI 36%-73%) in the high group and 79% (61%-89%) in the low group. Median PFS was 2.4 years in the high group and 4.1 years in the low group (p<0.01) with 5-year PFS of 21% (8.2%-37%) in the high group and 44% (27%-60%) in the low group.
Due to the similarities between the patients without cytogenetic data and those with a non-complex karyotype, a sensitivity analysis was performed in which those without cytogenetic data (an “unknown karyotype”) were included in the non-complex karyotype group (Supplemental Figure 1). In this sensitivity analysis, CK remained significantly associated with an inferior median OS (4.5 vs 11.6 years, p<0.01) and inferior median PFS (1.9 vs 4.2 years, p<0.01).
Effect of CK on survival stratified by prognostic markers
In the 52 patients with high-risk MIPI and cytogenetic data available, the CK and non-CK groups respectively had median OS of 3.8 years vs not reached (p=0.05) and median PFS of 1.7 years vs 3.8 years (p=0.07). In the 67 patients with intermediate-risk MIPI and cytogenetic data available, the CK and non-CK groups respectively had median OS of 4.7 years vs 11.6 years (p=0.21) and median PFS of 1.6 years vs 3.6 years (p=0.03). There were 49 patients with low-risk MIPI and cytogenetic data. Only 4 of these patients had a CK. There was no clear trend with respect to OS nor PFS given the small number of patients in the CK group.
In 66 patients (8 with CK) with Ki-67<30%, median OS in the CK vs non-CK groups were 4 years vs not reached (p<0.01). In this group of patients, the median PFS was 1.2 years vs 4.2 years (p<0.01). In 59 patients (11 with CK) with Ki-67≥30%, the median OS was not reached in the CK nor non-CK groups and no significant difference was seen in the 1, 2 nor 5 year OS (p=0.86), but this group of patients had limited follow-up. Median PFS in this same group of patients was 1.5 years vs 2.5 years respectively (p=0.11).
Effect of CK on survival stratified by Therapy Received
Among the 111 patients who underwent ASCT in CR1, median OS in the CK vs non-CK groups was 5.1 vs 8.6 years respectively (p=0.01), and the median PFS was 3.5 vs 6.7 years respectively (p<0.01). In 116 patients who did not undergo ASCT in CR1, median OS in the CK group was 4 years vs a median not reached in the non-CK group (p<0.01). The median PFS was 1.2 vs 2.9 years in the CK vs non-CK groups, respectively (p<0.01).
Among the 117 patients who underwent high-intensity cytarabine containing induction therapy, median OS in the CK vs non-CK groups was 4.5 vs 11.6 years respectively (p<0.01). In this group of patients, the median PFS was 2.3 vs 7.8 years respectively (p<0.01). In 127 patients who underwent low-intensity induction therapy, median OS in the CK group was 3.8 years vs not reached in the non-CK group (p<0.01). The median PFS was 1.5 vs 3.1 years in the CK vs non-CK groups, respectively (p<0.01).
In 91 patients who underwent high-intensity cytarabine containing induction followed by ASCT in CR1, CK was associated with poorer OS (5.1 vs 11.6 years, p=0.04) and PFS (3.6 vs 7.8 years, p<0.01) (Figure 2).
Figure 2. Survival in MCL patients who underwent high-intensity induction followed by consolidative ASCT.
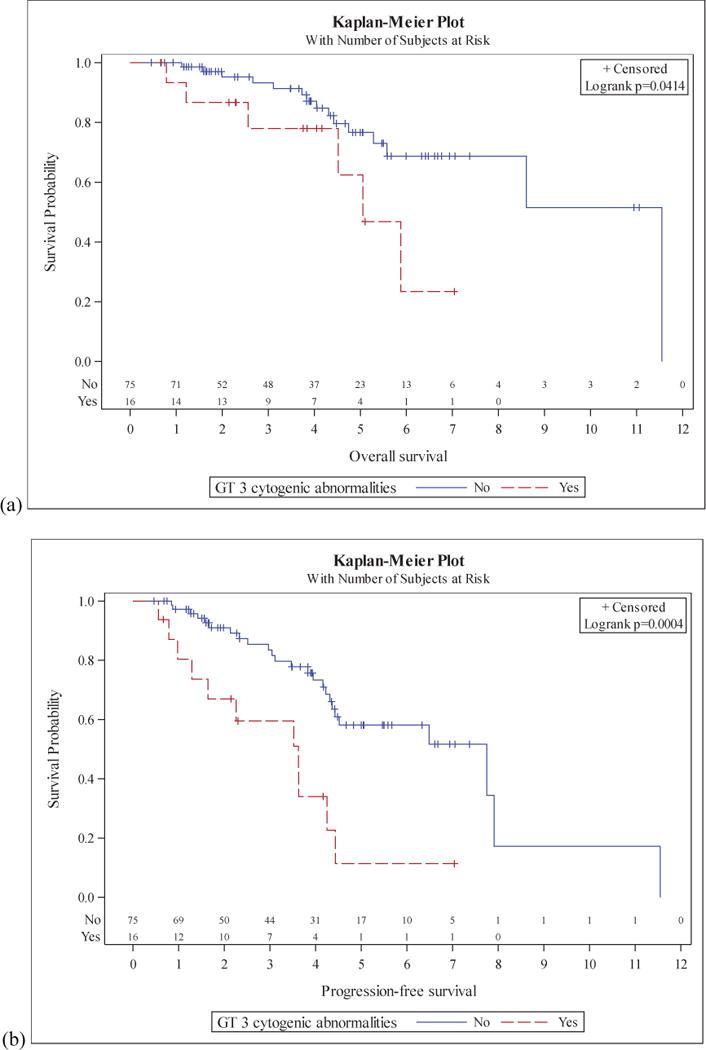
Overall survival (a) and progression free survival (b) are seen for those with and without a complex karyotype.
Effect of induction intensity and ASCT in CR1 on patients stratified by karyotypic complexity
In those patients with a CK, treatment with an intensive cytarabine containing induction regimen vs low-intensity regimen had no statistically significant effect on median OS (4.5 vs 3.8 years, p=0.77) nor median PFS (2.3 vs 1.5 years, p=0.45). Similarly, in those with CK, ASCT in CR1 did not improve PFS (3.5 vs 1.2 years, p=0.12), and there was no difference in OS (5.1 vs 4.0 years, p=0.27). In those patients with a non-CK, significant improvements in PFS were seen with high-intensity cytarabine containing induction (7.8 vs 3.1 years, p<0.01) as well as ASCT in CR1 (6.7 vs 2.9 years, p<0.01). However, there was no association with OS (Table 2).
Table 2.
Effect of high-intensity cytarabine containing regimen and ASCT in CR1 on survival stratified by karyotypic complexity
| Patients | Overall Survival | Progression Free Survival | ||||||
|---|---|---|---|---|---|---|---|---|
| High intensity | Low intensity | High intensity | Low intensity | p | High intensity | Low intensity | p | |
| CK | 28 | 24 | 4.5 | 3.8 | 0.8199 | 2.3 | 1.5 | 0.3495 |
| nonCK | 91 | 104 | 11.6 | NR | 0.8789 | 7.8 | 3.1 | 0.0025 |
| ASCT in CR1 | no ASCT | ASCT in CR1 | no ASCT | p | ASCT in CR1 | no ASCT | p | |
| CK | 21 | 13 | 5.1 | 4 | 0.2994 | 3.5 | 1.2 | 0.0823 |
| nonCK | 91 | 95 | 8.6 | NR | 0.9267 | 6.7 | 2.9 | 0.0044 |
High intensity: R-HCVAD, R-MCHOP, R-CHOP/R-DHAP, Nordic Regimin
Low intensity: All others
“Other” regimens not included in analysis
Presence of specific cytogenetic abnormalities is not associated with survival
In addition to karyotype complexity, the 11 most common specific cytogenetic abnormalities, seen in figure 3, underwent univariate and multivariable analysis with respect to survival.
Figure 3. Most common cytogenetic abnormalities seen in MCL patients.
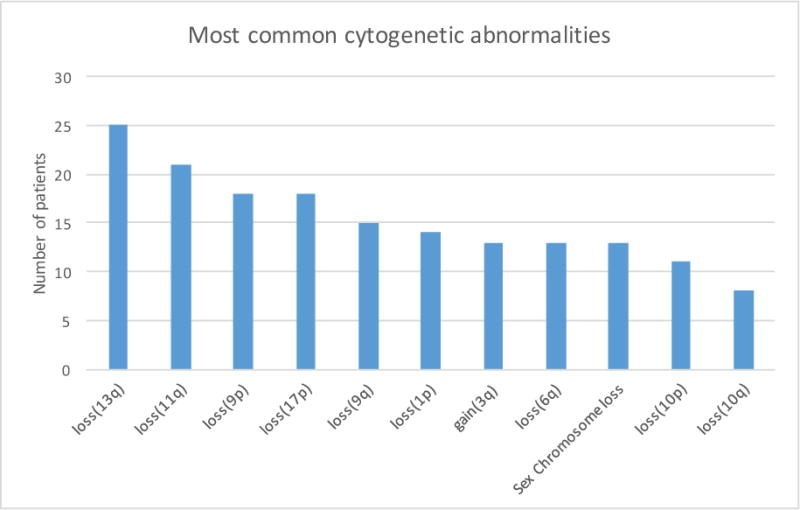
The 10 most common autosomal abnormalities in addition to sex chromosome losses and the number of patients each was seen in.
In univariate analysis, the specific abnormalities associated with inferior PFS were loss(9p), loss(9q), loss(10p), loss(10q), loss(13q), and loss(17p). Inferior OS was associated with loss(9p), loss(9q), and loss(13q). Notably, in univariate analysis, loss(17p) was associated with inferior PFS (HR 2.31, 95%CI 1.12 – 4.76, p=0.02), but not significantly associated with OS (HR 2.13, 95%CI 0.77 - 5.94, p=0.15).
In a multivariable model of CK and specific cytogenetic abnormalities, CK remained the only variable associated with PFS (HR 2.04, 95%CI 1.27 – 3.26, p=<0.01) and OS (HR 2.12, 95%CI 1.24 – 3.63, p<0.01). The only cytogenetic abnormality that remained in the PFS model after backwards elimination was loss(9q) (HR 2.00, 95%CI 0.99 – 4.01, p=0.05), while no cytogenetic abnormalities remained in the OS model. Of note, while loss(17p) was associated with inferior PFS in the univariate model, it was found not to be independently associated with survival when the backwards elimination process was applied to the multivariable model.
Complex karyotype remains associated with survival independent of MIPI and Ki67
In multivariable analysis of CK in addition to the well described prognostic factors of MIPI and Ki67, CK remained strongly associated with both PFS and OS. MIPI was not independently associated with survival, and Ki67 was only independently associated with PFS (Table 3).
Table 3.
Multivariable analysis of described prognostic factors in MCL
| PFS | OS | |||
|---|---|---|---|---|
| Cytogenetic abnormalitiy | HR (95%CI) | p | HR (95%CI) | p |
| Complex karyotype | 1.91 (1.17-3.12) | 0.01 | 1.98 (1.12-3.49) | 0.02 |
| MIPI Unknown | 1.06 (0.62-1.82) | 0.32 | 0.83 (0.41-1.68) | 0.87 |
| MIPI Low | 0.63 (0.32-1.22) | 0.73 (0.30-1.76) | ||
| MIPI Intermediate | 0.88 (0.49-1.58) | 0.73 (0.33-1.63) | ||
| MIPI High | - | |||
| Ki67 Unknown | 0.79 (0.47-1.33) | <0.01 | 1.06 (0.51-2.20) | 0.43 |
| Ki67 >30% | 1.86 (1.06-3.28) | 1.59 (0.68-3.72) | ||
| Ki67 ≤ 30% | - | - | ||
| loss (9q) | 1.83 (0.88-3.82) | 0.11 | - | - |
Only CK, but not any specific cytogenetic abnormalities, was included in the MIPI/Ki67 OS model due to the fact that no specific abnormality was independently associated with OS. In the CK/MIPI/Ki67 model, CK was associated with inferior survival (HR 1.98, 95%CI 1.12 – 3.49, p=0.02), while MIPI and Ki67 had no independent association.
Since both loss(9q) and CK remained in the PFS model above, they were both included in the MIPI/Ki67 PFS model. In multivariable analysis of CK/loss(9q)/MIPI/Ki67, CK remained independently associated with PFS (HR 1.91, 95%CI 1.17 – 3.12, p=0.01) as did Ki67>30% (HR 1.86, 95%CI 1.06 – 3.28, p=0.03) while loss(9q) was not.
Discussion
Initial management of MCL ranges from active observation to high-intensity induction therapy14. Additionally, post-induction consolidation with ASCT and/or maintenance therapy remains a consideration for patients. Given the broad clinical heterogeneity of MCL, improved characterization of disease behavior is important to define the optimal treatments for individual patients.
Our findings suggest an important role for cytogenetics in the risk-stratification of MCL patients. We show that a CK is associated with reduced OS (11.6 vs 4.5 years) and PFS (4.4 years vs 1.9 years) when compared to a non-CK in this large multi-institution population of MCL patients. These findings appear to apply regardless of intensity of the induction regimen and whether or not ASCT is included in the upfront treatment strategy suggesting that current aggressive therapy approaches do not abrogate the risk associated with presence of a CK. The poor outcomes in the CK population, in spite of intensive induction and ASCT in CR1 suggests a need for development of clinical trials to investigate novel/alternative treatment approaches for this population. Whether certain alternative approaches, such as allogeneic transplantation or specific novel agents and combination therapies will be effective in patients with CK will require carefully designed multi-center clinical trials to determine. Patients without a CK appeared to benefit significantly from receipt of an intensive cytarabine-containing regimen and ASCT in CR1 with marked differences in PFS but not OS, suggesting clinical trials investigating cytarabine-based approaches and ASCT in CR1 may benefit by focusing on this population or stratifying by CK.
As opposed to prior analyses, in this larger multi-institution study, CK but not MIPI score significantly predicted PFS and OS in multivariable analyses. In addition, presence of a CK was significantly associated with inferior outcome in both intermediate- and high-risk MIPI groups, and patients without CK had prolonged OS within the high-risk MIPI group, suggesting that cytogenetics may help to distinguish patients within MIPI subgroups. Additionally, in patients with a low Ki67, a CK was found to predict worse outcomes that those patients with a non-CK. The association of CK with inferior outcomes remained when accounting for specific cytogenetic abnormalities. Interestingly, while loss(17p) was initially hypothesized to be a possible driver of the CK-inferior survival association; in multivariable modeling only CK, but not loss(17p) was found to be associated with inferior survival. Loss(17p) was significant in the univariate PFS model. It had an elevated HR, but did not meet statistical significance in the univariate OS model. While surprising, the lack of association of loss(17p) has been described in multiple other analyses10,12,13,15. Espinet et al do describe loss(17p) being prognostic in a multivariable model9, and Sarkozy et al report that it is prognostic in univariable, but not multivariable modeling12.
More recently, Eskelund et al used data from the Nordic MCL trials to show that in patients younger than 66 years old, TP53 mutations were associated with inferior OS (HR 6.2, p<0.01) and time-to-relapse (HR 6.9, p<0.01) with a median OS of 1.8yrs16. Additionally, they demonstrated that TP53 mutated cases had an inferior response to intensive induction therapy. Their analysis used an 8-gene next-generation sequencing (NGS) panel as compared to our analysis using metaphase cytogenetics. The differences in sensitivity for single gene mutations using NGS likely contributes to their finding of an association of TP53 mutation with survival while we did not see this using metaphase cytogenetics. In addition, del(17p) may not necessarily imply a TP53 mutation. As a result, further investigation is required to better understand the relative importance of a TP53 deletion vs mutation. However, both analyses found that genomic aberrations play a more significant role in MCL prognosis than the traditional prognostic markers of MIPI and Ki6716.
Unfortunately, some subgroup analyses were limited due to small sample sizes and unavailable data. Other variables, such as SOX11 expression and blastoid variant histology have also been shown to have a prognostic impact8 but were not evaluated in this study. It is unknown as to whether these factors would correlate with karyotypic complexity and/or add to its prognostic impact. In addition, the specific cytogenetic abnormality analysis was considered exploratory, and a multiple comparisons adjustment with respect to outcomes was not utilized. Given the era in which these patients were treated, minimal residual disease was not routinely assessed. As a result, we cannot comment on the impact of a CK on the achievement of an MRD negative state nor the impact of CK on response duration for MRD negative patients.
In clinical practice, it is already possible to use biologic characteristics to evaluate disease phenotype. While MIPI score currently uses a combination of patient-related (age and PS) and laboratory (WBC and LDH) factors to predict prognosis, the Ki-67 proliferation index provides a prognostic marker that speaks more specifically to tumor biology. Adding to this, our data indicate that karyotypic complexity can also be a biologic measure used to predict outcomes until future methods with next generation sequencing and gene expression profiling are refined. Already, mutations in genes such as NOTCH1/2 are being correlated with prognosis, and recurrent mutations in ATM, CCND1, TP53, BIRC3, TLR2 and various chromatin modifiers have been identified in MCL by next generation sequencing17,18. Future studies will hopefully identify those mutations which predict a CK, and such mutations could then be evaluated prospectively or be used to develop risk-adapted approaches.
While cytogenetics are not formally a part of the standard work-up for MCL19,20, it is not unusual for a cytogenetic assessment to be available, and it can be easily implemented as this is a standard technique for multiple other hematologic malignancies. Since metaphase karyotyping cannot be performed once a diagnostic specimen (such as a lymph node biopsy) is placed in formalin, it is important to ensure that conventional cytogenetic analysis is ordered on fresh lymph node biopsies in patients suspected to have MCL. Fortunately, this analysis is readily available for the majority of patients with newly diagnosed lymphoma and can therefore be implemented easily into clinical practice. Our analysis has identified that a CK independently predicts for a poor prognosis overall in patients with MCL, regardless of additional prognostic markers such as the MIPI score, Ki-67, or specific cytogenetic abnormality. These data indicate that assessment of CK provides meaningful prognostic data independent of MIPI and Ki67 and should be collected in clinical trials and considered for incorporation into clinical practice. Unfortunately, outcomes in MCL patients with CK were still relatively poor, even with intensification of induction therapy and ASCT in CR1. As a result, we conclude that the development of novel/alternative approaches for MCL patients with a complex karyotype represents an unmet medical need that can be addressed with future clinical trials.
Supplementary Material
Overall survival (a) and progression free survival (b) in patients with a complex karyotype vs those with a non-complex karyotype OR an unknown karyotype.
Percentages only refer to patients for whom data on a given variable is available, missing data not included.
Acknowledgments
Dr. Jonathon B Cohen is a Lymphoma Research Foundation grantee and receives funding from the American Society of Hematology. Support for this publication was also supported in part by the Biostatistics and Bioinformatics Shared Resource of the Winship Cancer Institute of Emory University and NIH/NCI under award number P30CA138292. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Footnotes
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Contribution: I.B.G., J.M.S., and J.B.C. were responsible for conceptualization, formal analysis, writing-original draft preparation. I.B.G., A.D.S., M.J.L, J.M.S, D.S., J.J.M, K.A.B, N.S.G, S.M. M.J.G., A.V.D., N.E, T.S.F, M.H., S.I.P., C.R.F., J.B.C. were responsible for investigation, and writing – reviewing and editing.
References
- 1.Cohen JB, Han X, Jemal A, Ward EM, Flowers CR. Deferred therapy is associated with improved overall survival in patients with newly diagnosed mantle cell lymphoma. Cancer. 2016;122(15):2356–2363. doi: 10.1002/cncr.30068. [DOI] [PubMed] [Google Scholar]
- 2.Martin P, Chadburn A, Christos P, et al. Outcome of deferred initial therapy in mantle-cell lymphoma. J Clin Oncol. 2009;27(8):1209–1213. doi: 10.1200/JCO.2008.19.6121. [DOI] [PubMed] [Google Scholar]
- 3.Hoster E, Dreyling M, Klapper W, et al. A new prognostic index (MIPI) for patients with advanced-stage mantle cell lymphoma. Blood. 2008;111(2):558–565. doi: 10.1182/blood-2007-06-095331. [DOI] [PubMed] [Google Scholar]
- 4.Hoster E, Klapper W, Hermine O, et al. Confirmation of the mantle-cell lymphoma International Prognostic Index in randomized trials of the European Mantle-Cell Lymphoma Network. J Clin Oncol. 2014;32(13):1338–1346. doi: 10.1200/JCO.2013.52.2466. [DOI] [PubMed] [Google Scholar]
- 5.Budde LE, Guthrie KA, Till BG, et al. Mantle cell lymphoma international prognostic index but not pretransplantation induction regimen predicts survival for patients with mantle-cell lymphoma receiving high-dose therapy and autologous stem-cell transplantation. J Clin Oncol. 2011;29(22):3023–3029. doi: 10.1200/JCO.2010.33.7055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hoster E, Rosenwald A, Berger F, et al. Prognostic Value of Ki-67 Index, Cytology, and Growth Pattern in Mantle-Cell Lymphoma : Results From Randomized Trials of the European Mantle Cell Lymphoma Network. J Clin Oncol. 2016;34(12):1–10. doi: 10.1200/JCO.2015.63.8387. [DOI] [PubMed] [Google Scholar]
- 7.Husby S, Ralfkiaer U, Garde C, et al. miR-18b overexpression identifies mantle cell lymphoma patients with poor outcome and improves the MIPI-B prognosticator. Blood. 2015;125(17):2669–2677. doi: 10.1182/blood-2014-06-584193. [DOI] [PubMed] [Google Scholar]
- 8.Nordström L, Sernbo S, Eden P, et al. SOX11 and TP53 add prognostic information to MIPI in a homogenously treated cohort of mantle cell lymphoma - a Nordic Lymphoma Group study. Br J Haematol. 2014;166(1):98–108. doi: 10.1111/bjh.12854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Espinet B, Salaverria I, Beà S, et al. Incidence and prognostic impact of secondary cytogenetic aberrations in a series of 145 patients with mantle cell lymphoma. Genes, Chromosom Cancer. 2010;49:439–451. doi: 10.1002/gcc.20754. [DOI] [PubMed] [Google Scholar]
- 10.Cuneo A, Bigoni R, Rigolin GM, et al. Cytogenetic profile of lymphoma of follicle mantle lineage: correlation with clinicobiologic features. Blood. 1999;93(4):1372–1380. [PubMed] [Google Scholar]
- 11.Cohen JB, Ruppert AS, Heerema NA, et al. Complex Karyotype Is Associated With Aggressive Disease and Shortened Progression-Free Survival in Patients With Newly Diagnosed Mantle Cell Lymphoma. Clin Lymphoma Myeloma Leuk. 2014;15(5):278–285.e1. doi: 10.1016/j.clml.2014.12.012. [DOI] [PubMed] [Google Scholar]
- 12.Sarkozy C, Terré C, Jardin F, et al. Complex karyotype in mantle cell lymphoma is a strong prognostic factor for the time to treatment and overall survival, independent of the MCL international prognostic index. Genes, Chromosom Cancer. 2014;53(1):106–116. doi: 10.1002/gcc.22123. [DOI] [PubMed] [Google Scholar]
- 13.Katzenberger T, Kienle D, Stilgenbauer S, et al. Delineation of distinct tumour profiles in mantle cell lymphoma by detailed cytogenetic, interphase genetic and morphological analysis. Br J Haematol. 2008;142(4):538–550. doi: 10.1111/j.1365-2141.2008.07199.x. [DOI] [PubMed] [Google Scholar]
- 14.Cheah CY, Seymour JF, Wang ML. Mantle Cell Lymphoma. J Clin Oncol. 2016;34(11):1256–1269. doi: 10.1200/JCO.2015.63.5904. [DOI] [PubMed] [Google Scholar]
- 15.Parry-Jones N, Matutes E, Morilla R, et al. Cytogenetic abnormalities additional to t(11;14) correlate with clinical features in leukaemic presentation of mantle cell lymphoma, and may influence prognosis: A study of 60 cases by FISH. Br J Haematol. 2007;137:117–124. doi: 10.1111/j.1365-2141.2007.06526.x. [DOI] [PubMed] [Google Scholar]
- 16.Eskelund CW, Dahl C, Hansen JW, et al. TP53 mutations identify younger mantle cell lymphoma patients who do not benefit from intensive chemoimmunotherapy. Blood. 2017 doi: 10.1182/blood-2017-04-779736. Accepted, in print. [DOI] [PubMed] [Google Scholar]
- 17.Zhang J, Jima D, Moffitt AB, et al. The genomic landscape of mantle cell lymphoma is related to the epigenetically determined chromatin state of normal B cells. Blood. 2014;123(19):2988–2996. doi: 10.1182/blood-2013-07-517177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Beà S, Valdés-Mas R, Navarro A, et al. Landscape of somatic mutations and clonal evolution in mantle cell lymphoma. Proc Natl Acad Sci U S A. 2013;110(45):18250–18255. doi: 10.1073/pnas.1314608110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zelenetz AD, Gordon LI, Wierda WG, Abramson JS. (NCCN Guidelines).NCCN Guidelines: Non-Hodgkin’s Lymphomas Verson 3.2016. https://www.nccn.org/professionals/physician_gls/pdf/nhl.pdf.
- 20.Dreyling M, Thieblemont C, Gallamini A, et al. Esmo consensus conferences: Guidelines on malignant lymphoma. Part 2: Marginal zone lymphoma, mantle cell lymphoma, peripheral T-cell lymphoma. Ann Oncol. 2013;24(4):857–877. doi: 10.1093/annonc/mds643. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Overall survival (a) and progression free survival (b) in patients with a complex karyotype vs those with a non-complex karyotype OR an unknown karyotype.
Percentages only refer to patients for whom data on a given variable is available, missing data not included.