

Abstract
The axon initial segment (AIS) is located at the proximal axon and is the site of action potential initiation. This reflects the high density of ion channels found at the AIS. Adaptive changes to the location and length of the AIS can fine-tune the excitability of neurons and modulate plasticity in response to activity. AIS play an important role in maintaining neuronal polarity by regulating the trafficking and distribution of proteins that function in somatodendritic or axonal compartments of the neuron. In this review, we provide an overview of the AIS cytoarchitecture, mechanism of assembly, and recent studies revealing mechanisms of differential transport at the AIS that maintain axon and dendrite identities. We further discuss how genetic mutations in AIS components (ie. ankyrins, ion channels and spectrins) and injuries may cause neurological disorders.
Keywords: axon, cytoskeleton, ion channel, polarity
Graphical abstract
In this review, we provide an overview of the AIS cytoarchitecture, mechanism of assembly, and recent studies revealing mechanisms of differential transport at the AIS that maintain axon and dendrite identities. We further discuss how genetic mutations in AIS components (ie. ankyrins, ion channels and spectrins) and injuries may cause neurological disorders
Introduction
Neurons receive synaptic inputs that converge on their dendrites and cell bodies. The summation of synaptic inputs gives rise to action potentials at the axon initial segment (AIS), a 20-60 μm long domain located at the proximal axon/soma interface that has a high density of voltage-gated ion channels, membrane proteins and a unique repertoire of submembranous cytoskeletal scaffolds (Fig. 1). Although the AIS was originally defined ultrastructurally by fasciculated microtubules and an electron-dense undercoat beneath the plasma membrane 1, it is now defined molecularly by the master scaffolding protein ankyrinG (ankG) and the voltage-gated ion channels clustered by ankG. After action potentials are generated, they propagate along axons to synaptic terminals where they release neurotransmitters across the synaptic cleft to the next neuron or cell in the circuit. Axons may be myelinated by glial cells: oligodendrocytes in the CNS and Schwann cells in the PNS. In myelinated axons, action potentials are regenerated at the nodes of Ranvier. Nodes are small gaps in the myelin sheath where Na+ channels are clustered in high density (Fig. 1). The directionality of action potential propagation in the nervous system relies on the highly polarized compartments of neurons and the distribution and composition of ion channels within these compartments. In addition, the strategic location of the AIS at the proximal axon also confers physical and functional polarity on the distinct domains since the AIS acts as a gate between somatodendritic and axonal compartments. Thus, in addition to its role regulating action potential initiation, the AIS also functions to maintain axon-dendrite polarity 2,3. Although the molecular details for how ion channels are clustered at the AIS are mostly known, how the AIS restricts proteins to distinct compartments or how it excludes vesicles containing somatodendritic cargoes remains less understood. Varying lines of evidence supported both passive and active mechanisms that restrict the entrance of proteins/vesicles into the axon. In addition, recent work has revealed new details regarding retention and trafficking mechanisms. Here, we briefly review the molecular composition and assembly of the AIS and focus on its role in maintenance of neuronal polarity and its function as a major regulator of vesicle trafficking. Lastly, we consider how injuries and mutations in AIS-related genes can impact AIS structure and function.
Figure 1.
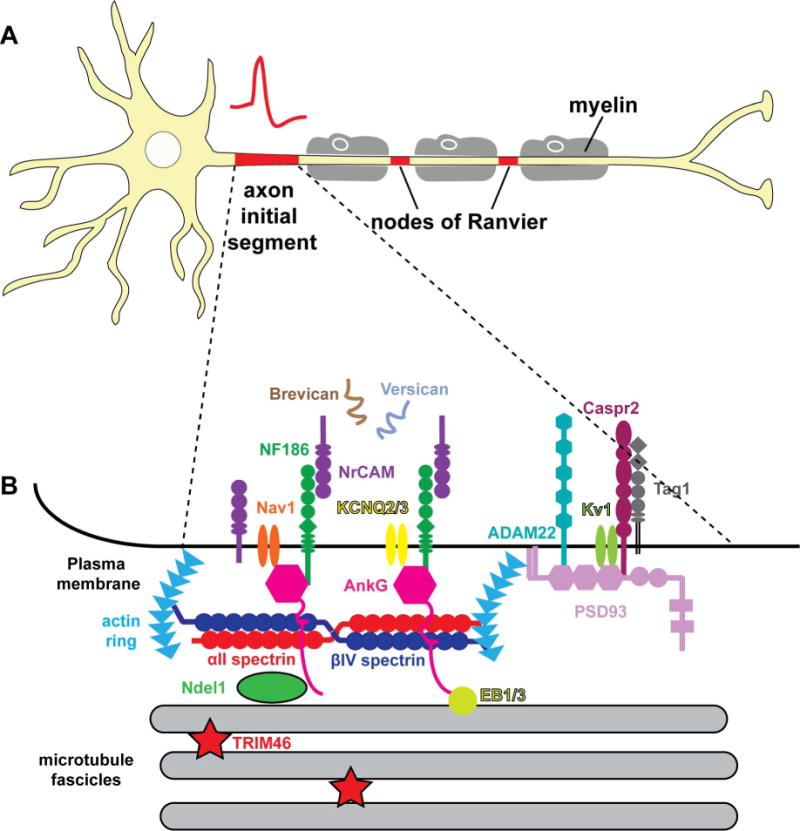
Cartoon of a multipolar neuron and the molecular composition of the AIS. (A) Neurons are polarized into somatodendritic and axonal domains. Axons can be myelinated by oligodendrocytes in the CNS and Schwann cells in the PNS. Nodes of Ranvier are gaps in the myelin sheath. Both axon initial segments and nodes of Ranvier have high densities of Na+ channels for action potential initiation and propagation, respectively. (B) The molecular organization of the axon initial segment.
AnkyrinG: the master regulator of AIS assembly
The submembranous scaffold protein ankyrin G (ankG) is the master organizer of the AIS and controls both ion channel clustering and maintenance of neuronal polarity. In mammals, there are three ankyrin genes: Ank1 (ankyrinR; ankR), Ank2 (ankyrinB; ankB) and Ank3 (ankyrinG; ankG) 4. The structures and functions of the ankyrins are similar but they have different subcellular distributions. AnkR was first discovered in erythrocytes and is also expressed in muscles and neurons. AnkB and ankG are found in a variety of tissues including brain, heart, muscle and epithelial cells. Ankyrins produce multiple splice variants in various cell types and these variants have different functions 5. In neurons, 270 kDa and 480 kDa ankG are the major isoforms specifically localized at the AIS and nodes of Ranvier 6,7 (Fig. 1B). Shortly after axon specification, ankG begins to cluster at the proximal axon and then recruits AIS components 8,9.
Most AIS constituents interact with ankG directly or indirectly. Na+ channels fail to cluster at the AIS and all the other AIS proteins lose their subcellular polarity after ankG expression is silenced by shRNA or ablated genetically. These results show that the clustering of all AIS components depends on ankG 10–12, whereas loss of NF186, NrCAM, Na+ channels, or βIV spectrin may affect the stability of other neuronal AIS proteins rather than their initial clustering 13. One notable exception to this is the NF186-dependent recruitment and clustering of the brevican-based AIS extracellular matrix which may function to further stabilize AIS proteins or synapses converging on the AIS 10.
In addition to ankG’s role as a master regulator of AIS protein clustering, upon loss of ankG, somatodendritic proteins can invade the former axon. Furthermore, with somatodendritic proteins in the axon, the axon begins to acquire the morphological and molecular characteristics of dendrites including spine-like protrusions and postsynaptic densities2,3. These results suggest that ankG is required to maintain neuronal polarity and more shall be said about this function of the AIS later.
AnkG interacting proteins
A defining feature of the AIS plasma membrane is the high density of voltage-gated ion channels, including Na+ channels, K+ channels and Ca2+ channels (Fig. 1B). The accumulation of voltage-gated Na+ channels (estimates range from about 5 to 50 fold higher at the AIS than in the distal axons or dendrites) lowers the threshold membrane potential required for action potential initiation 14. Mammalian Na+ channels consist of a pore-forming α subunit and accessory β subunits. The α subunit has a pseudotetrameric structure with domains (I–IV) consisting of six membrane-spanning segments. The cytoplasmic loop between the second and third subunits contains the AIS-targeting motif responsible for interacting with ankG 15,16. In addition, the clustering of Na+ channels at the AIS and nodes of Ranvier can be regulated by protein kinase CK2. Bréchet et al. 17 discovered that CK2 is enriched at the AIS and nodes of Ranvier both in vitro in cultured hippocampal neurons and in vivo. The serine residues (S1112, S1123, S1124, and S1126) were identified as the CK2 phosphorylation sites in the ankyrin binding domain and their phosphorylation increases the affinity of ankG for Na+ channels. CK2 inhibition reduces the density of Na+ channels at the AIS.
The major subtypes of Na+ channels at the AIS are Nav 1.1, Nav 1.2 and Nav 1.6 18,19. The differential expression and the asymmetric subcellular distribution of different Nav subtypes depends on neuron types and developmental stage. For example, during early development, Nav1.2 channels are initially found at the AIS. Later Nav1.6 channels appear in more mature neurons 18. Moreover, high-threshold Nav1.2 channels are preferentially located at the proximal AIS of cortical neurons while low-threshold Nav1.6 channels are found in the distal part of the AIS 20. This polarized distribution may reflect discrete functions of Na+ channel subtypes for action potential generation and backpropagation.
Besides Na+ channels, K+ channels at the AIS are also important to modulate the action potential. Kv7.2 and Kv7.3 (KCNQ2/3) channels can form homomeric or heteromeric complexes and accumulate at the AIS by interacting with ankG 21. The K+ channels Kv1.1 and Kv1.2 are also clustered at the AIS but do not bind to ankG 22,23. Instead, these channels may be clustered at the distal part of the AIS through binding to the scaffold protein postsynaptic density-93 (PSD93) 22. However, the mechanisms for Kv1 channel clustering remain poorly understood since acute shRNA knockdown of PSD93 in vitro blocks Kv1 channel clustering, but PSD93 knockout mice still have Kv1 channels located at their AIS, suggesting that there may be other, redundant mechanisms for Kv1 channel clustering in vivo 24.
Ca2+ channels have also been reported at the AIS 25, and these may be modulated by dopaminergic signaling 26. However, in contrast to the AIS Na+ and K+ channels, little is known about their molecular mechanisms of localization since the presence of these AIS channels has been inferred from electrophysiological studies rather than more direct immunolabeling approaches.
In addition to ion channels, several CAMs are also located at the AIS (Fig. 1B). For example, L1 CAMs are members of the immunoglobulin superfamily that contain: immunoglobulin and fibronectin repeats in their extracellular domain, a single transmembrane domain, and a cytoplasmic domain that binds to ankG through a highly conserved FIGQY motif. Phosphorylation of the tyrosine (Y) in the FIGQY motif inhibits the interaction of L1 CAMs with ankG 27,28. In Purkinje neurons of the cerebellum, NF186 gradients have been proposed to direct GABAergic innervation of the AIS by basket cells. In NF186-deficient mice, these AIS pinceau synapses are reduced and mis-targeted 29. NF186 also recruits extracellular matrix (ECM) molecules to the AIS, including aggrecan, brevican, versican and tenascin R, and thereby functions as a link between the AIS submembrane cytoskeleton and the ECM 10,30–32. Although many interactions among ECM components have been described, the functions are still poorly understood. Other potential roles for these ECMs include shaping synaptic plasticity through confining membrane surface receptors or molecules to distinct membrane domains 33. Other AIS CAMs include a disintegrin and metalloproteinase domain-containing protein 22 (ADAM22), Caspr2 (contactin-associated protein-like 2) and Tag-1 (transient axonal glycoprotein-1). These proteins are also clustered at the distal AIS through direct or indirect interaction with PSD-93 24. ADAM22 is a receptor for the secreted LGI (leucine-rich glioma inactivated) proteins that may also be part of the ECM proteins 34. However, it is not known if LGI proteins are also found at the AIS of some neurons and the extracellular binding partners and functions of most AIS CAMs remain poorly understood.
A submembranous cytoskeletal network consisting of the proteins ankG and βIV spectrin resides immediately underneath the AIS plasma membrane (Figs. 1B and 2A). Nearly all AIS components are recruited by and depend on direct or indirect interactions with ankG. AnkG is thought to be stabilized at the AIS through binding to the spectrin repeat 14–15 of βIV spectrin 35. βIV spectrin further interacts with the actin cytoskeleton at the inner shaft of the AIS. AIS βIV spectrin has two splice variants (Σ1 and Σ6); the longest isoform, βIVΣ1, is proposed to bind to the actin cytoskeleton via an actin-binding domain at the N terminus while the shorter truncated form (βIVΣ6) comprises only the C terminal half of the protein beginning with spectrin repeat 10 36,37. Because both isoforms include spectrin repeat 14-15, they both bind ankG and are located at the AIS. However, only the Σ1 splice variant harbors the canonical actin binding domain. Nevertheless, recent studies show that βIVΣ1 is most abundant during development whereas βIVΣ6 becomes the major splice variant after neurons mature 38. If and how βIVΣ6 interacts with actin remains unknown.
Figure 2.
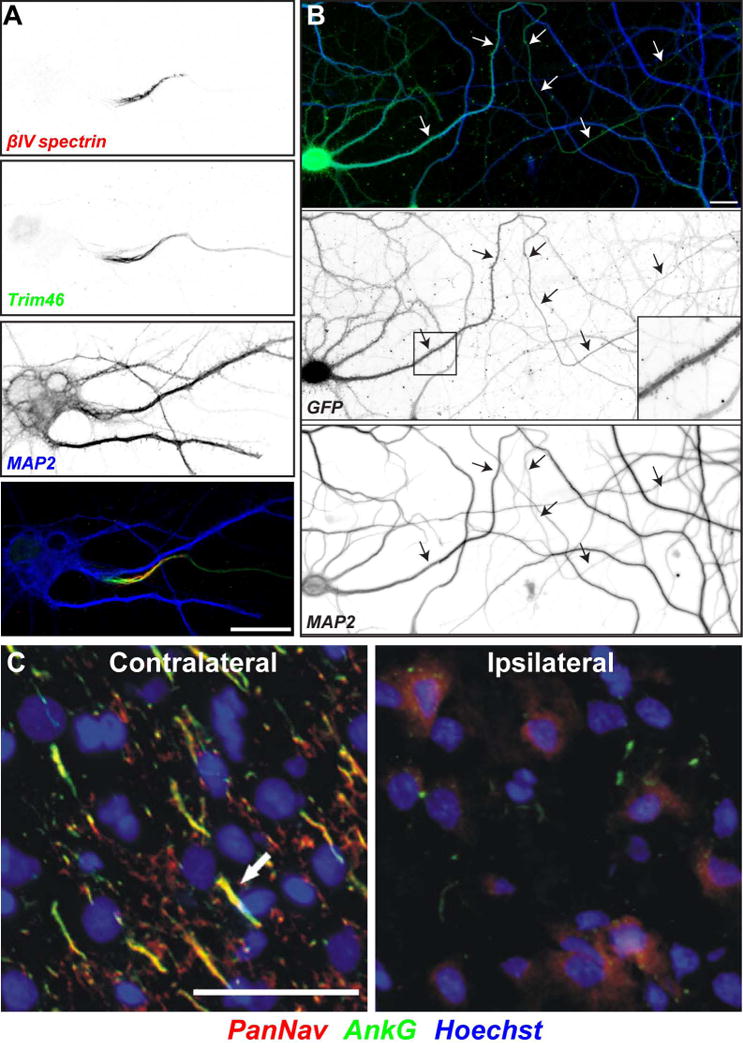
Axon initial segments in health and disease. (A) A cultured hippocampal neuron immunolabeled using antibodies against βIV spectrin (red), Trim46 (green), and MAP2 (blue). Scalebar = 10 μm. (B) A cultured hippocampal neuron infected with virus to express GFP and ankG shRNA. The neuron has lost neuronal polarity; the former axon (arrows) acquires dendritic features including MAP2 and spines (box and inset). Scalebar = 20 μm. Modified from Hedstrom et al.2 (C) Na+ channels are lost from the AIS following ischemic injury. Contralateral (left) and ipsilateral (right) regions of rat cortex were immunolabelled for Na+ channels (PanNav, red), ankG (green), and Hoechst to label nuclei (blue). The sections shown are from a brain collected 24 hr after MCAO. Arrows indicate labeled AIS in layer 2/3 cortex. Scale bar = 50 μm. Modified from Schafer et al.85
New super resolution microscopy techniques have provided new insights into the structure of this AIS cytoskeleton. For example, Stochastic Optical Reconstruction Microscopy (STORM) revealed that the actin/spectrin/ankyrin-based cytoskeleton forms a periodic structure with actin filaments organized in rings spaced 190 nm apart. These rings are connected by spectrin tetramers 39–41. It’s interesting to note that Yoshimura et al. 38 found that the distance between the periodic rings remains unchanged even though the shorter βIVΣ6 splice variant is incorporated into the pre-existing structure. STORM imaging in βIVΣ1-deficient mice might lend additional insights into how βIVΣ6 can participate in the AIS cytoskeleton.
Spectrin cytoskeletons are composed of two α and two β subunits to form heterotetramers 42. βI, βII, βIII, and βIV spectrin all participate in the assembly of spectrin cytoskeletons in the brain, but the only α spectrin found in brain is αII spectrin 43. Immunoprecipitation experiments from mouse brain homogenate suggested that αII spectrin interacts with both isoforms of βIV spectrin 44. However, direct evidence showing that αII spectrin is located at the AIS has remained elusive. We recently showed that αII and βIV spectrin interact and form a periodic cytoskeleton at the AIS 45 and at nodes of Ranvier 46. The importance of αII spectrin is emphasized by the observation that αII spectrin-deficient mice and zebrafish are embryonic and larval lethal, respectively, due to cardiac and nervous system malformations 47,48. Galiano et al. 9 reported that the spectrin cytoskeleton participates in AIS assembly by establishing an intra-axonal boundary. Using CNS αII spectrin conditional knockout (cko) mice we also showed that loss of αII spectrin results in a fragmented AIS 45. Thus, αII spectrin may have multiple roles in AIS structure and function depending on the β spectrin with which it is partnered.
Axonal microtubules are arranged in parallel and point their plus-ends towards the growth cone, while microtubules in dendrites have a mixed orientation 49. One distinctive feature of the AIS is that microtubules are fasciculated and cross-linked to one another, while at distal axons or dendrites they are evenly spaced or loosely-bundled 1. Mice lacking ankG are deficient in fasciculated microtubules, emphasizing their dependence on the AIS submembranous cytoskeleton 3. How are fasciculated microtubules linked to the ankyrin/spectrin cytoskeleton, and what are the molecular mechanisms responsible for this distinctive feature of AIS microtubules?
As described above, ankG binds to the actin cytoskeleton via βIV spectrin. STORM imaging revealed that ankG extends its C terminus ~26 nm below the submembranous cytoskeleton and into the AIS cytoplasm 41. There, it binds to microtubules via the microtubule plus-end-binding proteins EB1 and EB3 50. This link between ankG and EB proteins may stabilize microtubules and facilitate axonal trafficking. In addition, the extension and interaction of the ankG C terminal tail to microtubules and EB proteins may provide a partial mechanism for an ankG-dependent cytoplasmic filter 51.
The microtubule-associated protein called tripartite motif containing (TRIM) protein 46 was recently shown to be uniquely enriched at the proximal AIS 52 (Figs. 1B and 2A). Even though TRIM46 partially overlaps with ankG, it does not interact with ankG. However, TRIM46 interacts with microtubules and is both necessary and sufficient for fasciculation. MAP2 was also proposed to play important roles at the AIS to help regulate protein trafficking. Although in cortical and hippocampal neurons MAP2 is mainly a somatodendritic protein, in DRG sensory neurons it is found both in the soma and the proximal axon where it selectively controls vesicle entry via KIF1 and KIF5 53.
As described above STORM imaging revealed AIS actin is arranged as rings lining the inner cytoplasmic face of the axonal membrane. However, actin can also form highly dynamic patches oriented with the plus ends facing toward the cell body. These patches are proposed to function as ‘checkpoints’ for vesicles to enter the distal axon or reverse direction and be sorted back to the somatodendritic domain 54,55. However, these conclusions are controversial since Jones et al. 56 did not find any polarized actin patches or actin meshwork at the AIS using platinum replica electron microscopy. Instead, they found the actin cytoskeleton at the AIS mainly consists of sparse short, stable filaments and longer dynamic filaments with mixed orientation. More recently, highly dynamic AIS actin patches were shown to be present at the proximal axons of newly differentiated and mature neurons 54. Furthermore, live imaging revealed that vesicles containing dendritic cargoes halted and reversed direction at the actin patches, while vesicles containing axonal cargoes bypassed the patches. These results support a model where actin patches help regulate differential trafficking of somatodendritic and axonal cargoes. Exactly how this happens, which myosins participate in the process, and whether this is but one of many trafficking mechanisms the AIS uses to regulate protein distribution in the cell remain to be determined.
Ankyrin and spectrin cytoskeletons form an intra-axonal boundary
How is ankG restricted to the AIS during development? Since nodes of Ranvier share a molecular organization similar to the AIS, aspects of node assembly might apply to AIS assembly. The key insight came from the observation that the cytoskeletal proteins αII spectrin, βII spectrin, and ankB are located at paranodes flanking nodes of Ranvier57. Remarkably, while ankG and βIV spectrin are found at the AIS, ankB and βII spectrin are found in the distal axon 9. Furthermore, αII spectrin is found at both the AIS and in the distal axon, where it interacts with βIV spectrin and βII spectrin, respectively45. Since paranodes can function as a mechanism for nodal ion channel clustering 58, we considered whether the distal axon cytoskeleton might also function as a mechanism for clustering of ankG/βIV spectrin at the AIS. To determine the relationship between assembly of the distal axonal ankB/αII/βII spectrin cytoskeketon and ankG clustering, Galiano et al. 9 immunostained cultured hippocampal neurons at different developmental stages. Upon axon specification, a complex of ankB/αII/βII spectrin was transported to the growth cone via KIF3/KAP3. The ankB/αII/βII spectrin-based cytoskeleton was then observed to assemble from the distal tip of the axon towards the proximal axon; at this stage ankG/βIV spectrin were not detected. Finally, when ankG was detected at the AIS, ankB was exclusively located in the distal axon; ankB immunostaining was clearly reduced in the proximal axon with a ‘boundary’ between the two ankyrins and their spectrin binding parters. These results, suggested that ankB/αII/βII spectrin-based cytoskeleton may function as an intra-axonal barrier that restricts ankG/βIV spectrin to the proximal axon. The existence of intra-axonal boundaries had previously been observed in drosophila axons59. To directly test the hypothesis, Galiano et al. 9 manipulated the intra-axonal boundary by overexpressing ankG or the distal cytoskeleton components. Overexpressing distal axon components shortened the AIS, while overexpressing ankG lengthened the AIS and moved the distal boundary. Moreover, when ankB, αII spectrin or βII spectrin expression was silenced by shRNA in hippocampal neurons, AIS proteins and small clusters of ankG were found throughout the distal axon. Finally, the knockout of αII or βII spectrin also caused AIS to be fragmented and dispersed along the axons in the cerebral cortex 9,45. These results strongly support the notion of an intra-axonal boundary that limits the location of ankG in the axon.
AIS as the action potential initiation site
The physiological role of the AIS has been thoroughly discussed in several excellent recent reviews 60–62. Here, we will only give a brief overview. In the 1950s, while the structural and molecular composition of the AIS were almost completely unknown, the initial segment or axon hillock was proposed to be the site of action potential initiation based on intracellular recordings 63–65. The AIS has lower action potential threshold compared to the somatodendritic domain as its featured electrical property 66. Consistent with this observation, an enrichment of Na+ channels at the AIS was first revealed by autoradiography using the Na+ channel-binding 125I-scorpion toxin 67. Since that time, many studies have identified the AIS as the site of action potential initiation in diverse types of CNS neurons 14,68,69. Using quantitative freeze-fracture electron microscopy immunogold labeling, Lorincz et al. 70 confirmed that the density of Nav1.6 Na+ channels at the AIS and nodes of Ranvier, is about 40 fold higher than the soma in hippocampal neurons. Paradoxically, physiological studies by patch clamp suggested that the Na+ channel density at the AIS is no different than at the cell body or dendrites 71,72. Similarly, using high-resolution Na+ imaging, Fleidervish et al. 73 concluded that functional Na+ channel density is only three fold higher at the AIS than in the soma. The apparent discrepancy between these studies likely results from the different methodologies used to measure Na+ channel densities. Thus, there is a conflict between results obtained through direct imaging of the channels themselves, as compared to physiological methods that measure Na+ currents and physiological properties. Kole et al. 14 mostly resolved this controversy when they demonstrated that by disrupting the actin cytoskeleton, it was possible to record a greater Na+ current by patch clamp recording, and this current could be blocked by an actin stabilizer. Together, these results strongly supported a model where the tight coupling of channels to the actin cytoskeleton (presumably through ankG and βIV spectrin) causes Na+ channels to remain tethered to the cytoskeleton rather than in the membrane when drawn into the tip of the recording pipette. While a high density of Na+ channels is required to facilitate action potential generation, Na+ channel gating kinetics may also contribute to the low action potential threshold at the AIS 20,71,73. The differences in intrinsic excitability of various neuron types can be attributed to the different locations, densities, and types of Na+ and K+ channels.
AIS plasticity in response to activity
As the gatekeeper for action potential initiation, the AIS is perfectly positioned to modulate the excitability of the neuron. However, different types of neurons may have distinct AIS locations, lengths, and ion channels that tune the properties of each cell 23,74,75. Recent studies revealed that the anatomical properties of the AIS (i.e. length, position along the axon, etc.) can be dynamic and plastic in response to normal developmental activity or pathological activity. For example, during the critical period of visual system development, the AIS of cortical neurons undergoes dynamic changes in length 76. However, visual deprivation prevents these changes. Several studies suggest that AIS length or location may change to control the excitability of a cell in response to changes in activity. For example, when deprived of their inputs, the AIS of avian nucleus magnocellularis neurons elongate and shift closer to the soma 77. Physiological recordings show that the input deprived cells become more excitable, indicating that neurons adapt to the presynaptic activity by changing AIS properties to regulate neuronal excitability and to homeostatically maintain the output of the circuit. Consistent with the idea that activity can regulate the structural properties of the AIS, Grubb et al. 78 demonstrated that chronic depolarization of neurons shifted the AIS distally away from the soma, thereby reducing excitability (higher threshold for action potential initiation). Together, these results provided the first evidence that synaptic input can shape neuronal excitability and fine-tune neuronal networks by altering AIS location and size.
Differences in AIS ion channel expression and subcellular distribution may also contribute to the distinct neuronal excitability among cell types 23. For example, during development, AIS Na+ channels undergo a subtype switch and may alter the firing properties of the neuron. In retinal ganglion cells Nav1.2 channels are the major Na+ channel subtype at the AIS during early development, while Nav1.6 channels are expressed later and become dominant when the neuron matures 18. Nav1.2 channels have a higher threshold for firing action potentials compared to Nav1.6 channels, suggesting that these expression changes may impact the firing properties of these cells 20. Previous studies showed that Nav1.6 expression is decreased and Nav1.2 increased in demyelinated axons 79,80. Na+ channel isoform expression patterns may underlie the pathophysiology of some diseases or injuries. Whether Na+ channel subtypes and/or their subcellular distribution change during plastic alteration of the AIS remains unclear and an area of active study. Finally, little is known about the transcriptional and translational regulation of ion channel expression in pathological conditions.
What mechanisms control the structural changes observed during homeostatic AIS plasticity? It is worth noting that AIS homeostatic plasticity is a very protracted process, occurring over the course of hours or days 78. The slow change in AIS properties and position along the axon may reflect rearrangement or disassembly of the very stable AIS cytoskeleton 81. The evidence supports a role for Ca2+ signaling in this process 78. Specifically, a Ca2+- and calmodulin-activated phosphatase, calcineurin, acts downstream of L-type Ca2+ channels to regulate AIS plasticity 82. Phosphorylation of proteins might be another pathway downstream of Ca2+ signaling. Casein kinase 2 (CK2) phosphorylates Na+ channels. This increases their affinity for ankG and facilitates Na+ channel clustering at the AIS 17, while cyclin-dependent kinase 5 (Cdk5) promotes Kv1 channel targeting to the axonal domain 83. Calmodulin also regulates Kv7 channel KCNQ2/3 heteromer assembly and its clustering at the AIS 84. Since AIS relocation involves cytoskeletal protein disassembly and reorganization of AIS components, Ca2+-dependent proteolysis by calpain may also be crucial for AIS plasticity, especially in response to injury or disease 85.
AIS maintains neuronal polarity
In addition to its function controlling action potential initiation in axons, the AIS also functions as a gatekeeper to separate somatodendritic molecules from entering axonal domains86. In recent years this remarkable function of the AIS has captured the attention of molecular neurobiologists. What mechanisms could exclude somatodendritic proteins from the axon, while permitting axonal molecules to enter? The regulation of polarity occurs at both the level of the plasma membrane, and at the level of regulating vesicular transport. It is likely that unique mechanisms exist for each of these functions, but all mechanisms apparently depend on ankG, the master regulator of AIS assembly and function. Loss of ankG from hippocampal neurons causes all major AIS components (i.e., ion channels, cytoskeletal proteins and CAMs) to disperse 10. More related to regulation of polarity though, loss of ankG caused somatodendritic proteins (e.g. MAP2, KCC2, PSD95) to redistribute and enter the former axon, and spine-like structures and postsynaptic densities even develop along the former axon2 (Fig. 2B). These observations were coroborated using a cerebellum-specific ankG knockout mouse. In those animals, Purkinje cell axons lacking AIS ankG developed spines enriched with postsynaptic molecules 3. Using electron microscopy, they found that many of the AIS specific ultrastructrual features (e.g. an electron-dense undercoat beneath the plasma membrane and the microtubule fascicles) could not be detected. These results emphasize that ankG-dependent AIS integrity is crucial to maintain neuronal polarity in vivo. Instead of maintenance of neuronal polarity, the AIS-specific microtubule-associated protein TRIM46 contributes to the development of polarity. In cultured hippocampal neurons, TRIM46 was reported in the newly specified axon before the clustering of ankG 52. Knockdown of TRIM46 in cortical neurons using in utero electroporation of shRNA, showed that TRIM46-depleted neurons are unable to migrate properly and fail to properly form axons. Together, these observations show that ankG and TRIM46 play key roles in establishing and maintaining neuronal polarity through their functions at the AIS.
That the AIS functions as a diffusion barrier was first proposed by Kobayashi et al. 87. They reported that fluorescent phospholipids in the axon were unable to enter the somatodendritic compartment in cultured hippocampal neurons. Later, Winckler et al. 88 showed that the membrane proteins L1 and Thy1 have very low lateral mobility at the AIS compared to the distal axon. However, disruption of F-actin allowed these membrane proteins to diffuse freely throughout axonal and somatodendritic domains. These results suggested that the actin cytoskeleton is a key structural component of the AIS and functions to restrict membrane diffusion. The diffusion barrier forms during AIS assembly when various transmembrane proteins accumulate and are tethered to the ankG/βIV spectrin/actin-based cytoskeleton 89. Together, these results suggested a “picket fence” model where the high density of transmembrane proteins (pickets) impede the lateral diffusion of membrane proteins by steric hindrance. Later studies confirmed that the AIS membrane proteins, including ion channels and CAMs, are anchored to ankG and thus immobilized at the AIS 8,15,89,90.
Newly developed imaging techniques also support a role for the AIS in limiting the diffusion of proteins. For example, STORM imaging revealed axonal actin is organized in periodic rings connected by spectrin tetramers with a ~190 nm spacing between adjacent rings; ankG and βIV spectrin are also arranged with the same periodicity39–41. Albrecht et al. 91 used super-resolution and single particle tracking to show that after the AIS forms, GPI-anchored GFP molecules located at the proximal axon begin to show reduced diffusion, while the GPI-GFP can diffuse freely at all times at the distal axon. Remarkably, further analysis of the particle trajectory revealed that the GPI-anchored molecules were confined to a ~190 nm long region, consistent with the spacing between adjacent actin rings. The periodic pattern of the restricted GPI-GFP molecules overlapped with the spectrin cytoskeleton and was mutually exclusive from the actin rings. These observations suggested a model where the actin rings function as submembranous diffusion barriers that restrict lateral diffusion of the GPI molecules 92. However, this alternative “actin fence” model needs additional experimental support since Albrecht et al. 91 observed GPI molecules were only confined to the AIS, but the periodic actin/spectrin/ankyrin-based cytoskeleton exists throughout the entire axon.
Besides constituting a barrier to limit diffusion of lipids and membrane proteins, Song et al. 51 proposed that AIS actin can also function as a size-selective sieve to filter intracellular molecules. In support of this idea, they showed that large cytoplasmic molecules are excluded from the axon, but when the actin cytoskeleton was depolymerized these same macromolecules were able to freely pass through the AIS and enter axons. However, a variety of imaging techniques reveal that AIS actin mostly exists as rings lining the plasma membrane or as actin patches 41,55,56. Furthermore, if actin functioned to impede the transit of proteins through the AIS, a reduction in trafficking velocities should be observed in the AIS. However, this is not the case; axonal vesicles traverse the AIS without impediment93. Thus, an actin-based filter that functions solely based on size is difficult to reconcile with the available data. Another potential molecular basis for an axonal filter includes ankG itself. The C terminus of ankG extends into the core of the axon shaft where it binds to microtubules via EB1 and EB3. These interactions may constitute a barrier to the transport of cytoplasmic molecules through the AIS 41,50.
Actin and its associated myosin motors may contribute to differential vesicle trafficking at the AIS through active mechanisms rather than simply functioning as a passive sieve. Since live imaging of the vesicles carrying dendritic cargoes halt or reverse direction when going through the AIS, Al-Bassam et al. 94 proposed that this phenomenon depends on the actin-associated plus-end-directed motor, myosinVa. The interaction between dendritic protein vesicles and myosin Va is sufficient for their somatodendritic localization 54,95. Balasanyan et al., 54 suggested that actin patches at the proximal axon form a filter that allows the minus-end-directed myosinVI motor to enter the axon but these same patches block the entry for the plus-end-directed myosin Va motor. The molecular mechanisms that cause these patches to have differential responses to the myosin motors remain unknown.
Besides actin, spectrin, and ankyrins, microtubules are also a prominent component of the AIS. Microtubules function as a directed track for long distance anterograde and retrograde vesicle trafficking via microtubule-based motors (i.e., kinesins and dyneins) 96. Most kinesins transport cargos toward the distal tip of the axon while dyneins carry vesicles back to the soma. Several microtubule-associated proteins have been shown to regulate polarized trafficking at the AIS through their actions on their motors. For example, MAP2 defines a pre-axonal region in sensory DRG neurons, where it controls selective cargo entry by preventing KIF5 binding to microtubules. In addition, MAP2 may regulate vesicle distribution by balancing KIF1 and KIF5 motor activities along the axon 53. However, since sensory neurons have a special pseudo-unipolar morphology (i.e., a central and a peripheral axon, but no dendrites) most lack an AIS. Thus, whether a MAP2-dependent trafficking mechanism is universal for more complex neurons with somatodendritic and axonal domains remains unknown. Nevertheless, depletion of MAP2 in hippocampal neurons results in fewer vesicle entries into the axon, suggesting MAP2 may also contribute to polarized trafficking in CNS neurons. In contrast to the somatodendritic localization of MAP2, the microtubule-associated protein MAP6 is selectively enriched at the proximal axon of mature neurons. Recent studies indicate that MAP6 stabilizes microtubules and also participates in neuronal polarization, partly by functioning as a cofactor to promote vesicle transport together with KIF5B 97.
Although most research on the mechanisms controlling microtubule-dependent sorting of vesicules has focused on kinesins and anterograde axonal transport through the AIS, the role of dynein in sorting somatodendritic vesicles has been much more enigmatic. A major breakthrough in our understanding of how vesicles may be excluded at the AIS occurred with the discovery that the dynein regulator nuclear distribution element-like 1 (Ndel1) and its binding partner Lissencephaly 1 (LIS1) are both found at the AIS after neurons polarize 98. In a series of elegant experiments they showed that Ndel1 interacts directly with ankG and dynein whereas LIS1 only binds to dynein. Furthermore, in an ankG-dependent manner, Ndel1 and LIS1 facilitate the reversal of transport for somatodendritic cargos at the AIS, suggesting Ndel1 functions as a checkpoint at the AIS, to switch the direction of vesicle trafficking and exclude entry into the axon. How Ndel1 recognizes somatodendritic cargos remains an active area of investigation. Microtubules may also regulate retrograde trafficking of cytoplasmic proteins through the AIS. Li et al. 99 observed that the axonal microtubule-binding protein Tau could diffuse freely in an anterograde fashion to the distal axon, but it was restricted to the axon and never passed from the axon back through the AIS to enter the somatodendritic domain. This barrier depends on the phosphorylation of Tau and its binding to microtubules.
Diseases and injuries that affect the AIS
Action potential initiation depends on a high density of voltage-gated Na+ channels clustered at the AIS to lower firing threshold100. K+ and Ca2+ channels further modulate action potential properties. Many mutations in ion channel subtypes (ie. Nav1.1, Nav1.2, Nav1.6, Nav β subunit and KCNQ2/3 K+ channels) normally located at the AIS have been discovered and shown to cause epilepsy; indeed, the idea that epilepsy may result from dysregulated AIS ion channels has been discussed elsewhere 101. Many pathogenic variants of AIS-associated proteins, as well as disruptions or alterations to AIS proteins or AIS structure have been reported (Table I).
Table I.
Diseases and injuries associated with the altered AIS structure and function.
| Gene name | Associated disorders | Reference |
|---|---|---|
| SCN1A | GEFS+ (generalized epilepsy with febrile seizures plus) | 127 |
| Dravet syndrome-severe myoclonic epilepsy in infancy (SMEI) | 128 | |
|
| ||
| SCN1B | GEFS+ | 129,130 |
| Dravet syndrome | 131 | |
|
| ||
| SCN2A | Dravet syndrome | 132 |
| Benign familial infantile seizures (BFIS) | 133 | |
| Ohtahara syndrome (OS) | 134 | |
|
| ||
| SCN8A | Early onset epileptic encephalopathies (EOEEs) | 135 |
|
| ||
| KCNQ2/3 | BFIS | 136 |
|
| ||
| ANK3 | Bipolar disorder | 102,103 |
|
| ||
| ANK3 exon1b | Epilepsy & bipolar disorder | 104 |
|
| ||
| ANK3 480 kDa | Intellectual disability | 105 |
|
| ||
| ANK3 | Schizophrenia | 137 |
|
| ||
| ANK3 | Autism spectrum disorder | 138 |
|
| ||
| SPNB4 | (Spectrinopathy?) Congenital myopathy, neuropathy and deafness | 111 |
|
| ||
| SPTAN1 | West syndrome | 112–114 |
| Diseases or injuries could alter AIS structure | ||
|
| ||
| UBE3A | Angelman syndrome | 116 |
| Focal cortical and white matter stroke | 126 | |
| Hypoxia-induced ischemic injury | 85 | |
| Traumatic brain injuries | 124,125 | |
| Alzheimer’s disease | 121 | |
| Multiple Sclerosis | 122 | |
In addition to channelopathies, mutations in other AIS proteins also cause neurological disorders. For example, genome-wide association studies (GWAS) implicate ANK3 (gene encoding ankG) as a risk factor gene for bipolar disorder 102. Reductions in the levels of ankG also result in psychiatric disease-related behaviors in mice103. AnkG undergoes extensive alternative splicing, and different splice variants have been proposed to contribute to disease. For example, parvalbumin (PV)-positive GABAergic interneurons express the ankG exon 1b variant while excitatory principal cells express the exon 1e splice variant 104. Na+ channels fail to cluster at the AIS of PV interneurons in ankG exon 1b-deficient mice, and the mutant mice also have bipolar-like behaviors, epilepsy and even sudden death. These observations led to the suggestion that bipolar-related phenotypes may result from disrupted inhibitory and excitatory balance. Iqbal et al. 105 reported that some human ANK3 variants, predicted to truncate the 480 kDa splice form, cause intellectual disability. ANK3 has also been associated with other psychiatric disorders including schizophrenia and autism spectrum disorder 106,107. Overall, several different neuropsychiatric developmental disorders and epilepsy converge on ANK3, highlighting the importance of the AIS for these diseases.
Mutations in α and β spectrins also cause nervous system dieases. For example, βIV spectrin loss-of-function mutant mice (Quivering mice) have ataxia and central auditory deafness likely due to aberrant ion channel clustering or maintenance of clusters108–110. Recently, the first pathogenic variant of SPNB4 (the gene encoding human βIV spectrin) was reported 111. Remarkably, the patient had myopathies and auditory neuropathies similar to those seen in the quivering mice. It will be interesting to determine how human and Quivering variants of βIV spectrin cause nervous system dysfunction, and if the structure and function of AIS and nodes are affected in human βIV spectrin loss-of-function patients.
We recently showed that αII spectrin interacts with βIV spectrin and is also enriched at the AIS 45. Pathogenic SPTAN1 (human gene encoding αII spectrin) variants cause early infantile epileptic encephalopathy type 5, an infantile epileptic encephalopathy with diffuse hypomyelination, brain atrophy and developmental retardation 112–115. Subsequent studies of these human variants, as well as studies using mice with conditional knockout of αII spectrin all show disruptions in AIS structure and neuronal excitability 45,114,115.
Altered AIS structure also appears to be a common feature of many nervous system diseases. For example, in ubiquitin ligase UBE3A gene knockout mice, a model for Angelman syndrome, ankG and Nav1.6 are up-regulated and mutant mice have longer AIS 116. The change in AIS structure may be a homeostatic response since genetic reduction in the levels of Na+/K+-ATPase rescued hippocampal abnormalities in these same mice 117.
AIS structure is also affected in neurodegenerative diseases. For example, Alzheimer’s disease (AD) is associated with the aberrant accumulation of β-amyloid (Aβ) plaques in the cerebral cortex 118. High levels of Aβ induce seizures in the brain and can cause cognitive decline in AD mouse models. The prevailing models suggest Aβ disrupts synaptic inputs to cause seizures. However, an association between AD and AIS related-genes has been reported 119,120. Thus, AIS may be involved in the pathogenesis of AD. Recently, Marin et al. 121 showed that AIS number and length decrease when in close proximity to Aβ plaques, suggesting that loss or disruption of AIS may contribute to the hyperexcitability and impaired brain function in AD.
Autoimmune diseases may also disrupt the AIS. For example, mice with inflammatory demyelination show profound disruption and even loss of the AIS 122. Remarkably, AIS were preserved when microglia activity was inhibited. Furthermore, autoantibodies against AIS proteins (e.g., NF186) have been implicated in the pathogenesis of multiple sclerosis, and the passive transfer of antibodies against NF186 is sufficient to dramatically worsen the clinical course of demyelinating diseases 123. Whether or not these autoantibodies affect the AIS directly remains unknown.
AIS structure is also altered after traumatic and ischemic nervous system injuries. For example, axotomy has been reported at the AIS following mild traumatic brain injury (TBI) 124, while in a blast-induced mild TBI model, the AIS shortened in response to injury with concomitant memory impairment 125. The AIS of neurons in stroked brain are significantly shorter or are completely lost due to proteolysis of the AIS cytoskeleton by the Ca2+-dependendent protease calpain 85,126 (Fig. 2C). These results suggest that one component of any therapy must also include preservation of the AIS cytoskeleton. Consistent with this idea, Schafer et al., 85 showed that calpain inhibitors can preserve the AIS after ischemic injury. Whether this approach will also work for the myriad of other injuries and diseases that affect the AIS remains to be determined.
Conclusion
The AIS is a remarkable structure highly enriched with voltage-gated ion channels and a supporting ankyrin/spectrin-based cytoskeleton. The precise molecular organization of the AIS ensures proper neuronal excitability by regulating action potential generation. The dynamic properties of the AIS fine-tune the excitability of neurons in response to activity, and thereby maintain homeostatic output of a circuit. The AIS also functions as a ‘gate’ to exclude somatodendritic molecules from axons, while allowing cargos with axonal proteins to pass through. Previous models viewed the AIS as a passive filter, but more recent studies suggest polarized sorting of vesicles is an active process that depends on microtubule and actin-associated proteins. Nevertheless, our understanding of the mechanisms controlling trafficking remain in their infancy. Future studies aimed at determining the mechanisms controlling trafficking of distinct cargos will no doubt yield many new interesting mechanisms. We expect that part of any effort to understand AIS trafficking mechanisms will require the identification of more AIS proteins through proteomic approaches. Finally, another important area requiring additional attention is the consequence of disease and injury for AIS structure and function, and how an altered AIS impacts the course of disease. We speculate that restoring or preserving AIS structure and function will be an important component of any therapy aimed at treating these diseases and injuries.
Acknowledgments
Supported by NIH grants NS044916 and NS069688 to MNR.
Footnotes
Competing interests
The authors declare no competing interests.
References
- 1.Palay SL, Sotelo C, Peters A, Orkand PM. The axon hillock and the initial segment. J Cell Biol. 1968;38:193–201. doi: 10.1083/jcb.38.1.193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hedstrom KL, Ogawa Y, Rasband MN. AnkyrinG is required for maintenance of the axon initial segment and neuronal polarity. J Cell Biol. 2008;183:635–40. doi: 10.1083/jcb.200806112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sobotzik JM, et al. AnkyrinG is required to maintain axo-dendritic polarity in vivo. Proc Natl Acad Sci U S A. 2009;106:17564–9. doi: 10.1073/pnas.0909267106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bennett V, Baines AJ. Spectrin and ankyrin-based pathways: metazoan inventions for integrating cells into tissues. Physiol Rev. 2001;81:1353–92. doi: 10.1152/physrev.2001.81.3.1353. [DOI] [PubMed] [Google Scholar]
- 5.Cunha SR, Mohler PJ. Ankyrin protein networks in membrane formation and stabilization. J Cell Mol Med. 2009;13:4364–76. doi: 10.1111/j.1582-4934.2009.00943.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kordeli E, Lambert S, Bennett V, Ankyrin G. A new ankyrin gene with neural-specific isoforms localized at the axonal initial segment and node of Ranvier. Journal of Biological Chemistry. 1995;270:2352–9. doi: 10.1074/jbc.270.5.2352. [DOI] [PubMed] [Google Scholar]
- 7.Jenkins PM, et al. Giant ankyrin-G: a critical innovation in vertebrate evolution of fast and integrated neuronal signaling. Proc Natl Acad Sci U S A. 2015;112:957–64. doi: 10.1073/pnas.1416544112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Boiko T, et al. Ankyrin-dependent and -independent mechanisms orchestrate axonal compartmentalization of L1 family members neurofascin and L1/neuron-glia cell adhesion molecule. J Neurosci. 2007;27:590–603. doi: 10.1523/JNEUROSCI.4302-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Galiano MR, et al. A distal axonal cytoskeleton forms an intra-axonal boundary that controls axon initial segment assembly. Cell. 2012;149:1125–39. doi: 10.1016/j.cell.2012.03.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hedstrom KL, et al. Neurofascin assembles a specialized extracellular matrix at the axon initial segment. J Cell Biol. 2007;178:875–86. doi: 10.1083/jcb.200705119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jenkins SM, Bennett V. Ankyrin-G coordinates assembly of the spectrin-based membrane skeleton, voltage-gated sodium channels, and L1 CAMs at Purkinje neuron initial segments. J Cell Biol. 2001;155:739–46. doi: 10.1083/jcb.200109026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhou D, et al. AnkyrinG is required for clustering of voltage-gated Na channels at axon initial segments and for normal action potential firing. J Cell Biol. 1998;143:1295–304. doi: 10.1083/jcb.143.5.1295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Leterrier C, et al. Ankyrin G Membrane Partners Drive the Establishment and Maintenance of the Axon Initial Segment. Front Cell Neurosci. 2017;11:6. doi: 10.3389/fncel.2017.00006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kole MH, et al. Action potential generation requires a high sodium channel density in the axon initial segment. Nat Neurosci. 2008;11:178–86. doi: 10.1038/nn2040. [DOI] [PubMed] [Google Scholar]
- 15.Garrido JJ, et al. A targeting motif involved in sodium channel clustering at the axonal initial segment. Science. 2003;300:2091–4. doi: 10.1126/science.1085167. [DOI] [PubMed] [Google Scholar]
- 16.Lemaillet G, Walker B, Lambert S. Identification of a conserved ankyrin-binding motif in the family of sodium channel alpha subunits. J Biol Chem. 2003;278:27333–9. doi: 10.1074/jbc.M303327200. [DOI] [PubMed] [Google Scholar]
- 17.Bréchet A, et al. Protein kinase CK2 contributes to the organization of sodium channels in axonal membranes by regulating their interactions with ankyrin G. J Cell Biol. 2008;183:1101–14. doi: 10.1083/jcb.200805169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Boiko T, et al. Functional specialization of the axon initial segment by isoform-specific sodium channel targeting. J Neurosci. 2003;23:2306–13. doi: 10.1523/JNEUROSCI.23-06-02306.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Duflocq A, Le Bras B, Bullier E, Couraud F, Davenne M. Nav1.1 is predominantly expressed in nodes of Ranvier and axon initial segments. Mol Cell Neurosci. 2008 doi: 10.1016/j.mcn.2008.06.008. [DOI] [PubMed] [Google Scholar]
- 20.Hu W, et al. Distinct contributions of Na(v)1.6 and Na(v)1.2 in action potential initiation and backpropagation. Nat Neurosci. 2009;12:996–1002. doi: 10.1038/nn.2359. [DOI] [PubMed] [Google Scholar]
- 21.Pan Z, et al. A common ankyrin-G-based mechanism retains KCNQ and Nav channels at electrically active domains of the axon. Journal of neuroscience. 2006;26:2599–2613. doi: 10.1523/JNEUROSCI.4314-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ogawa Y, et al. Postsynaptic density-93 clusters Kv1 channels at axon initial segments independently of Caspr2. J Neurosci. 2008;28:5731–9. doi: 10.1523/JNEUROSCI.4431-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lorincz A, Nusser Z. Cell-type-dependent molecular composition of the axon initial segment. J Neurosci. 2008;28:14329–40. doi: 10.1523/JNEUROSCI.4833-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ogawa Y, et al. ADAM22, a Kv1 channel-interacting protein, recruits membrane-associated guanylate kinases to juxtaparanodes of myelinated axons. Journal of Neuroscience. 2010;30:1038–1048. doi: 10.1523/JNEUROSCI.4661-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bender KJ, Trussell LO. Axon initial segment Ca2+ channels influence action potential generation and timing. Neuron. 2009;61:259–71. doi: 10.1016/j.neuron.2008.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bender KJ, Ford CP, Trussell LO. Dopaminergic modulation of axon initial segment calcium channels regulates action potential initiation. Neuron. 2010;68:500–11. doi: 10.1016/j.neuron.2010.09.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jenkins SM, et al. FIGQY phosphorylation defines discrete populations of L1 cell adhesion molecules at sites of cell-cell contact and in migrating neurons. J Cell Sci. 2001;114:3823–35. doi: 10.1242/jcs.114.21.3823. [DOI] [PubMed] [Google Scholar]
- 28.Tuvia S, Garver TD, Bennett V. The phosphorylation state of the FIGQY tyrosine of neurofascin determines ankyrin-binding activity and patterns of cell segregation. Proc Natl Acad Sci U S A. 1997;94:12957–62. doi: 10.1073/pnas.94.24.12957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ango F, et al. Ankyrin-based subcellular gradient of neurofascin, an immunoglobulin family protein, directs GABAergic innervation at purkinje axon initial segment. Cell. 2004;119:257–72. doi: 10.1016/j.cell.2004.10.004. [DOI] [PubMed] [Google Scholar]
- 30.Brückner G, Szeöke S, Pavlica S, Grosche J, Kacza J. Axon initial segment ensheathed by extracellular matrix in perineuronal nets. Neuroscience. 2006;138:365–75. doi: 10.1016/j.neuroscience.2005.11.068. [DOI] [PubMed] [Google Scholar]
- 31.Susuki K, et al. Three mechanisms assemble central nervous system nodes of Ranvier. Neuron. 2013;78:469–82. doi: 10.1016/j.neuron.2013.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.John N, et al. Brevican-containing perineuronal nets of extracellular matrix in dissociated hippocampal primary cultures. Mol Cell Neurosci. 2006;31:774–84. doi: 10.1016/j.mcn.2006.01.011. [DOI] [PubMed] [Google Scholar]
- 33.Frischknecht R, et al. Brain extracellular matrix affects AMPA receptor lateral mobility and short-term synaptic plasticity. Nat Neurosci. 2009;12:897–904. doi: 10.1038/nn.2338. [DOI] [PubMed] [Google Scholar]
- 34.Kegel L, et al. Functional phylogenetic analysis of LGI proteins identifies an interaction motif crucial for myelination. Development. 2014;141:1749–56. doi: 10.1242/dev.107995. [DOI] [PubMed] [Google Scholar]
- 35.Yang Y, Ogawa Y, Hedstrom KL, Rasband MN. βIV spectrin is recruited to axon initial segments and nodes of Ranvier by ankyrinG. J Cell Biol. 2007;176:509–19. doi: 10.1083/jcb.200610128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Berghs S, et al. betaIV spectrin, a new spectrin localized at axon initial segments and nodes of ranvier in the central and peripheral nervous system. J Cell Biol. 2000;151:985–1002. doi: 10.1083/jcb.151.5.985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Komada M, Soriano P. βIV-spectrin regulates sodium channel clustering through ankyrin-G at axon initial segments and nodes of Ranvier. J Cell Biol. 2002;156:337–48. doi: 10.1083/jcb.200110003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yoshimura T, Stevens SR, Leterrier C, Stankewich MC, Rasband M. Developmental Changes in Expression of βIV Spectrin Splice Variants and Axon Initial Segments and Nodes of Ranvier. Front Cell Neurosci. 2017;10:1–8. doi: 10.3389/fncel.2016.00304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Xu K, Zhong G, Zhuang X. Actin, Spectrin, and Associated Proteins Form a Periodic Cytoskeletal Structure in Axons. Science. 2013;339:30495–501. doi: 10.1126/science.1232251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhong G, et al. Developmental mechanism of the periodic membrane skeleton in axons. Elife. 2014;3 doi: 10.7554/eLife.04581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Leterrier C, et al. Nanoscale Architecture of the Axon Initial Segment Reveals an Organized and Robust Scaffold. Cell Rep. 2015;13:2781–93. doi: 10.1016/j.celrep.2015.11.051. [DOI] [PubMed] [Google Scholar]
- 42.Bennett V, Lorenzo DN. Spectrin- and ankyrin-based membrane domains and the evolution of vertebrates. Curr Top Membr. 2013;72:1–37. doi: 10.1016/B978-0-12-417027-8.00001-5. [DOI] [PubMed] [Google Scholar]
- 43.Zhang Y, et al. An RNA-sequencing transcriptome and splicing database of glia, neurons, and vascular cells of the cerebral cortex. J Neurosci. 2014;34:11929–47. doi: 10.1523/JNEUROSCI.1860-14.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Uemoto Y, et al. Specific role of the truncated betaIV-spectrin Sigma6 in sodium channel clustering at axon initial segments and nodes of ranvier. J Biol Chem. 2007;282:6548–55. doi: 10.1074/jbc.M609223200. [DOI] [PubMed] [Google Scholar]
- 45.Huang CY, et al. alphaII Spectrin Forms a Periodic Cytoskeleton at the Axon Initial Segment and Is Required for Nervous System Function. J Neurosci. 2017;37:11311–11322. doi: 10.1523/JNEUROSCI.2112-17.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Huang CY, Zhang C, Zollinger DR, Leterrier C, Rasband MN. An alphaII Spectrin-Based Cytoskeleton Protects Large-Diameter Myelinated Axons from Degeneration. J Neurosci. 2017;37:11323–11334. doi: 10.1523/JNEUROSCI.2113-17.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Stankewich MC, et al. Cell organization, growth, and neural and cardiac development require alphaII-spectrin. J Cell Sci. 2011;124:3956–66. doi: 10.1242/jcs.080374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Voas MG, et al. alphaII-spectrin is essential for assembly of the nodes of Ranvier in myelinated axons. Curr Biol. 2007;17:562–8. doi: 10.1016/j.cub.2007.01.071. [DOI] [PubMed] [Google Scholar]
- 49.Baas PW, Deitch JS, Black MM, Banker GA. Polarity orientation of microtubules in hippocampal neurons: uniformity in the axon and nonuniformity in the dendrite. Proc Natl Acad Sci U S A. 1988;85:8335–9. doi: 10.1073/pnas.85.21.8335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Leterrier C, et al. End-binding proteins EB3 and EB1 link microtubules to ankyrin G in the axon initial segment. Proc Natl Acad Sci U S A. 2011;108:8826–31. doi: 10.1073/pnas.1018671108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Song AH, et al. A selective filter for cytoplasmic transport at the axon initial segment. Cell. 2009;136:1148–60. doi: 10.1016/j.cell.2009.01.016. [DOI] [PubMed] [Google Scholar]
- 52.van Beuningen SF, et al. TRIM46 Controls Neuronal Polarity and Axon Specification by Driving the Formation of Parallel Microtubule Arrays. Neuron. 2015;88:1208–26. doi: 10.1016/j.neuron.2015.11.012. [DOI] [PubMed] [Google Scholar]
- 53.Gumy LF, et al. MAP2 Defines a Pre-axonal Filtering Zone to Regulate KIF1- versus KIF5-Dependent Cargo Transport in Sensory Neurons. Neuron. 2017;94:347–362 e7. doi: 10.1016/j.neuron.2017.03.046. [DOI] [PubMed] [Google Scholar]
- 54.Balasanyan V, et al. Structure and Function of an Actin-Based Filter in the Proximal Axon. Cell Rep. 2017;21:2696–2705. doi: 10.1016/j.celrep.2017.11.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Watanabe K, et al. Networks of polarized actin filaments in the axon initial segment provide a mechanism for sorting axonal and dendritic proteins. Cell Rep. 2012;2:1546–53. doi: 10.1016/j.celrep.2012.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Jones SL, Korobova F, Svitkina T. Axon initial segment cytoskeleton comprises a multiprotein submembranous coat containing sparse actin filaments. J Cell Biol. 2014;205:67–81. doi: 10.1083/jcb.201401045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ogawa Y, et al. Spectrins and ankyrinB constitute a specialized paranodal cytoskeleton. J Neurosci. 2006;26:5230–9. doi: 10.1523/JNEUROSCI.0425-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Rasband MN, et al. Dependence of nodal sodium channel clustering on paranodal axoglial contact in the developing CNS. Journal of Neuroscience. 1999;19:7516–28. doi: 10.1523/JNEUROSCI.19-17-07516.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Katsuki T, Ailani D, Hiramoto M, Hiromi Y. Intra-axonal patterning: intrinsic compartmentalization of the axonal membrane in Drosophila neurons. Neuron. 2009;64:188–99. doi: 10.1016/j.neuron.2009.08.019. [DOI] [PubMed] [Google Scholar]
- 60.Bender KJ, Trussell LO. The Physiology of the Axon Initial Segment. Annu Rev Neurosci. 2012 doi: 10.1146/annurev-neuro-062111-150339. [DOI] [PubMed] [Google Scholar]
- 61.Kole MH, Stuart GJ. Signal processing in the axon initial segment. Neuron. 2012;73:235–47. doi: 10.1016/j.neuron.2012.01.007. [DOI] [PubMed] [Google Scholar]
- 62.Sasaki T. The axon as a unique computational unit in neurons. Neurosci Res. 2013;75:83–8. doi: 10.1016/j.neures.2012.12.004. [DOI] [PubMed] [Google Scholar]
- 63.Araki T, Otani T. Response of single motoneurons to direct stimulation in toad’s spinal cord. J Neurophysiol. 1955;18:472–85. doi: 10.1152/jn.1955.18.5.472. [DOI] [PubMed] [Google Scholar]
- 64.Coombs JS, Eccles JC, Fatt P. The electrical properties of the motoneurone membrane. J Physiol. 1955;130:291–325. doi: 10.1113/jphysiol.1955.sp005411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Fuortes MG, Frank K, Becker MC. Steps in the production of motoneuron spikes. J Gen Physiol. 1957;40:735–52. doi: 10.1085/jgp.40.5.735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Coombs JS, Curtis DR, Eccles JC. The generation of impulses in motoneurones. J Physiol. 1957;139:232–49. doi: 10.1113/jphysiol.1957.sp005888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Catterall WA. Localization of sodium channels in cultured neural cells. J Neurosci. 1981;1:777–83. doi: 10.1523/JNEUROSCI.01-07-00777.1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Khaliq ZM, Raman IM. Relative contributions of axonal and somatic Na channels to action potential initiation in cerebellar Purkinje neurons. J Neurosci. 2006;26:1935–44. doi: 10.1523/JNEUROSCI.4664-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Foust A, Popovic M, Zecevic D, McCormick DA. Action potentials initiate in the axon initial segment and propagate through axon collaterals reliably in cerebellar Purkinje neurons. J Neurosci. 2010;30:6891–902. doi: 10.1523/JNEUROSCI.0552-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Lorincz A, Nusser Z. Molecular identity of dendritic voltage-gated sodium channels. Science. 2010;328:906–9. doi: 10.1126/science.1187958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Colbert CM, Pan E. Ion channel properties underlying axonal action potential initiation in pyramidal neurons. Nat Neurosci. 2002;5:533–8. doi: 10.1038/nn0602-857. [DOI] [PubMed] [Google Scholar]
- 72.Colbert CM, Johnston D. Axonal action-potential initiation and Na+ channel densities in the soma and axon initial segment of subicular pyramidal neurons. J Neurosci. 1996;16:6676–86. doi: 10.1523/JNEUROSCI.16-21-06676.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Fleidervish IA, Lasser-Ross N, Gutnick MJ, Ross WN. Na+ imaging reveals little difference in action potential-evoked Na+ influx between axon and soma. Nat Neurosci. 2010;13:852–60. doi: 10.1038/nn.2574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Kuba H, Ishii TM, Ohmori H. Axonal site of spike initiation enhances auditory coincidence detection. Nature. 2006;444:1069–72. doi: 10.1038/nature05347. [DOI] [PubMed] [Google Scholar]
- 75.Fried SI, Lasker AC, Desai NJ, Eddington DK, Rizzo JF., 3rd Axonal sodium-channel bands shape the response to electric stimulation in retinal ganglion cells. J Neurophysiol. 2009;101:1972–87. doi: 10.1152/jn.91081.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Gutzmann A, et al. A period of structural plasticity at the axon initial segment in developing visual cortex. Front Neuroanat. 2014;8:11. doi: 10.3389/fnana.2014.00011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Kuba H, Oichi Y, Ohmori H. Presynaptic activity regulates Na(+) channel distribution at the axon initial segment. Nature. 2010;465:1075–8. doi: 10.1038/nature09087. [DOI] [PubMed] [Google Scholar]
- 78.Grubb MS, Burrone J. Activity-dependent relocation of the axon initial segment fine-tunes neuronal excitability. Nature. 2010:1070–4. doi: 10.1038/nature09160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Rasband MN, Kagawa T, Park EW, Ikenaka K, Trimmer JS. Dysregulation of axonal sodium channel isoforms after adult-onset chronic demyelination. J Neurosci Res. 2003;73:465–70. doi: 10.1002/jnr.10675. [DOI] [PubMed] [Google Scholar]
- 80.Craner MJ, Hains BC, Lo AC, Black JA, Waxman SG. Co-localization of sodium channel Nav1.6 and the sodium-calcium exchanger at sites of axonal injury in the spinal cord in EAE. Brain. 2004;127:294–303. doi: 10.1093/brain/awh032. [DOI] [PubMed] [Google Scholar]
- 81.Saifetiarova J, Taylor AM, Bhat MA. Early and Late Loss of the Cytoskeletal Scaffolding Protein, Ankyrin G Reveals Its Role in Maturation and Maintenance of Nodes of Ranvier in Myelinated Axons. J Neurosci. 2017;37:2524–2538. doi: 10.1523/JNEUROSCI.2661-16.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Evans MD, et al. Calcineurin signaling mediates activity-dependent relocation of the axon initial segment. J Neurosci. 2013;33:6950–63. doi: 10.1523/JNEUROSCI.0277-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Vacher H, et al. Cdk-mediated phosphorylation of the Kvbeta2 auxiliary subunit regulates Kv1 channel axonal targeting. J Cell Biol. 2011;192:813–24. doi: 10.1083/jcb.201007113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Liu W, Devaux JJ. Calmodulin orchestrates the heteromeric assembly and the trafficking of KCNQ2/3 (Kv7.2/3) channels in neurons. Mol Cell Neurosci. 2014;58:40–52. doi: 10.1016/j.mcn.2013.12.005. [DOI] [PubMed] [Google Scholar]
- 85.Schafer DP, et al. Disruption of the axon initial segment cytoskeleton is a new mechanism for neuronal injury. J Neurosci. 2009;29:13242–54. doi: 10.1523/JNEUROSCI.3376-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Rasband MN. The axon initial segment and the maintenance of neuronal polarity. Nat Rev Neurosci. 2010;11:552–62. doi: 10.1038/nrn2852. [DOI] [PubMed] [Google Scholar]
- 87.Kobayashi T, Storrie B, Simons K, Dotti CG. A functional barrier to movement of lipids in polarized neurons. Nature. 1992;359:647–50. doi: 10.1038/359647a0. [DOI] [PubMed] [Google Scholar]
- 88.Winckler B, Forscher P, Mellman I. A diffusion barrier maintains distribution of membrane proteins in polarized neurons. Nature. 1999;397:698–701. doi: 10.1038/17806. [DOI] [PubMed] [Google Scholar]
- 89.Nakada C, et al. Accumulation of anchored proteins forms membrane diffusion barriers during neuronal polarization. Nat Cell Biol. 2003;5:626–32. doi: 10.1038/ncb1009. [DOI] [PubMed] [Google Scholar]
- 90.Brachet A, et al. Ankyrin G restricts ion channel diffusion at the axonal initial segment before the establishment of the diffusion barrier. J Cell Biol. 2010;191:383–95. doi: 10.1083/jcb.201003042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Albrecht D, et al. Nanoscopic compartmentalization of membrane protein motion at the axon initial segment. J Cell Biol. 2016;215:37–46. doi: 10.1083/jcb.201603108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Huang YM, Rasband MN. Organization of the axon initial segment: Actin like a fence. J Cell Biol. 2016;215:9–11. doi: 10.1083/jcb.201609084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Petersen JD, Kaech S, Banker G. Selective microtubule-based transport of dendritic membrane proteins arises in concert with axon specification. J Neurosci. 2014;34:4135–47. doi: 10.1523/JNEUROSCI.3779-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Al-Bassam S, Xu M, Wandless TJ, Arnold DB. Differential trafficking of transport vesicles contributes to the localization of dendritic proteins. Cell Rep. 2012;2:89–100. doi: 10.1016/j.celrep.2012.05.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Lewis TL, Jr, Mao T, Svoboda K, Arnold DB. Myosin-dependent targeting of transmembrane proteins to neuronal dendrites. Nat Neurosci. 2009;12:568–76. doi: 10.1038/nn.2318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Kapitein LC, Hoogenraad CC. Building the Neuronal Microtubule Cytoskeleton. Neuron. 2015;87:492–506. doi: 10.1016/j.neuron.2015.05.046. [DOI] [PubMed] [Google Scholar]
- 97.Tortosa E, et al. Dynamic Palmitoylation Targets MAP6 to the Axon to Promote Microtubule Stabilization during Neuronal Polarization. Neuron. 2017;94:809–825 e7. doi: 10.1016/j.neuron.2017.04.042. [DOI] [PubMed] [Google Scholar]
- 98.Kuijpers M, et al. Dynein Regulator NDEL1 Controls Polarized Cargo Transport at the Axon Initial Segment. Neuron. 2016;89:461–71. doi: 10.1016/j.neuron.2016.01.022. [DOI] [PubMed] [Google Scholar]
- 99.Li X, et al. Novel diffusion barrier for axonal retention of Tau in neurons and its failure in neurodegeneration. Embo J. 2011;30:4825–37. doi: 10.1038/emboj.2011.376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Kole MH, Stuart GJ. Is action potential threshold lowest in the axon? Nat Neurosci. 2008;11:1253–5. doi: 10.1038/nn.2203. [DOI] [PubMed] [Google Scholar]
- 101.Buffington SA, Rasband MN. The axon initial segment in nervous system disease and injury. Eur J Neurosci. 2011;34:1609–19. doi: 10.1111/j.1460-9568.2011.07875.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Leussis MP, Madison JM, Petryshen TL. Ankyrin 3: genetic association with bipolar disorder and relevance to disease pathophysiology. Biol Mood Anxiety Disord. 2012;2:18. doi: 10.1186/2045-5380-2-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Leussis MP, et al. The ANK3 bipolar disorder gene regulates psychiatric-related behaviors that are modulated by lithium and stress. Biol Psychiatry. 2013;73:683–90. doi: 10.1016/j.biopsych.2012.10.016. [DOI] [PubMed] [Google Scholar]
- 104.Lopez AY, et al. Ankyrin-G isoform imbalance and interneuronopathy link epilepsy and bipolar disorder. Mol Psychiatry. 2016 doi: 10.1038/mp.2016.233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Iqbal Z, et al. Homozygous and heterozygous disruptions of ANK3: at the crossroads of neurodevelopmental and psychiatric disorders. Hum Mol Genet. 2013;22:1960–70. doi: 10.1093/hmg/ddt043. [DOI] [PubMed] [Google Scholar]
- 106.Codina-Sola M, et al. Integrated analysis of whole-exome sequencing and transcriptome profiling in males with autism spectrum disorders. Mol Autism. 2015;6:21. doi: 10.1186/s13229-015-0017-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Schizophrenia Psychiatric Genome-Wide Association Study, C. Genome-wide association study identifies five new schizophrenia loci. Nat Genet. 2011;43:969–76. doi: 10.1038/ng.940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Parkinson NJ, et al. Mutant beta-spectrin 4 causes auditory and motor neuropathies in quivering mice. Nat Genet. 2001;29:61–5. doi: 10.1038/ng710. [DOI] [PubMed] [Google Scholar]
- 109.Yang Y, Lacas-Gervais S, Morest DK, Solimena M, Rasband MN. BetaIV spectrins are essential for membrane stability and the molecular organization of nodes of Ranvier. J Neurosci. 2004;24:7230–40. doi: 10.1523/JNEUROSCI.2125-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Devaux JJ. The C-terminal domain of betaIV-spectrin is crucial for KCNQ2 aggregation and excitability at nodes of Ranvier. J Physiol. 2010;588:4719–30. doi: 10.1113/jphysiol.2010.196022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Knierim E, et al. A recessive mutation in beta-IV-spectrin (SPTBN4) associates with congenital myopathy, neuropathy, and central deafness. Hum Genet. 2017;136:903–910. doi: 10.1007/s00439-017-1814-7. [DOI] [PubMed] [Google Scholar]
- 112.Tohyama J, et al. SPTAN1 encephalopathy: distinct phenotypes and genotypes. J Hum Genet. 2015;60:167–73. doi: 10.1038/jhg.2015.5. [DOI] [PubMed] [Google Scholar]
- 113.Writzl K, et al. Early onset West syndrome with severe hypomyelination and coloboma-like optic discs in a girl with SPTAN1 mutation. Epilepsia. 2012;53:e106–10. doi: 10.1111/j.1528-1167.2012.03437.x. [DOI] [PubMed] [Google Scholar]
- 114.Saitsu H, et al. Dominant-negative mutations in alpha-II spectrin cause West syndrome with severe cerebral hypomyelination, spastic quadriplegia, and developmental delay. Am J Hum Genet. 2010;86:881–91. doi: 10.1016/j.ajhg.2010.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Wang Y, et al. Critical roles of alphaII spectrin in brain development and epileptic encephalopathy. J Clin Invest. 2018 doi: 10.1172/JCI95743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Kaphzan H, Buffington SA, Jung JI, Rasband MN, Klann E. Alterations in intrinsic membrane properties and the axon initial segment in a mouse model of angelman syndrome. J Neurosci. 2011;31:17637–48. doi: 10.1523/JNEUROSCI.4162-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Kaphzan H, et al. Genetic reduction of the alpha1 subunit of Na/K-ATPase corrects multiple hippocampal phenotypes in Angelman syndrome. Cell Rep. 2013;4:405–12. doi: 10.1016/j.celrep.2013.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Palop JJ, Mucke L. Epilepsy and cognitive impairments in Alzheimer disease. Arch Neurol. 2009;66:435–40. doi: 10.1001/archneurol.2009.15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Sanchez-Mut JV, et al. DNA methylation map of mouse and human brain identifies target genes in Alzheimer’s disease. Brain. 2013;136:3018–27. doi: 10.1093/brain/awt237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Santuccione AC, et al. Active vaccination with ankyrin G reduces beta-amyloid pathology in APP transgenic mice. Mol Psychiatry. 2013;18:358–68. doi: 10.1038/mp.2012.70. [DOI] [PubMed] [Google Scholar]
- 121.Marin MA, Ziburkus J, Jankowsky J, Rasband MN. Amyloid-beta plaques disrupt axon initial segments. Exp Neurol. 2016;281:93–8. doi: 10.1016/j.expneurol.2016.04.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Clark KC, et al. Compromised axon initial segment integrity in EAE is preceded by microglial reactivity and contact. Glia. 2016 doi: 10.1002/glia.22991. [DOI] [PubMed] [Google Scholar]
- 123.Ng JK, et al. Neurofascin as a target for autoantibodies in peripheral neuropathies. Neurology. 2012;79:2241–8. doi: 10.1212/WNL.0b013e31827689ad. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Greer JE, Hanell A, McGinn MJ, Povlishock JT. Mild traumatic brain injury in the mouse induces axotomy primarily within the axon initial segment. Acta Neuropathol. 2013;126:59–74. doi: 10.1007/s00401-013-1119-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Baalman KL, Cotton RJ, Rasband SN, Rasband MN. Blast wave exposure impairs memory and decreases axon initial segment length. J Neurotrauma. 2013;30:741–51. doi: 10.1089/neu.2012.2478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Hinman JD, Rasband MN, Carmichael ST. Remodeling of the axon initial segment after focal cortical and white matter stroke. Stroke. 2013;44:182–9. doi: 10.1161/STROKEAHA.112.668749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Ogiwara I, et al. Na(v)1.1 localizes to axons of parvalbumin-positive inhibitory interneurons: a circuit basis for epileptic seizures in mice carrying an Scn1a gene mutation. J Neurosci. 2007;27:5903–14. doi: 10.1523/JNEUROSCI.5270-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Claes L, et al. De novo mutations in the sodium-channel gene SCN1A cause severe myoclonic epilepsy of infancy. Am J Hum Genet. 2001;68:1327–32. doi: 10.1086/320609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Meadows LS, et al. Functional and biochemical analysis of a sodium channel beta1 subunit mutation responsible for generalized epilepsy with febrile seizures plus type 1. J Neurosci. 2002;22:10699–709. doi: 10.1523/JNEUROSCI.22-24-10699.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Wallace RH, et al. Febrile seizures and generalized epilepsy associated with a mutation in the Na+-channel beta1 subunit gene SCN1B. Nat Genet. 1998;19:366–70. doi: 10.1038/1252. [DOI] [PubMed] [Google Scholar]
- 131.Patino GA, et al. A functional null mutation of SCN1B in a patient with Dravet syndrome. J Neurosci. 2009;29:10764–78. doi: 10.1523/JNEUROSCI.2475-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Shi X, et al. Missense mutation of the sodium channel gene SCN2A causes Dravet syndrome. Brain Dev. 2009;31:758–62. doi: 10.1016/j.braindev.2009.08.009. [DOI] [PubMed] [Google Scholar]
- 133.Heron SE, et al. Sodium-channel defects in benign familial neonatal-infantile seizures. Lancet. 2002;360:851–2. doi: 10.1016/S0140-6736(02)09968-3. [DOI] [PubMed] [Google Scholar]
- 134.Nakamura K, et al. Clinical spectrum of SCN2A mutations expanding to Ohtahara syndrome. Neurology. 2013;81:992–8. doi: 10.1212/WNL.0b013e3182a43e57. [DOI] [PubMed] [Google Scholar]
- 135.Ohba C, et al. Early onset epileptic encephalopathy caused by de novo SCN8A mutations. Epilepsia. 2014;55:994–1000. doi: 10.1111/epi.12668. [DOI] [PubMed] [Google Scholar]
- 136.Biervert C, et al. A potassium channel mutation in neonatal human epilepsy. Science. 1998;279:403–6. doi: 10.1126/science.279.5349.403. [DOI] [PubMed] [Google Scholar]
- 137.Athanasiu L, et al. Gene variants associated with schizophrenia in a Norwegian genome-wide study are replicated in a large European cohort. J Psychiatr Res. 2010;44:748–53. doi: 10.1016/j.jpsychires.2010.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Awadalla P, et al. Direct measure of the de novo mutation rate in autism and schizophrenia cohorts. Am J Hum Genet. 2010;87:316–24. doi: 10.1016/j.ajhg.2010.07.019. [DOI] [PMC free article] [PubMed] [Google Scholar]