

Abstract
The use of N-methylpyrrolidone (NMP) as a co-solvent in ferric salt catalyzed cross-coupling reactions is crucial for achieving the highly selective, preparative scale formation of cross-coupled product in reactions utilizing alkyl Grignard reagents. Despite the critical importance of NMP, the molecular level effect of NMP on in situ formed and reactive iron species that enables effective catalysis remains undefined. Herein, we report the isolation and characterization of a novel trimethyliron(II) ferrate species, [Mg(NMP)6][FeMe3]2 (1), which forms as the major iron species in situ in reactions of Fe(acac)3 and MeMgBr under catalytically relevant conditions where NMP is employed as a co-solvent. Importantly, combined GC analysis and 57Fe Mössbauer spectroscopic studies identified 1 as a highly reactive iron species for the selective formation generating cross-coupled product. These studies demonstrate that NMP does not directly interact with iron as a ligand in catalysis but, alternatively, interacts with the magnesium cations to preferentially stabilize the formation of 1 over [Fe8Me12]− cluster generation, which occurs in the absence of NMP.
Keywords: cross-coupling, iron, MCD spectroscopy, Mössbauer spectroscopy, N-methylpyrrolidone
Iron-catalyzed cross-coupling reactions are of significant synthetic interest as they can provide more sustainable alternatives to precious metal cross-coupling catalysis while achieving complementary reactivity.[1] Simple ferric salt catalyzed cross-couplings involving alkyl nucleophiles are especially attractive due to their simplicity and tolerance of nucleophiles containing β-hydrogens.[2] Originally developed by Kochi in the 1970s, recent studies have identified [Fe8Me12]− as the key reactive iron species formed in these reactions with MeMgBr (Scheme 1).[3] In 1998, Cahiez and Avedissian overcame many of the limitations of the Kochi ferric salt system through the use of N-methylpyrrolidone (NMP) as a co-solvent.[4] With NMP present, cross-couplings of various vinyl halides and alkyl Grignard reagents could be achieved with high chemoselectivity without the requirement of excess alkenyl halide (required in the absence of NMP), enabling preparative scale iron-catalyzed cross-coupling reactions (Scheme 1). Furthermore, NMP has been demonstrated to be critically important for extending the use of ferric salt-catalyzed cross-coupling to substrates such as aryl halides, alkenyl triflates, and acyl chlorides.[5] While NMP has been employed extensively in iron-catalyzed cross-coupling methodology,[5b,6] its role in enabling improved catalytic performance remains poorly defined. For example, while a NMP-ligated iron complex has previously been isolated under non-catalytically relevant conditions, it remains ambiguous as to whether NMP is a ligand to iron during cross-coupling or serves another effect.[7] The ability to define the role of NMP in simple ferric salt catalyzed cross-coupling, a mystery for twenty years, would be a significant step forward in our molecular-level understanding of the nature of reactive iron species required for effective cross-coupling catalysis.
Scheme 1.

Iron-catalyzed alkyl-alkenyl cross-coupling reactions with ferric salts reported by Kochi and Cahiez and the molecular structure of [Fe8Me12]−.
Herein, we report the use of 57Fe Mössbauer spectroscopy, electron paramagnetic resonance (EPR), and magnetic circular dichroism (MCD) to evaluate the effects of NMP on in situ generated iron species from reactions of simple ferric salts with a methyl Grignard reagent (MeMgBr). This study represents the first direct evaluation of the effect of NMP on iron speciation and reactivity with electrophile in cross-coupling. Critically, these studies demonstrate that the presence of NMP as an additive results in the stabilization of an unprecedented and reactive, three-coordinate homoleptic iron(II)-methyl species under catalytically relevant conditions, where NMP acts as a ligand to the MgII counter-ion.
While previous studies demonstrated that generation of [Fe8Me12]− could be accomplished by reacting Fe(acac)3 with 20 equiv of MeMgBr,[3] it was important to directly evaluate if the Cahiez protocol involving NMP has any effect on cluster formation. The addition of 20 equiv of MeMgBr to a 3 mM solution of 57Fe(acac)3 in 1:1 THF/2-MeTHF with 180 equiv of NMP (9 equiv of NMP relative to Grignard reagent; Cahiez protocol ratio[4]) at RT led to the rapid formation of a pale yellow solution. Note that the drop-wise addition of Grignard reagent using a syringe pump (12 equiv of Grignard reagentmin−1 relative to iron) was utilized to mimic the use of an addition funnel as described in the original Cahiez report.[4] The 80 K 57Fe Mössbauer spectrum of the in situ formed iron species freeze-trapped 30 s after completion of the Grignard reagent addition indicates the formation of a single major iron species (97% of total iron, green component) with 57Fe Mössbauer parameters of δ = 0.25 mms−1 and ΔEQ = 1.36 mms−1 (Figure 1A), whereas in the absence of NMP exclusive formation of a broad doublet signal associated with [Fe8Me12]− was previously observed (δ =0.30 mms−1 and ΔEQ = 0.85 mms−1). A minor additional iron species is also observed by 57Fe Mössbauer spectroscopy with parameters δ =−0.28 mms−1 and ΔEQ = 0.73 mms−1 (3% of total iron, pink component) that corresponds to a S =3/2 species (axial, g ≈ 4.3, 2.0, ≈ 3% of total iron by spin quantitation), which is also observed by 10 K EPR spectroscopy (Figure 1A, inset). There is also a minor amount of an additional broad S =1/2 (isotropic, g ≈ 2) species observed in the EPR spectrum, which is consistent with the previously identified iron cluster [Fe8Me12]− though present in too minor an amount to be observed by 57Fe Mössbauer spectroscopy. The 5 K, 7 T near-infrared (NIR) MCD spectrum of the in situ formed iron species shows two intense ligand field (LF) transitions at ≈10200 cm−1 with a shoulder at ≈9200 cm−1 (Figure 1C), assigned to the major iron species formed in solution. While most studies were performed in 1:1 THF/2-MeTHF for glassing purposes, the same iron speciation was also observed in pure THF by 57Fe Mössbauer spectroscopy (see Supporting Information, Figure S1).
Figure 1.

80 K 57Fe Mössbauer spectra of a frozen solution of 57Fe-(acac)3 (9 equiv of NMP relative to MeMgBr) with (A) 20 equiv and (B) 5 equiv of MeMgBr (black dots), total fit (black line) and individual fit components are shown. (C) 5 K, 7 T NIR MCD spectrum of 20 equiv of MeMgBr reacted with Fe(acac)3 and 180 equiv of NMP at RT in 1:1 THF/2-MeTHF.
It was important for subsequent single-turnover reactivity studies to determine the minimum quantity of MeMgBr required to form the major iron species. In fact, it was determined that 5 equiv of MeMgBr is required for the formation of this species. The reaction of 57Fe(acac)3 with 5 equiv of MeMgBr and 45 equiv of NMP at RT generates the same major iron species as determined by 80 K 57Fe Mössbauer spectroscopy (Figure 1B, δ =0.25 mms−1 and ΔEQ = 1.36 mms−1, 82% of total iron, green component). The same additional minor iron species is observed (δ = −0.28 mms−1 and EQ = 0.73 mms−1, 3% of total iron, pink component) in addition to a new iron species with parameters δ =0.64 mms−1 and ΔEQ = 1.27 mms−1 (15% of total iron, blue component) which, from reactivity studies (see below), is assigned to an under-transmetallated iron complex that forms prior to the major iron species (Figure 4). The 10 K EPR spectrum indicates minor contributions from a S =3/2 species (≈3%) and a S =1/2 cluster (Figure 1B, inset).
Figure 4.
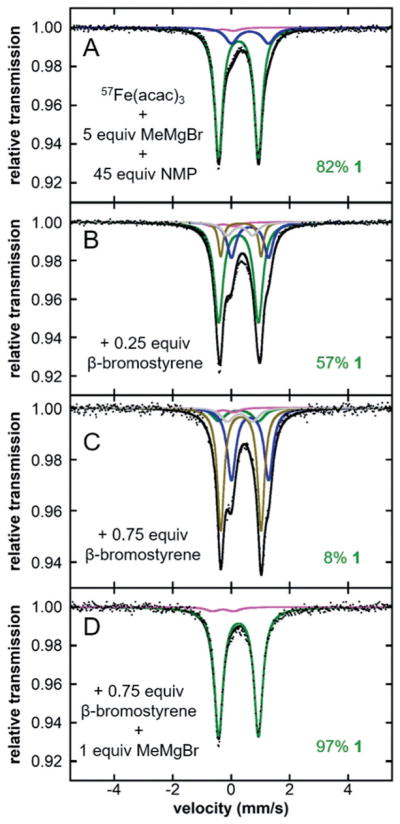
80 K 57Fe Mössbauer spectra of a frozen solution of 57Fe-(acac)3 (9 equiv of NMP relative to MeMgBr) with (A) 5 equiv of MeMgBr and subsequent addition of (B) 0.25 equiv, (C) 0.75 equiv of β-bromostyrene and (D) 0.75 equiv of β-bromostyrene followed by 1 equiv of MeMgBr (black dots), total fit (black line) and individual fit components are shown.
Spectroscopic investigations of the in situ generated iron species indicated a limited window of thermal stability at 23°C (t ≈ 3 min) before iron species associated with decomposition were formed (Figure S2). Thus, efforts were directed toward isolating the major iron species. The treatment of a red solution containing Fe(acac)3 and 9 equiv of NMP in THF with 5 equiv of MeMgBr at −5°C provided a yellow colored solution. Rapid cooling of the solution to −80°C (to disfavor decomposition) followed by layering with diethyl ether rendered large pale-yellow crystals. A single crystal X-ray diffraction study (XRD) revealed three independent instances of [Mg(NMP)6][FeMe3]2, 1, in the asymmetric unit in general positions, with the cations and anions well separated (Figure 2A). Although the crystals diffracted very weakly, precluding a detailed discussion of the structural parameters, the overall connectivity, geometry, and chemical formulation of this species are unambiguous. Notably, the magnesium ion is coordinated through the carbonyl oxygens of the six NMP molecules (Note: NMP O-coordination mode for a magnesium ion and other metals has been previously reported[6f,7–8]) and no NMP coordination to iron is present. A previous report showed similar effects of co-solvent on iron speciation in which ligation of tetramethylethylenediamine (TMEDA) to magnesium cations in homoleptic triaryl-ferrate complexes was observed.[9]
Figure 2.

(A) Structure of [Mg(NMP)6][FeMe3]2 (1), drawn at 50% probability level. Hydrogen atoms omitted for clarity. (B) 80 K 57Fe Mössbauer spectrum of a frozen solution of 1 (black dots), total fit (green line). (C) 5 K, 7 T NIR MCD of 1 dissolved in 1:1 THF/2-MeTHF. (D) Saturation magnetization data (dots) and best fit (lines) collected at 10486 cm−1.
To date, very few examples of homoleptic alkyl- and aryl-iron complexes devoid of stabilizing ligands exist in the literature.[8] The first example incorporating methyl ligands was reported by Fürstner in which a distorted tetrahedral homoleptic tetramethyliron(II) ferrate complex, [(Me4Fe)-(MeLi)][Li(OEt2)]2, was identified.[10] This inspired later studies from Neidig and co-workers in which a distorted square-planar tetramethyliron(III) ferrate, [MgCl(THF)5]-[FeMe4], was identified.[11] The unique coordination geometry of the iron species was determined to be strongly influenced by the choice of solvent and cation. This assertion is further supported by the generation of 1 instead of [Fe8Me12]− when utilizing NMP as a co-solvent demonstrating the powerful effect of NMP coordination to cations versus other solvents. Both Bedford and Fürstner have independently reported the isolation of triaryl-ferrate complexes which are reactive with electrophile, but unlike 1 lack high selectivity in producing cross-coupled product.[9,12]
While 1 represents an interesting, three-coordinate homoleptic iron-methyl structure, spectroscopic characterization of 1 in solution was necessary to verify its identity as the major iron species formed under catalytically relevant conditions. The 5 K, 7 T NIR MCD spectrum of 1 in solution (Figure 2C), prepared from dissolution of crystals of 1 in 1:1 THF/2-MeTHF at −80°C to prevent thermal decomposition, is consistent with the analogous spectrum (Figure 1C) of the iron species generated from reaction of Fe(acac)3 with 20 equiv of MeMgBr in 180 equiv of NMP. Saturation magnetization data collected at 10846 cm−1 is well-fit to S = 2 positive zero-field split (+ZFS) non-Kramers doublet model with ground-state spin Hamiltonian parameters of D =13 ±3 cm−1, | E/D | =0.07 ± 0.03 with giso = 2.20 ± 0.10 (Figure 2D). Additionally, the solid 80 K 57Fe Mössbauer spectrum of crystals of 1 (Figure 2B) yields identical parameters (δ =0.25 mms−1 and ΔEQ = 1.36 mms−1, green component) to those obtained from in situ reaction of Fe(acac)3 with MeMgBr in the presence of NMP (Figure 1A). Together, this data demonstrates that 1 is the major iron species formed in situ under the catalytic conditions previously reported by Cahiez when using NMP as an additive.
Spin-unrestricted DFT calculations were performed on 1 with a focus on the ferrous trimethyl anion subunit, excluding the [Mg(NMP)6] 2+ dication in order to gain further insight into the electronic structure and bonding of this novel low-coordinate iron(II) species. The PBE0 functional and def2-TZVP basis set were employed to yield calculated structures for the S =2 system, which are in good agreement with crystallographic data in both the gas phase and THF solvent models (see SI). Additionally, B3LYP/TZVP was used to evaluate the molecular orbitals (see SI).
The ground-state electronic structure can be described by examination of the frontier molecular orbitals of the β-manifold (Figure 3), which shows the highest occupied (β25) and the four lowest unoccupied MOs (β26, β27, β28 and β29) with significant d-orbital character. Strong Fe s-orbital character is also present in β26 and modest antibonding interactions are observed between the methyl carbon atoms in β26, β28, and β29.
Figure 3.

Calculated molecular orbital energy diagram for 1.
In order to understand the role of 1 in cross-coupling, reactions of 1 with sub-stoichiometric amounts of electrophile, β-bromostyrene, were tracked by 80 K 57Fe Mössbauer, with concurrent reaction aliquots quenched to determine product yields by gas chromatography (GC). These experiments demonstrate that 1 is consumed within 10 s in the presence of electrophile to generate exclusively cross-coupled product (kobs > 6 min−1, Table 1 and Figure 4). Furthermore, the minor iron side-products (S =3/2 species and iron cluster (S =1/2)), monitored by EPR spectroscopy, remain unchanged when reacted with sub-stoichiometric amounts of electrophile suggesting they are unreactive or less reactive than 1 (Figure S3). While the iron products formed upon reaction with electrophile could not be isolated, it was demonstrated that the addition of 1 equiv of MeMgBr leads to rapid regeneration of 1 (Figure 4D) suggesting that these are under-transmetallated iron species (blue and brown components). These results indicate that 1 is a very reactive species for the productive formation of cross-coupled product.
Table 1.
GC analysis of the products formed upon reaction of in situ generated 1 with β-bromostyrene at 23°C
Yields are with respect to iron. No other side products were observed.
Reactions are stereoselective and retain the same conformation in the cross-coupled product as in the starting reagent (E- and Z-isomers present). Yields account for both observed isomers.
Importantly, unlike [Fe8Me12]− which has been implicated as the major reactive species in Kochi’s catalysis, 1 reacts with electrophile without the requirement of an additional equivalent of MeMgBr to produce cross-coupled product.[3] From reactivity studies it appears that 1 produces less reactive under-transmetallated iron species (Figure 4), whereas [Fe8Me12]− generates an intermediate iron species intimately associated with electrophile, which was apparent from the disappearance of its S =1/2 EPR signal and the lack of formation of cross-coupled product. The requirement of additional MeMgBr for turnover from this intermediate species may be the reason for the excess electrophile required in Kochi’s protocol, as this intermediate species is expected to be unstable and, hence, susceptible to unproductive reaction pathways. Therefore, this observed difference in reactivity is likely a key contributing factor to the improved catalysis observed by Cahiez when using NMP.[4]
While a previously identified tetramethyliron(III) ferrate, [MgCl(THF)5][FeMe4], was implicated as an intermediate along the reduction pathway of FeCl3 and MeMgBr to generate [Fe8Me12]−, it is interesting to consider whether a trimethyliron(II) ferrate such as 1, is accessible along this pathway in the absence of NMP as co-solvent since 57Fe Mössbauer parameters consistent with [Fe8Me12]− are generated upon prolonged mixing of 1 (δ =0.30 mms−1 and ΔEQ = 0.87 mms−1, Figure S2).[11] To explore this possibility, we attempted to synthesize a [FeMe3]− at low temperature without NMP (see SI).
Crystals of [Mg3Cl3(OMe)2(THF)6][FeMe3]·2THF were identified by XRD. Note that the methoxy-substituents of the magnesium cation are attributed to a methanol impurity in the Grignard reagent. Synthesis of this analogous structure suggests the possibility of 2 being an intermediate species between the tetramethyliron(III) ferrate and iron-cluster. However, the consistent preparation of 2 proved difficult, further demonstrating the strong influence of NMP on the stability of 1 leading to greater ease of preparation and handling. Future studies will be aimed at exploring the stability of NMP analogs of the tetramethyliron(III) ferrate and its effect on the overall reduction pathway of ferric salts with MeMgBr.
Supplementary Material
Acknowledgments
This work was supported by a grant from the National Institutes of Health (R01GM111480 to M.L.N.), an NSF Graduate Research Fellowship (T.M.B.), an Alfred P. Sloan Fellowship (M.L.N.) and a Ruth L. Kirchstein National Research Service award (F32GM120823 to S.B.M. III).
Footnotes
Conflict of interest
The authors declare no conflict of interest.
Supporting information and the ORCID identification number(s) for the author(s) of this article can be found under: https://doirg/10.1002/anie.201802087.
References
- 1.a) Sherry BD, Fürstner A. Acc Chem Res. 2008;41:1500–1511. doi: 10.1021/ar800039x. [DOI] [PubMed] [Google Scholar]; b) Fürstner A, Martin R. Chem Lett. 2005;34:624–629. [Google Scholar]; c) Mako TL, Byers JA. Inorg Chem Front. 2016;3:766–790. [Google Scholar]; d) Martin R, Fürstner A. Angew Chem Int Ed. 2004;43:3955–3957. doi: 10.1002/anie.200460504. [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2004, 116, 4045 – 4047; e) Carpenter SH, Neidig ML. Isr J Chem. 2017;57:1106–1116. doi: 10.1002/ijch.201700036. [DOI] [PMC free article] [PubMed] [Google Scholar]; f) Bauer I, Knçlker HJ. Chem Rev. 2015;115:3170–3387. doi: 10.1021/cr500425u. [DOI] [PubMed] [Google Scholar]; g) Fürstner A. ACS Cent Sci. 2016;2:778–789. doi: 10.1021/acscentsci.6b00272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.a) Tamura M, Kochi JK. J Am Chem Soc. 1971;93:1487–1489. [Google Scholar]; b) Neumann SM, Kochi JK. J Org Chem. 1975;40:599–606. [Google Scholar]
- 3.Muñoz SB, III, Daifuku SL, Brennessel WW, Neidig ML. J Am Chem Soc. 2016;138:7492–7495. doi: 10.1021/jacs.6b03760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cahiez G, Avedissian H. Synthesis. 1998;1998:1199–1205. [Google Scholar]
- 5.a) Fürstner A, Leitner A, Méndez M, Krause H. J Am Chem Soc. 2002;124:13856–13863. doi: 10.1021/ja027190t. [DOI] [PubMed] [Google Scholar]; b) Seidel G, Laurich D, Fürstner A. J Org Chem. 2004;69:3950–3952. doi: 10.1021/jo049885d. [DOI] [PubMed] [Google Scholar]
- 6.a) Ottesen LK, Ek F, Olsson R. Org Lett. 2006;8:1771–1773. doi: 10.1021/ol0600234. [DOI] [PubMed] [Google Scholar]; b) Cahiez G, Gager O, Buendia J, Patinote C. Chem Eur J. 2012;18:5860–5863. doi: 10.1002/chem.201200184. [DOI] [PubMed] [Google Scholar]; c) Gotta M, Lehnemann BW, Jacobi von Wangelin A, Guelak S. 9,024,045 B2. US. 2015; d) Gülak S, Gieshoff TN, Jacobi von Wangelin A. Adv Synth Catal. 2013;355:2197–2202. [Google Scholar]; e) Malhotra S, Seng PS, Koenig SG, Deese AJ, Ford KA. Org Lett. 2013;15:3698–3701. doi: 10.1021/ol401508u. [DOI] [PubMed] [Google Scholar]; f) Gärtner D, Stein AL, Grupe S, Arp J, Jacobi von Wangelin A. Angew Chem Int Ed. 2015;54:10545–10549. doi: 10.1002/anie.201504524. [DOI] [PubMed] [Google Scholar]; Angew Chem. 2015;127:10691–10695. [Google Scholar]
- 7.Ding K, Zannat F, Morris JC, Brennessel WW, Holland PL. J Organomet Chem. 2009;694:4204–4208. [Google Scholar]
- 8.a) Williamson MM, Prosser-McCartha CM, Mukundan S, Hill CL. Inorg Chem. 1988;27:1061–1068. [Google Scholar]; b) Zhou Q, Hambley TW, Kennedy BJ, Lay PA, Turner P, Warwick B, Biffin JR, Regtop HL. Inorg Chem. 2000;39:3742–3748. doi: 10.1021/ic991477i. [DOI] [PubMed] [Google Scholar]; c) Evans WJ, Fujimoto CH, Johnston MA, Ziller JW. Organometallics. 2002;21:1825–1831. [Google Scholar]
- 9.Bedford RB, Brenner PB, Carter E, Cogswell PM, Haddow MF, Harvey JN, Murphy DM, Nunn J, Woodall CH. Angew Chem Int Ed. 2014;53:1804–1808;. doi: 10.1002/anie.201308395. [DOI] [PubMed] [Google Scholar]; Angew Chem. 2014;126:1835–1839. [Google Scholar]
- 10.Fürstner A, Martin R, Krause H, Seidel G, Goddard R, Lehmann CW. J Am Chem Soc. 2008;130:8773–8787. doi: 10.1021/ja801466t. [DOI] [PubMed] [Google Scholar]
- 11.Al-Afyouni MH, Fillman KL, Brennessel WW, Neidig ML. J Am Chem Soc. 2014;136:15457–15460. doi: 10.1021/ja5080757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sun CL, Krause H, Fürstner A. Adv Synth Catal. 2014;356:1281–1291. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.