

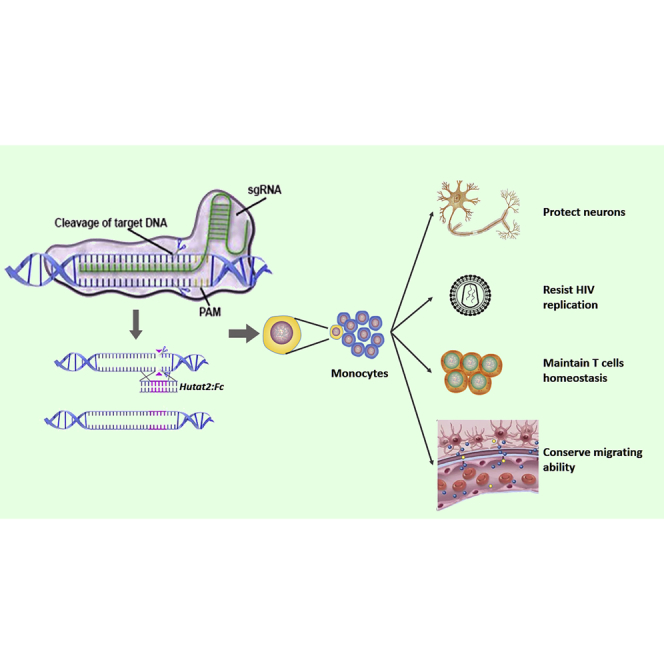
Abstract
The ability of monocytes to travel through the bloodstream, traverse tissue barriers, and aggregate at disease sites endows these cells with the attractive potential to carry therapeutic genes into the nervous system. However, gene editing in primary human monocytes has long been a challenge. Here, we applied the CRISPR/Cas9 system to deliver the large functional Hutat2:Fc DNA fragment into the genome of primary monocytes to neutralize HIV-1 transactivator of transcription (Tat), an essential neurotoxic factor that causes HIV-associated neurocognitive disorder (HAND) in the nervous system. Following homology-directed repair (HDR), ∼10% of the primary human monocytes exhibited knockin of the Hutat2:Fc gene in the AAVS1 locus, the “safe harbor” locus of the human genome, without selection. Importantly, the release of Hutat2:Fc by these modified monocytes protected neurons from Tat-induced neurotoxicity, reduced HIV replication, and restored T cell homeostasis. Moreover, compared with lentiviral transfection, CRISPR-mediated knockin had the advantage of maintaining the migrating function of monocytes. These results establish CRISPR/Cas9-mediated Hutat2:Fc knockin monocytes and provide a potential method to cross the blood-brain barrier for HAND therapy.
Keywords: anti-Tat antibody, CRISPR/Cas9, knockin, primary monocytes, HIV-1, HIV-associated neurocognitive disorder, gene therapy
Graphical Abstract
Introduction
In recent years, the lifespan of HIV-infected patients has been prolonged because of the advancement of combination antiretroviral therapy (cART).1 However, approximately 50% of patients suffer from varying levels of HIV-associated neurocognitive disorder (HAND) ranging from undetectable neurocognitive impairments to severe dementia, which greatly affects their survival rate and quality of life.2 The progression of HAND is related to several factors, and HIV transactivator of transcription (Tat) is one of the most important. On the one hand, HIV-Tat contributes to the release of several inflammatory factors that induce oxidative stress and excitotoxicity, ultimately resulting in neurodegeneration.3, 4 On the other hand, HIV-Tat increases the levels of viral transcription in HIV-infected astrocytes and microglial cells, forming a positive feedback loop.5 Given the role of HIV-Tat in the progression of HAND, efforts to block its effects on the CNS might be exploited as an adjuvant strategy to reduce HIV-induced neuropathogenesis and improve patients’ quality of life.
The humanized anti-Tat intrabody Hutat2 has previously been shown to exhibit high-affinity binding to residues 1–20 in the N-terminal activation domain of Tat.6 Furthermore, Hutat2 was additionally modified with a C-terminal human Κ-domain to increase its cytoplasmic stability and N-terminal signal peptides to direct the antibody toward the secretory pathway.7, 8 Previous studies attempting to transfer the Hutat2:Fc gene into CD4+ T cells using retroviruses showed a protective effect against HIV-1 replication.6, 9, 10 Although these results are encouraging, the difficulties of combating HAND in the CNS have not yet been resolved because of the lack of cell-based delivery tools to traverse the blood-brain barrier (BBB).
Previous studies have revealed the potential for blood-circulating monocytes to act as “Trojan horses” to penetrate the BBB for drug and gene delivery.11 In contrast with other cell vectors, monocytes have the particular ability to differentiate into resident microglial cells, ensuring their long-term function in the brain.12 Thus, considerable efforts have been devoted to exploring the use of this cell type as a cure for neurological disorders. However, low editing efficiency in primary monocytes has long been a limitation because these cells are difficult to expand and genetically manipulate in vitro.13, 14 Improved technology is needed to effectively modify monocytes while maintaining their migrating capacity. Recently, the development of CRISPR/Cas9, an effective genome-editing system, has enabled the precise engineering of various mammalian cells.15, 16, 17 Through homology-directed repair (HDR) of the double-strand breaks (DSBs) created by CRISPR/Cas9, therapeutic DNA templates are knocked into the AAVS1 locus, which has been widely studied because this locus enables the insertion of gene cassettes into almost all human cell types without known adverse effects.18, 19 However, it has not yet been verified if this system is applicable in primary human monocytes.
In the present study, we generated Hutat2:Fc knockin (KI) primary human monocytes using CRISPR/Cas9 and investigated the biological activity of secreted Hutat2:Fc to neutralize HIV-Tat in vitro. Based on our data, Hutat2:Fc was successfully inserted into the AAVS1 locus and effectively expressed in primary monocytes. Secreted Hutat2:Fc protected mouse primary cortical neurons, limited viral replication and spread, and restored T cell homeostasis. Moreover, compared with traditional lentiviral transduction methods, CRISPR-mediated KI did not affect the normal functionality of monocytes, particularly their migrating ability. Thus, CRISPR-mediated Hutat2:Fc KI monocytes (KI-Ms) may represent a promising strategy for HAND therapy in the future.
Results
CRISPR/Cas9-Mediated KI of the Hutat2:Fc Gene into the AAVS1 Locus of HeLa, 293T, and U937 Cell Lines and Primary Human Monocytes
We designed six single guide RNAs (sgRNAs) (Table S1) targeting the AAVS1 locus to generate specific DSBs (Figure S1A). T7E I and universal CRISPR activity assays were both employed to select the most active guide RNA, which was identified as sgRNA2 (Figures S1B and S1C). Notably, the activity of sgRNA2 was dose dependent, and 2 μg of sgRNA2 achieved the highest knockout efficiencies of up to ∼20% in HeLa cells (Figure S1D) and ∼12% in primary monocytes (Figure S3B).
A donor plasmid containing the CAG promoter, Hutat2:Fc fragment, GFP, the selective marker Puro, and flanking sequences of ∼1.5 kb on each side homologous to the sgRNA2 targeting region was designed to achieve high HDR efficiency (Figure 1A). Although the expression of Hutat2:Fc could be driven by the PPP1R12C promoter itself, the CAG promoter was included to enhance its expression.18 The genotyping of HeLa cells after co-transfection and selection with Puro revealed that donor templates were successfully knocked into the genome (Figures 1B–1D).
Figure 1.

CRISPR-Mediated KI of Hutat2:Fc Gene Fragments into the AAVS1 Locus in Various Cell Lines and Primary Human Monocytes
(A) Schematic of the construction of the donor plasmid and primers design. The homology arms of the donor vector are labeled LR and RR. (B–D) After co-transfection of HeLa cells with sgRNA2 with or without donor plasmids, genomic templates were amplified by PCR using the primers, GT-F/Ai3-2781R (B), Ai3-2737F/Puro-GT-R (C), and Neo-F/GT-R (D), shown in (A). (E) Following selection with Puro, PCR using the primers GT-F/GT-R shown in (A) was performed to semiquantitatively analyze the KI efficiency in HeLa cells transfected with different ratios of sgRNA2 to donor plasmids. The positions of wild-type and transgene chromatids are indicated below and above the gel, respectively. In this assay, HDR resulted in an additional weak PCR band above the wild-type chromatid band. The editing frequencies were calculated and shown below the gel. The percentage of GFP+ cells was quantified by FACS, and representative FACS plots are shown directly below the same genotype. (F) As in (E), 293T cells were transfected with the maximum ratio of sgRNA2 and donor plasmids. (G and H) As in (F), U937 cells (G) and primary monocytes (H) were transfected by electroporation but not subjected to selection. KI-293T, CRISPR/Cas9-mediated KI in 293T cells; KI-HeLa, CRISPR/Cas9-mediated KI in HeLa cells; KI-M, CRISPR/Cas9-mediated KI in primary human monocyte; KI-U937, CRISPR/Cas9-mediated KI in U937 cells.
Furthermore, we performed PCR using a pair of primers, GT-F and GT-R, which were located at a region of the chromosome outside of the homology arms, to determine the optimal ratio of sgRNA2 to donor plasmids. With the optimized ratio of 1:4, the highest KI efficiencies obtained were ∼35% in HeLa cells (Figure 1E, upper panel) and ∼45% in 293T cells (Figures 1F and S2A, upper panel) after selection with Puro. Additionally, without screening, ∼20% of U937 cells (Figures 1G and S2B, upper panel) and ∼10% of monocytes (Figures 1H and S3C, upper panel) exhibited Hutat2:Fc insertion following electroporation.
The proportion of GFP+ cells was analyzed by FACS to confirm the insertion of the target fragments. Consistent with the semiquantitative PCR results, up to 32.40% of GFP+ cells were detected in HeLa cells (Figure 1E, lower panel). Although the donor plasmids themselves might produce GFP in cells without insertion, less than 1% of cells treated with donor plasmids alone expressed GFP (Figure 1E, lower left panel), and no transgenic chromatid bands were detected in that sample by PCR (Figure 1E, upper left panel). It is likely that long-term culture might eliminate such interference.20 Similarly, with optimal sgRNA2 to donor plasmid ratios, the percentages of GFP+ cells were 40.20% in 293T cells (Figures 1F and S2A, lower panel), 21.00% in U937 cells (Figures 1G and S2B, lower panel), and 9.12% in monocytes (Figures 1H and S3C, lower panel). Furthermore, Sanger sequencing of both boundaries confirmed that the full-length DNA template was completely inserted into the AAVS1 locus targeted by sgRNA2 (Figure S4). Based on these data, CRISPR/Cas9 mediated successful insertion of the Hutat2:Fc gene into the AAVS1 locus of the HeLa, 293T, and U937 cell lines and monocytes. Moreover, specific KI was achieved in primary human monocytes without selection, albeit at relatively low rates.
Finally, the off-target (OT) effects on these transduced monocytes were evaluated. Recent studies suggested that CRISPR/Cas9 frequently induces unwanted OT mutations.21 These mismatches might exert negative effects on the normal functions of the targeted cells. Here, we examined the top four potential OT sites for sgRNA2 (Table S1). Indeed, only one OT mutation, OT-3, was detected in the T7E I assay (Figure S5). Due to the specific design of the homology arms, no misinsertion of the DNA template was observed (Figure S5).
Evaluation of the Stable Expression and Secretion of Hutat2:Fc
Although successful insertion was confirmed, we needed to ensure that the functional Hutat2:Fc fragment was effectively expressed and secreted from the transduced cells. To this end, we conducted a preliminary panel of experiments to detect intracellular Hutat2:Fc expression. First, in HeLa cells, GFP was localized to both the nucleus and cytoplasm, whereas Hutat2:Fc was mainly distributed in the cytoplasm (Figure 2A). In monocytes, the expression of GFP was shown in Figure 6A. The levels of Hutat2:Fc and GFP transcription among the transformed cell lines and monocytes were compared by RT-PCR. Consistent with the KI efficiency, 293T cells exhibited the highest levels of expression, whereas monocytes exhibited the lowest expression levels (Figure 2B). This finding was confirmed by western blot analyses using cell lysates (Figure 2E).
Figure 2.

Transduced Cell Lines and Primary Monocytes Co-express Hutat2:Fc and GFP
(A) Co-localization of Hutat2:Fc and GFP expression in HeLa cells. Hutat2:Fc (red) was located in the cytoplasm, whereas GFP (green) was located in both the nucleus and cytoplasm. Nuclei were counterstained with DAPI (blue). Scale bars, 100 μm. (B) Detection of the relative expression of Hutat2:Fc and GFP in transduced HeLa cells, 293T cells, U937 cells, and monocytes. (C) Kinetics of Hutat2:Fc secretion into the culture media of primary monocytes 7 days after transfection (n = 4). (D) Comparison of the concentration of Hutat2:Fc in the culture media from HeLa, 293T, and U937 cell lines (n = 5). Dunnett’s t test was used to compare cells cultured under different experimental conditions; *p < 0.05; **p < 0.01. (E and F) Detection of Hutat2:Fc in both cell lysates (E) and culture media (F) through western blot analysis. The results represent the means from independent experiments. The error bars denote the SEM. KI-293T-Hutat2, culture supernatant of KI-293T cells; KI-HeLa-Hutat2, culture supernatant of KI-HeLa cells; KI-M-Hutat2, culture supernatant of KI-Ms; KI-U937-Hutat2, culture supernatant of KI-U937 cells; RTase (−), RTase-negative control; U937, non-transduced U937.
Figure 6.

Evaluation of the Efficiency and Adverse Effects of CRISPR- and Lentivirus-Mediated Transduction of Primary Monocytes or MDMs
(A) The cells were transfected and sorted for suspension monocytes. Representative images of monocytes or MDMs transduced with Hutat2:Fc using different gene-editing methods are shown. Scale bars, 100 μm. (B) Detection of Hutat2:Fc in KI-Ms, LT-Ms, and LT-MDMs by western blotting. Monocyte, non-transfected monocytes. (C) Ratios of the mean values for Hutat2:Fc to β-actin in each cell type shown in (B) (n = 5). One-way ANOVA was employed for the statistical analyses. (D) Comparison of the levels of 15 monocyte-related regulatory genes in each group by real-time PCR (n = 6). (E–G) Comparison of the IL-10 (E), TNF-α (Φ), and IL-1β (G) levels in the culture supernatants of each group by ELISAs (n = 5). (H) Comparison of the cellular transmigration rates of each group using TEM assays (n = 6). Dunnett’s t tests were used for the statistical analyses of the data shown in (D)–(H); *p < 0.05; **p < 0.01. The results are presented as the means from independent experiments. The error bars denote the SEM. KI-M, CRISPR/Cas9-mediated Hutat2:Fc KI monocytes; LT-M, lentivirus-transfected monocytes; LT-MDM, lentivirus-transfected monocyte-derived macrophages.
Second, the release of Hutat2:Fc from the HeLa, 293T, and U937 cell lines and monocytes was quantified by ELISA. The analysis of HeLa and 293T cells revealed that the concentration of secreted Hutat2:Fc increased exponentially over time and reached a plateau on day 4 (704.23 ± 44.09 ng/mL for HeLa cells and 923.27 ± 43.54 ng/mL for 293T cells). For U937 cells, the concentration peaked on day 5 (194.30 ± 17.76 ng/mL) (Figure 2D), and similarly to the findings for U937 cells, a delay in the peak concentration was observed in monocytes on day 5 (113.30 ± 8.82 ng/mL) (Figure 2C). Thus, the cell culture supernatants were collected on day 5 and subjected to western blot analysis to confirm the release of Hutat2:Fc (Figure 2F).
Hutat2:Fc secretion was detected significantly earlier than GFP, which was apparent on day 9 and maximal on day 12, indicating that co-expression through the internal ribosome entry site (IRES) element was weaker than the expression of promoter-proximal genes.22
Effects of Hutat2:Fc on Tat-Induced Neurotoxicity, HIV-1 Challenge, and T Cell Homeostasis
Even though Hutat2:Fc was released extracellularly and accumulated to a detectable concentration in the culture medium, we wanted to ascertain whether it had biological activity for HAND therapy. Therefore, we subsequently determined the effects of Hutat2:Fc on Tat-induced neurotoxicity, HIV-1 replication, and T cell homeostasis in the presence of HIV-1 infection.
First, based on dot-immunobinding assay (DIBA) data, Hutat2:Fc secreted in the conditioned media specifically and sufficiently bound to Tat and acted as a functional antibody (Figure S6A). Cell Counting Kit-8 (CCK8) and apoptosis assays were subsequently performed to measure the viability and apoptotic status of HTB-11 cells after exposure to Tat in the presence or absence of conditioned media for 3 days. Despite the low levels of secreted Hutat2:Fc, the media collected from KI-Ms significantly increased the relative cell viability (p < 0.05) and reduced the percentages of 7-aminoactinomycin D (7-AAD+) and Annexin V+ cells (p < 0.05) (Figures S6B–S6D). Moreover, primary mouse cortical neurons were isolated and treated using the same approach as HTB-11 cells to more directly assess the protective effects of Hutat2:Fc. As shown in Figure 3A, Tat-treated mouse neurons exhibited an increase of cellular atrophy and apoptosis, as well as a shorter length of dendrite. However, treatment with the media of transduced monocytes partly reversed the conditions and resulted in a higher proportion of survival neurons than treated with Tat alone (p < 0.01) (Figure 3B).
Figure 3.

Protective Effects of Secreted Hutat2:Fc on HIV-1 Tat-Induced Neurotoxicity in Mouse Primary Cortical Neurons
(A) Representative images of mouse primary neurons exposed to HIV-Tat in the presence or absence of KI-M-Hutat2. Cells were stained with MAP-2 (red), TUNEL (green), and a nuclear stain (blue). Scale bar, 100 μm. (B) Comparison of the relative neuronal survival rates of the cells shown in (A) after treatment with each type of conditioned media (n = 3). The calculation method is described in the Materials and Methods. Dunnett’s t test was used for statistical comparisons; **p < 0.01. The results are presented as the means from independent experiments. The error bars denote the SEM. KI-293T-Hutat2, culture supernatant of KI-293T cells; KI-HeLa-Hutat2, culture supernatant of KI-HeLa cells; KI-M-Hutat2, culture supernatant of KI-Ms; KI-U937-Hutat2, culture supernatant of KI-U937 cells.
Second, we examined the ability of secreted Hutat2:Fc to restrict HIV-1 spread and replication. Both KI-Ms and their culture media (KI-M-Hutat2) were added to peripheral blood mononuclear cells (PBMCs) isolated from healthy donors after exposure to full-length infectious HIV-1Ba-L. The HIV-1 p24 levels were quantified for 8 days to determine the viral titer in the culture media. On day 8, p24 levels were dramatically decreased in KI-M and KI-M-Hutat2 groups (by 9- to 20-fold) compared with those in the negative control group (Figure 4A). At the same time, total HIV DNA copies per 106 cells were determined to reflect the size of HIV reservoirs. Consistent with the results of p24 levels, a reduction in the total HIV DNA levels was also observed in KI-M and KI-M-Hutat2 groups (p < 0.01) (Figure 4B). Notably, the levels of both p24 and total HIV-DNA in the KI-M group increased faster than that in the KI-M-Hutat2 group within the first 4 days. This result might be attributed to the relatively low levels of Hutat2:Fc secreted in the early period of time posttransfection (Figure 2C).
Figure 4.

Evaluation of the Suppressive Effects of Secreted Hutat2:Fc and KI-M on HIV-1 Replication in PBMCs after Exposure to HIV-1Ba-L
(A) Kinetics of HIV p24 levels, measured over a period of 8 days after HIV-1Ba-L infection. A significant reduction in the p24 levels was observed when the cells were treated with KI-M-Hutat2 and KI-M compared with normal media (p < 0.01) (n = 5). No difference was observed between the two groups. (B) Detection of the concentration of total HIV-DNA over a period of eight days after HIV-1Ba-L infection. As in (A), the HIV-DNA levels were decreased by treatment with KI-M-Hutat2 and KI-M (p < 0.01) (n = 5). One-way ANOVA was employed for the statistical analyses. The results are presented as the means from independent experiments. The error bars denote the SEM.
Third, to investigate the regulatory effects of secreted Hutat2:Fc on T cell stability in the presence of HIV-1Ba-L, the distributions of T cell subsets in six healthy donors were assessed. Overall, compared with the HIV-1Ba-L-infected group, the distributions of both CD4+ T cells and CD8+ T cells subsets in the KI-M-Hutat2 group were much closer to the normal control. For CD4+ T cells, treatment with KI-M-Hutat2 led to a lower percentage of effector memory (EM) CD4+ T cells and a higher percentage of naive CD4+ T cells than the merely HIV-1Ba-L-infected group (p < 0.05) (Figure 5A). But for CD8+ T cells, an increase in the proportion of naive CD8+ T cells and a reciprocal reduction of the proportion of terminally differentiated (TD) CD8+ T cells were observed (p < 0.05) (Figure 5B).
Figure 5.

Effects of Secreted Hutat2:Fc on the Distributions of T Cell Subsets and Exhaustion Status after Exposure to HIV-1Ba-L
(A) Representative FACS plots of the distributions of CD4+ and CD8+ T cell subsets after HIV-1Ba-L infection, with or without KI-M-Hutat2. Commercial anti-Tat antibodies were used as a positive control. (B) Comparison of CD4+ and CD8+ T distributions following treatment with or without each type of conditioned media (n = 6). (C) Expression of CD38 and HLA-DR in CD4+ and CD8+ T cells treated with or without each type of conditioned media (n = 6). (D) Expression of PD-1, PD-L1, and CTLA-4 in CD4+ and CD8+ T cells treated with or without each type of conditioned media (n = 6). Wilcoxon matched-pairs signed-ranks tests were used to statistically compare the data shown in (B)–(D); *p < 0.05. The results are presented as the medians from independent experiments.
Because HIV is characterized as inducing global T cell dysfunction and death, we further evaluated the effects of secreted Hutat2:Fc on T cell exhaustion and apoptosis after exposure to HIV-1Ba-L. The exhaustion status was determined by FACS to examine the frequency of the activation markers CD38 and human leukocyte antigen-antigen D related (HLA-DR), as well as the inhibitory markers, programmed cell death protein 1 (PD-1), programmed cell death-ligand 1 (PD-L1), and cytotoxic T-lymphocyte-associated protein 4 (CTLA-4), expressed on the surface of CD4+ and CD8+ T cells. As shown in Figures 5C and 5D, after exposure to HIVBa-L, PBMCs treated with Kl-M-Hutat2 group exerted a notable reduction in the percentage of CD4+ T cells expressing PD-1 and PD-L1, as well as CD8+ T cells expressing CD38, HLA-DR, PD-1, and CTLA-4 compared with the HIV-1Ba-L exposure alone group (p < 0.05). Moreover, the apoptotic status of PBMCs was also detected by FACS. Despite a different degree of loss in cell viability on 3 days after sustained exposure to HIV-1Ba-L, KI-M-Hutat2 supplementation slightly decreased the percentage of Annexin V+ cells, as compared with HIV-1Ba-L-treated alone (p < 0.05) (Figures S7A and S7B). Because apoptosis was partly suppressed, KI-M-Hutat2 slowed the reduction in the number of CD4+ T cells and facilitated the restoration of the balance between CD4+ T and CD8+ T cells (Figures S7C–S7E).
Based on the data shown above, secreted Hutat2:Fc partially protected neurons from Tat-induced neurotoxicity, limited HIV replication, and maintained T cell homeostasis, although not as effectively as the commercial anti-Tat antibody. These results revealed that Hutat2:Fc exhibits excellent biological activity and has the potential to exert therapeutic effects on HAND.
Comparison of the Transduction Efficiency and Potential Adverse Effects of Different Gene-Editing Strategies on Primary Monocyte Cells
Both the transduction efficiency and monocyte functionality, including migration, differentiation, and cytokine secretion, should be considered to ensure that Hutat2:Fc functions in the CNS. Therefore, we compared the efficiencies and potential adverse effects between two transfection methods, CRISPR/Cas9 and traditional lentiviral transduction, the latter of which has been used extensively to transfect hard-to-transduce monocytes and monocyte-derived macrophages (MDMs).
First, we initially compared the transduction efficiency among CRISPR/Cas9-mediated Hutat2:Fc KI-Ms, lentivirus-mediated transfected monocytes (LT-Ms), or MDMs (LT-MDMs). GFP+ monocytes (not LT-MDMs) were sorted by flow cytometry 9 days after transduction to eliminate interference from non-transfected cells. After sorting, the percentages of GFP+ cells were calculated to equal 96.42% ± 3.07% for KI-Ms, 95.36% ± 2.81% for LT-Ms, and 51.28% ± 1.42% for LT-MDMs (Figure 6A). A prominent decrease in the number of cells was observed in the KI-M group, and this effect can be attributed to the transduction method of electroporation, in which approximately 40% of cells are killed by the high-intensity discharge. The levels of Hutat2:Fc produced by these three transduced cell types were further measured by western blot analysis (Figures 6B and 6C). Although slightly higher than those of LT-MDM, the levels of Hutat2:Fc secreted by KI-Ms were approximately half of the levels produced by LT-M.
Second, the potential adverse effects of gene editing on monocytes were assessed. Fifteen monocyte-related genes, including molecular signal genes, apoptosis-induced genes, inflammatory genes, and chemokine receptor genes, were quantified by real-time PCR to evaluate the function of monocytes. Overall, the LT-MDM group was quite different from the other groups. Notably, the expression levels of P53 (2.65% ± 0.68%) and STAT-1 (3.19% ± 0.82%) were significantly increased in LT-M compared with those in normal monocytes, whereas no differences were observed between KI-Ms and non-transduced monocytes (Figure 6D). Next, the levels of IL-10, TNF-α, and IL-1β in culture media were subsequently measured by ELISAs to confirm the changes in cytokine secretion among the four groups. Consistent with the real-time PCR results, the release of both IL-10 and IL-1β was greatly increased while the release of TNF-α was declined in the LT-MDM group compared with other groups. No variations were observed among KI-Ms, LT-Ms, and non-transduced monocytes (Figures 6E–6G). A transendothelial migration (TEM) assay was subsequently employed to evaluate the migrating ability of monocytes. The percentage of migrated cells in the KI-M group (82.30% ± 2.31%) was similar to that in normal monocytes (86.49% ± 2.75%) (Figure 6H). However, the percentage of transmigrated cells was decreased to different extents in LT-M (71.46% ± 1.67%) and LT-MDM (43.64% ± 1.88%) groups (Figure 6H). Moreover, we noticed that, compared with MDM, the percentage of transmigrated cells in LT-MDM was much less (Figure 6H).
Thus, despite the inferior transduction efficiency, KI-Ms were superior to LT-Ms and LT-MDMs in terms of their conservation of normal monocyte functionality, which is critical for modified monocytes to function in the CNS.
Discussion
CRISPR/Cas9 technology has been widely employed to edit various eukaryotic cells because of its convenient manipulation and relatively high efficiency. Here, we constructed Hutat2:Fc KI-M via HDR induced by CRISPR/Cas9, and ∼10% of monocytes exhibited successful insertion into the AAVS1 locus without selection. Furthermore, secreted Hutat2:Fc neutralized Tat-induced neurotoxicity, suppressed HIV replication, and restored T cell homeostasis without inhibiting the migrating ability of monocytes. These data collectively provide support for the wide application of monocytes for the delivery of therapeutic genes to treat HAND.
Although gene editing has been effective in various cell lines, induced pluripotent stem cells (iPSCs), and primary T cells, little attention has been paid to modifying the genomes of monocytes because of their poor ability to proliferate and express genes in vitro.13, 14, 15, 23 In the present study, we introduced a successful multi-kilobase functional gene KI method in primary human monocytes using CRISPR/Cas9. We identified the best of six sgRNAs and assayed multiple concentrations to determine the experimental conditions with optimal editing efficiency. In addition, due to the use of electroporation with a maximum amount of donor plasmids, including extended homology arms, Hutat2:Fc was successfully inserted into monocytes, despite at a low proportion. The efficiency of this strategy was consistent with that observed in the previous studies.24, 25 Recently, there is a wonderful double-cut donor gene-editing strategy based on CRISPR/Cas9, which is most likely to yield a higher KI efficiency in hard-to-transduce cells, such as astrocytes, neurons, as well as mouse and monkey embryos both in vitro and in vivo.26, 27 More attempts will be performed to increase the efficiency of the new strategy in the future.
Additionally, the non-specific mutation, OT-3, was identified using T7E I assay (Figure S5). OT-3 is located on the Chromosome 13: +106612910. Neither long non-coding RNA (LncRNA) or protein had been found to be associated with this locus until now. Additionally, based on the comparison of the expression of 15 monocyte-related functional genes, secretion of 3 cytokines, as well as the migrating ability (Figures 6D–6H) between KI-Ms and normal monocytes, we considered that OT-3 did not appear to cause any adverse effects on primary monocytes.
As a class of lymphocytes, monocytes migrate through the bloodstream to tissues and then differentiate into resident macrophages. These characteristics endow these cells with the ability to deliver therapeutic genes to locations that are out of reach to drugs, such as the CNS, and to function persistently at disease sites.12 Based on the results of the present study, Hutat2:Fc KI-M exhibited excellent biological activity in vitro, although merely ∼10% cells contained an insertion. Notably, because Hutat2:Fc expression was enhanced by the exogenous CAG promoter, the selection of modified monocytes was avoided, which greatly increased the suitability of utilizing these cells for therapeutic applications, such as auto-transplantation.
Although the expression was clearly enhanced, Hutat2:Fc was still secreted at a relatively low level in the monocytes medium, with levels nearly 8- or 11-fold lower than those obtained with engineered HeLa or 293T cell lines (Figures S6B and S6C). However, this concentration was sufficient to protect neurons, restrict the virus, and balance the immune system, despite not being as effective as cell lines or commercial anti-Tat antibodies. Nevertheless, the low levels of Hutat2:Fc released from monocytes were considered beneficial as a therapeutic strategy to treat HAND in vivo. On the one hand, given the effective viral suppression achieved by cART, the residual Tat in the CNS is so little that these modified monocytes might be sufficient to neutralize them.28 On the other hand, the low levels of secreted Hutat2:Fc might minimize the disturbance to the CNS microenvironment, improving the safety of this strategy.
Traditional editing strategies, such as lentiviral-vector-mediated gene transfer, have also been widely utilized in monocytes and MDMs.29, 30 Compared with CRISPR/Cas9, transduction mediated by lentiviral vectors induced higher levels of Hutat2:Fc expression but inhibited the normal function of monocytes to varying extents. Several factors might contribute to this phenomenon. First, the genomic integration sites of HIV-based lentiviral vectors are random but are more likely to occur in actively transcribed chromatin regions.31 Interactions between exogenous DNA fragments and self-genomic sequences might affect normal gene transcription and expression in monocytes. Second, lentivirus-mediated integration of multiple gene copies at multiple loci might influence the stability of the whole genome. Thus, the single-copy KI induced by CRISPR/Cas9 at the AAVS1 locus presents an advantage over lentiviral strategies for maintenance of the normal function of monocytes.
The ability to edit specific DNA sequences in the AAVS1 locus of monocytes using the CRISPR/Cas9 system, as demonstrated in the present study, provides the possibility of delivering target genes to the CNS. However, owing to the lack of animal models of HAND, as well as the specificity of genomic sequences among each species, we did not examine whether the edited human monocytes were able to reach the injured sites of the brain tissue to alleviate HIV-Tat-induced neurotoxicity in an animal model.32, 33 Next step, we are going to generate Hutat2:Fc KI mouse monocytes in Rosa26 locus and detect in an HIV-Tat-induced brain inflammation mouse model. Looking forward, the present study on Hutat2:Fc editing in primary monocytes using CRISPR/Cas9 represents a potentially powerful strategy as an adjuvant to cART for the treatment of HAND.
Materials and Methods
Plasmid Preparation
N20NGG motifs in the AAVS1 locus were scanned, and candidate sgRNAs that fit the rules for U6 Pol III transcription and the PAM recognition domain of Streptococcus pyogenes Cas9 were identified.34 The top six sgRNA candidates were selected from the MIT CRISPR Design Tool (http://crispr.mit.edu). Using the same procedure, potential OT sites were also predicted. The sequences are listed in Table S1. Oligonucleotides were annealed and cloned into Bbs I-digested pX330-U6-Chimeric_BB-CBh-hSpCas9 (Addgene plasmid #42230). The resulting plasmids containing sgRNAs were further confirmed by Sanger sequencing.
The donor plasmid was generated by cloning the Hutat2:Fc and GFP genes from the pHR-Hutat2:Fc-IRES-GFP plasmid, which was kindly provided by Dr. Yuanan Lu,29 into the PTA2 vector. Additionally, ∼1.5-kb homology arms were ligated on both sides of the target fragments (Hutat2:Fc and GFP) using T4 ligase (Thermo Scientific).
Generation of Lentiviral Vectors
The pHR-Hutat2:Fc-IRES-GFP plasmid, which co-expresses the Hutat2:Fc fusion protein and GFP from the CMV promoter, was co-transfected with pCMV-ΔR8.2 and pCMV-VSV-G plasmids into 293T cells using Lipofectamine 2000 (Invitrogen). The vector concentrations were determined as previously described.35 Finally, high-titer HR-Hutat2 lentiviral vectors (∼5 × 108 IU/mL) were prepared for transduction.
Cell Culture
All cell lines were kindly provided by the Stem Cell Bank, Chinese Academy of Science. HeLa, 293T, and HTB-11 cell lines were maintained in DMEM (Corning Life Science) supplemented with 100 IU/mL penicillin (Sigma-Aldrich), 0.1 mg/mL streptomycin (Sigma-Aldrich), and 10% fetal bovine serum (FBS) (HyClone). U937 cells were maintained in RPMI 1640 (Corning Life Science) supplemented with 100 IU/mL penicillin, 0.1 mg/mL streptomycin, and 10% FBS. Human umbilical vein endothelial cells (HUVECs) were cultured in endothelial cell medium (ECM) (ScienCell). The culture media were replaced every 2–3 days, and the cells were passaged using EDTA solution containing 0.25% trypsin (Corning).
Primary Human Monocytes
Total PBMCs were obtained from healthy donors (serum-negative for HIV-1, hepatitis B, and hepatitis C). CD14+ cells were purified by negative selection using EasySep magnetic beads (STEMCELL Technologies) (Figure S3A). The isolated monocytes were maintained in RPMI 1640 supplemented with 100 IU/mL penicillin, 0.1 mg/mL streptomycin, and 10% FBS. Half of the culture media was replaced every 3–5 days.
Human MDMs
PBMCs were cultured in RPMI 1640 supplemented with 100 IU/mL penicillin, 0.1 mg/mL streptomycin, 10% FBS, and 20 ng/mL M-CSF (PeproTech). The culture medium was replaced every 3 days, and non-adherent cells were removed.36
Mouse Primary Neurons
Primary neurons were isolated from the cortices of early post-natal (P0) C57BL/6j mice using a previously described method.37 The cells were cultured in Neurobasal medium (GIBCO) supplemented with B27 (Invitrogen), L-glutamine (Sigma-Aldrich), 100 IU/mL penicillin, and 0.1 mg/mL streptomycin. Half of the culture medium was replaced every three days.
Cell Transduction
HeLa and 293T cells (1 × 105 cells) were transfected with different amounts of plasmids using Lipofectamine 2000 (Invitrogen), according to the manufacturer’s protocol. The efficiency of CRISPR-mediated knockout was analyzed 3 days after transfection with sgRNA1-6 plasmids. For CRISPR-mediated KI, the cells were co-transfected with sgRNA2 and donor plasmids for 3 days, and selected with 400 ng/mL puromycin for 9 days prior to the final analyses.
U937 cells and monocytes (5 × 105 cells) were transfected with plasmids by electroporation (Bio-Rad). in brief, the cells were suspended in 200 μL of electroporation buffer (Bio-Rad) and then electroporated (1,000 V, 50 ms, one pulse for U937 cells; 1200 V, 20 ms, three pulses for monocytes). The efficiency of CRISPR-mediated knockout was analyzed 9 days after electroporation. For CRISPR-mediated KI, the cells were co-electroporated with sgRNA2 and donor plasmids and cultured for 9 days, and GFP+ monocytes were then sorted by flow cytometry using a FACSAria II cell sorter (BD Biosciences).
MDMs (1 × 105 cells) were infected with HR-Hutat2 vectors at an MOI of 10 in the presence of 8 μg/mL polybrene for 4 hr. The transduction efficiency was evaluated 9 days after transfection.
In addition, 5 × 105 monocytes were infected with HR-Hutat2 vectors at an MOI of 10 in the presence of 8 μg/mL polybrene with rotation at 1,500 × g for 4 hr. GFP+ monocytes were then sorted by flow cytometry 9 days after transfection.
Genomic DNA Extraction, PCR, Purification, and Sequence Analysis
Genomic DNA was extracted from different groups of cells using a QIAamp DNA Blood Mini Kit (QIAGEN) according to the manufacturer’s recommended protocol. Genomic DNA was subjected to PCR (Takara) using primers listed in Table S1. The general reaction conditions were 95°C for 10 min followed by 30 cycles of 95°C for 30 s, annealing at 61°C for 30 s, and extension at 72°C for 30 s. After each reaction, 200 ng of the PCR products was purified using a QIAquick PCR Purification Kit (QIAGEN), subjected to T7E I assays, and then analyzed by agarose gel electrophoresis. The interest bands were extracted and cloned to pUC118 vector by Mighty cloning Reagent (Takara) and confirmed by Sanger Sequencing.
T7 Endonuclease I Assay
As described before,38 200 ng of the purified PCR products was denatured and re-annealed in 1× NEB Buffer 2 with or without T7E I (New England Biolabs). The reaction mixtures were then separated by 2% agarose gel electrophoresis. The knockout efficiency (KO%) was determined using the following formula: KO% = 100 × (1−[1−b]/[a+b])1/2, where a is the integrated intensity of the undigested PCR product and b is the combined integrated intensity of the cleavage products.
Universal CRISPR Activity Assay
Universal CRISPR activity assays were performed according to the manufacturer’s protocol. In brief, PCR amplicons of the genomic DNA templates encompassing the target sites of sgRNA1-6 were cloned into pUCA vectors (Luc) (Biocytogen). After 24 hr of co-transfection with sgRNA and its pUCA (Luc) into 293T cells, the luciferase activity was detected to reflect the ability of sgRNA to cut the target site.
Western Blot
For the western blot assays, 5 × 105 cells with or without transduction and their supernatants were harvested and resuspended in 1 × SDS buffer. The samples were separated by 10% SDS-PAGE, transferred to polyvinylidene difluoride membranes, and incubated with immunoglobulin G (IgG) (H+L) (Rockland) overnight at 4°C. After washing, the membranes were treated with horseradish peroxidase (HRP)-conjugated goat anti-rabbit IgG (Abcam) for 2 hr at room temperature (RT). Finally, the membranes were exposed to 3,3'-diaminobenzidine (DAB) substrate (Pierce) for detection. As a control, β-actin was detected with rabbit anti-β-actin antibodies (Rockland).
ELISA
Plates were coated with goat anti-human IgG Fc capture antibodies (Rockland) overnight at 4°C. After washing, diluted supernatant samples were added for 1 hr, and the plates were then incubated with goat anti-human IgG Fc biotin-conjugated detection antibodies (Abcam) for 1 hr. Finally, streptavidin-HRP and tetramethylbenzidine (R&D Systems) were added, and the plates were then incubated for 20 min. The optical density at 450 nm was read with a microplate reader (Bio-Rad).
The levels of IL-10, TNF-α, and IL-1β in the supernatants of monocytes, KI-Ms, LT-Ms, and LT-MDMs were all determined using ELISA Kits (R&D Systems).
Dot-Immunobinding Assay
For each DIBA, 200 ng of recombinant HIV-Tat protein (Abcam) was spotted onto a nitrocellulose membrane and incubated with conditioned media collected from KI-HeLa, KI-293T, KI-U937, or KI-M. The membrane was incubated with rabbit anti-human IgG (H+L) (Rockland) and goat anti-rabbit IgG HRP (Abcam) before exposure to DAB substrate. Anti-Tat antibodies (Abcam) served as a positive control.
Real-Time PCR
Total mRNA was extracted from KI cells using TriPure (Roche) according to the manufacturer’s instructions. cDNA was synthesized with a Transcriptor First-Strand cDNA Synthesis Kit (Roche) and then used for PCR using the following primers: Hutat2, 5′-ACATCTGTGGTTCTTCCTTCTCCT-3′/5′-TCACTCCATATCACTCCCAGCCACTC-3′; EGFP, 5′-GGTGAGCAAGGGCGAGGAG-3′/5′-GCCGGTGGTGCAGATGAACT-3′; and ACTB, 5′-AGGTGACACTATAGAATAGGCATCCTCACCCTGAAGTA-3′/5′-GTACGACTCACTATAGGGACAGAGGCGTACAGGGATAGC-3′. The PCR products were analyzed by 2.5% agarose gel electrophoresis. The primers that were used to profile the expression of 15 genes in monocytes have been described as previously29 except that ACTB was listed above. Three reference genes, ACTB, GK and Ezrin, were used for normalization.
Real-time PCR was performed on a LightCycler z480 instrument (Roche) using FastStart Universal SYBR Green Master Mix (Roche) according to the manufacturers’ instructions. Fold changes were determined using the arithmetic comparative method (2−ΔΔCt).
Fluorescent Immunocytochemistry
Cells were fixed with 4% paraformaldehyde and blocked with 1% BSA in PBS containing 0.1% Triton X-100 (Sigma-Aldrich). IgG Fc was stained by primary rabbit anti-human IgG Fc (Rockland) and secondary goat anti-rabbit IgG Cy3-conjugated antibodies (Abcam). For mouse neuronal staining, the cells were incubated with rabbit anti-MAP2 antibodies (Abcam) followed by the same secondary antibody. To detect neuronal apoptosis, terminal deoxynucleotidyl transferase-mediated dUTP nick end labeling (TUNEL) assays were performed using an In Situ Cell Death Detection Fluorescein Kit (Roche) according to the manufacturer’s instructions. Finally, all cells were incubated with DAPI to stain the nuclei. Images were captured using an Olympus DP70 immunofluorescence microscope and analyzed using ImageJ.
Primary Neuron Protection Assay
As previously described,29 mouse primary neurons were treated with 500 ng/mL HIV-Tat and conditioned media from KI-HeLa, KI-293T, KI-U937, or KI-M for 3 days. Neurons treated with normal medium were used as a normal control, cells treated with Tat alone were used as a negative control, and cells treated with Tat plus commercial anti-Tat antibodies (Abcam) were used as a positive control. The cells were subjected to fluorescence immunocytochemistry. The surviving neurons, which displayed intact cell bodies (red) and nuclei (blue), but not TUNEL labeling (green), were counted to determine the survival rate. The rate observed in the normal control was defined as 100%. The relative neuronal survival rate was evaluated for each treatment.
HIV-1 Challenge
HIV-1Ba-L was obtained from the NIH AIDS Reagent Program. PBMCs from six healthy donors were isolated, and after HIV-1Ba-L (final concentration of p24 was 8 ng/mL) was added, the cells were incubated at 37°C for 4 hr. Subsequently, the cells were washed three times, and KI-M-Hutat2, KI-M, or anti-Tat antibodies were added. Half of the volume of each culture’s supernatant was replaced every 3 days. Viral replication was evaluated by monitoring the p24 levels in the culture supernatants using an HIV-1 p24 Antigen ELISA 2.0 (Petrotec), according to the manufacturer’s instructions. The viral reservoirs were evaluated by quantifying the total HIV-DNA levels in the genomic DNA of each cell sample using an HIV-DNA Detection Kit (Supbio) as previously described.39
Cell Viability Assay
HTB-11 cells were treated with HIV-Tat (500 ng/mL) and conditioned media from KI-HeLa, KI-293T, KI-U937, or KI-Ms. PBMCs isolated from healthy donors and HIV-1Ba-L (final concentration of p24 was 8 ng/mL) were added, and the mixtures were incubated at 37°C for 4 hr. Subsequently, the cells were washed three times, and KI-M-Hutat2, KI-Ms, or anti-Tat antibodies were added. Normal medium was used as a negative control, whereas anti-Tat antibody was used as a positive control.
HTB-11 and PBMCs were harvested 3 days after treatment. At the time of harvest, the cells were stained using a PE Annexin V Apoptosis Detection Kit I (BD Biosciences) and then analyzed immediately on a FACSCalibur flow cytometer (BD Biosciences). HTB-11 cells were further analyzed using a CCK8 (MCE), according to the manufacturer’s instructions.
Flow Cytometry
The percentages of GFP+ HeLa cells, 293T cells, U937 cells, and monocytes were evaluated by suspending the cells in PBS and fixing them with 2% paraformaldehyde for 10 min prior to flow cytometry analysis using a FACSAria II cell sorter (BD Biosciences). Intact cells were gated using forward scatterplots versus side scatterplots, and further gating for KI cells was performed based on the presence of control samples expressing GFP using fluorescein isothiocyanate (FITC) versus side scatterplots. The number of GFP+ cells in control samples ranged from 0% to 1%.
At the time of harvest, PBMCs were stained with antibodies against the following proteins: CD45-PerCp (2D1), CD4-phycoerythrin (PE)/Cy7 (OKT4), CD8-allophycocyanin (APC)/Cy7 (SK1), CD45RA-APC (HI100), CCR7-FITC (G043H7), CD38-APC (HIT2), HLA-DR-PE (L243), PD-1-PE (EH12.2H7), PD-L1-APC (29E.2A3), and CTLA-4-FITC (A3.4H3.H12) (all from BioLegend). The lymphocyte gate was based on CD45. Among lymphocytes, T cells were sub-gated based on their expression of CD4 or CD8. Memory subsets of CD4+ and CD8+ T cells were defined by the expression of CCR7 and CD45RA, respectively. The activation status was determined based on CD38 and HLA-DR expression, whereas the inhibitory status was determined based on PD-1, PD-L1, and CTLA-4. The gates for activation markers and inhibitory markers were determined using isotype control antibodies.
TEM Assay
TEM assays were performed as described previously.40 In brief, three-dimensional collagen matrices containing 1.25 mg/mL Cultrex type I bovine collagen (R&D Systems), 35.7 mM NaOH, and 10% ECM were co-incubated for 4 hr to allow polymerization. HUVECs were seeded onto the gels at a density of 2 × 104 cells/mL and cultured for 48 hr until a confluent HUVEC monolayer was formed. Normal monocytes, KI-Ms, LT-Ms, and LT-MDMs were plated on the HUVECs at a density of 2 × 105 cells/mL, and the mixtures were incubated for 1.5 hr at 37°C and then washed three times to remove unattached cells. After 48 hr, the collagen gels were digested with collagenase D (Roche). The proportion of GFP+ cells was analyzed by flow cytometry. The proportion of migrated monocytes in the MDM population was calculated using the following formula: transmigrated cells (%) = GFP+ cells (%)/transduction efficiency. The transduction efficiency was determined by immunofluorescence staining (Figure 4A).
Statistics
Statistical analyses were performed using SPSS Version 16.0. The results from T cell exhaustion studies were treated as nonparametric data and assessed using the Wilcoxon signed-rank test. For independent data, Student’s t test was performed to determine the statistical significance. For multiple comparisons, one-way ANOVA was performed and confirmed using the Kruskal-Wallis test. Comparisons of each group with the control were performed using Dunnett’s t test. Two-tailed p values less than 0.05 were considered statistically significant.
Author Contributions
B.W., Wen Kang, and Y.S. designed the research; B.W. and J.Z. performed the research; B.W., Q.W., J.L., and C.W. analyzed the data; Wenzhen Kang, Z.L., Y.Z., Y.L., and B.D. contributed with the reagents/materials/analysis tools; B.W., Wen Kang, Y.L., and Y.S. wrote the paper. All authors read and approved the final manuscript.
Acknowledgments
This study was supported by grants from the National Natural Science Foundation of China (81501041) and the Major Program of National Natural Science Foundation of China (2017ZX10202203-008-003, 2017ZX10202101-003-007, 2017ZX10202101-004-005, and 2014ZX10001002). pHR-Hutat2:Fc-IRES-GFP plasmid was kindly provided by Dr. Yuanan Lu. HIV-1Ba-L was obtained from Dr. Suzanne Gartner, Dr. Mikulas Popovic, and Dr. Robert Gallo (Cat #510) through the NIH AIDS Reagent Program, Division of AIDS, NIAID, NIH.
Footnotes
Supplemental Information includes seven figures and one table and can be found with this article online at https://doi.org/10.1016/j.omtn.2018.01.012.
Contributor Information
Wen Kang, Email: kangwenkevin@gmail.com.
Yongtao Sun, Email: yongtaos@hotmail.com.
Supplemental Information
References
- 1.Dahabieh M.S., Battivelli E., Verdin E. Understanding HIV latency: the road to an HIV cure. Annu. Rev. Med. 2015;66:407–421. doi: 10.1146/annurev-med-092112-152941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Heaton R.K., Clifford D.B., Franklin D.R., Jr., Woods S.P., Ake C., Vaida F., Ellis R.J., Letendre S.L., Marcotte T.D., Atkinson J.H., CHARTER Group HIV-associated neurocognitive disorders persist in the era of potent antiretroviral therapy: CHARTER Study. Neurology. 2010;75:2087–2096. doi: 10.1212/WNL.0b013e318200d727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rahn K.A., Slusher B.S., Kaplin A.I. Glutamate in CNS neurodegeneration and cognition and its regulation by GCPII inhibition. Curr. Med. Chem. 2012;19:1335–1345. doi: 10.2174/092986712799462649. [DOI] [PubMed] [Google Scholar]
- 4.Huang W., Chen L., Zhang B., Park M., Toborek M. PPAR agonist-mediated protection against HIV Tat-induced cerebrovascular toxicity is enhanced in MMP-9-deficient mice. J. Cereb. Blood Flow Metab. 2014;34:646–653. doi: 10.1038/jcbfm.2013.240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Selby M.J., Bain E.S., Luciw P.A., Peterlin B.M. Structure, sequence, and position of the stem-loop in tar determine transcriptional elongation by tat through the HIV-1 long terminal repeat. Genes Dev. 1989;3:547–558. doi: 10.1101/gad.3.4.547. [DOI] [PubMed] [Google Scholar]
- 6.Mhashilkar A.M., Bagley J., Chen S.Y., Szilvay A.M., Helland D.G., Marasco W.A. Inhibition of HIV-1 Tat-mediated LTR transactivation and HIV-1 infection by anti-Tat single chain intrabodies. EMBO J. 1995;14:1542–1551. doi: 10.1002/j.1460-2075.1995.tb07140.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Poznansky M.C., Foxall R., Mhashilkar A., Coker R., Jones S., Ramstedt U., Marasco W. Inhibition of human immunodeficiency virus replication and growth advantage of CD4+ T cells from HIV-infected individuals that express intracellular antibodies against HIV-1 gp120 or Tat. Hum. Gene Ther. 1998;9:487–496. doi: 10.1089/hum.1998.9.4-487. [DOI] [PubMed] [Google Scholar]
- 8.Morgan R.A., Walker R., Carter C.S., Natarajan V., Tavel J.A., Bechtel C., Herpin B., Muul L., Zheng Z., Jagannatha S. Preferential survival of CD4+ T lymphocytes engineered with anti-human immunodeficiency virus (HIV) genes in HIV-infected individuals. Hum. Gene Ther. 2005;16:1065–1074. doi: 10.1089/hum.2005.16.1065. [DOI] [PubMed] [Google Scholar]
- 9.Marasco W.A., LaVecchio J., Winkler A. Human anti-HIV-1 tat sFv intrabodies for gene therapy of advanced HIV-1-infection and AIDS. J. Immunol. Methods. 1999;231:223–238. doi: 10.1016/s0022-1759(99)00159-3. [DOI] [PubMed] [Google Scholar]
- 10.Braun S.E., Taube R., Zhu Q., Wong F.E., Murakami A., Kamau E., Dwyer M., Qiu G., Daigle J., Carville A. In vivo selection of CD4(+) T cells transduced with a gamma-retroviral vector expressing a single-chain intrabody targeting HIV-1 tat. Hum. Gene Ther. 2012;23:917–931. doi: 10.1089/hum.2011.184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Garcia-Bonilla L., Faraco G., Moore J., Murphy M., Racchumi G., Srinivasan J., Brea D., Iadecola C., Anrather J. Spatio-temporal profile, phenotypic diversity, and fate of recruited monocytes into the post-ischemic brain. J. Neuroinflammation. 2016;13:285. doi: 10.1186/s12974-016-0750-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tong H.I., Kang W., Davy P.M., Shi Y., Sun S., Allsopp R.C., Lu Y. Monocyte trafficking, engraftment, and delivery of nanoparticles and an exogenous gene into the acutely inflamed brain tissue—evaluations on monocyte-based delivery system for the central nervous system. PLoS ONE. 2016;11:e0154022. doi: 10.1371/journal.pone.0154022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sharma S., Patnaik S.K., Taggart R.T., Kannisto E.D., Enriquez S.M., Gollnick P., Baysal B.E. APOBEC3A cytidine deaminase induces RNA editing in monocytes and macrophages. Nat. Commun. 2015;6:6881. doi: 10.1038/ncomms7881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jamburuthugoda V.K., Chugh P., Kim B. Modification of human immunodeficiency virus type 1 reverse transcriptase to target cells with elevated cellular dNTP concentrations. J. Biol. Chem. 2006;281:13388–13395. doi: 10.1074/jbc.M600291200. [DOI] [PubMed] [Google Scholar]
- 15.Hsu P.D., Lander E.S., Zhang F. Development and applications of CRISPR-Cas9 for genome engineering. Cell. 2014;157:1262–1278. doi: 10.1016/j.cell.2014.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dai W.J., Zhu L.Y., Yan Z.Y., Xu Y., Wang Q.L., Lu X.J. CRISPR-Cas9 for in vivo gene therapy: promise and hurdles. Mol. Ther. Nucleic Acids. 2016;5:e349. doi: 10.1038/mtna.2016.58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kang H., Minder P., Park M.A., Mesquitta W.T., Torbett B.E., Slukvin I.I. CCR5 disruption in induced pluripotent stem cells using CRISPR/Cas9 provides selective resistance of immune cells to CCR5-tropic HIV-1 virus. Mol. Ther. Nucleic Acids. 2015;4:e268. doi: 10.1038/mtna.2015.42. [DOI] [PubMed] [Google Scholar]
- 18.DeKelver R.C., Choi V.M., Moehle E.A., Paschon D.E., Hockemeyer D., Meijsing S.H., Sancak Y., Cui X., Steine E.J., Miller J.C. Functional genomics, proteomics, and regulatory DNA analysis in isogenic settings using zinc finger nuclease-driven transgenesis into a safe harbor locus in the human genome. Genome Res. 2010;20:1133–1142. doi: 10.1101/gr.106773.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.He X., Tan C., Wang F., Wang Y., Zhou R., Cui D., You W., Zhao H., Ren J., Feng B. Knock-in of large reporter genes in human cells via CRISPR/Cas9-induced homology-dependent and independent DNA repair. Nucleic Acids Res. 2016;44:e85. doi: 10.1093/nar/gkw064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Leong-Poi H., Kuliszewski M.A., Lekas M., Sibbald M., Teichert-Kuliszewska K., Klibanov A.L., Stewart D.J., Lindner J.R. Therapeutic arteriogenesis by ultrasound-mediated VEGF165 plasmid gene delivery to chronically ischemic skeletal muscle. Circ. Res. 2007;101:295–303. doi: 10.1161/CIRCRESAHA.107.148676. [DOI] [PubMed] [Google Scholar]
- 21.Kleinstiver B.P., Pattanayak V., Prew M.S., Tsai S.Q., Nguyen N.T., Zheng Z., Joung J.K. High-fidelity CRISPR-Cas9 nucleases with no detectable genome-wide off-target effects. Nature. 2016;529:490–495. doi: 10.1038/nature16526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mizuguchi H., Xu Z., Ishii-Watabe A., Uchida E., Hayakawa T. IRES-dependent second gene expression is significantly lower than cap-dependent first gene expression in a bicistronic vector. Mol. Ther. 2000;1:376–382. doi: 10.1006/mthe.2000.0050. [DOI] [PubMed] [Google Scholar]
- 23.Zhang Z., Zhang Y., Gao F., Han S., Cheah K.S., Tse H.F., Lian Q. CRISPR/Cas9 genome-editing system in human stem cells: current status and future prospects. Mol. Ther. Nucleic Acids. 2017;9:230–241. doi: 10.1016/j.omtn.2017.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Milano F., van Baal J.W., Rygiel A.M., Bergman J.J., Van Deventer S.J., Kapsenberg M.L., Peppelenbosch M.P., Krishnadath K.K. An improved protocol for generation of immuno-potent dendritic cells through direct electroporation of CD14+ monocytes. J. Immunol. Methods. 2007;321:94–106. doi: 10.1016/j.jim.2007.01.004. [DOI] [PubMed] [Google Scholar]
- 25.Byrne S.M., Ortiz L., Mali P., Aach J., Church G.M. Multi-kilobase homozygous targeted gene replacement in human induced pluripotent stem cells. Nucleic Acids Res. 2015;43:e21. doi: 10.1093/nar/gku1246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhang J.P., Li X.L., Li G.H., Chen W., Arakaki C., Botimer G.D., Baylink D., Zhang L., Wen W., Fu Y.W. Efficient precise knockin with a double cut HDR donor after CRISPR/Cas9-mediated double-stranded DNA cleavage. Genome Biol. 2017;18:35. doi: 10.1186/s13059-017-1164-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yao X., Wang X., Hu X., Liu Z., Liu J., Zhou H., Shen X., Wei Y., Huang Z., Ying W. Homology-mediated end joining-based targeted integration using CRISPR/Cas9. Cell Res. 2017;27:801–814. doi: 10.1038/cr.2017.76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Clifford D.B., Ances B.M. HIV-associated neurocognitive disorder. Lancet Infect. Dis. 2013;13:976–986. doi: 10.1016/S1473-3099(13)70269-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kang W., Marasco W.A., Tong H.I., Byron M.M., Wu C., Shi Y., Sun S., Sun Y., Lu Y. Anti-tat Hutat2:Fc mediated protection against tat-induced neurotoxicity and HIV-1 replication in human monocyte-derived macrophages. J. Neuroinflammation. 2014;11:195. doi: 10.1186/s12974-014-0195-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Dullaers M., Breckpot K., Van Meirvenne S., Bonehill A., Tuyaerts S., Michiels A., Straetman L., Heirman C., De Greef C., Van Der Bruggen P., Thielemans K. Side-by-side comparison of lentivirally transduced and mRNA-electroporated dendritic cells: implications for cancer immunotherapy protocols. Mol. Ther. 2004;10:768–779. doi: 10.1016/j.ymthe.2004.07.017. [DOI] [PubMed] [Google Scholar]
- 31.Marini B., Kertesz-Farkas A., Ali H., Lucic B., Lisek K., Manganaro L., Pongor S., Luzzati R., Recchia A., Mavilio F. Nuclear architecture dictates HIV-1 integration site selection. Nature. 2015;521:227–231. doi: 10.1038/nature14226. [DOI] [PubMed] [Google Scholar]
- 32.Saylor D., Dickens A.M., Sacktor N., Haughey N., Slusher B., Pletnikov M., Mankowski J.L., Brown A., Volsky D.J., McArthur J.C. HIV-associated neurocognitive disorder—pathogenesis and prospects for treatment. Nat. Rev. Neurol. 2016;12:234–248. doi: 10.1038/nrneurol.2016.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Li W., Li G., Steiner J., Nath A. Role of Tat protein in HIV neuropathogenesis. Neurotox. Res. 2009;16:205–220. doi: 10.1007/s12640-009-9047-8. [DOI] [PubMed] [Google Scholar]
- 34.Ran F.A., Hsu P.D., Wright J., Agarwala V., Scott D.A., Zhang F. Genome engineering using the CRISPR-Cas9 system. Nat. Protoc. 2013;8:2281–2308. doi: 10.1038/nprot.2013.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wu C., Lu Y. High-titre retroviral vector system for efficient gene delivery into human and mouse cells of haematopoietic and lymphocytic lineages. J. Gen. Virol. 2010;91:1909–1918. doi: 10.1099/vir.0.020255-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Jobe O., Trinh H.V., Kim J., Alsalmi W., Tovanabutra S., Ehrenberg P.K., Peachman K.K., Gao G., Thomas R., Kim J.H. Effect of cytokines on Siglec-1 and HIV-1 entry in monocyte-derived macrophages: the importance of HIV-1 envelope V1V2 region. J. Leukoc. Biol. 2016;99:1089–1106. doi: 10.1189/jlb.2A0815-361R. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Beaudoin G.M., 3rd, Lee S.H., Singh D., Yuan Y., Ng Y.G., Reichardt L.F., Arikkath J. Culturing pyramidal neurons from the early postnatal mouse hippocampus and cortex. Nat. Protoc. 2012;7:1741–1754. doi: 10.1038/nprot.2012.099. [DOI] [PubMed] [Google Scholar]
- 38.Shen B., Zhang J., Wu H., Wang J., Ma K., Li Z., Zhang X., Zhang P., Huang X. Generation of gene-modified mice via Cas9/RNA-mediated gene targeting. Cell Res. 2013;23:720–723. doi: 10.1038/cr.2013.46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Jones R.B., O’Connor R., Mueller S., Foley M., Szeto G.L., Karel D., Lichterfeld M., Kovacs C., Ostrowski M.A., Trocha A. Histone deacetylase inhibitors impair the elimination of HIV-infected cells by cytotoxic T-lymphocytes. PLoS Pathog. 2014;10:e1004287. doi: 10.1371/journal.ppat.1004287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Westhorpe C.L., Zhou J., Webster N.L., Kalionis B., Lewin S.R., Jaworowski A., Muller W.A., Crowe S.M. Effects of HIV-1 infection in vitro on transendothelial migration by monocytes and monocyte-derived macrophages. J. Leukoc. Biol. 2009;85:1027–1035. doi: 10.1189/jlb.0808501. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.