

Abstract
Endometrial cancer is one of the most common cancers of the female reproductive system. CHD4, a core subunit of the nucleosome remodeling and deacetylation (NuRD) complex, is frequently mutated in these patients. However the role it plays in promoting endometrial tumorigenesis is poorly understood. Here, we use genetic engineering approaches to modulate CHD4 expression levels and functionally evaluate hot spot mutations R975H and R1162W. We find that CHD4 depletion induces up-regulation of the cancer stem cell (CSC) marker CD133. This CSC phenotype was verified functionally by invasion ability, spheroid formation, and tumorigenicity in vivo. While cells expressing mutated CHD4 did not display impaired CHD4 DNA recruitment or NuRD complex formation, the mutations did reduce the stability of CHD4 protein to phenocopy CHD4 depletion. Consistently, patients with mutant CHD4 showed overexpression of CD133. Network analysis indicated activation of the TGFβ signaling pathway may drive this CSC phenotype, and chemical blockade of TGFβ abrogated the ability CHD4 knockdown cells to form spheroids. Taken together, these results indicate that mutations in CHD4 can promote endometrial tumorigenesis by increasing CSC character through TGFβ signaling pathway.
Keywords: CHD4, endometrial cancer, TGFβ, CD133, cancer stem cell
Introduction
Uterine (endometrial) cancer is one of the most common cancers of the female reproductive system among American women. It is estimated that about 61,380 new cases and 10,920 death of this disease are projected to occur in 2018 in the United States. Recently, the mapping of the genomic landscape of endometrial cancer showed that the various mutations of genes are associated with the progression of endometrial cancer. Thus, it is critical to identify key gene aberrations to develop the novel and effective methods for endometrial cancer therapy. So far the mutations of TP53 (71%), PIK3CA (31%) and PPP2R1A (25%) have been well reported and established roles in the pathogenesis of endometrial. Other genes such as CHD4, SPOP, FBXW7, ABCC9, CYP4X1 and MAP3K4 also have a high mutations without well-understanding [1]. In present study, we focused on Chromodomain-helicase-DNA-binding protein 4 (CHD4) and its two mutations with high frequency, to explore if they are critical and uncover their molecular mechanism in the endometrial cancer.
CHD4 belongs to the SNF2/RAD54 helicase family, and it is high conserved in animals and plants [2]. This protein consists of two PHD figures, two chromodomains, an ATPase domain, and a C-terminal bridge [3]. PHD figures are shown to interact with HDAC1 to modify histone tails [4,5]. Chromodomains have the binding activity of DNA and nucleosome [6]. ATPase domains are involved in ATP hydrolysis, providing energy for binding and mobilizing nucleosomes such as Histone 3 along DNA [7,8].
CHD4 is considered as one of the core subunits of nucleosome remodeling and deacetylation (NuRD) complex, composed of HDAC1/2 (histone deacetylases), CHD3/4 (chromodmain-helicase-DNA binding proteins), MBD2/3 (methyl-CpG-binding domain protein), MTA1/2/3 (Metastasis-associated proteins), Rbbp4/7 (Histone-binding proteins), and Gata2a/b (Nuclear zinc-finger protein) [3]. NuRD complex has been viewed as an ATP-dependent chromatin remodeling complexes according to characteristics of its components. By the use of energy releasing from ATP, this chromatin remodeler can restructure nucleosomes and affect chromatin accessibility, therefore it is an important player for many DNA dependent biological activities [9]. This complex was firstly identified to suppress gene transcription by binding to gene promoter with other transcriptional factors, and recently it was also found to protect genome integrity by controlling cell cycle and DNA repair [10].
CHD4 as a component of NuRD complex has been demonstrated to control the gene expression in Embryonic stem (ES) cells and to balance the self-renewal and differentiation of ES cells [3,10]. CHD4 activity is also closely associated with cancer disease. Loss of CHD4 can be found in gastric and colorectal cancer [11]. It is known that CHD4 can maintain genome stability by controlling HR repair, and its deficiency in cancer cells impairs the recruitment of DNA repair proteins such as BRIT1 at early steps of HR repair. Therefore CHD4-depleted cancer cells are more sensitive to poly(ADP-ribose) polymerase inhibitor treatment [12]. A high frequency of CHD4 mutation has been detected in endometrial cancer (17% in serous tumors, 7% in endometrioid, 4% in clear-cells, and 11% in mixed-histology tumors). And it is estimated that most of mutations are missense mutations and affect highly conserved residues clustered in ATP domain, PHD fingers, and C-terminal Bridge [1,13,14].
In our study, we confirmed that there is a high frequency mutations of CHD4 in endometrial cancer using bioinformatics tools, and two point mutations of CHD4 (R975H and R1162W) were further chosen because their high frequency of occurrence and potential damage to CHD4 function. We demonstrated for the first time that TGF-beta/CD133 pathway activation was associated with the knockdown or the two mutations of CHD4 in the endometrial cancer cells. Our results provide new insights into the molecular mechanism by which CHD4 mutants can contribute to endometrial cancer progression.
Methods
Cell culture and transfection
293T cell line and SKU-T2 cell line were cultured in Dulbecco’s Modified Eagle Medium (CORNING, Life Sciences, Tewksbury, MA, USA) with 10% fetal bovine serum (GIBCO, Life Technologies, Grand Island, NY, USA). EFE184 cell line was cultured in RPMI 1640 Medium with 10% fetal bovine serum. 293T cells cultured into 60 mm cell culture dishes were transfected with CHD4R975H/CHD4R1162W/CHD4WT-pcDNA3.1 plasmids using Lipofectamine 2000 (Life Techologies, Grand Island, NY, USA).
Lentiviral production and generation of CHD4 variants
293T cells were used to produce lentiviral particles containing cDNA for CHD4R975H, CHD4R1162W, CHD4WT, or shRNA against CHD4. EFE184 and SKU-T2 cells were cultured with viral particles containing shRNA against CHD4 to generate CHD4 knockdown cell lines. And then the SKU-T2 cells with stable knockdown of CHD4 were infected with viral particles containing CHD4R975H, CHD4R1162W, or CHD4WT to generate variants with the expression of them.
Protein degradation assay
Protein degradation assays were performed as described previously (6). Briefly, 293T cells were transfected with CHD4R975H, CHD4R1162W or CHD4WT. Cycloheximide (50 ug/ml) was added to the medium after transfection for 48 hours, and the cells were harvested at different time points. The cell lysates have been collected for Western blotting with anit-CHD4 antibody.
In vitro invasion assay
SKU-T2 cells and EFE 184 cells in the 6-well plates were starved overnight with serum-free medium. A total of 5×104 cells with in 300 ul of serum-free medium were placed in the upper compartment of a transwell chamber (Corning, New York, USA; 24-well insert, pore size: 8 um) with pre-coated matrix gel (200-300 ug/ml). The lower chamber was filled with 750 ul medium with 10% FBS. These transwell chambers were incubated overnight for the cell invasion. In the next day cells on the upper surface of the membrane were removed, and those on the lower surface were fixed and stained with 0.1% crystal violet. Stained cells were counted with image J software.
Sphere formation assay
For non-adherent sphere culture, SKUT-T2 cells (7500 cells) or EFE 184 cells (20000 cells) were plated onto ultra-low attachment 6-well plates with medium containing 1xB27, 20 ng/ml EGF, and 10 ng/ml FGF. After 2 weeks, the spheroid culture from every well was harvested for quantification. After the pelleting spheroids, pellets were suspended with 500 ul medium, and 50 ul of the suspension was transferred to a well of 96-well plates for cell sphere counting (diameter > 20 um) under microscope. In some experiments, 10 um TGFβ receptor I (TβRI) inhibitor (LY2157299) or equivolume DMSO vehicle control was added into medium.
Gene expression microarray
Microarray analysis was conducted as described previously [15]. Specifically, total RNA was extracted from the cells 48 hours after plating in presence or absence of doxycycline using a QIAGEN RNeasy RNA isolation kit. Complementary RNA was generated using a TotalPrep RNA amplification kit and loaded onto a Human HT-12 v4 Expression BeadChip for analysis using the manufacturer’s procedure. Patient gene expression data was acquired through GDC (https://portal.gdc.cancer.gov/), and CHD4 mutation information was downloaded from cBioPortal (http://www.cbioportal.org/).
Flow cytometry
Cells (106 cells/mL) were re-suspend in cold FACS staining buffer (PBS, 10% FBS, 10 mM EDTA). Cells were stained on ice with PE Mouse Anti-Human CD133 (clone W6B3C1, BD Biosciences) or IgG control for 60 minutes, washed three times with FACS buffer, and then analyzed by flow cytometry. CD133 positivity was determined relative to IgG-stained control.
Western blotting and immunoprecipitation
Whole-cell lysates were separated by SDS-PAGE, and proteins were blotted onto nitrocellulose membranes. The membranes were incubated with primary antibody at 4°C overnight followed by incubation with a secondary antibody for 1 hour at room temperature. Beta-actin was used as a loading control. Signals were visualized using the West-Q Pico ECL Solution (GenDepot, Katy, TX, USA) by exposure to film. Primary antibodies used in this study for Western, immunoprecipitation, and FACS are as follows: anti-CHD4 (Cell Signaling), anti-TGFB1 (Cell Signaling), anti-CD133 (Miltenyi Biotec), anti-HDAC1/2 (Cell Signaling), anti-MTA1 (Cell Signaling), anti-RBAP46 (Cell Signaling), and anti-MBD (Cell Signaling). As to immunoprecipitation, whole-cell lysates were collected by lysis buffer (20 mM Tris-HCl (pH 8.0), 100 mM NaCl, 1 mM EDTA, and 0.5% Nonidet P-40) and then incubated with ANTI-FLAG M2 affinity gel overnight at 4°C for binding. In the next day, after washing proteins were eluted from gel by 3X FLAG peptide and transferred to a new tube. SDS loading buffer was loaded into the tube, and samples were boiled for Western blotting.
Statistical analysis
The cell number in the invasion assay and the sphere number in the sphere formation assay were analyzed by t-test. Data were presented as means ± SD of three independent experiments. P < 0.05 was considered statistically significant.
Results
Prediction of CHD4 mutation from endometrial cancer
Through analyzing gene sequencing data from The Cancer Genome Atlas (TCGA), we have found a high somatic mutation rate of CHD4 reaching 16% in endometrial cancer, which is consistent with the previous publications, in which the whole-exome sequencing was used to screen 53 tumor samples. Notably, this high CHD4 mutation rate was not seen in other tumor types (Figure 1A). In the further screening from TCGA, two point mutations of CHD4 (CHD4R975H and CHD4R1162W) were found to recur four times and twice respectively from 24 samples (Figure 1B). Both of them are missense mutations and localized in the ATPase domain (Figure 1C). To predict the functional effect of the two mutations, two widely used functional annotation tools were employed for prediction. The analysis from polymorphism phenotyping-2 (polyphen-2) showed R975H with the score of 1.000 and R1162W with score of 0.998 (Figure 1D). The analysis from sorting intolerant from tolerant (SIFT) showed that substitution at pos 975 from R to H is with a score of 0.01 and substitution at position 1162 from R to W is with a score of 0.00. These predictions suggests that these two mutations may potentially disrupt the function of CHD4 protein.
Figure 1.
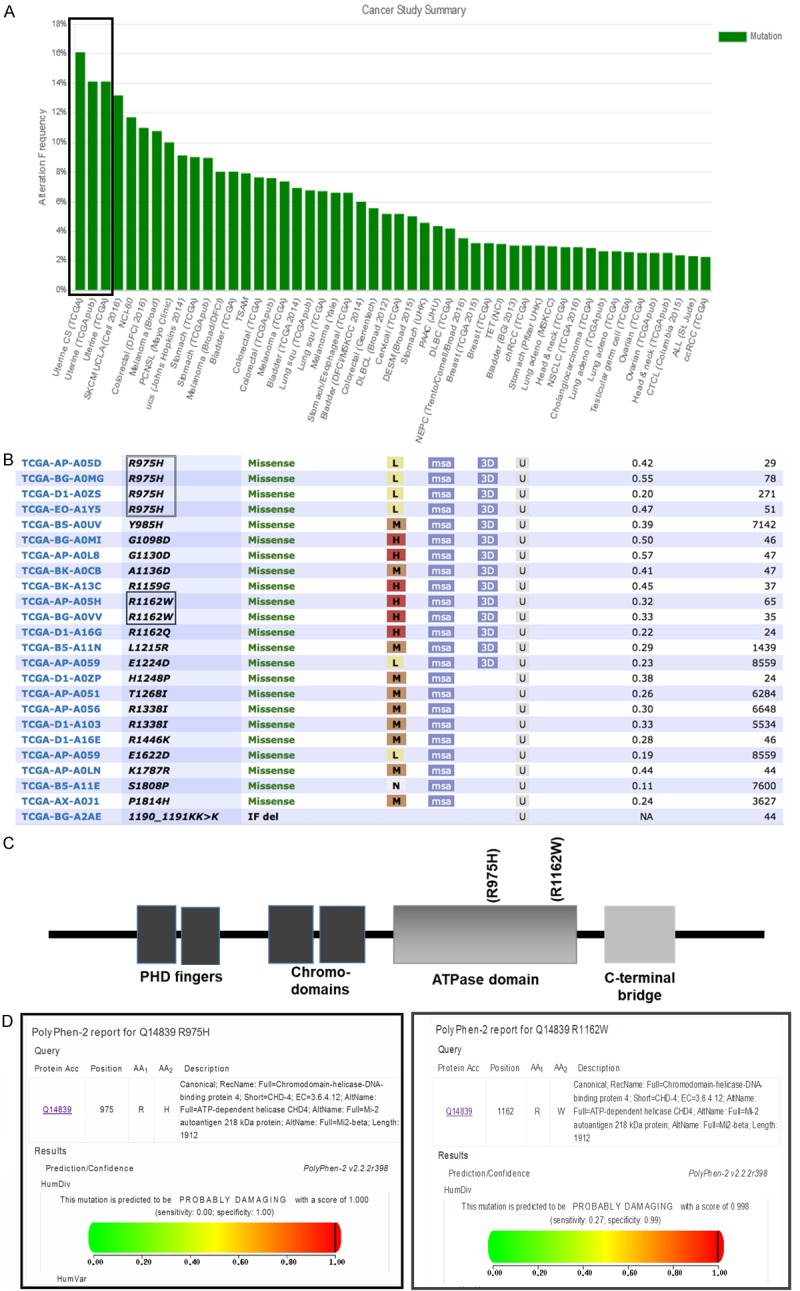
High frequency of CHD4 mutation in endometrial cancer. A. The alternation frequency of CHD4 in different type of cancers from TCGA samples. The black rectangular box displays a high somatic mutation rate (about 16%) of CHD4 in uterine cancer. B. CHD4R975H and CHD4R1162W are screened with a high occurrence in endometrial cancer patients from TCGA. C. The protein structure of CHD4. The two mutations of CHD4 are localized in the ATPase domain. D. Polymorphism phenotyping-2 (polyphen-2) predict that CHD4R975H and CHD4R1162W will affect the function of CHD4 seriously. In polyphen-2 reporter, the scaled score range between 0 and 1, in which scores closer to 1 indicate that amino acid substitution is damaging, and scores closer to 0 indicate that it is neutral.
CHD4R975H and CHD4R1162W accelerated the protein degradation
It is generally known that a mutation can be a loss of function mutation resulting in reduced or abolished protein function, or it is can be a gain of function mutation getting a new or enhanced activity of the protein. CHD4 is the key component of NuRD complex which associates with chromatin for chromatin remodeling. We first tested if CHD4R975H and CHD4R1162W reduced their binding affinity to other components in the NuRD complex and/or their binding affinity to chromatin. As shown in Figure 2, we found, by immunoprecipitation, that WT and mutant CHD4 could pull down equal amount of other compoents in the NuRD complex including MTA1, HDAC1, HDAC2, MBD, and RBAP46 (Figure 2A), and no difference of chromation association was found between WT or mutant CHD4 (Figure 2B). These data indicated that CHD4R975H or CHD4R1162W did not affect NuRD complex formation and CHD4 chromatin binding activity. Interetingly, we found that both CHD4R975H and CHD4R1162W displayed reduced protein half-life, suggesting these two point mutations might distablized the CHD4 protein, and consequencely, diminished the function of CHD4 in cells (Figure 2C).
Figure 2.

The effect of CHD4R975H and CHD4R1162W in 293T cells. (A) CHD4R975H and CHD4R1162W do not affect the interaction with other subunits in the NuRD complex. Plasmids with 3*flag containing CHD4R975H, CHD4R1162W and CHD4WT were transfected into 293T cells. The same amount of proteins, including MTA1, HDAC1/2, RBAP46, and MBD, was pulled down with CHD4 together by CHD4 antibody. (B) The two CHD4 mutants do not affect the binding activity of CHD4. The same amount of CHD4 was found for binding in the chromatin fractions. (C) CHD4R975H and CHD4R1162W proteins reduce CHD4 stability. Transfected cells were cultured with 50 ug/ml cycloheximide (CHX) for different incubation time. CHD4 protein intensity (Western blotting result) was quantified by image J software (C). And representative Western blotting results of CHD4R975H, CHD4R1162W and CHD4WT after the CHX treatment in every 4 hours were also shown in (D).
Gene expression profile of EFE184 endometrial cells with shCHD4
To better understand how CHD4 functions in endometrial cancer, we analyzed genes differentially expressed between EFE184 control cells and CHD4 knockdown cells by microarray. We found 37 genes up-regulated 2-fold or greater in shCHD4 cells, and 18 genes were found down-regulated. Based on unsupervised hierarchical clustering analysis, we found a grouping of co-expressed genes expression (MMP9, PROM1, PROL1, LCP1, ID2, CXADR, C5RF13, COL8A1, COL5A1, COL11A1, and TGFBI). Among them, PROM1, which is also known as the cancer stem cell (CSC) marker CD133, was of great interest (Figure 3A). In our microarray analysis, CD133 is upregulated 2.5 fold. We verified this result using flow cytometry, showing that while only ~30% shCTRL cells were positive for CD133 nearly the entire population (99.5%) of the shCHD4 cells were positive for CD133 (Figure 3B). To verify this phenoptype was relevant in patients, we analyzed TCGA patients with mutations in CHD4 and found they showed 2.15 fold up-regulation of CD133 compared to patients with wild type CHD4 (Figure 3C).
Figure 3.
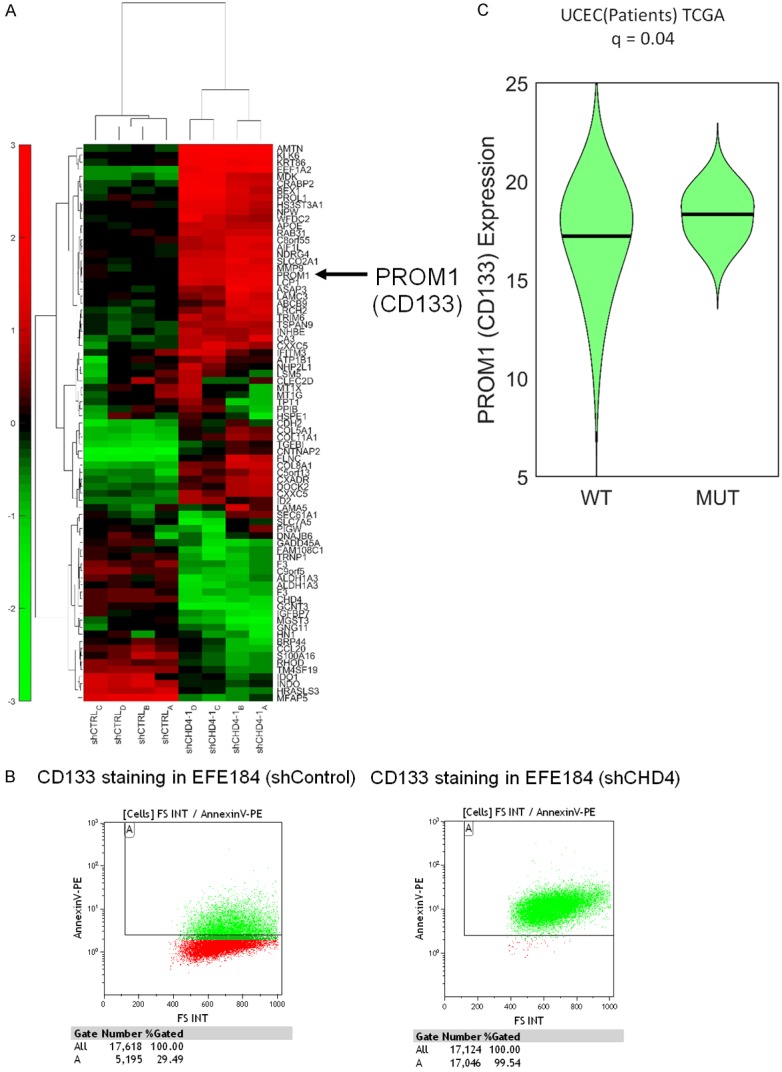
Gene expression profiling. A. Hierarchical Combined Tree in EFE184 cells with knockdown of CHD4. B. EFE184 cells with knockdown of CHD4 showed enhanced CD133 staining by Flow cytometry (Green: CD133 positive cells. Red: CD133 negative cells). C. Analysis of RNAseq expression data from TCGA patients with matched DNA sequencing shows patients with CHD4 mutations overexpress PROM1/CD133.
Knockdown of CHD4, and its mutations can contribute the characteristics of endometrial cancer stem cell
CD133 positive cell population is thought to be a cancer stem cell (CSC) population with the self-renewal which can drive the tumorigenesis [16], so we hypothesized that EFE184 and SKU-T2 cells with knockdown of CHD4 may gain a CSC-like phenotype. This hypothesis was verified by the tumor sphere formation assay and cell invasion assay. Tumor sphere generation is an indicative of self-renewal potential [17]. Results showed that the tumor sphere increased significantly in the EFE184 and SKU-T2 cells with knockdown of CHD4 compared to control cells (Figure 4A, 4B). Cell invasion was examined through by Matrigel-coated transwells, finding increased invasion in CHD4 knockdown cells compared to control cells (Figure 4C, 4D).
Figure 4.

Knockdown of CHD4 or mutations increase cancer cell malignancy in vitro and in vivo. Representative microscopic images of sphere formation and invasion ability of cells are shown in EFE184 and SKU-T2 cells. Knockdown of CHD4 (A, B), CHD4R975H and CHD4R1162W (E) formed more spheres in the comparison with control cells and CHD4WT cells. Tumor spheres in each well with the diameter above 20 μm are counted. In transwell assay, more invading cells are observed in cells with knockdown of CHD4 (C, D) or with CHD4 mutations (F). (G) Tumorigenicity of CHD4 knockdown cell line (SKU-T2) after subcutaneous injection into mice. Representative photograph of tumor tissues and the measurement of tumor volumes are showed that loss of CHD4 has higher tumorigenicity. The t-test was used to analyze the result. *P < 0.05 when compared with control cells. Values represent the mean ± SD.
We found that CHD4R975H and CHD4R1162W can reduce the protein stability and diminish its function (Figure 2C), so the two CHD4 mutations can function like the knockdown of CHD4. To determine if the CSC phenotype was conserved with CHD4 mutations, we re-expressed the CHD4 variants CHD4R975H, CHD4R1162W or wild type (CHD4WT) in SKU-T2 cells for comparison. In cells with CHD4R975H or CHD4R1162W, the number of tumor sphere (Figure 4E) and the number of invasive cells (Figure 4F) were increased dramatically in comparison with CHD4WT, as previously observed with CHD4 knockdown. To test this in vivo, nude mice xenograft model was subsequently applied to evaluate the effect of CHD4 on tumor growth. Statistical analysis of tumor volume indicated that knockdown of CHD4 can support the tumor growth (Figure 4G). All of these findings demonstrated that the two CHD4 mutants function like the loss of CHD4 in endometrial cancer cells, which induces a cancer stem cell phenotype to promote cancer progression.
The activated TGF-beta pathway in variants with CHD4 knockdown cells and CHD4 mutant cells
Biological Functional Analysis using Ingenuity Pathway Analysis (IPA) predicted that TGF-beta pathway was activated by knockdown of CHD4 in EFE184 cells. Figure 5A shows the genes associated with the TGF-beta activation in EFE184 cells, including CD133 which is upregulated with the expression of TGF-beta. Protein expression of TGF-beta was evaluated in EFE184 and SKUT-2 cells by Western blotting, and the increased expression can be detected in cells with knockdown of CHD4, or those expressing CHD4 mutatants (Figure 5B). To test if TGF-beta may be driving this CSC phenotype, we repeated the CSC sphere forming assaying following supplementation with TGFβ receptor I (TβRI) inhibitor (LY2157299/Galunisertib), it repressed growth of tumor spheres dramatically (Figure 5C). Taken together, this demonstrates that loss of CHD4 function can promote endometrial cancer by activating the TGF-beta pathway inducing a CSC phenotype.
Figure 5.
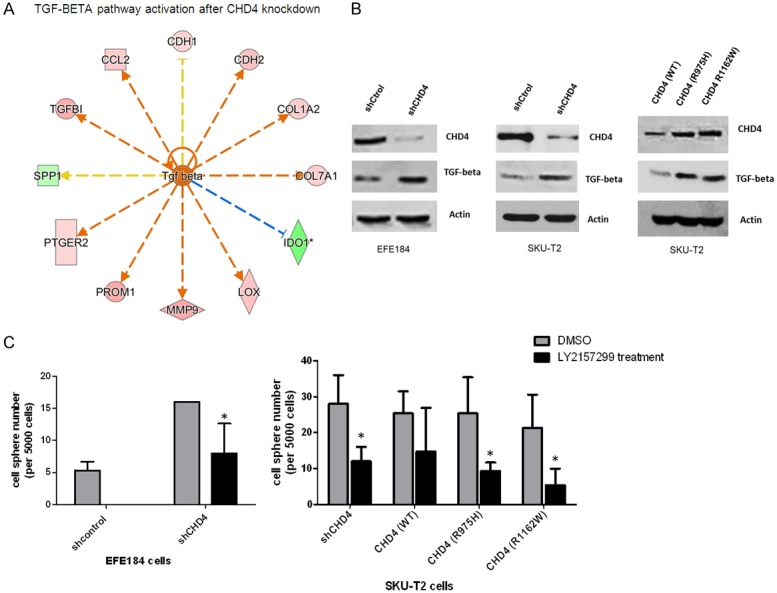
Activated TGF-Beta pathway in the cells with knockdown of CHD4 or mutation. A. TGF-Beta/CD133 pathway is activated based on IPA software. B. The expression of TGF-beta were evaluated by Western blotting in EFE184 cells and SKU-T2 cells with knockdown of CHD4 or CHD4 mutations. C. Inhibition of TGF-beta can suppress the sphere formation in CHD4R975H, CHD4R1162W and knockdown of CHD4 cells. The t-test was used to analyze the result. *P < 0.05 when compared with control cells. Values represent the mean ± SD.
Discussion
In our present study, we demonstrate that CHD4, a protein recurrently mutatated in the TCGA endometrial cancer patient cohort as well as other endometrial cohorts induces a CSC-like phenotype via a TGF-beta dependent mechanism. In addition to endometrial cancer, other types of cancer in the TCGA were analyzed, but we did not found any mutations as high as in endometrial cancer, suggesting CHD4 mutation is specific to the endometrial cancer. Half of CHD4 mutations are in conserved residues and involved in ATP domain required for catalysis of ATP hydrolysis and helicase activity [14]. In this study we worked on two point mutations in ATP domain, CHD4R975H and CHD4R1162W. Although the two mutations have been reported, there is no description about the role of the two mutations in endometrial cancer progression. By the transfection of CHD4R975H, CHD4R1162W, or CHD4WT in 293T cells, we firstly found that both mutants can decrease CHD4 stability without effecting NuRD complex formation and DNA binding. These results indicated that the two point mutations do not change the function of CHD4, but can cause loss of function of CHD4. To verify this, we generated variants with knockdown of CHD4, CHD4R975H, CHD4R1162W, or CHD4WT in endometrial cancer cells. Across multiple assays, data showed that cells with knockdown of CHD4, CHD4R975H, or CHD4R1162W exhibited the same phenotype. The three types of variants are able to elevate cell invasion ability and increase the cancer stem-like cell population compared to the control cells and cells with CHD4WT. All of the results demonstrate that CHD4 acts as a tumor suppressor in endometrial cancer.
Currently, some literature also reports that CHD4 has a function in impeding cancer progression. Firstly CHD4 can bind transcriptional repressors to inhibit tumor-associated gene expression. It is known that CHD4 can interact with ZIP to suppress breast carcinogenesis [8]. Rajini Srinivasan et al. also reported that CHD4 can interact with NAB2 in prostate cancer to suppress EGR activity and its targets [18]. Secondly CHD4 can respond to DNA damage, maintain genomic integrity, and prevent cell malignancy. CHD4 can be recruited to the sites of damaged DNA by two modes. The first mode is PARP dependent, where PARP induces CHD4 and other subunits of NuRD to the site of double strand breaks for the further repair [19,20]. The second mode is CHD4 can relax chromatin, so in this way RNF8/RNF168 facilitates histone ubiquitination and helps RNF168 and BRCA1 to be localized to damaged site for DNA repair [21-23]. To explore how CHD4 works as a tumor suppressor in endometrial cancer, we found loss of CHD4 can promote endometrial cancer progression by activating TGFB1 pathway through the microarray and IPA analysis to induce a CSC-phenotype. The increased expression of TGFB1 and CD133 can be observed in our variants with knockdown of CHD4, CHD4R975H, or CHD4R1162W. Both published evidence and our data support that CHD4 mutation is closely associated with TGFB1/CD133 pathway. Cancer stem cells are known to play a large role in the progression of endometrial cancer [24]. Others have found that the CD133-positive, CSC-like subpopulation of endometrial cancer cells express high levels of TGFB1 [25]. Overexpression of TGFB1 is recurrent in many types of cancers, including lung cancer, breast cancer, prostate cancer, and endometrial cancer. TGFB1 triggers the EMT to enhance metastasis along with the tumor growth. CD133 is an important marker of cancer stem cells and target of TGFB1 [26]. The TGFB1/CD133 pathway can induce cell invasion, stem-like characteristics, and drug resistance [27]. And TGFB1 also can promote genomic instability in the way of reducing DNA double strand breaks (DSB) repair [28-32]. Therefore loss of function of CHD4 can activate TGFB1 pathway to expand cancer stem-like cell population, reduce genome integrity, and then enhance the malignance.
Others have suggested that CHD4 can also act as an oncogene to promote cancer progression. In colorectal cancer, CHD4 has been shown to facilitate DNA hypermethylation and interact with other proteins to silence tumor suppressor genes [33]. As one of core components of the NuRD complex, CHD4 can interact with other different subunits such as MTA1/2/3 or MBD2/3 to perform various functions It will be critical to consider the cellular context when evaluating the role of CHD4 in future studies.
In conclusion, we found that CHD4 mutations can reduce CHD4 stability to promote endometrial cancer progression by activating TGFB1 pathway, promoting formation of -CSC-like cells that can be abrogated by TGF-beta blockade. Because CSCs have important roles in the cancer progression, targeting CHD4-driven CSCs by TGF-beta inhibition may be an effective strategy for treatment of endometrial cancer.
Acknowledgements
This work was supported by a grant from the NCI (R01CA172490) to SYL. Additional funding was provided by NCI (T32CA186892) to DJM.
Disclosure of conflict of interest
None.
References
- 1.Le Gallo M, O’Hara AJ, Rudd ML, Urick ME, Hansen NF, O’Neil NJ, Price JC, Zhang S, England BM, Godwin AK, Sgroi DC NIH Intramural Sequencing Center (NISC) Comparative Sequencing Program. Hieter P, Mullikin JC, Merino MJ, Bell DW. Exome sequencing of serous endometrial tumors identifies recurrent somatic mutations in chromatin-remodeling and ubiquitin ligase complex genes. Nat Genet. 2012;44:1310–1315. doi: 10.1038/ng.2455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.O’Shaughnessy A, Hendrich B. CHD4 in the DNA-damage response and cell cycle progression: not so NuRDy now. Biochem Soc Trans. 2013;41:777–782. doi: 10.1042/BST20130027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lai AY, Wade PA. Cancer biology and NuRD: a multifaceted chromatin remodelling complex. Nat Rev Cancer. 2011;11:588–596. doi: 10.1038/nrc3091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Watson AA, Mahajan P, Mertens HD, Deery MJ, Zhang W, Pham P, Du X, Bartke T, Zhang W, Edlich C, Berridge G, Chen Y, Burgess-Brown NA, Kouzarides T, Wiechens N, Owen-Hughes T, Svergun DI, Gileadi O, Laue ED. The PHD and chromo domains regulate the ATPase activity of the human chromatin remodeler CHD4. J Mol Biol. 2012;422:3–17. doi: 10.1016/j.jmb.2012.04.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Nishioka K, Chuikov S, Sarma K, Erdjument-Bromage H, Allis CD, Tempst P, Reinberg D. Set9, a novel histone H3 methyltransferase that facilitates transcription by precluding histone tail modifications required for heterochromatin formation. Genes Dev. 2002;16:479–489. doi: 10.1101/gad.967202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Denslow SA, Wade PA. The human Mi-2/NuRD complex and gene regulation. Oncogene. 2007;26:5433–5438. doi: 10.1038/sj.onc.1210611. [DOI] [PubMed] [Google Scholar]
- 7.Stanley FK, Moore S, Goodarzi AA. CHD chromatin remodelling enzymes and the DNA damage response. Mutat Res. 2013;750:31–44. doi: 10.1016/j.mrfmmm.2013.07.008. [DOI] [PubMed] [Google Scholar]
- 8.Becker PB, Horz W. ATP-dependent nucleosome remodeling. Annu Rev Biochem. 2002;71:247–273. doi: 10.1146/annurev.biochem.71.110601.135400. [DOI] [PubMed] [Google Scholar]
- 9.Xue Y, Wong J, Moreno GT, Young MK, Cote J, Wang W. NURD, a novel complex with both ATP-dependent chromatin-remodeling and histone deacetylase activities. Mol Cell. 1998;2:851–861. doi: 10.1016/s1097-2765(00)80299-3. [DOI] [PubMed] [Google Scholar]
- 10.Basta J, Rauchman M. Translating Epigenetics to the Clinic. 2017. The nucleosome remodeling and deacetylase complex in development and disease; pp. 37–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kim MS, Chung NG, Kang MR, Yoo NJ, Lee SH. Genetic and expressional alterations of CHD genes in gastric and colorectal cancers. Histopathology. 2011;58:660–668. doi: 10.1111/j.1365-2559.2011.03819.x. [DOI] [PubMed] [Google Scholar]
- 12.Pan MR, Hsieh HJ, Dai H, Hung WC, Li K, Peng G, Lin SY. Chromodomain helicase DNAbinding protein 4 (CHD4) regulates homologous recombination DNA repair, and its deficiency sensitizes cells to poly(ADP-ribose) polymerase (PARP) inhibitor treatment. J Biol Chem. 2012;287:6764–6772. doi: 10.1074/jbc.M111.287037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhao S, Bellone S, Lopez S, Thakral D, Schwab C, English DP, Black J, Cocco E, Choi J, Zammataro L, Predolini F, Bonazzoli E, Bi M, Buza N, Hui P, Wong S, Abu-Khalaf M, Ravaggi A, Bignotti E, Bandiera E, Romani C, Todeschini P, Tassi R, Zanotti L, Odicino F, Pecorelli S, Donzelli C, Ardighieri L, Facchetti F, Falchetti M, Silasi DA, Ratner E, Azodi M, Schwartz PE, Mane S, Angioli R, Terranova C, Quick CM, Edraki B, Bilguvar K, Lee M, Choi M, Stiegler AL, Boggon TJ, Schlessinger J, Lifton RP, Santin AD. Mutational landscape of uterine and ovarian carcinosarcomas implicates histone genes in epithelial-mesenchymal transition. Proc Natl Acad Sci U S A. 2016;113:12238–12243. doi: 10.1073/pnas.1614120113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhao S, Choi M, Overton JD, Bellone S, Roque DM, Cocco E, Guzzo F, English DP, Varughese J, Gasparrini S, Bortolomai I, Buza N, Hui P, AbuKhalaf M, Ravaggi A, Bignotti E, Bandiera E, Romani C, Todeschini P, Tassi R, Zanotti L, Carrara L, Pecorelli S, Silasi DA, Ratner E, Azodi M, Schwartz PE, Rutherford TJ, Stiegler AL, Mane S, Boggon TJ, Schlessinger J, Lifton RP, Santin AD. Landscape of somatic singlenucleotide and copy-number mutations in uterine serous carcinoma. Proc Natl Acad Sci U S A. 2013;110:2916–2921. doi: 10.1073/pnas.1222577110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Peng G, Chun-Jen Lin C, Mo W, Dai H, Park YY, Kim SM, Peng Y, Mo Q, Siwko S, Hu R, Lee JS, Hennessy B, Hanash S, Mills GB, Lin SY. Genome-wide transcriptome profiling of homologous recombination DNA repair. Nat Commun. 2014;5:3361. doi: 10.1038/ncomms4361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bao B, Ahmad A, Azmi AS, Ali S, Sarkar FH. Overview of cancer stem cells (CSCs) and mechanisms of their regulation: implications for cancer therapy. Curr Protoc Pharmacol. 2013 doi: 10.1002/0471141755.ph1425s61. Chapter 14: Unit 14.25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rybak AP, He L, Kapoor A, Cutz JC, Tang D. Characterization of sphere-propagating cells with stem-like properties from DU145 prostate cancer cells. Biochim Biophys Acta. 2011;1813:683–694. doi: 10.1016/j.bbamcr.2011.01.018. [DOI] [PubMed] [Google Scholar]
- 18.Srinivasan R, Mager GM, Ward RM, Mayer J, Svaren J. NAB2 represses transcription by interacting with the CHD4 subunit of the nucleosome remodeling and deacetylase (NuRD) complex. J Biol Chem. 2006;281:15129–15137. doi: 10.1074/jbc.M600775200. [DOI] [PubMed] [Google Scholar]
- 19.Polo SE, Kaidi A, Baskcomb L, Galanty Y, Jackson SP. Regulation of DNA-damage responses and cell-cycle progression by the chromatin remodelling factor CHD4. EMBO J. 2010;29:3130–3139. doi: 10.1038/emboj.2010.188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chou DM, Adamson B, Dephoure NE, Tan X, Nottke AC, Hurov KE, Gygi SP, Colaiacovo MP, Elledge SJ. A chromatin localization screen reveals poly (ADP ribose)-regulated recruitment of the repressive polycomb and NuRD complexes to sites of DNA damage. Proc Natl Acad Sci U S A. 2010;107:18475–18480. doi: 10.1073/pnas.1012946107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Smeenk G, Wiegant WW, Vrolijk H, Solari AP, Pastink A, van Attikum H. The NuRD chromatinremodeling complex regulates signaling and repair of DNA damage. J Cell Biol. 2010;190:741–749. doi: 10.1083/jcb.201001048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Larsen DH, Poinsignon C, Gudjonsson T, Dinant C, Payne MR, Hari FJ, Rendtlew Danielsen JM, Menard P, Sand JC, Stucki M, Lukas C, Bartek J, Andersen JS, Lukas J. The chromatin-remodeling factor CHD4 coordinates signaling and repair after DNA damage. J Cell Biol. 2010;190:731–740. doi: 10.1083/jcb.200912135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Luijsterburg MS, Acs K, Ackermann L, Wiegant WW, Bekker-Jensen S, Larsen DH, Khanna KK, van Attikum H, Mailand N, Dantuma NP. A new non-catalytic role for ubiquitin ligase RNF8 in unfolding higher-order chromatin structure. EMBO J. 2012;31:2511–2527. doi: 10.1038/emboj.2012.104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kato K. Endometrial cancer stem cells: a new target for cancer therapy. Anticancer Res. 2012;32:2283–2293. [PubMed] [Google Scholar]
- 25.Rutella S, Bonanno G, Procoli A, Mariotti A, Corallo M, Prisco MG, Eramo A, Napoletano C, Gallo D, Perillo A, Nuti M, Pierelli L, Testa U, Scambia G, Ferrandina G. Cells with characteristics of cancer stem/progenitor cells express the CD133 antigen in human endometrial tumors. Clin Cancer Res. 2009;15:4299–4311. doi: 10.1158/1078-0432.CCR-08-1883. [DOI] [PubMed] [Google Scholar]
- 26.Lebrun JJ. The dual role of TGFbeta in human cancer: from tumor suppression to cancer metastasis. ISRN Mol Biol. 2012;2012:381428. doi: 10.5402/2012/381428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tabu K, Taga T, Tanaka S. Tumor stem cells: CD133 gene regulation and tumor stemness. Stem Cells and Cancer Stem Cells. 2012;Volume 2:145–153. [Google Scholar]
- 28.Comaills V, Kabeche L, Morris R, Buisson R, Yu M, Madden MW, LiCausi JA, Boukhali M, Tajima K, Pan S, Aceto N, Sil S, Zheng Y, Sundaresan T, Yae T, Jordan NV, Miyamoto DT, Ting DT, Ramaswamy S, Haas W, Zou L, Haber DA, Maheswaran S. Genomic instability is induced by persistent proliferation of cells undergoing epithelial-to-mesenchymal transition. Cell Rep. 2016;17:2632–2647. doi: 10.1016/j.celrep.2016.11.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Aivaliotis IL, Pateras IS, Papaioannou M, Glytsou C, Kontzoglou K, Johnson EO, Zoumpourlis V. How do cytokines trigger genomic instability? J Biomed Biotechnol. 2012;2012:536761. doi: 10.1155/2012/536761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pal D, Pertot A, Shirole NH, Yao Z, Anaparthy N, Garvin T, Cox H, Chang K, Rollins F, Kendall J, Edwards L, Singh VA, Stone GC, Schatz MC, Hicks J, Hannon GJ, Sordella R. TGF-beta reduces DNA ds-break repair mechanisms to heighten genetic diversity and adaptability of CD44+/CD24- cancer cells. Elife. 2017:6. doi: 10.7554/eLife.21615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Dubrovska A, Kanamoto T, Lomnytska M, Heldin CH, Volodko N, Souchelnytskyi S. TGFbeta1/Smad3 counteracts BRCA1dependent repair of DNA damage. Oncogene. 2005;24:2289–2297. doi: 10.1038/sj.onc.1208443. [DOI] [PubMed] [Google Scholar]
- 32.Krishnan V, Chong YL, Tan TZ, Kulkarni M, Bin Rahmat MB, Tay LS, Sankar H, Jokhun DS, Ganesan A, Chuang LSH, Voon DC, Shivashankar GV, Thiery JP, Ito Y. TGFbeta promotes genomic instability after loss of RUNX3. Cancer Res. 2018;78:88–102. doi: 10.1158/0008-5472.CAN-17-1178. [DOI] [PubMed] [Google Scholar]
- 33.Xia L, Huang W, Bellani M, Seidman MM, Wu K, Fan D, Nie Y, Cai Y, Zhang YW, Yu LR, Li H, Zahnow CA, Xie W, Chiu Yen RW, Rassool FV, Baylin SB. CHD4 has oncogenic functions in initiating and maintaining epigenetic suppression of multiple tumor suppressor genes. Cancer Cell. 2017;31:653–668. e657. doi: 10.1016/j.ccell.2017.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]