

Abstract
Natural killer (NK) cells have exhibited promising efficacy in inhibiting cancer growth. We aimed to explorer the effect of NK cells on oxaliplatin-resistant colorectal cancer and the underlying molecular mechanism. Oxaliplatin-resistant colorectal cancer cell lines were co-cultured with NK cells to evaluate the effect on viability, proliferation, migration and invasion in vitro. Oxaliplatin-resistant colorectal cancer cells were also co-injected with NK cells into mice to establish xenograft tumor model, to assess the in vivo effect of NK cells on tumorigenesis of the oxaliplatin-resistant colorectal cancer cells. Expression of WBSCR22 gene was assessed in the oxaliplatin-resistant colorectal cancer cells following NK cell treatment to elucidate the mechanism. NK cell treatment significantly reduces growth of oxaliplatin-resistant colorectal cancer cells both in vitro and in vivo, as well as reduced WBSCR22 expression. MicroRNAs potentially targeting WBSCR22 were analyzed, and microRNA-146b-5p was found to be significantly upregulated following NK cell treatment. MicroRNA-146b-5p directly targeted WBSCR22 mRNA 3’-UTR to inhibit its expression, which was required for NK cell-induced inhibition of oxaliplatin-resistant colorectal cancer cell lines. NK cells inhibit oxaliplatin-resistant colorectal cancer by repressing WBSCR22 via upregulating microRNA-146b-5p, both of which could serve as candidates for targeted therapy against oxaliplatin-resistant colorectal cancer.
Keywords: Natural killer cells, oxaliplatin, colorectal cancer, WBSCR22, microRNA-146b-5p
Introduction
Colon cancer is among the most common types of cancer in China, for which an important treatment is Chemotherapy [1]. Oxaliplatin, one of the first line drugs in the treatment for colon cancer, is a third generation platinum based drug. It was reported in several clinical trials that as the standard first line treatment of advanced colon cancer, the FOLFOX regimen [LOHP, 5fluorouracil (5FU) plus leucovorin (LV)] significantly increases the median survival to nearly two years and the response rate up to 54%. However, the response rate reduces to only 4% with second line treatment when the first line chemotherapy fails [2]. Unsuccessful chemotherapy is predominantly resulted from the congenital or acquired multidrug resistance (MDR) against a variety of chemotherapeutic agents in a small population of cancer cells [3]. Such cancer cells can survive the chemotherapy and proliferate continuously, eventually leading to the death of the patient. Prior studies have shown that cancer stem cells likely play a critical role in the course of drug resistant [4]. Therefore, investigations of the mechanism of oxaliplatin resistance in colon cancer cells are necessary to provide novel strategies of overcoming such resistance in clinical practice.
Natural Killer (NK) cells are a type of cytotoxic lymphocytes that are crucial in the rapid response of the innate immune system to virally infected cells and tumor development [5]. NK cells are differentiated from the common lymphoid progenitor cells, which can generate T and B lymphocytes as well [6]. It is best known that NK cells differentiate and mature in the bone marrow [6]. In addition to the innate immune response, NK cells are also involved in adaptive immune responses [5], which are becoming increasingly important in suppressing a number of tumors progression [7-9]. NK cells function in tumor immuno-surveillance by directly inducing the death of tumor cells even in the absence of antigenic peptides and the surface adhesion molecules [8]. In comparison to the cytotoxic T cells, NK cells appear more important for the tumor surveillance particularly because T cells are incapable of recognizing pathogens without surface antigens [6,8,10]. Lysis of tumor cells by NK cells is mediated through alternative receptors such as NKp46, NKp44, DNAM, and NKG2D [11]. NKG2D forms a disulfide-linked homodimer and recognizes various ligands, including MICA and ULBP, both of which are commonly expressed in tumor cells [12].
In the current study, we explored NK cells with the aim to investigate their effect on tumorigenesis of colorectal cancer, in particular, oxaliplatin-resistant (OR) colorectal cancer cells both in vitro and in vivo. Following evaluation of the anti-tumor effect of NK cells, we further elucidated the underlying molecular mechanism, and revealed a novel regulatory cascade involving WBSCR22/microRNA-146b-5p.
Materials and methods
Oxaliplatin-resistant (OR) colorectal cancer cell lines
RKO and DLD1 human colorectal cancer cell lines were obtained from the American Type Culture Collection (ATCC, Manassas, VA, USA) and cultured in RPMI-1640 medium containing 10% fetal bovine serum (Gibco, Waltham, MA, USA), 100 mg/mL streptomycin and 100 U/mL penicillin. To generate the oxaliplatin-resistant colorectal cancer cell lines, RKO or DLD1 cells were treated with increasing doses of oxaliplatin, up to 10 μM or 40 μM, respectively. The surviving oxaliplatin-resistant cells were named OR-RKO and OR-DLD1, which were then maintained in medium supplemented with 8 μM or 20 μM oxaliplatin. All cultures were kept at 37°C in 5% CO2.
Cologenic assay
Cells were digested using a trypsin-EDTA solution (Sigma, St. Louis, MI, USA) and a single-cell suspension was prepared at the density of 200 cells/ml in culture medium. A total of 2 ml cell suspension (400 cells/well) was seeded into six-well plates, two wells per cell group. Cells were treated with oxaliplatin or vehicle for 24 hrs, then rinsed twice with PBS, fixed by 4% paraformaldehyde, and stained using hematoxylin solution. The number of colonies containing 50 or more cells was counted under a microscope.
Proliferation assay
The cell proliferation was determined by CCK-8 kit (Dojindo, Kumamoto, Japan). Briefly, 10 ul of CCK-8 solution was added to each well followed by the chromogenic reaction for 15 min at 37°C. To calculate the relative cell viability, the absorption at 450 nm was recorded using a microplate reader (Molecular Devices, Sunnyvale, CA, USA).
Cell migration assay
To examine the capability of cell migration, a wound healing assay was performed. In brief, a linear cut was made across confluent cells with the tip of a 200 μL pipette, which was defined as the boundary of cells at time zero. The migration of cells into the scratches was assessed 24 hours after the generation of the wound under an inverted phase-contrast microscope (Olympus, Japan), and the distance the growing edge migrated was measured using the ImageJ software (National Institute of Health, USA).
Cell invasion assay
Resuspension of 2×105 cells in 200 mL serum-free RPMI 1640 was added to 24-well transwell Matrigel-coated insert (8 μm pore size, Corning, USA) with 600 mL complete culture medium in the lower compartment. Cultures were maintained at 37°C in a 5% CO2 humidified incubator for 24 hours, followed by the treatment with 5-ethynyl-2’-deoxyuridine (EdU, 20 μM) for another 4 h at 37°C. Inserts were then detached from the plate and visualized using the ENU kit (Invitrogen, Carlsbad, CA, USA). For each well, cells were counted from six random representative fields. Invasion rate was calculated and normalized to the appropriate control.
Xenograft mouse model
All animal experiments were approved by the Animal Ethics Committee at The Affiliated Hospital of Inner Mongolia Medical University in compliance with the National Institutes of Health Guide for Care and Use of Laboratory Animals. BALB/C nude mice (6-8 weeks old) were obtained from Shanghai SLAC Laboratory Animal Co. Ltd (Shanghai, China) and maintained in a specific pathogen-free environment at a constant room temperature with a 12:12 h light/dark cycle and ad libitum access to standard rodent diet and water. Each mouse received a subcutaneous injection of 100 μl sterile PBS containing 5×106 colorectal cancer cells, with or without 2.5×106 NK cells, into the right flank. After the euthanization of mice, tumors were isolated and weighed for a tumor size analysis (tumor volume and weight). Tumor volumes were calculated using the following formula: V=0.5×L×W2, where V is the volume, L is the length, and W is the width.
Quantitative real-time PCR
Total RNA was extracted with TRIzol (Invitrogen, Carlsbad, CA, USA), and cDNA was synthesized with the use of a SuperScript RT-PCR kit (Invitrogen, Carlsbad, CA, USA). The primers used in the current study are as follows: WBSCR22 sense 5’-CAT TTG ATG GTT GCA TCA GC-3’, anti-sense 5’-CTT GGC AGG GTT TTC AGA CT-3’; Actin sense 5’-CAT CGA GCA CGG CAT CGT CA-3’, anti-sense 5’-TAG CAC AGC CTG GAT AGC AAC-3’. Actin was used as the internal control.
Western blot
Total protein was prepared from cultured cells using lysis buffer containing 2% NP-40, 0.5% sodium deoxycholate, 0.2% sodium dodecyl sulfate, 150 mM NaCl, 50 mM Tris (pH 8.0), and 10 mM phenylmethylsulfonyl fluoride. The protein concentration was measured with the BCA kit (Byeotime Biotechnology, Nantong, China). Aliquots of 20 μg total protein was boiled in loading buffer for 3 min and then separated on a 12% SDS-PAGE. The proteins were transferred to a nitrocellulose membrane (Pall, Corp., Pensacola, FL, USA), blocked with 5% non-fat milk in Tris-buffered saline containing 0.5% Tween-20 (TBST), and then incubated with primary antibodies against human WBSCR22 (GeneTex, Irvine, CA, USA) or actin (Cell Signaling Technology, Inc., Danvers, MA, USA) overnight at 4°C. After five rinses with TBST, the membranes were incubated with horseradish peroxidase-conjugated secondary antibody (Cell Signaling Technology, Inc.) for 1 h at room temperature. Finally, after two rinses with TBST, the labeled proteins were visualized using enhanced chemiluminescence (Santa Cruz Biotechnology, Inc., Santa Cruz, CA, USA) on enhanced chemiluminescence films (Eastman Kodak, Co., Rochester, NY, USA).
Dual-luciferase reporter assay
Wild type (WBSCR22-UTR) and mutated (WBSCR22-mut) miR-146b-5p targeting sequences from WBSCR22 3’-UTR were cloned into the downstream of a luciferase reporter gene on the pGL3-enhancer plasmid with the pMir-Report vector kit (Applied Biosystems, Carlsbad, CA, USA). A total of 105 cells per well were placed in 24 well plate and transfected with indicated luciferase constructs using Lipofectamine 2000 (Invitrogen, Carlsbad, CA, USA). Cells were allowed to grow for 24 hrs after the transfection before various experiments. The luciferase activities were measured using a dual-luciferase reporter assay kit (Promega, Madison, WI, USA) following the manufacturer’s protocol.
MiR-146b-5p assays
MystiCq microRNA assay kits (Sigma-Aldrich, St. Louis, MO, USA) were used to measure expressions of microRNAs, normalized against RNU6 control microRNA assay (MIRCP00001; Sigma-Aldrich, USA). The Mission miRNA lentiviral inhibitor for miR-146b-5p (HLTUD0230; Sigma-Aldrich, USA) and control inhibitor (HLTUD001C; Sigma-Aldrich, USA) were packaged for transduction to create stable cell lines.
Statistical analysis
All experiments were repeated independently for at least three times. Data was analyzed using one-way or two-way analysis of variance, followed by Tukey’s post-hoc test. Statistical analysis was performed with IBM SPSS Statistics 19.0 Software (SPSS Inc., Chicago, Illinois, USA). Data is presented as the mean ± SD. P values less than 0.05 were considered statistically significant.
Results
Oxaliplatin-resistant colorectal cancer cell lines
We started by treating two colorectal cancer cell lines, DLD1 and RKO1, with oxaliplatin to establish resistance (see materials and methods), namely OR-DLD1 and OR-RKO, respectively. Next, cell viability was assessed to evaluate the oxaliplatin-resistance of OR-DLD1 and OR-RKO cells. Compared with their parental cell lines, OR-DLD1 or OR-RKO cells exhibited increased resistance to oxaliplatin, as shown in Figure 1A and 1B.
Figure 1.
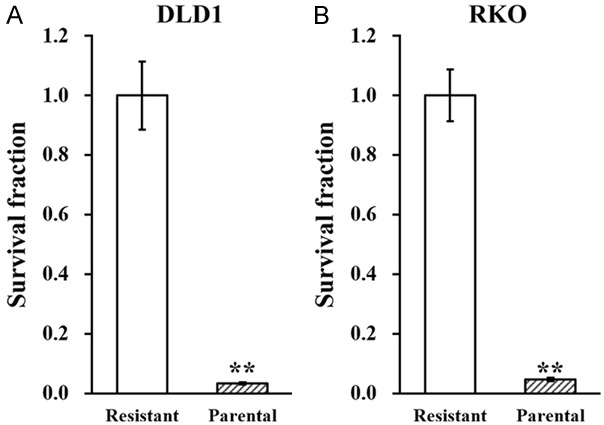
Establishing oxaliplatin-resistant colorectal cancer cell lines. Colorectal cell lines DLD1 and RKO were exposed to long term oxaliplatin treatment to establish oxaliplatin-resistant cell lines (see materials and methods). (A) Resistant DLD1 and (B) RKO, respectively with their parental cell lines, were incubated in the presence of oxaliplatin, followed by clonogenic assay to assess their survival. Data are presented as mean ± SD from at least three independent experiments. **P<0.01, resistant vs parental.
NK cell co-culture reduces growth of OR colorectal cancer cell lines in vitro
Next, to assess the effect of NK cells on these established OR-DLD1 and OR-RKO cells, a co-culture system of employed using previously reported protocol [13]. Viability and proliferation of the OR-DLD1 and OR-RKO cells, in the presence or absence of NK cell co-culture, were then assessed by clonogenic assay and CCK8 kit, respectively. As shown in Figure 2A and 2B, both viability and proliferation of OR-DLD1 and OR-RKO cells were significantly reduced when they are co-cultured with NK cells, compared to the control. Moreover, wound healing assay and transwell invasion assay were also used to assess migration and invasion capacities of the OR-DLD1 and ORRKO cells. Similarly, both migration and invasion were significantly inhibited when these two cell lines were co-cultured with NK cells (Figure 3A and 3B).
Figure 2.
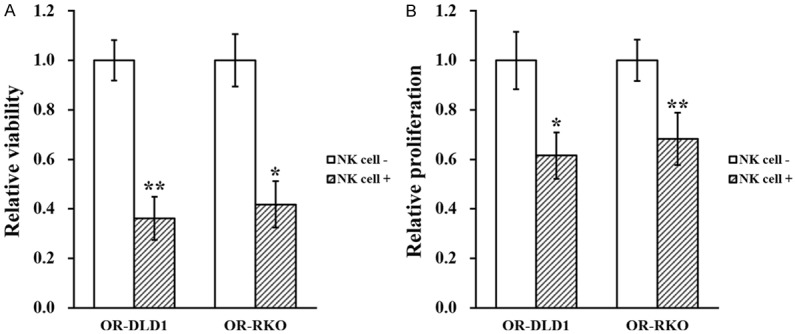
NK cell co-culture reduces viability and proliferation of oxaliplatin-resistant colorectal cancer cell lines. Oxaliplatin-resistant (OR)-DLD1 and OR-RKO cells were subjected to (A) viability and (B) proliferation assays, in the absence (NK cell -) or presence (NK cell +) of co-cultured natural killer cells, respectively. Data are presented as mean ± SD from at least three independent experiments. **P<0.01, *P<0.05, NK cell - vs NK cell +.
Figure 3.
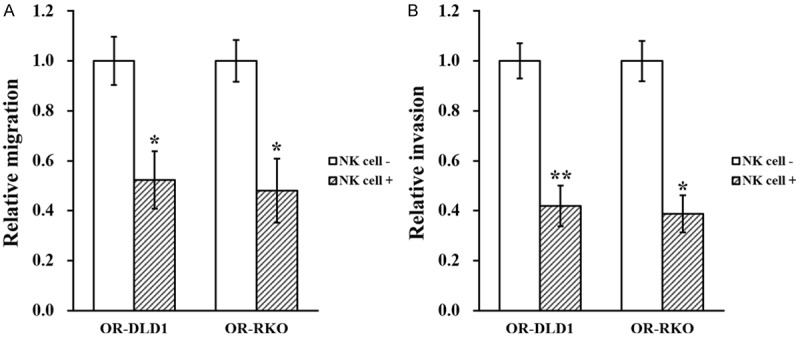
NK cell co-culture reduces migration and invasion capacities of oxaliplatin-resistant colorectal cancer cell lines. Oxaliplatin-resistant (OR)-DLD1 and OR-RKO cells were subjected to (A) wound healing and (B) transwell invasion assays, in the absence (NK cell -) or presence (NK cell +) of co-cultured natural killer cells, respectively. Data are presented as mean ± SD from at least three independent experiments. **P<0.01, *P<0.05, NK cell - vs NK cell +.
NK cell co-culture reduces growth of OR colorectal cancer cell lines in vivo
To further investigate whether NK cells could exert inhibitory effect on OR-DLD1 and OR-RKO cells in vivo, we established xenograft tumor model in nude mice, by co-injecting them with NK cells. Growth of xenograft tumors were measured on day 7, 14 and 21 after injection, and the sizes of tumors in the NK cell co-injected group were significantly bigger than those without co-injected NK cells (Figure 4A and 4B). At the end of day 21, tumors were extracted and their weight was measured, and we found the weight of tumors in the NK cell co-injected mice were also significantly heavier than those without co-injected NK cells (Figure 4C and 4D).
Figure 4.
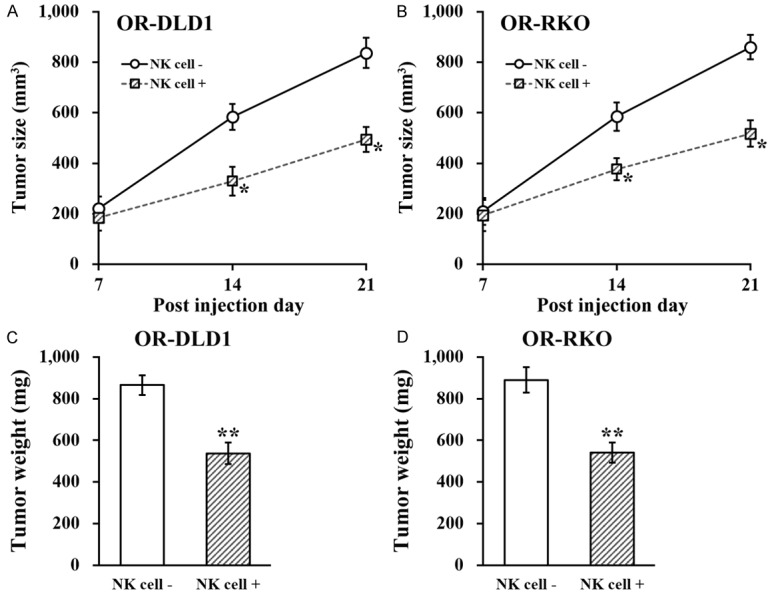
NK cell co-culture reduces migration and invasion capacities of oxaliplatin-resistant colorectal cancer cell lines. A and B. Oxaliplatin-resistant (OR)-DLD1 and OR-RKO cells were injected into mice (n=8 each group), without (NK cell -) or with co-injection of natural killer cells (NK cell +), respectively, followed by growth curve evaluation on day 7, 14 and 21 after injection. C and D. At the end of day 21, xenograft tumors from all mice were extracted and weighed. Data are presented as mean ± SD, n=8 each group. **P<0.01, *P<0.05, NK cell - vs NK cell +.
NK cell co-culture reduces WBSCR22 expression in oxaliplatin-resistant colorectal cancer cell lines
Since WBSCR22 was very recently reported to confer oxaliplatin resistance in human colorectal cancer [14], we wondered whether NK cells could regulate its expression in the OR-DLD1 and OR-RKO cells. In this context, OR-DLD1 and OR-RKO cells co-cultured in the presence and absence of NK cells were subjected to RT-PCR and Western blot analyses to assess the expression levels of both WBSCR22 mRNA and protein, respectively. As shown in Figure 5A, expressions of WBSCR22 mRNA and protein were both significantly reduced by NK cell co-culture. Similarly, tumor tissues on day 21 from the xenograft mice were subjected to the same analysis, and mRNA and protein levels of WBSCR22 were also consistently downregulated in the tumors extracted from NK co-injected mice (Figure 5B).
Figure 5.
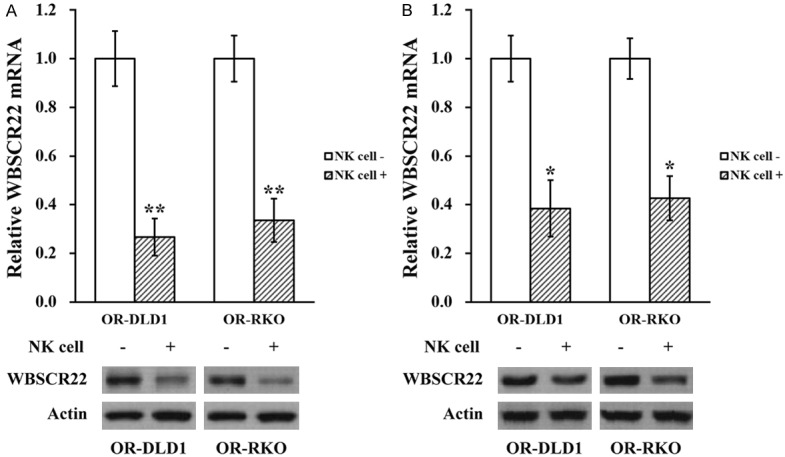
NK cell co-culture reduces WBSCR22 expression in oxaliplatin-resistant colorectal cancer cell lines. A. Oxaliplatin-resistant (OR)-DLD1 and OR-RKO cells in the absence (NK cell -) or presence (NK cell +) of co-cultured natural killer cells, respectively, were subjected to RT-PCR and Western blot analyses to assess mRNA and protein levels of WBSCR22. Data are presented as mean ± SD from at least three independent experiments. B. At the end of day 21, OR-DLD1 and OR-RKO xenograft tumors from all mice were extracted and subjected to RT-PCR and Western blot analyses to assess mRNA and protein levels of WBSCR22. Data are presented as mean ± SD, n=8 each group. **P<0.01, *P<0.05, NK cell - vs NK cell +.
NK cell co-culture elevates miR-146b-5p in oxaliplatin-resistant colorectal cancer cell lines
MicroRNAs (miRNAs, miRs) was previously reported to play pivotal roles in several biological processes including tumorigenesis of colorectal cancer [15], we therefore analyzed sequence of WBSCR22 mRNA to identify miRs that could potentially target its expression, using the miRanda algorithm [16]. Several miRs were predicted to have targeting sequence on the 3’ untranslated region (UTR) of WBSCR22, among which only miR-146b-5p expression was found to be upregulated by NK cell co-culture (Figure 6A, 6B, and data not shown). This upregulated was also verified in xenograft tumors extract from the mouse model, where miR-146b-5p expression was greatly elevated in tumor tissues from NK cell co-injected mice (Figure 6C).
Figure 6.

MiR-146b-5p directly targets WBSCR22 mRNA 3’-UTR to inhibit its expression. A. Predicted miR-146b-5p targeting sequence on the 3’-UTR of WBSCR22 mRNA. B. Oxaliplatin-resistant (OR)-DLD1 and OR-RKO cells in the absence (NK cell -) or presence (NK cell +) of co-cultured natural killer cells, respectively, were subjected to microRNA assay to assess miR-146b-5p levels. C. At the end of day 21, OR-DLD1 and OR-RKO xenograft tumors from all mice were extracted and subjected to microRNA assay to assess miR-146b-5p levels. D. Wild type (WBSCR22-UTR) targeting sequence of miR-146b-5p on 3’-UTR of WBSCR22 mRNA, and the mutated version (WBSCR22-mut), were cloned at the downstream of the luciferase open reading frame (Luc). E. Luciferase activities of WBSCR22-UTR or WBSCR22-mut constructs were examined after stable expression of miR-NC or miR-146b-5p, respectively. F. Luciferase activities of WBSCR22-UTR or WBSCR22-mut constructs were examined in the absence (NK cell -) or presence (NK cell +) of co-cultured natural killer cells, respectively. Data are presented as mean ± SD, from at least three independent in vitro experiments, or n=8 each group for animal experiments. **P<0.01, *P<0.05, ns not significant, NK cell - vs NK cell +, or miR-NC vs miR-146b-5p.
Next, in order to establish direct targeting between miR-146b-5p and WBSCR22 3’-UTR, wild type (WBSCR22-UTR) targeting sequence of miR-146b-5p on 3’-UTR of WBSCR22 mRNA, and the mutated version (WBSCR22-mut), were cloned at the downstream of the luciferase open reading frame (Luc) (Figure 6D). Luciferase constructs were transfected into OR colorectal cancer cells stably expressing miR-146b-5p mimic (Supplementary Figure 1A), followed by luciferase activity assay. Indeed, activity of WBSCR22-UTR was greatly reduced in cells overexpressing miR-146b-5p, whereas activity of WBSCR22-mut was unaltered under the same condition (Figure 6E). To confirm the effect of NK cell co-culture on miR-146b-5p targeting WBSCR22 3’-UTR, Luciferase constructs were transfected into OR colorectal cancer cells in the presence or absence of NK cell co-culture as well. The results indicated that with NK cell co-culture, activity of WBSCR22-UTR was significantly repressed, whereas activity of WBSCR22-mut was remained unchanged (Figure 6F).
MiR-146b-5p is required for NK cell-induced inhibition of oxaliplatin-resistant colorectal cancer both in vitro and in vivo
Finally, to evaluate the role of miR-146b-5p in NK cell-induced inhibition of oxaliplatin-resistant colorectal cancer, stable expression of miR-146b-5p inhibitor was introduced in ORDLD1 and OR-RKO cells, respectively (Supplementary Figure 1B), which were then co-cultured with NK cells in vitro to assess their growth. We found that the NK cell-induced suppressive effect on viability (Figure 7A), proliferation (Figure 7B), migration (Figure 7C) and invasion (Figure 7D) were all markedly attenuated by miR-146b-5p inhibition. The same miR-146b-5p inhibited OR-DLD1 and OR-RKO cells were also co-injected with NK cells into mice, where the similar attenuating effects were observed (Figure 8A and 8B), with tumor size and weight both restored to indistinguishable levels as the control (Figure 8C).
Figure 7.
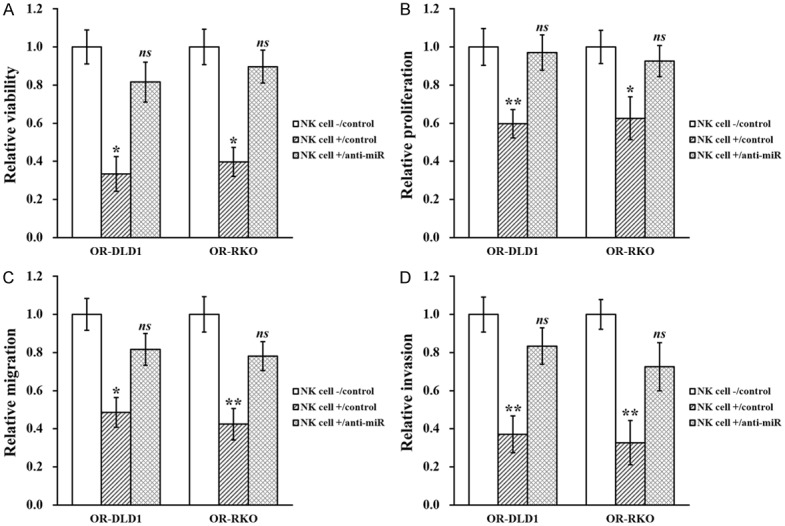
MiR-146b-5p is required for NK cell-induced inhibition of oxaliplatin-resistant colorectal cancer cell lines in vitro. Oxaliplatin-resistant (OR)-DLD1 and OR-RKO cells stably transduced with control miR inhibitor (control) or miR-146b-5p inhibitor (anti-miR) in the absence (NK cell -) or presence (NK cell +) of co-cultured natural killer cells, respectively, were subjected to (A) viability, (B) proliferation, (C) wound healing and (D) transwell invasion assays. Data are presented as mean ± SD from at least three independent experiments. **P<0.01, *P<0.05, NK cell +/control vs both NK cell -/control and NK cell +/anti-miR. ns not significant, NK cell -/control vs NK cell +/anti-miR.
Figure 8.
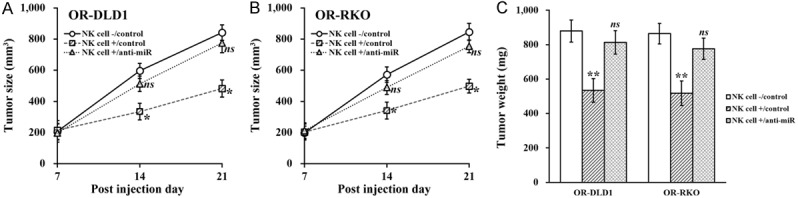
MiR-146b-5p is required for NK cell-induced inhibition of oxaliplatin-resistant colorectal cancer cell lines in vivo. A and B. Oxaliplatin-resistant (OR)-DLD1 and OR-RKO cells stably transduced with control miR inhibitor (control) or miR-146b-5p inhibitor (anti-miR) were injected into mice (n=8 each group), without (NK cell -) or with co-injection of natural killer cells (NK cell +), respectively, followed by growth curve evaluation on day 7, 14 and 21 after injection. C. At the end of day 21, xenograft tumors from all mice were extracted and weighed. Data are presented as mean ± SD, n=8 each group. **P<0.01, *P<0.05, NK cell +/control vs both NK cell -/control and NK cell +/anti-miR. ns not significant, NK cell -/control vs NK cell +/anti-miR.
Discussion
In this study, we first established two colorectal cancer cell lines that were resistant to oxaliplatin, namely OR-DLD1 and OR-RKO. In addition, we have presented data supporting that co-culturing OR-DLD1 and OR-RKO cells with NK cells could significantly reduce the growth of these two oxaliplatin resistant colorectal cancer cell lines both in vitro and in vivo using mouse xenograft model. Importantly, since WBSCR22 was previously reported to be implicated in oxaliplatin resistance of human colorectal cancer [14], we examined its expression in OR-DLD1 and OR-RKO cells following NK cell co-culture and found it downregulated. Further investigation revealed that expression of WBSCR22 mRNA was a direct target of a novel miR-146b-5p, which was upregulated by NK cell co-culture in in OR-DLD1 and OR-RKO cells. Finally, to highlight the critical role of miR-146b-5p, specific inhibitor of miR-146b-5p attenuated NK cell-induced growth repression of oxaliplatin-resistant colorectal cancer cells.
Human WBSCR22 gene was initially identified as one of 26 genes mutated in Williams-Beuren syndrome with characteristics such as congenital heart and vascular disease, dysmorphic facial features, and unique cognitive dysfunction [17-19]. While mRNA of WBSCR22 was ubiquitously expressed in all tissues, its protein was specifically detected in heart, skeletal muscle and kidney [20,21]. WBSCR22 contains a nuclear localization signal and a common S-adenosyl-L-methionine binding motif evolutionarily conserved in methyltransferases [22]. WBSCR22 was upregulated in invasive breast cancer, and its ectopic expression in non-metastatic cells strongly promoted the metastasis formation through suppression of Zac1/p53-dependent apoptosis, but did not alter cell growth and motility [23]. In multiple myeloma, WBSCR22 was necessary for the survival of tumor cells [24], and its product increased in both primary plasma cells and primary multiple myeloma tumor cells, implying its roles in plasma cell biology [24]. On the other hand, WBSCR22 knockdown attenuated invasive capacities and cell growth in various cells [25,26].
Of particular interest to our current study, WBSCR22 was very recently reported to confer oxaliplatin resistance in human colorectal cancer [14]. Yan et al. analyzed the TCGA cohort and found WBSCR22 expression to be markedly elevated in human colorectal cancer tissue. Knockdown of WBSCR22 dramatically sensitized colorectal cancer cells to oxaliplatin in vitro and in vivo, whereas overexpression of WBSCR22 resulted in cellular resistance to oxaliplatin treatment [14]. This result is agreeable with discoveries in our current study, where WBSCR22 expression was correlated with tumorigenesis of oxaliplatin-resistant colorectal cancer cells.
Besides WBSCR22, another novel factor that has been identified in our study is miR-146b-5p. MiRNAs was previously reported to be involved in several biological processes such as tumorigenesis, including colorectal cancer [15]. MiRNAs are a group of small non-protein-coding RNAs (~22 nucleotides), majority of which are negative regulators of gene expression via binding to the three-prime untranslated regions (3’-UTRs) of target mRNAs. Recent progress in cancer biology revealed that miRNAs are often deregulated in different types of human cancers, including colorectal cancer, thereby promoting cancer development and induction of drug resistance [15,27]. In the context of our study, miR-146b-5p has been widely reported as a tumor-suppressor miR in several human cancers. For instance, down-regulation of miR-146b-5p in hepatocellular carcinoma promotes cancer growth and metastasis [28]. In addition, miR-146b was also able to induce apoptosis and suppress cell proliferation, migration and invasion of the prostate cancer cell lines [29]. In terms of involvement in resistance to cancer therapies, overexpression of miR-146b-5p attenuated stemness and radioresistance of glioma stem cells [30], whereas low expression of miR-146b-5p predicted poor outcome of therapies using cyclophosphamide, doxorubicin, vincristine and prednisone to treat large B-cell lymphoma [31]. However, to date, there has been no investigation implicating miR-146b-5p in colorectal cancer. Our current study, for the first time, identified miR-146b-5p as a critical player in the tumorigenesis and oxaliplatin resistance of colorectal cancer.
Conclusions
In conclusion, NK cells inhibit oxaliplatin-resistant colorectal cancer by repressing WBSCR22 via upregulating microRNA-146b-5p. Our study revealed a novel regulatory cascade involving WBSCR22/microRNA-146b-5p, both of which could serve as candidates for targeted therapy against oxaliplatin-resistant colorectal cancer.
Disclosure of conflict of interest
None.
Supporting Information
References
- 1.Segal NH, Saltz LB. Evolving treatment of advanced colon cancer. Annu Rev Med. 2009;60:207–219. doi: 10.1146/annurev.med.60.041807.132435. [DOI] [PubMed] [Google Scholar]
- 2.Tournigand C, Andre T, Achille E, Lledo G, Flesh M, Mery-Mignard D, Quinaux E, Couteau C, Buyse M, Ganem G, Landi B, Colin P, Louvet C, de Gramont A. FOLFIRI followed by FOLFOX6 or the reverse sequence in advanced colorectal cancer: a randomized GERCOR study. J. Clin. Oncol. 2004;22:229–237. doi: 10.1200/JCO.2004.05.113. [DOI] [PubMed] [Google Scholar]
- 3.Wu DL, Huang F, Lu HZ. [Drug-resistant proteins in breast cancer: recent progress in multidrug resistance] . Ai Zheng. 2003;22:441–444. [PubMed] [Google Scholar]
- 4.Hong SP, Wen J, Bang S, Park S, Song SY. CD44-positive cells are responsible for gemcitabine resistance in pancreatic cancer cells. Int J Cancer. 2009;125:2323–2331. doi: 10.1002/ijc.24573. [DOI] [PubMed] [Google Scholar]
- 5.Vivier E, Raulet DH, Moretta A, Caligiuri MA, Zitvogel L, Lanier LL, Yokoyama WM, Ugolini S. Innate or adaptive immunity? The example of natural killer cells. Science. 2011;331:44–49. doi: 10.1126/science.1198687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sun JC, Lanier LL. NK cell development, homeostasis and function: parallels with CD8(+) T cells. Nat Rev Immunol. 2011;11:645–657. doi: 10.1038/nri3044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yan Y, Steinherz P, Klingemann HG, Dennig D, Childs BH, McGuirk J, O’Reilly RJ. Antileukemia activity of a natural killer cell line against human leukemias. Clin Cancer Res. 1998;4:2859–2868. [PubMed] [Google Scholar]
- 8.Ljunggren HG, Malmberg KJ. Prospects for the use of NK cells in immunotherapy of human cancer. Nat Rev Immunol. 2007;7:329–339. doi: 10.1038/nri2073. [DOI] [PubMed] [Google Scholar]
- 9.Cheng M, Zhang J, Jiang W, Chen Y, Tian Z. Natural killer cell lines in tumor immunotherapy. Front Med. 2012;6:56–66. doi: 10.1007/s11684-012-0177-7. [DOI] [PubMed] [Google Scholar]
- 10.Vivier E, Ugolini S, Blaise D, Chabannon C, Brossay L. Targeting natural killer cells and natural killer T cells in cancer. Nat Rev Immunol. 2012;12:239–252. doi: 10.1038/nri3174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Vacca P, Cantoni C, Prato C, Fulcheri E, Moretta A, Moretta L, Mingari MC. Regulatory role of NKp44, NKp46, DNAM-1 and NKG2D receptors in the interaction between NK cells and trophoblast cells. Evidence for divergent functional profiles of decidual versus peripheral NK cells. Int Immunol. 2008;20:1395–1405. doi: 10.1093/intimm/dxn105. [DOI] [PubMed] [Google Scholar]
- 12.Farooqi AA, Fayyaz S, Qureshi MZ, Rashid S. Prostate cancer and immunoproteome: awakening and reprogramming the guardian angels. Arch Immunol Ther Exp (Warsz) 2012;60:191–198. doi: 10.1007/s00005-012-0169-y. [DOI] [PubMed] [Google Scholar]
- 13.Lin SJ, Chou FJ, Li L, Lin CY, Yeh S, Chang C. Natural killer cells suppress enzalutamide resistance and cell invasion in the castration resistant prostate cancer via targeting the androgen receptor splicing variant 7 (ARv7) Cancer Lett. 2017;398:62–69. doi: 10.1016/j.canlet.2017.03.035. [DOI] [PubMed] [Google Scholar]
- 14.Yan D, Tu L, Yuan H, Fang J, Cheng L, Zheng X, Wang X. WBSCR22 confers oxaliplatin resistance in human colorectal cancer. Sci Rep. 2017;7:15443. doi: 10.1038/s41598-017-15749-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Thomas J, Ohtsuka M, Pichler M, Ling H. MicroRNAs: clinical relevance in colorectal cancer. Int J Mol Sci. 2015;16:28063–28076. doi: 10.3390/ijms161226080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Enright AJ, John B, Gaul U, Tuschl T, Sander C, Marks DS. MicroRNA targets in drosophila. Genome Biol. 2003;5:R1. doi: 10.1186/gb-2003-5-1-r1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pober BR. Williams-Beuren syndrome. N Engl J Med. 2010;362:239–252. doi: 10.1056/NEJMra0903074. [DOI] [PubMed] [Google Scholar]
- 18.Morris CA, Demsey SA, Leonard CO, Dilts C, Blackburn BL. Natural history of Williams syndrome: physical characteristics. J Pediatr. 1988;113:318–326. doi: 10.1016/s0022-3476(88)80272-5. [DOI] [PubMed] [Google Scholar]
- 19.Bellugi U, Lichtenberger L, Mills D, Galaburda A, Korenberg JR. Bridging cognition, the brain and molecular genetics: evidence from Williams syndrome. Trends Neurosci. 1999;22:197–207. doi: 10.1016/s0166-2236(99)01397-1. [DOI] [PubMed] [Google Scholar]
- 20.Merla G, Ucla C, Guipponi M, Reymond A. Identification of additional transcripts in the Williams-Beuren syndrome critical region. Hum Genet. 2002;110:429–438. doi: 10.1007/s00439-002-0710-x. [DOI] [PubMed] [Google Scholar]
- 21.Doll A, Grzeschik KH. Characterization of two novel genes, WBSCR20 and WBSCR22, deleted in Williams-Beuren syndrome. Cytogenet Cell Genet. 2001;95:20–27. doi: 10.1159/000057012. [DOI] [PubMed] [Google Scholar]
- 22.Petrossian TC, Clarke SG. Uncovering the human methyltransferasome. Mol Cell Proteomics. 2011;10:M110.000976. doi: 10.1074/mcp.M110.000976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nakazawa Y, Arai H, Fujita N. The novel metastasis promoter Merm1/Wbscr22 enhances tumor cell survival in the vasculature by suppressing Zac1/p53-dependent apoptosis. Cancer Res. 2011;71:1146–1155. doi: 10.1158/0008-5472.CAN-10-2695. [DOI] [PubMed] [Google Scholar]
- 24.Tiedemann RE, Zhu YX, Schmidt J, Shi CX, Sereduk C, Yin H, Mousses S, Stewart AK. Identification of molecular vulnerabilities in human multiple myeloma cells by RNA interference lethality screening of the druggable genome. Cancer Res. 2012;72:757–768. doi: 10.1158/0008-5472.CAN-11-2781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ounap K, Kasper L, Kurg A, Kurg R. The human WBSCR22 protein is involved in the biogenesis of the 40S ribosomal subunits in mammalian cells. PLoS One. 2013;8:e75686. doi: 10.1371/journal.pone.0075686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Stefanska B, Cheishvili D, Suderman M, Arakelian A, Huang J, Hallett M, Han ZG, AlMahtab M, Akbar SM, Khan WA, Raqib R, Tanvir I, Khan HA, Rabbani SA, Szyf M. Genome-wide study of hypomethylated and induced genes in patients with liver cancer unravels novel anticancer targets. Clin Cancer Res. 2014;20:3118–3132. doi: 10.1158/1078-0432.CCR-13-0283. [DOI] [PubMed] [Google Scholar]
- 27.Clancy C, Joyce MR, Kerin MJ. The use of circulating microRNAs as diagnostic biomarkers in colorectal cancer. Cancer Biomark. 2015;15:103–113. doi: 10.3233/CBM-140456. [DOI] [PubMed] [Google Scholar]
- 28.Li C, Miao R, Liu S, Wan Y, Zhang S, Deng Y, Bi J, Qu K, Zhang J, Liu C. Down-regulation of miR-146b-5p by long noncoding RNA MALAT1 in hepatocellular carcinoma promotes cancer growth and metastasis. Oncotarget. 2017;8:28683–28695. doi: 10.18632/oncotarget.15640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ding HY, Qian WQ, Xu J. MicroRNA-146b acts as a potential tumor suppressor in human prostate cancer. J BUON. 2016;21:434–443. [PubMed] [Google Scholar]
- 30.Yang W, Yu H, Shen Y, Liu Y, Yang Z, Sun T. MiR-146b-5p overexpression attenuates stemness and radioresistance of glioma stem cells by targeting HuR/lincRNA-p21/beta-catenin pathway. Oncotarget. 2016;7:41505–41526. doi: 10.18632/oncotarget.9214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wu PY, Zhang XD, Zhu J, Guo XY, Wang JF. Low expression of microRNA-146b-5p and microRNA320d predicts poor outcome of large B-cell lymphoma treated with cyclophosphamide, doxorubicin, vincristine, and prednisone. Hum Pathol. 2014;45:1664–1673. doi: 10.1016/j.humpath.2014.04.002. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.