

Abstract
Background: Mitochondrial flashes (mitoflashes) are transient signals from transient bursts of reactive oxygen species (ROS) and changes in pH that occur in certain physiological or pathological conditions. Mitoflashes are closely related to metabolism, cell differentiation, stress response, diseases, and aging. Sepsis can trigger mitochondrial dysfunction in myocardial cells, which leads to ROS overproduction, while uncoupling protein 2 (UCP2) can reduce ROS production. This study aims to observe whether UCP2 overexpression can regulate the frequency of mitoflashes in cardiomyocytes during sepsis and thereby play a protective role. Methods: A cell model for sepsis-induced myocardial damage was established using lipopolysaccharide (LPS). UCP2 overexpression in cardiomyocytes was achieved by adenovirus transfection. Creatinine kinase (CK), lactate dehydrogenase (LDH), tumor necrosis factor (TNF-α), and interleukin (IL-6) activities were detected, and mitochondrial membrane potentials (MMP) were measured. The frequency of mitoflashes in cardiomyocytes was observed. Results: With LPS stimulation, mitoflashes in cardiomyocytes increased significantly, and the MMP was damaged. Additionally, significant increases in CK, LDH, TNF-α, and IL-6 expression levels were observed. UCP2 overexpression can significantly reverse myocardial cell injuries that result from LPS stimulation. Compared with the LPS group, the LPS+UCP2 overexpression group showed a decrease in mitoflash frequency, an improved MMP, and decreases in CK, LDH, TNF-α, and IL-6 expression levels. Conclusion: This study is the first to demonstrate the function of UCP2 overexpression in protecting the myocardium by regulating mitoflash frequency and reversing sepsis-induced myocardial injuries.
Keywords: Sepsis, myocardial injury, mitochondrial flashes, uncoupling protein 2, reactive oxygen species
Introduction
The occurrence of sepsis has gradually increased over the years, and it has become one of the top 10 causes of death [1-3]. Sepsis-induced myocardial injury was first recognized during the 1990s [4]. Compared with the 20% mortality rate among myocardial injured patients without sepsis complications, the mortality rate is as high as 70-90% for those complicated by sepsis. Therefore, improving sepsis-induced myocardial injuries has become the key for effective sepsis treatment.
Sepsis can trigger mitochondrial dysfunction in the myocardium and is considered a critical factor in this process [5-8]. Under sepsis conditions, the activity of one or more membrane protein complexes in the mitochondrial respiratory chain is reduced. The reduced activity causes mitochondrial respiratory dysfunction in the myocardium impairs oxidative phosphorylation and ATP synthesis and triggers cardiac dysfunction. Myocardial energy metabolism disorder results in the inability of cytochrome oxidase to restore oxygen to H2O and is associated with excessive reactive oxygen species (ROS) production and insufficient ROS elimination. The excessive ROS can not only directly attack mitochondria by damaging the integrity of the membrane also cause irreversible damage to mitochondria through calcium overload. Furthermore, excessive ROS then leads to reductions in myocardial contractility and cardiac dysfunction [9,10]. Therefore, reduced mitochondrial ROS production and treatments for oxidative damage could be effective methods to address sepsis-induced myocardial injury [11].
A mitoflash is considered a transient quantized burst of superoxide in the mitochondrial matrix of a resting cell. In other words, a mitoflash is an elementary event in ROS production caused by an individual mitochondrion [12]. A Mitoflash is a transient signal from transient bursts of ROS and changes in pH that occurs in certain physiological or pathological conditions. Mitoflashes randomly occurs in space and time and can be detected using circular permuted yellow fluorescent protein (cpYFP) localized in the mitochondrial matrix. The mitoflash phenomenon can be observed in various cells, including myocardial cells, neurons, and non-excitable cells [13]. Several studies have indicated that the mitochondrial permeability transition pore (mPTP) opening is critical for the occurrence of mitoflashes, as the stochastic mPTP transient opening may trigger mitoflashes [12,14,15]. Many research groups have performed studies on mitoflashes [16-21]. It is commonly agreed that each mitoflash event involves multiple interconnected signals that constitute bursts of superoxide anion formation, conversion from a reduction state to an oxidation state, matrix alkylation, and an action potential-like membrane potential depolarization [22,23]. Mitochondrial ROS is typically determined by extrapolation from quantified ROS metabolites. Mitoflash detection is an intuitive method for the real-time monitoring of mitochondrial ROS production bursts. Mitoflash regulation in myocardial cells under the influence of sepsis has been seldom reported.
Uncoupling protein 2 (UCP2) is a member of the uncoupling protein family that localizes on the inner mitochondrial membrane. When a UCP2 ion carrier opens, protons can leak through the mitochondrial intermembrane space into the mitochondrial matrix without ATP synthase [24]. The function of UCP2 in inhibiting ROS production is known; excessive ROS can activate UCP2 and regulate ROS production through negative feedback [25,26]. When hyperpolarization of the inner mitochondrial membrane potential occurs, electron transport chain function decreases and the half-lives of semiquinone anions increase. These changes then lead to stagnant electron transport chain intermediate functions and an increased chance for electron escape; these escaped electrons are then accepted by oxygen to form ROS. The high proton transport activity of UCP2 allows for the consumption of free energy from the proton electrochemical potential in the form of heat. UCP2 can reduce the membrane potential and inhibit ROS formation [27,28]. We hypothesized that when sepsis-induced myocardial injury occurs, UCP2 overexpression could reduce ROS production and regulate mitoflash frequency, resulting in myocardial protection.
In our study, endogenous UCP2 was the putative target for intervention. UCP2 overexpression was induced in lipopolysaccharide (LPS)-stimulated primary myocardial cells from neonatal rats. Mitoflash regulation was observed, and indicators of mitochondrial function were monitored. The protective function of overexpressed UCP2 in sepsis-induced myocardial injury was explored with the aim of revealing the protection mechanism. The results from this study could constitute a novel support method for treating sepsis-induced myocardial injury.
Materials and methods
Animals
This study was approved by the First Affiliated Hospital of Zunyi Medical University Animal Ethics Committees. One-day-old Sprague-Dawley (SD) rats were provided by the Laboratory Animal Center of the Third Military Medical University (Chongqing, China); the accreditation number was SCXK (Yu) 2012-0005. The rats were cared for in accordance with the Policy on the Humane Care and Use of Laboratory Animals published by the U.S. Public Health Service (NIH publication No.23-85, revised 1996) and the Policy on Laboratory Animal Care and Use of Zunyi Medical University.
Experimental groupings
Primary myocardial cells from neonatal rats were randomly grouped into 6 groups: normal (given saline stimulation), UCP2 overexpressed (myocardial cells were infected with UCP2-overexpressing adenovirus), empty vector (myocardial cells infected with empty vector), LPS (cells were given LPS stimulation), LPS+UCP2 overexpressed (myocardial cells infected with UCP2-overexpressing adenovirus and then given LPS stimulation), and LPS+empty vector (myocardial cells infected with empty vector and then given LPS stimulation). The final concentration of LPS in the abovementioned transfections was 2 µg/ml and stimulation lasted for 24 h. An adenovirus that induces UCP2 overexpression and the empty vector was used. The adenovirus that induces UCP2 overexpression was purchased from HanBio (Shanghai, China).
Cell culture
Ventricle tissues from one day old SD neonatal rats were sectioned into 1-mm3 cubes. The cubes were transferred into digestion fluid containing 0.08% collagenase and 0.1% trypsin. Fractional digestion was performed 12 times at 37°C (5 min per digestion). The supernatants were collected and the digestion was terminated by the addition of serum-containing culture medium. The collected supernatants were centrifuged at 800 rpm for 5 min. Cells were re-suspended in DMEM/F12 culture medium containing 10% fetal bovine serum and 10 nM bromodeoxyuridine (BrdU). Samples were passed through a 100-mesh filter. Fibroblasts were removed by a 90-min differential velocity adherent technique. Myocardial cells were seeded on a 6-well plate. After 48 hrs and following myocardial cell adhesion, the culture medium was replaced with serum-free medium along with LPS for stimulation.
Determination of LDH and CK levels
The levels of LDH and CK were determined using colorimetry with a multifunctional microplate reader. Detection was performed using an LDH and CK detection kit purchased from the Nanjing Jiancheng Bioengineering Institute according to the manufactures’s protocol.
Determination of IL-6 and TNF-α levels
In our study, IL-6 and TNF-α levels were determined using an enzyme linked immunosorbent assay (ELISA). The levels were determined using the RAT IL-6 ELISA KIT purchased from Shanghai HuDing Biotechnology (Shanghai, China) according to the manufactures’s protocol.
Western blot analysis
RIPA lysis buffer was employed to lyse the cells. Total protein was extracted and its concentration was determined by bicinchoninic acid (BCA) assay. A fixed amount of extracted protein was subjected to SDS-PAGE (10% gel) and then transferred to a PVDF membrane. Samples were then blocked for 2 hrs with blocking buffer. Subsequently, the samples were treated with UCP2/tubulin primary antibody and kept at 4°C overnight. Detection was performed with HRP secondary antibody; the samples were incubated at room temperature for 2 h followed by ECL illumination. The membranes were then scanned with an absorbance image scanner. ImageJ was employed for the grayscale image analysis. Data analysis was performed using tubulin as an internal standard, and the results were expressed relative to the amount of protein expressed in the control group.
Mitoflash measurements
Expression of cpYFP in the cultured myocardial cells from neonatal rat was detected by adding AAV-mito-cpYFP adenovirus. Mitoflash data analysis was performed utilizing FlashSniper, a program developed based on the Interactive Data Language (IDL) environment. The main steps of the workflow include reading the LSM image data, image pretreatment (to correct for photoconversion, optical blurring, and photobleaching), auto-detection of the mitoflashes, manual confirmation of the detection, automatic analysis of the characteristic mitoflash parameters, manual verification of the analysis, and storage of the analysis results. Subsequent statistical analysis and graphing were performed in GraphPad.
Mitochondrial membrane potential (MMP)
Samples were treated with 100 nM tetramethylrhodamine methyl ester (TMRM) for 10 min and rinsed 3 times with Tyrode’s solution. The fluorescence intensity was measured in a randomly selected field of view using confocal laser scanning microscopy. The standardized MMP was obtained by inter-group comparison of the relative fluorescence intensity.
Statistical analysis
Statistical analysis was performed utilizing SPSS 17.0. Quantitative data are expressed as the means ± SD. When the quantitative data were normally distributed, they were subjected to oneway analysis of variance (ANOVA). If the differences between groups were statistically significant, the least significant difference (LSD) was employed to perform multi-sample pairwise comparisons of means for data with homogeneous variance, while Dunnett’s T3 test was used for data with heterogeneous variance. Differences with P<0.05 were considered statistically significant. Differences in the mitoflash frequency between the two groups were compared using the t-test.
Results
Effects of overexpressed UCP2 on IL-6 and TNF-α levels in the myocardial cells with sepsis
IL-6 and TNF-α levels in the LPS group were significantly higher than in the normal group (P<0.05). However, IL-6 and TNF-α levels in the LPS+UCP2 overexpressed group were significantly lower than the LPS group (P<0.05), suggesting that UCP2 overexpression provided protection for the LPS-stimulated myocardial cells. When comparing the UCP2 overexpressed group vs. the empty vector group and the LPS group vs. the LPS+empty vector group, there were no significant differences in IL-6 and TNF-α levels (P>0.05) (Figures 1 and 2).
Figure 1.
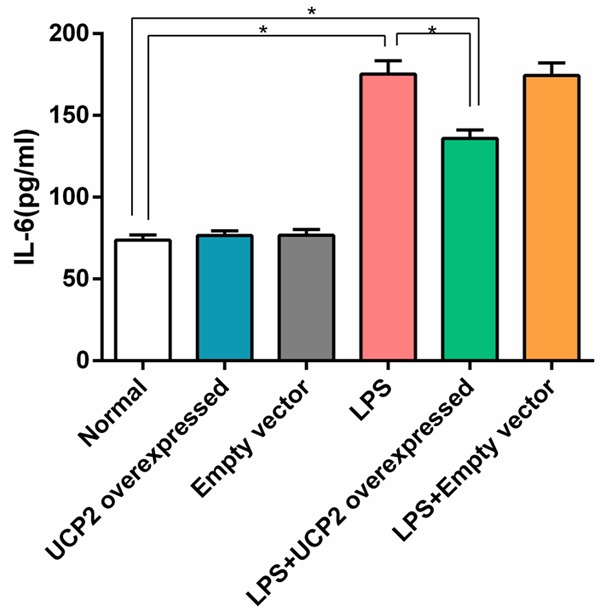
UCP2 overexpression reverses the elevated expression of IL-6 in cardiomyocytes that is induced by LPS stimulation. The IL-6 level of the LPS group was significantly higher than that of the normal group. However, the IL-6 level in the LPS+UCP2 overexpressed group was significantly lower than that of the LPS group. N=6. *P<0.05.
Figure 2.
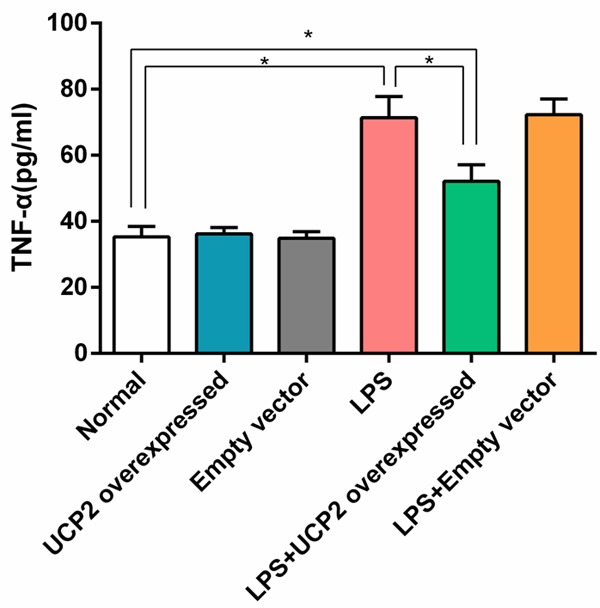
UCP2 overexpression reverses the elevated expression of TNF-α in cardiomyocytes that is induced by LPS stimulation. The TNF-α level of the LPS group was significantly higher than that of the normal group. However, the TNF-α level in the LPS+UCP2 overexpressed group was significantly lower than that of the LPS group, but were higher than normal group. N=6. *P<0.05.
LDH and CK release
The levels of LDH and CK in the LPS group were significantly higher than in the normal group (P<0.05). Interestingly, LDH and CK levels in the LPS+UCP2 overexpressed group were significantly lower than in the LPS group (P<0.05). There were no significant differences in the LDH and CK levels when comparing the UCP2 overexpressed group vs. the empty vector group or the LPS group vs. the LPS+empty vector group (P>0.05) (Figures 3 and 4).
Figure 3.
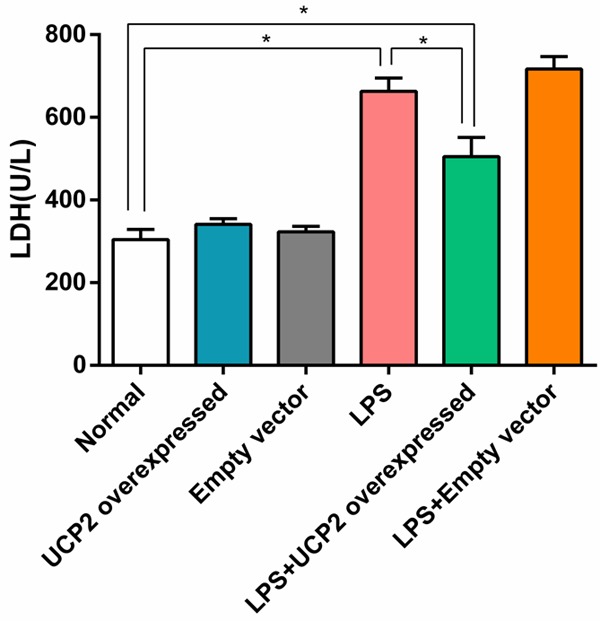
UCP2 overexpression reverses the elevated expression of LDH in cardiomyocytes that is induced by LPS stimulation. The level of LDH in the LPS group was significantly higher than that of the normal group. Interestingly, the level of LDH in the LPS+UCP2 overexpressed group was significantly lower than that of the LPS group, but were higher than normal group (P<0.05). N=6. *P<0.05.
Figure 4.
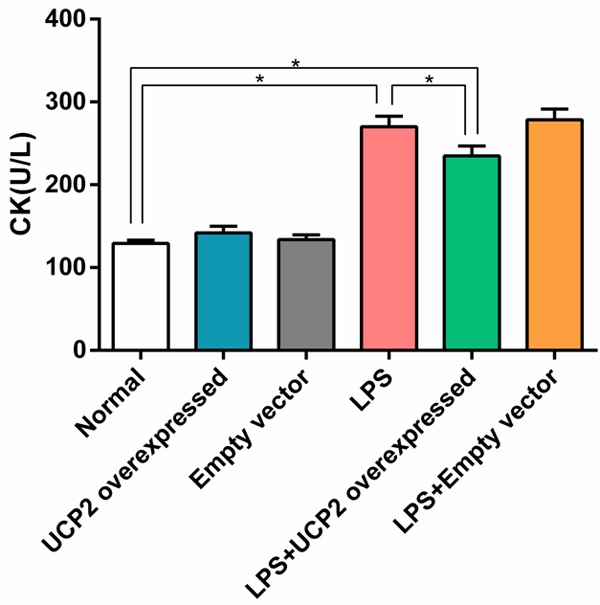
UCP2 overexpression reverses the elevated expression of CK in cardiomyocytes that is induced by LPS stimulation. The level of CK in the LPS group was significantly higher than that of the normal group, and the level of CK in the LPS+UCP2 overexpressed group was significantly lower than that of the LPS group, but were higher than normal group. N=6. *P<0.05.
Regulation of mitoflash in the myocardial cells with sepsis by UCP2 overexpression
In neonatal rat cardiac myocytes stably expressing the mitochondrial superoxide biosensor cpYFP, time-lapse confocal imaging visualized mitoflash activity that occurred upon challenge by LPS induced (2 μg/ml, 24 h). UCP2 overexpression reversed the LPS challenge, leading to enhanced mitoflash frequency. Individual flashes were identified as sudden, discrete ROS bursts that were randomly ignited over space and time (Figure 5A-D, Supplementary Movies 1, 2, 3, 4, 5, 6). After stimulating myocardial cells with LPS, the mitoflash frequency was significantly increased compared with the normal group (P<0.05). UCP2 overexpression significantly reversed the increased mitoflash frequency resulting from LPS stimulation (LPS group vs. LPS+UCP2 overexpressed group, P<0.05). Transfection of the empty vector had no effect on the mitoflash frequency; there were no significant differences in the mitoflash frequencies when comparing the normal group to the empty vector group or the LPS group to the LPS+empty vector group (P>0.05) (Figure 5A-D).
Figure 5.
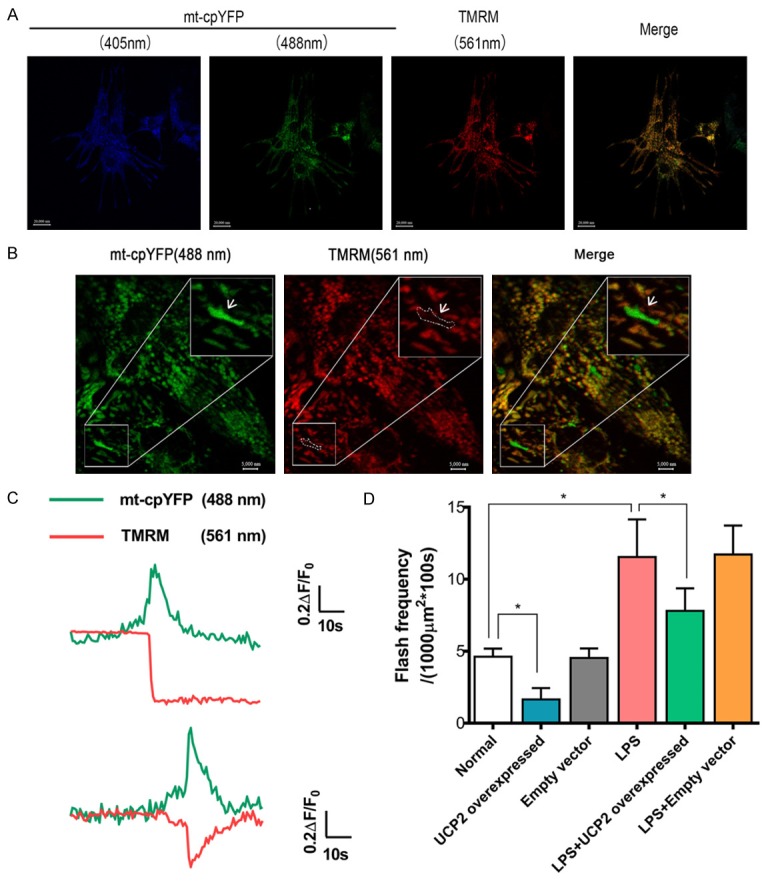
Single mitochondrial flashes detected in cardiomyocytes. (A) Representative images of the intact myocardium showing the colocalization of cpYFP and TMRM. Scale bar = 20000 nm. (B) Image of a single cardiomyocyte in the intact myocardium. The area highlighted by the white boxes is enlarged to show a mitochondrial flash accompanied with a decrease in the membrane potential (white arrows). Scale bars = 20000 and 5000 nm for the full size and enlarged images, respectively. (C) Traces showing the fluorescence changes during the single mitochondrial flash highlighted in (B). (D) Effects of UCP2 overexpression or LPS treatment (2 μg/ml, 24 h) on mitoflash activity in cardiomyocytes. 40/oil immersion field per group. N=6. *P<0.05.
Changes in the mitochondrial membrane potential of each group
To further explore the effects of overexpressed UCP2 on the MMP of cardiomyocytes with sepsis, TMRM (100 nM, 10 min) was employed. After staining, quantitative analysis was performed utilizing confocal laser scanning microscopy, and the relative fluorescence intensity was analyzed to explore the changes in the MMP. Compared with the normal group, the fluorescence intensities from the LPS and LPS+empty vector groups were significantly lower (P<0.05), while the fluorescence from the LPS+UCP2 overexpressed group was significantly higher than that the LPS group (P<0.05). The differences between the normal, UCP2 overexpressed, and empty vector groups were not statistically significant (P>0.05). The differences between the LPS group and the LPS+empty vector were also not significant (P>0.05) (Figure 6).
Figure 6.
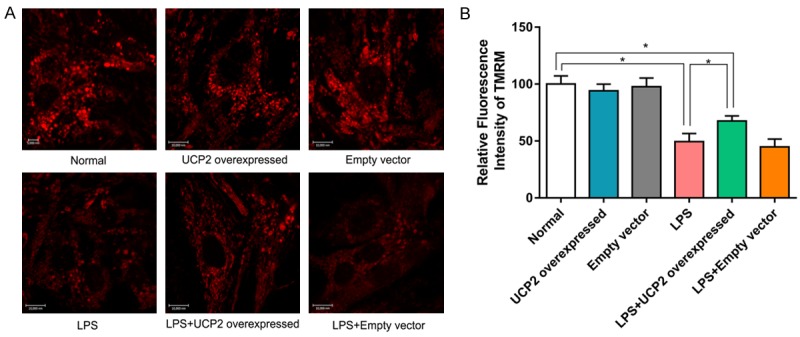
Cardiomyocytes were stained with TMRM and a confocal microscope was used to observe the fluorescence intensity at 561 nm. The contrast in relative fluorescence intensity reflects the magnitude of the mitochondrial membrane potentials of cardiomyocytes from various groups. LPS stimulation causes the mitochondrial membrane potential to decrease significantly, which is reversed by UCP2 overexpression. N=6. *P<0.05.
UCP2 protein expression
In the UCP2 and LPS+UCP2 overexpressed groups, UCP2 protein expression levels were significantly increased (P<0.05 vs. the normal group, the empty vector group, the LPS group, or the LPS+empty vector). Compared with the normal group, UCP2 expression in the LPS group was significantly upregulated. However, no significant differences were detected in UCP2 protein expression between the LPS and LPS+empty vector groups (P>0.05) (Figure 7).
Figure 7.
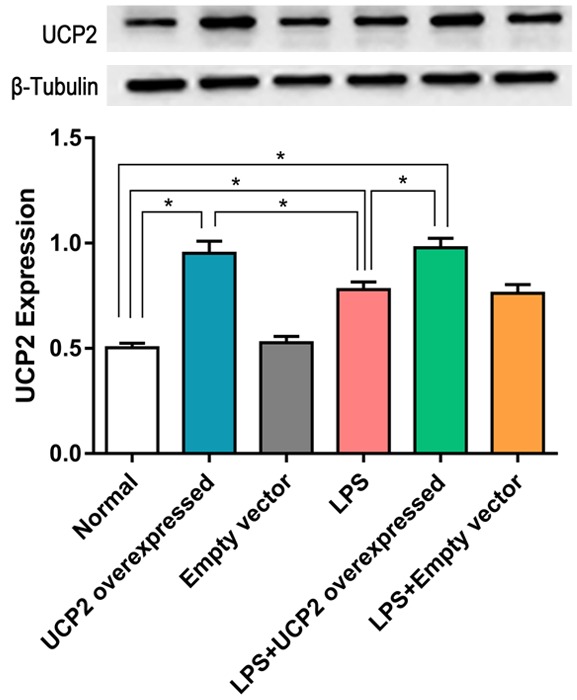
UCP2 protein expression in the LPS+UCP2 overexpression group is significantly higher than the in the normal, LPS, or empty viral vector groups. UCP2 protein expression in the LPS group is significantly higher than in the normal group. N=3. *P<0.05.
Discussion
The results from this study suggest that UCP2 overexpression functions to protect myocardial cells injured by LPS stimulation. We also observed that the mitoflash frequency in LPS-stimulated myocardial cells was significantly increased, suggesting an overproduction of mitochondrial ROS. UCP2 overexpression can reverse this process; the reversal mechanism could be due to the leakage of hydrogen ions from the intermembrane space of the mitochondria into the mitochondrial matrix through the UCP2 ion carrier. Thus, the excessive potential resulted from the decreased function of the mitochondrial complex induced by LPS. Therefore, ROS production decreases by reducing the interactions between electrons and oxygen.
LPS is a components in the cell walls of Gram-negative bacteria and a trigger for sepsis [29]. It is also a mediator of shock and inflammation. LPS can enter the lymphatic circulation system and trigger a systemic inflammatory response [30]. Pro-inflammatory cytokines, such as TNF-α and IL-6, participate in the occurrence and regulation of this inflammatory response [31]. CK, LDH, and MMP are commonly recognized indicators of myocardial injuries. In our study, we found that CK, LDH, IL-6, and TNF-α expression levels in primary myocardial cells increased significantly with LPS stimulation, while a decrease in the MMP was observed.
ROS have both positive and negative functions in the cell environment. An appropriate level of ROS is beneficial for defending the body from bacterial infection. However, excessive ROS can result in cellular damage [32,33]. In the context of sepsis, excessive ROS damages the functional proteins and enzymes on the mitochondrial membrane and further worsens mitochondrial dysfunction [7,34,35]. LPS can increase NADH oxidase expression and oxide levels, which can lead to increased ROS production [36,37]. Studies have reported that the oxidative stress response can result in mitochondrial dysfunction, the opening of mPTPs, and the release of pro-apoptotic proteins. All of these changes can cause cell death in the form of either apoptosis or necrosis [38,39]. In our study, the frequency of mitoflashes in LPS-stimulated myocardial cells was significantly increased, suggesting a significant increase in ROS production in the presence of LPS stimulation [12]. A possible mechanism for these changes is the abnormal increase in mPTP opening in myocardial cells caused by LPS stimulation. These openings allow for the transfer of water and cations into the mitochondrial matrix, membrane potential depolarization, and inner membrane edema. This mechanical pressure could cause the dislocation of proteins in the respiratory electron transport chain, and these electrons could leak through protein molecules interfaces and interact with oxygen, resulting in bursts of ROS. The vicious cycle perpetuated by excessive ROS production can cause severe myocardial injury. Membrane depolarization and bursts of ROS formation stimulate the operation of the respiratory electron transport chains, increasing transmembrane proton pumping and causing matrix alkylation [23].
UCP2 is a mitochondrial anion carrier protein that achieves oxidative phosphorylation uncoupling by clearing the proton gradients on the two sides of the inner mitochondrial membrane during ATP production, which then reduces the electrochemical gradients on the two sides of the inner mitochondrial membrane [40]. Kizaki T et al. transfected macrophages with UCP2 cDNA and found that ROS production by macrophages decreased [41]. Blanc J found that the lack of UCP2 in blood cells can lead to increased ROS synthesis and can cause the worsening of atherosclerosis [42]. The results from these studies are similar to those found in our study. With LPS stimulation and UCP2 overexpression, the frequency of mitoflash occurrence was significantly lower compared with cells without UCP2 overexpression. Additionally, CK, LDH, IL-6, and TNF-α expression levels decreased, and the decreased MMP was reversed. These results suggested that UCP2 overexpression effectively reduced ROS production and provided a certain degree of protection for myocardial cells from sepsis-induced injury. A possible mechanism for these effects is an enhanced uncoupling function resulting from UCP2 overexpression. This enhanced function allows protons to leak from the intermembrane space into the mitochondrial matrix, which partially relieves the mitochondrial complex dysfunction triggered by LPS stimulation, reduces excessive potential, decreases the interactions between electrons and oxygens, and therefore reduces ROS production. Additionally, transmembrane proton pumping and matrix alkylation are also reduced. The mitochondrial homeostasis then approaches a natural state, resulting in a certain degree of protection for the myocardium.
The results from our study indicated that sepsis can cause ROS overproduction in the mitochondria of myocardial cells. ROS overproduction then leads to increases in mitoflash frequency and pro-inflammatory cytokine release and decreases in myocardial enzymatic activity and MMP. These changes then cause myocardial injury. UCP2 overexpression may decrease ROS production by applying its uncoupling function and facilitating the decrease in mitoflash frequency. Thus the myocardial injury caused by sepsis is then reversed.
There are some limitations in our study. First, the function of UCP2 overexpression in protecting the myocardium by reducing ROS production under LPS stimulation was only supported by in vitro experiments. Additionally, the observed significant regulation of mitoflash frequency by UCP2 overexpression could be due to the uncoupling function of UCP2. However, the mechanism of this effect is unclear and requires further investigation.
In conclusion, this study is the first demonstrating the function of UCP2 overexpression in protecting the myocardium by regulating mitoflash frequency and reversing sepsis-induced myocardial injuries.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (No. 81560308). We wish to thank Prof. Heping Cheng, Institute of Molecular Medicine, Peking University, Beijing, China, People’s Republic, who provided technical support to us. We appreciate Prof. Huaping Liang, Army Medical University (Third Military Medical University), who provided laboratory and instrument supporting our experiments.
Disclosure of conflict of interest
None.
Supplementary Movie 1. Mitoflash activity under basal condition (Normal group)
Supplementary Movie 2. Mitoflash activity in response to UCP2 overexpressed condition
Supplementary Movie 3. Mitoflash activity in response to empty vector
Supplementary Movie 4. Mitoflash activity in response to LPS induced (2 μg/ml, 24 h)
Supplementary Movie 5. Mitoflash activity in LPS+UCP2 overexpressed group (2 μg/ml, 24 h)
Supplementary Movie 6. Mitoflash activity in LPS+empty vector
References
- 1.Iwashyna TJ, Cooke CR, Wunsch H, Kahn JM. Population burden of long-term survivorship after severe sepsis in older Americans. J Am Geriatr Soc. 2012;60:1070–1077. doi: 10.1111/j.1532-5415.2012.03989.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gaieski DF, Edwards JM, Kallan MJ, Carr BG. Benchmarking the incidence and mortality of severe sepsis in the United States. Crit Care Med. 2013;41:1167–1174. doi: 10.1097/CCM.0b013e31827c09f8. [DOI] [PubMed] [Google Scholar]
- 3.Hawiger J, Veach RA, Zienkiewicz J. New paradigms in sepsis: from prevention to protection of failing microcirculation. J Thromb Haemost. 2015;13:1743–1756. doi: 10.1111/jth.13061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Suffredini AF, Fromm RE, Parker MM, Brenner M, Kovacs JA, Wesley RA, Parrillo JE. The cardiovascular response of normal humans to the administration of endotoxin. N Engl J Med. 1989;321:280–287. doi: 10.1056/NEJM198908033210503. [DOI] [PubMed] [Google Scholar]
- 5.Soriano FG, Nogueira AC, Caldini EG, Lins MH, Teixeira AC, Cappi SB, Lotufo PA, Bernik MM, Zsengeller Z, Chen M, Szabo C. Potential role of poly (adenosine 5’-diphosphate-ribose) polymerase activation in the pathogenesis of myocardial contractile dysfunction associated with human septic shock. Crit Care Med. 2006;34:1073–1079. doi: 10.1097/01.CCM.0000206470.47721.8D. [DOI] [PubMed] [Google Scholar]
- 6.Harrois A, Huet O, Duranteau J. Alterations of mitochondrial function in sepsis and critical illness. Curr Opin Anaesthesiol. 2009;22:143–149. doi: 10.1097/ACO.0b013e328328d1cc. [DOI] [PubMed] [Google Scholar]
- 7.Vanasco V, Saez T, Magnani ND, Pereyra L, Marchini T, Corach A, Vaccaro MI, Corach D, Evelson P, Alvarez S. Cardiac mitochondrial biogenesis in endotoxemia is not accompanied by mitochondrial function recovery. Free Radic Biol Med. 2014;77:1–9. doi: 10.1016/j.freeradbiomed.2014.08.009. [DOI] [PubMed] [Google Scholar]
- 8.Drosatos K, Khan RS, Trent CM, Jiang H, Son NH, Blaner WS, Homma S, Schulze PC, Goldberg IJ. Peroxisome proliferator-activated receptor-gamma activation prevents sepsis-related cardiac dysfunction and mortality in mice. Circ Heart Fail. 2013;6:550–562. doi: 10.1161/CIRCHEARTFAILURE.112.000177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Smeding L, Plotz FB, Groeneveld AB, Kneyber MC. Structural changes of the heart during severe sepsis or septic shock. Shock. 2012;37:449–456. doi: 10.1097/SHK.0b013e31824c3238. [DOI] [PubMed] [Google Scholar]
- 10.Zang QS, Wolf SE, Minei JP. Sepsis-induced Cardiac Mitochondrial Damage and Potential Therapeutic Interventions in the Elderly. Aging Dis. 2014;5:137–149. doi: 10.14336/AD.2014.0500137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ferrer R, Artigas A, Levy MM, Blanco J, Gonzalez-Diaz G, Garnacho-Montero J, Ibanez J, Palencia E, Quintana M, de la Torre-Prados MV Edusepsis Study Group. Improvement in process of care and outcome after a multicenter severe sepsis educational program in Spain. JAMA. 2008;299:2294–2303. doi: 10.1001/jama.299.19.2294. [DOI] [PubMed] [Google Scholar]
- 12.Wang W, Fang H, Groom L, Cheng A, Zhang W, Liu J, Wang X, Li K, Han P, Zheng M, Yin J, Wang W, Mattson MP, Kao JP, Lakatta EG, Sheu SS, Ouyang K, Chen J, Dirksen RT, Cheng H. Superoxide flashes in single mitochondria. Cell. 2008;134:279–290. doi: 10.1016/j.cell.2008.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Feng G, Liu B, Hou T, Wang X, Cheng H. Mitochondrial flashes: elemental signaling events in eukaryotic cells. Handb Exp Pharmacol. 2017;240:403–422. doi: 10.1007/164_2016_129. [DOI] [PubMed] [Google Scholar]
- 14.Hou T, Zhang X, Xu J, Jian C, Huang Z, Ye T, Hu K, Zheng M, Gao F, Wang X, Cheng H. Synergistic triggering of superoxide flashes by mitochondrial Ca2+ uniport and basal reactive oxygen species elevation. J Biol Chem. 2013;288:4602–4612. doi: 10.1074/jbc.M112.398297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Shang W, Gao H, Lu F, Ma Q, Fang H, Sun T, Xu J, Ding Y, Lin Y, Wang Y, Wang X, Cheng H, Zheng M. Cyclophilin D regulates mitochondrial flashes and metabolism in cardiac myocytes. J Mol Cell Cardiol. 2016;91:63–71. doi: 10.1016/j.yjmcc.2015.10.036. [DOI] [PubMed] [Google Scholar]
- 16.Wei-LaPierre L, Gong G, Gerstner BJ, Ducreux S, Yule DI, Pouvreau S, Wang X, Sheu SS, Cheng H, Dirksen RT, Wang W. Respective contribution of mitochondrial superoxide and pH to mitochondria-targeted circularly permuted yellow fluorescent protein (mt-cpYFP) flash activity. J Biol Chem. 2013;288:10567–10577. doi: 10.1074/jbc.M113.455709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Santo-Domingo J, Giacomello M, Poburko D, Scorrano L, Demaurex N. OPA1 promotes pH flashes that spread between contiguous mitochondria without matrix protein exchange. EMBO J. 2013;32:1927–1940. doi: 10.1038/emboj.2013.124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Schwarzlander M, Logan DC, Johnston IG, Jones NS, Meyer AJ, Fricker MD, Sweetlove LJ. Pulsing of membrane potential in individual mitochondria: a stress-induced mechanism to regulate respiratory bioenergetics in Arabidopsis. Plant Cell. 2012;24:1188–1201. doi: 10.1105/tpc.112.096438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wei L, Salahura G, Boncompagni S, Kasischke KA, Protasi F, Sheu SS, Dirksen RT. Mitochondrial superoxide flashes: metabolic biomarkers of skeletal muscle activity and disease. FASEB J. 2011;25:3068–3078. doi: 10.1096/fj.11-187252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fang H, Chen M, Ding Y, Shang W, Xu J, Zhang X, Zhang W, Li K, Xiao Y, Gao F, Shang S, Li JC, Tian XL, Wang SQ, Zhou J, Weisleder N, Ma J, Ouyang K, Chen J, Wang X, Zheng M, Wang W, Zhang X, Cheng H. Imaging superoxide flash and metabolism-coupled mitochondrial permeability transition in living animals. Cell Res. 2011;21:1295–1304. doi: 10.1038/cr.2011.81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Azarias G, Chatton JY. Selective ion changes during spontaneous mitochondrial transients in intact astrocytes. PLoS One. 2011;6:e28505. doi: 10.1371/journal.pone.0028505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hou T, Wang X, Ma Q, Cheng H. Mitochondrial flashes: new insights into mitochondrial ROS signalling and beyond. J Physiol. 2014;592:3703–3713. doi: 10.1113/jphysiol.2014.275735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wang X, Zhang X, Huang Z, Wu D, Liu B, Zhang R, Yin R, Hou T, Jian C, Xu J, Zhao Y, Wang Y, Gao F, Cheng H. Protons Trigger Mitochondrial Flashes. Biophys J. 2016;111:386–394. doi: 10.1016/j.bpj.2016.05.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Azzu V, Brand MD. The on-off switches of the mitochondrial uncoupling proteins. Trends Biochem Sci. 2010;35:298–307. doi: 10.1016/j.tibs.2009.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Douette P, Sluse FE. Mitochondrial uncoupling proteins: new insights from functional and proteomic studies. Free Radic Biol Med. 2006;40:1097–1107. doi: 10.1016/j.freeradbiomed.2005.12.010. [DOI] [PubMed] [Google Scholar]
- 26.Echtay KS. Mitochondrial uncoupling proteins-what is their physiological role? Free Radic Biol Med. 2007;43:1351–1371. doi: 10.1016/j.freeradbiomed.2007.08.011. [DOI] [PubMed] [Google Scholar]
- 27.Hoang T, Smith MD, Jelokhani-Niaraki M. Toward understanding the mechanism of ion transport activity of neuronal uncoupling proteins UCP2, UCP4, and UCP5. Biochemistry. 2012;51:4004–4014. doi: 10.1021/bi3003378. [DOI] [PubMed] [Google Scholar]
- 28.Dikov D, Aulbach A, Muster B, Drose S, Jendrach M, Bereiter-Hahn J. Do UCP2 and mild uncoupling improve longevity? Exp Gerontol. 2010;45:586–595. doi: 10.1016/j.exger.2010.03.011. [DOI] [PubMed] [Google Scholar]
- 29.Zhang K, Liu J, You X, Kong P, Song Y, Cao L, Yang S, Wang W, Fu Q, Ma Z. P2X7 as a new target for chrysophanol to treat lipopolysaccharideinduced depression in mice. Neurosci Lett. 2016;613:60–65. doi: 10.1016/j.neulet.2015.12.043. [DOI] [PubMed] [Google Scholar]
- 30.Chen T, Guo Q, Wang H, Zhang H, Wang C, Zhang P, Meng S, Li Y, Ji H, Yan T. Effects of esculetin on lipopolysaccharide (LPS)-induced acute lung injury via regulation of RhoA/Rho Kinase/NF-small ka, CyrillicB pathways in vivo and in vitro. Free Radic Res. 2015;49:1459–1468. doi: 10.3109/10715762.2015.1087643. [DOI] [PubMed] [Google Scholar]
- 31.Chang X, He H, Zhu L, Gao J, Wei T, Ma Z, Yan T. Protective effect of apigenin on Freund’s complete adjuvant-induced arthritis in rats via inhibiting P2X7/NF-kappaB pathway. Chem Biol Interact. 2015;236:41–46. doi: 10.1016/j.cbi.2015.04.021. [DOI] [PubMed] [Google Scholar]
- 32.Pisarenko O, Shulzhenko V, Studneva I, Pelogeykina Y, Timoshin A, Anesia R, Valet P, Parini A, Kunduzova O. Structural apelin analogues: mitochondrial ROS inhibition and cardiometabolic protection in myocardial ischaemia reperfusion injury. Br J Pharmacol. 2015;172:2933–2945. doi: 10.1111/bph.13038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Xiang P, Chen T, Mou Y, Wu H, Xie P, Lu G, Gong X, Hu Q, Zhang Y, Ji H. NZ suppresses TLR4/NF-kappaB signalings and NLRP3 inflammasome activation in LPS-induced RAW264.7 macrophages. Inflamm Res. 2015;64:799–808. doi: 10.1007/s00011-015-0863-4. [DOI] [PubMed] [Google Scholar]
- 34.Chen HW, Hsu C, Lu TS, Wang SJ, Yang RC. Heat shock pretreatment prevents cardiac mitochondrial dysfunction during sepsis. Shock. 2003;20:274–279. doi: 10.1097/00024382-200309000-00013. [DOI] [PubMed] [Google Scholar]
- 35.Rocha M, Herance R, Rovira S, Hernandez-Mijares A, Victor VM. Mitochondrial dysfunction and antioxidant therapy in sepsis. Infect Disord Drug Targets. 2012;12:161–178. doi: 10.2174/187152612800100189. [DOI] [PubMed] [Google Scholar]
- 36.Peng T, Lu X, Feng Q. Pivotal role of gp91phox-containing NADH oxidase in lipopolysaccharide-induced tumor necrosis factor-alpha expression and myocardial depression. Circulation. 2005;111:1637–1644. doi: 10.1161/01.CIR.0000160366.50210.E9. [DOI] [PubMed] [Google Scholar]
- 37.Zhang ZH, Yu Y, Wei SG, Felder RB. Centrally administered lipopolysaccharide elicits sympathetic excitation via NAD(P)H oxidase-dependent mitogen-activated protein kinase signaling. J Hypertens. 2010;28:806–816. doi: 10.1097/HJH.0b013e3283358b6e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Baines CP. The mitochondrial permeability transition pore and the cardiac necrotic program. Pediatr Cardiol. 2011;32:258–262. doi: 10.1007/s00246-010-9880-9. [DOI] [PubMed] [Google Scholar]
- 39.Chen JK, Chow SE. Antioxidants and myocardial ischemia: reperfusion injuries. Chang Gung Med J. 2005;28:369–377. [PubMed] [Google Scholar]
- 40.Toda C, Diano S. Mitochondrial UCP2 in the central regulation of metabolism. Best Pract Res Clin Endocrinol Metab. 2014;28:757–764. doi: 10.1016/j.beem.2014.02.006. [DOI] [PubMed] [Google Scholar]
- 41.Kizaki T, Suzuki K, Hitomi Y, Taniguchi N, Saitoh D, Watanabe K, Onoe K, Day NK, Good RA, Ohno H. Uncoupling protein 2 plays an important role in nitric oxide production of lipopolysaccharide-stimulated macrophages. Proc Natl Acad Sci U S A. 2002;99:9392–9397. doi: 10.1073/pnas.142206299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Blanc J, Alves-Guerra MC, Esposito B, Rousset S, Gourdy P, Ricquier D, Tedgui A, Miroux B, Mallat Z. Protective role of uncoupling protein 2 in atherosclerosis. Circulation. 2003;107:388–390. doi: 10.1161/01.cir.0000051722.66074.60. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.