

ABSTRACT
The actin cytoskeleton plays key roles in every eukaryotic cell and is essential for cell adhesion, migration, mechanosensing, and contractility in muscle and non-muscle tissues. In higher vertebrates, from birds through to mammals, actin is represented by a family of six conserved genes. Although these genes have evolved independently for more than 100 million years, they encode proteins with ≥94% sequence identity, which are differentially expressed in different tissues, and tightly regulated throughout embryogenesis and adulthood. It has been previously suggested that the existence of such similar actin genes is a fail-safe mechanism to preserve the essential function of actin through redundancy. However, knockout studies in mice and other organisms demonstrate that the different actins have distinct biological roles. The mechanisms maintaining this distinction have been debated in the literature for decades. This Review summarizes data on the functional regulation of different actin isoforms, and the mechanisms that lead to their different biological roles in vivo. We focus here on recent studies demonstrating that at least some actin functions are regulated beyond the amino acid level at the level of the actin nucleotide sequence.
KEY WORDS: Actin, Isoform, Regulation
Summary: We propose a new concept, the actin code, which encompasses the regulation of the essential functions of mammalian actins at the nucleotide level, rather than at the level of amino acids.
Introduction
Actin is one of the most abundant intracellular proteins that is present in every eukaryotic cell and plays key roles in many essential biological processes (Box 1). In vertebrates, the six actin isoforms (Table 1) are nearly identical in their amino acid sequences, which are also conserved between species, and yet they are encoded by a set of different evolutionarily conserved genes (see Ampe and Van Troys, 2017; Simiczyjew et al., 2017 for reviews). Despite this high similarity, knockout studies demonstrate that the different actins have distinct biological roles (Perrin and Ervasti, 2010; Wagner et al., 2002). The mechanisms maintaining this distinction have been debated in the literature for decades.
Box 1. Actin isoforms.
Actin was first observed in muscle extracts as a protein that can induce coagulation of the muscle plasma by W. D. Halliburton in 1887 (Halliburton, 1887). Later, muscle actin was purified by Brunó Ferenc Straub in 1942, who named it ‘actin’ because of its ability to activate the motor protein myosin (Straub, 1942). Along with microtubules and intermediate filaments, actin is a major component of the cytoskeleton that defines global and local cellular architecture. Decades of research on the actin cytoskeleton has revealed a plethora of essential roles of actin in cell migration, cell adhesion, mitochondrial dynamics, muscle contraction, protein trafficking, cell division, membrane organization, mechanotransduction and tension sensing, nuclear matrix association, chromatin remodeling and regulation of transcription (Ampe and Van Troys, 2017; Bruser and Bogdan, 2017; Galkin et al., 2012; Lehtimaki et al., 2017; Luxenburg and Geiger, 2017; Pollard, 2017; Sanger et al., 2017; Sheterline et al., 1998; Viita and Vartiainen, 2017).
In the late 1970s, mammalian actins were sequenced by amino acid hydrolysis (Vandekerckhove and Weber, 1978a,b, 1981), revealing that actin in vivo is represented by multiple isoforms, which are nearly identical, except for a few amino acid substitutions. These isoforms were classified as α-, β- and γ-actins, based on their isoelectric points, named in ascending order of their acidity (Vandekerckhove and Weber, 1978a). A further sub-classification was later made based on their tissue-specific expression. Six different mammalian actin isoforms are known to date, four of them predominant in different types of muscle (α-cardiac, α-skeletal, α-smooth muscle and γ-smooth muscle actins), and two majorly non-muscle isoforms, β- and γ-actin (Erba et al., 1988; Gunning et al., 1983; Miwa et al., 1991; Vandekerckhove and Weber, 1978a,b) (Table 1). Each of these isoforms is encoded by a unique gene (Pollard, 2001), and has a highly regulated spatial and temporal expression during development and homeostasis (Andrade, 2015; McHugh et al., 1991; Tondeleir et al., 2009).
Table 1.
Tissue-specific expression patterns of actin isoforms in higher mammals

This Review summarizes data on the functional regulation of different actin isoforms and the mechanisms that lead to their different biological roles in vivo. We place a special focus on β- and γ-non-muscle actins, which are the most similar in amino acid sequence and are both ubiquitously present in all cell types, but play vastly different biological roles (Ampe and Van Troys, 2017; Perrin and Ervasti, 2010; Simiczyjew et al., 2017; see also Box 1). We specifically emphasize recent data highlighting the essential role of the nucleotide sequence in actin function and introduce a new concept – the ‘actin code’ – that reflects regulation of the essential functions of β- and γ-actin at the nucleotide level, a mechanism that is potentially broadly applicable to other members of the actin family.
Actin isoforms
Actin isoforms are among the most conserved proteins at the level of amino acid sequence, not only within the same organism, but also between vertebrate species from birds to mammals (Erba et al., 1986). Such high conservation makes it clear that the actin amino acid sequence can tolerate very little change, in contrast to the bacterial actin homologs MreB, FtsA and ParM, which are highly divergent in their amino acid sequence. In line with these observations is the evolutionary history of actin. It has been proposed that different degrees of sequence identity within the actin family are due to different levels of selective pressure imposed by actin-interacting partners. The bacterial actins have very few interacting partners, and thus exist under very low selective pressure. Vertebrate actins interact with over 100 different proteins through virtually every exposed residue on their surface, resulting in very high selective pressure for sequence conservation (Gunning et al., 2015). Another level of evolution in vertebrate actins arose through proposed gene duplication events prior to avian evolution. One round of gene duplication is proposed to have given rise to the highly similar non-muscle β- and γ-actin, while two rounds of gene duplication generated the more divergent set of the muscle actins (Erba et al., 1988; Erba et al., 1986; Miwa et al., 1991). Notably, Erba et al. (1986) observed and characterized the ‘anomalous’ evolution of β- and γ-actin genes following their divergence in birds, with a greater conservation in their nucleotide sequence than would be predicted based on the evolutionary time – an indicator of selection pressure above and beyond regular conservation of the amino acid sequence.
Functionality of amino acid-based differences in actin isoforms
Structure and biochemical properties
All actin isoforms differ markedly within the three or four residues at their extreme N-terminus; a region of the actin molecule that is exposed on the surface of actin monomers and filaments (Fig. 1; see Ampe and Van Troys, 2017; Simiczyjew et al., 2017 for reviews). Notably, however, these substitutions are largely conservative, and result in only a small overall change in the negative charge in this region. Changes in other residues located in both the buried and exposed sites of the folded actin molecule are predicted to lead to subtle structural changes that can potentially affect a number of the properties and functional interactions of actin (McKane et al., 2006; Mounier and Sparrow, 1997).
Fig. 1.

Structural comparison of mouse actin isoforms. (A) Primary sequence alignment of mouse actins. Internal residue differences are shown in bold; blue indicates substitutions unique to one isoform, green for those found in two or three isoforms and black for the residues found in the majority of the isoforms. Differences in N-terminal residues are denoted in red. See Table 1 for the protein names that correspond to the gene symbols listed on the top left. (B) Structural maps of the actin isoforms. Top and bottom rows show the ‘front’ and ‘back’ views of the same molecule, with the positions containing different residues in different isoforms denoted in yellow over the maroon color for the residues identical in all the compared isoforms. Nt, N-terminus (shown partially due to the lack of structural data in this region). Images were prepared using Cn3D macromolecular structure viewer (NIH), based on the structure of cardiac α-actin (PDB identifier 1J6Z).
Actin isoforms are reported to exhibit different biophysical and biochemical properties in vitro (Allen et al., 1996; Gordon et al., 1977; Just et al., 1994). However, such studies have been limited, largely because of a shortage of readily available methods to purify specific actin isoforms. Each cell type contains more than one actin isoform, and these isoforms always co-purify due to their high similarities in primary amino acid sequences and biochemical properties. Some of these problems can be alleviated by using the tissue specificity of actin isoforms. For example, cardiac and skeletal muscles contain predominantly α-cardiac or α-skeletal actin, respectively, and thus these two actins have been studied in vitro more extensively than others. Actin purified from platelets contains 80-90% β-actin, providing a source for enriched β-actin preparations. However, no procedure for isolation of the other actin isoforms is available. An early study reported differential binding of complexes between non-muscle β- and γ-actin and profilin to poly-proline, suggesting a potential method for isolation of these two actin isoforms from the same cells (Lindberg et al., 1988); but to date, no consistent biochemical differences between β- and γ-actin prepared by this method have been reported.
Since actins require an ensemble of modifying enzymes and chaperones for proper folding and maturation, their expression in native form in bacteria has proved difficult. Yeast cells lack a methyltransferase, required for the essential His73 methylation in mammalian actin isoforms (Nyman et al., 2002), in addition to incompletely processing the N-terminus (Cook et al., 1991; Kalhor et al., 1999). Expression in the baculovirus-Sf9 insect cell system produces correctly folded and processed actin (Bai et al., 2014); however, these preparations still contain ∼30% endogenous Sf9 actin, which is difficult to detect or quantify as a result of high similarities in sequence and electrophoretic mobility.
Using baculovirus-expressed β- and γ-actin, it was found that these actin isoforms can form heteropolymers, but exhibit differences in their polymerization kinetics, Pi release and treadmilling (faster for β-actin than for γ-actin) (Bergeron et al., 2010). The ability of non-muscle actins to copolymerize was later verified by Förster resonance energy transfer (FRET; Müller et al., 2013). Interestingly, biochemical differences between β- and γ-actin were exacerbated when starting with Ca2+ actin rather than Mg2+ actin (Bergeron et al., 2010). While Mg2+ is the more physiologically relevant ion, the authors propose that the differences observed with Ca2+ ions may reflect local calcium fluxes in vivo, which would allow the cells to rapidly respond to signals by using a specific actin isoform (Bergeron et al., 2010). More recently, it was shown that both platelet actin (85% β-actin and 15% γ-actin) and chicken gizzard actin (75% γ-actin and 25% β-actin), when polymerized in vitro, yield a mixture of homopolymers of β- and γ-actin (Chen et al., 2017), rather than heteropolymers, as revealed by staining the polymerized actin filaments on a coverslip with antibodies against β- and γ-actin. This could be because gizzard γ-actin is composed predominantly of γ-smooth muscle rather than the γ-cytoplasmic actin isoform, and thus this result by Chen et al. (2017) is likely consistent with prior observations that muscle actins are not interchangeable with non-muscle actins in intracellular structures (Kaech et al., 1997; Mounier et al., 1997). Overall, the question of homo- and heteropolymer formation by the actin isoforms remains open, since in vitro studies have not fully recapitulated the mechanism of in vivo polymerization and in vivo studies have not produced convincing data for isoform mixing within a single filament.
Binding to actin-associated proteins
Reports in the literature show preferential binding of β-actin to myosin 2B and tropomyosin (Pathan-Chhatbar et al., 2018), as well as the existence of the β-actin-specific capping protein betacap73 (Shuster et al., 1996; Welch et al., 2005) and nucleator DIAPH3 (Chen et al., 2017). It has been found that depletion of intracellular cofilin strongly affects the β-actin filament to monomer ratio, but has a much weaker effect on γ-actin (Kapustina et al., 2016). β-actin performs better than γ-actin in activation of non-muscle myosin 2C1, and worse than γ-actin in activation assays with myosin 7a (Müller et al., 2013). Finally, a number of studies showed a broader difference between muscle and non-muscle actins binding to profilin (Ohshima et al., 1989), thymosin β4 (Weber et al., 1992), ezrin (Shuster and Herman, 1995; Yao et al., 1995) and plastin (Prassler et al., 1997). It has been assumed that all of these preferences are mediated directly by the differences in the primary amino acid sequences between actin isoforms. However, it is also possible that some of these interactions are mediated through indirect regulatory mechanisms, for instance, unique post-translational modifications differentially targeting β- and γ-actin. These mechanisms need to be further explored.
Differential post-translational modifications of actin isoforms
Actin is known to be extensively post-translationally modified (see Terman and Kashina, 2013). Nearly all of the intracellular actins, both muscle and non-muscle isoforms, have their N-terminus acetylated in a multi-step process that is closely coupled to actin synthesis (Martin and Rubenstein, 1987; Rubenstein and Martin, 1983; Sheff and Rubenstein, 1989; Strauch and Rubenstein, 1984). Physiological actin preparations have been found to contain nearly every known type of post-translational modification, including phosphorylation, oxidation, sumoylation and methylation, affecting every potentially reactive residue of the actin surface (Terman and Kashina, 2013). Because of high sequence similarity between actin isoforms, it is still unknown whether these modifications are specific to certain actin isoforms or whether they target all actins uniformly. One known exception is N-terminal arginylation, a post-translational modification that targets β-actin and not the highly similar γ-actin (Karakozova et al., 2006; Zhang et al., 2010). This differential arginylation is regulated at the level of the actin coding sequence (Zhang et al., 2010). However, very little is known about the specific role of arginylation, or any other actin modification, in its biological functions.
Actin isoforms play divergent roles in cells
It was observed relatively early that muscle actin isoforms cannot replace the cytoplasmic actins in certain cellular structures (Kaech et al., 1997; Mounier et al., 1997) and conversely, cytoplasmic actins cannot replace the muscle actins (Antin and Ordahl, 1991; Fyrberg et al., 1998; von Arx et al., 1995). Immunolocalization studies indicate a differential enrichment of the muscle and non-muscle actin isoforms in motile pericytes (DeNofrio et al., 1989). These results suggest that the functional distinction between muscle and non-muscle actins is higher than expected based on their sequence and structural similarity.
For decades, functional similarities between non-muscle β- and γ-actin have been extensively debated in the literature. Recurring studies report prominent differences in intracellular localization of these two actins (Dugina et al., 2016; Dugina et al., 2009), whereas others have observed their similar distribution in cells (Bunnell et al., 2011; Otey et al., 1988; Otey et al., 1986; Vedula et al., 2017). β- and γ-actin are both ubiquitously expressed and coexist at comparable levels in many cell types, even though their relative abundances tend to vary (Khaitlina, 2001; Patrinostro et al., 2017; Vedula et al., 2017).
Some of the differences in the localization of β- and γ-actin have been partially explained by the finding that β-actin mRNA is specifically targeted to the cell periphery through a zipcode-binding sequence located in its 3′ untranslated region (UTR) (Kislauskis et al., 1993; Kislauskis et al., 1994). A substantial fraction of β-actin mRNA (an estimated 10%) is localized to the leading edge in motile cells through this mechanism, and this localization is required for directional cell motility (Condeelis and Singer, 2005; Katz et al., 2012; Lawrence and Singer, 1986). γ-actin mRNA shows no such targeting (Hill and Gunning, 1993). Consequently, staining of motile non-muscle cells with antibodies against β-actin tends to highlight a prominent zone at the leading edge (Dugina et al., 2009; Karakozova et al., 2006), regardless of its distribution in the rest of the cell. Overall, these results are somewhat inconsistent between different studies, probably as a result of highly dynamic actin localization in the cells, as well as variables such as different actin visualization tools, different tissue and cell types, cell migratory activity and physiological state.
Studies of cultured cells derived from β- and γ-actin knockout mice showed that β-actin, unlike γ-actin, is specifically required for cell migration and proliferation (Bunnell et al., 2011; Tondeleir et al., 2012). β-actin is more essential than γ-actin in cell-matrix and cell-cell adhesions (Baranwal et al., 2012; Bunnell et al., 2011; Sheterline et al., 1998), likely as a consequence of its mRNA targeting to the cell periphery and local translation (Gutierrez et al., 2014; Katz et al., 2012; Rodriguez et al., 2006). A recent report demonstrated that β-actin filaments preferentially nucleate at the cytokinetic ring (Chen et al., 2017). Disruption of the β-actin gene causes defects in neural crest cell migration, vasculature and erythropoiesis (Shawlot et al., 1998; Tondeleir et al., 2013, 2014), as well as impairments in gene expression and cell signaling (Patrinostro, et al., 2017). β-actin protein and mRNA localize to highly dynamic structures in neurons, where it is required for growth cone extension and remodeling of dendritic spines, both in vivo and in culture (Bassell et al., 1998; Buxbaum et al., 2014; Micheva et al., 1998; Zhang et al., 1999). Only one study so far, using siRNA-mediated knockdowns, suggested a potential involvement of γ-actin, rather than β-actin, in cell motility (Dugina et al., 2009) – a discrepancy that may be due to cell-type-specific effects. It should also be noted that repression of one of the actin isoforms normally leads to compensatory upregulation of the other(s), to maintain constant total actin levels in cells (see Perrin and Ervasti, 2010), and this potential cross-compensation needs to be considered in the interpretation of these studies.
In addition to its cytoplasmic localization, actin is also targeted to the nucleus (Falahzadeh et al., 2015). Most of the studies report this targeting to be β-actin specific, linked to transcription by RNA polymerase (Pol) II and III (Hofmann et al., 2004; Hu et al., 2004). Cells derived from β-actin knockout mice show permanent changes in gene expression and have defects in transcription and epigenetic regulation (Almuzzaini et al., 2016; Tondeleir et al., 2012). However, a recent report described a small amount of γ-actin in the nucleus of melanoma cells, which, although considerably lower than nuclear β-actin levels, also showed partial co-localization with RNA Pol II and heterogeneous nuclear ribonucleoprotein (hnRNP) U (Migocka-Patrzalek et al., 2015). Thus, it is possible that γ-actin also plays a minor nuclear role, at least in some cell types.
Non-redundant roles of actin isoforms at the organismal level
The most decisive demonstration of unique functions for the different actin isoforms has been provided by using gene knockouts in mice (see Perrin and Ervasti, 2010) and Drosophila – another eukaryotic organism with six actin genes that play equivalent roles in muscle and non-muscle systems (Fyrberg et al., 1998). Below, we focus on the mammalian actins and the data obtained in mouse models (see Table 2).
Table 2.
Non-redundant biological roles of mouse actin isoforms

Mice lacking α-cardiac actin show embryonic and/or perinatal lethality and disorganized myofibril organization in the heart tissue (Kumar et al., 1997). Interestingly, heart-specific expression of γ-smooth muscle actin in these mice, driven by the heart-specific α-myosin heavy chain promoter, rescues the perinatal lethality and enables these mice to survive to adulthood (Kumar et al., 1997). However, the hearts in these mice still show hypertrophy and are enlarged and hypodynamic. These experiments suggest that while γ-smooth muscle actin can compensate for the loss of α-cardiac actin, the extent of such functional compensation is not complete (Kumar et al., 1997). In humans, mutations in α-cardiac actin have been linked to dilated cardiomyopathy (DCM) (Olson et al., 1998) and hypertrophic cardiomyopathy (HCM) (Arad et al., 2005; Mogensen et al., 1999, 2004; Olson et al., 2000; Van Driest et al., 2003). Although an extensive biochemical characterization of these mutations has not been performed, the two mutations associated with DCM have been proposed to affect force transmission (Mogensen et al., 1999), whereas the seven mutations found to be associated with HCM are thought to affect sarcomere assembly and acto-myosin interactions (Mogensen et al., 2004; Olson et al., 2000).
Gene knockout of α-skeletal actin in mice causes muscle weakness and death within the first nine days of birth (Crawford et al., 2002). Mice lacking α-skeletal actin have increased levels of α-smooth and α-cardiac actin expressed in the skeletal muscle; however, these other actin isoforms do not completely compensate for the lack of functional α-skeletal actin (Crawford et al., 2002). Intriguingly, transgenic overexpression of γ-cytoplasmic actin in skeletal muscle results in a corresponding decrease in α-skeletal actin, without significant changes to muscle morphology or function (Jaeger et al., 2009). However, this transgenic expression does not rescue the α-skeletal muscle actin knockout. Thus, while γ-cytoplasmic actin is able to at least partially replace α-skeletal muscle actin in sarcomeres, it cannot substitute for all α-actin functions. Mutations in α-skeletal muscle actin gene have been linked to three congenital myopathic disorders: congenital fiber type disproportion (CFTD), Nemaline myopathy (NM) and central core disease (CCD) (Agrawal et al., 2004; Goebel et al., 2006; Nowak et al., 2013; Wallefeld et al., 2006). NM is the most well studied, and is a complex disease with over 200 mutations. Even though only 20% of these are in α-skeletal muscle actin, they account for over half of the severe NM cases (Nowak et al., 2013; Wallgren-Pettersson et al., 2011). There is, however, a large gap in our understanding of the effects of these mutations on the biochemical properties of actin and the mechanisms of disease progression.
Mice with a knockout of α-smooth muscle actin are viable and morphologically normal, but show defects in vascular contractility and blood pressure regulation (Schildmeyer et al., 2000). Mutations in α-smooth muscle actin have been linked to thoracic aortic aneurysms and dissections (TAADs) (Guo et al., 2007). Nine missense mutations have been identified in the α-smooth muscle actin gene that lead to vascular defects, accompanied by a reduced fraction of actin filaments and an increase in monomeric actin in vascular tissues, but the underlying mechanisms are unknown.
Knockout of γ-cytoplasmic actin in mice leads to growth defects, reduced viability and progressive deafness (Belyantseva et al., 2009; Bunnell and Ervasti, 2010). Conditional γ-cytoplasmic actin knockout in the skeletal muscle results in progressive myopathy (Sonnemann et al., 2006) and affects muscle relaxation (O'Rourke et al., 2018). Seven different autosomal dominant missense mutations in the γ-actin gene have been linked to Baraitser-Winter syndrome (Kemerley et al., 2017) and progressive hearing loss in humans: T278I, T89I, K118M, P264L, P332A, I122V and V370A (Bryan et al., 2006; Liu et al., 2008; Rendtorff et al., 2006; van Wijk et al., 2003; Zhu et al., 2003). Together, these studies indicate an essential role for γ-actin in the maintenance of stereocilia, even though this actin isoform appears to be largely dispensable for the viability of an organism.
Disruption of the β-actin gene in mice causes by far the most severe phenotype, leading to early embryonic lethality. This has been recapitulated by using two different approaches: insertion of reporter genes (Shawlot et al., 1998; Shmerling et al., 2005; Strathdee et al., 2008; Tondeleir et al., 2013, 2014) and the targeted deletion of protein-coding exons by using floxed alleles (Bunnell et al., 2011). Mice lacking β-actin die in early embryogenesis, prior to embryonic day 8.5 (E8.5) (Bunnell et al., 2011). Conditional knockout of β-actin in the skeletal muscle leads to quadriceps myopathy (Prins et al., 2011) and impairs muscle relaxation (O'Rourke et al., 2018). Two mutations in the human β-actin gene have been characterized: E364 K mutation, which has been correlated to neutrophil dysfunction (Nunoi et al., 1999), and R183W, linked to developmental malformations, dystonia and deafness (Procaccio et al., 2006). Human β-actin mutations, along with those in γ-cytoplasmic actin, have been linked to Baraitser-Winter syndrome (Kemerley et al., 2017). The severity of these mutations, as well as the lack of any observed homozygous mutations in the β-actin gene, present additional evidence of its highly essential biological role.
Notably, the mouse knockout phenotypes discussed above, are not a result of a reduction in overall actin levels. In all cases, knockout of a single actin isoforms leads to the upregulation of the other isoforms to maintain a constant amount of actin protein (Table 2), suggesting the existence of global ‘dosage-sensing’ mechanisms that ensure the constitutive abundance of the total actin. This dosage, however, does not result in total functional compensation in most cases. The most striking example is non-muscle β- and γ-actin, which have dramatically different mouse knockout phenotypes, despite the fact that these two actin isoforms always coexist in the same cells and are highly similar in their biochemical properties. None of the actin isoforms, despite their upregulation in β-actin knockout mice, can functionally compensate for its loss. The mechanisms that maintain this functional distinction between actin isoforms are unknown.
Nucleotide sequence as a determinant of actin function
In all the studies discussed so far, it has been consistently assumed that the underlying mechanisms conferring different functions to the actin isoforms arise at the amino acid level (with the exception of 3′UTR zipcode targeting of β-actin mRNA; Kislauskis et al., 1993). Researchers have not tended to look beyond these differences, with the exception of just two studies: one demonstrating a role for intron III in the γ-actin gene in γ-actin-dependent effects on myoblast morphology (Lloyd and Gunning., 1993) and the other showing that the nucleotide-coding sequence, rather than the amino acid sequence, can define the differential post-translational arginylation of β- and γ-actin (Zhang et al., 2010). Is it possible that the determinants of actin isoform function reside at the nucleotide level and have little to do with their specific amino acid variations?
The nucleotide sequence of β-actin, rather than its amino acid sequence, determines its essential role in vivo
To directly test the contribution of the actin nucleotide sequence to its function, a recent study (Vedula et al., 2017) utilized CRISPR/Cas9 to introduce five point mutations into the mouse β-actin gene (which is essential for mouse embryogenesis and viability) that changed four divergent codons in its 5′ end into those that encode the amino acids specific for γ-actin (which is much more dispensable for mouse survival) (Fig. 2). In the resulting mice, which express the so-called ‘β-coded γ-actin’ (termed Actbc-g based on the gene nomenclature), no β-actin protein is present, but the nucleotide sequence of the β-actin gene remains nearly intact, with only five nucleotide substitutions, and this gene produces functional γ-actin protein. Thus, this mouse model enables a definitive answer to the question of whether the β-actin-specific amino acid sequence, or the nucleotide sequence of its gene, is required for its essential in vivo function.
Fig. 2.
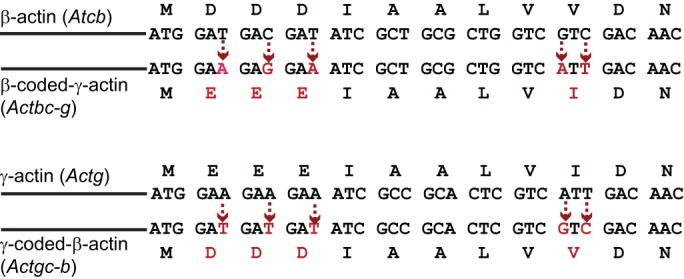
Gene editing strategy for generation of Actbc-g and Actgc-b mice, producing β-coded γ-actin and γ-coded β-actin, respectively. Sequence alignment of the 5′ regions of β- and γ-actin coding sequences, starting with the first ATG. The nucleotides, replaced during the editing to generate the amino acid sequence of the other actin isoform, are indicated in red. Dotted arrows show the direction of the replacement in each experiment.
Remarkably, Actbc-g mice have no defects in viability, survival, fertility or cell migration, and no detectable phenotype (Vedula et al., 2017) – in contrast to the embryonic lethality seen in β-actin knockout mice (Bunnell et al., 2011). Therefore, it is the intact actin gene, not the protein, that determines essential functions of β-actin in the survival of an organism, while the differences in the amino acid sequences between β- and γ-actin are functionally dispensable for animal survival and cell migration. An equivalent experiment was also performed for γ-actin, with a similar result (Vedula et al., 2017). Thus, nucleotide sequence, rather than the amino acid sequence, determines the in vivo function of non-muscle actin isoforms.
Taken together, this work significantly shifts our understanding of the essential determinants that govern the functions of actin isoforms, and shows that some of their key functional determinants, if not all, reside at the nucleotide level. Below, we discuss the potential contribution of each of the nucleotide-based gene elements to the essential function of β-actin.
Promoter and gene expression regulation
Gene promoters interact with regulatory elements to control the tissue specificity and relative abundance of mRNA for each encoded protein in a cell. This is reflected very well for the muscle actins, which are dominantly expressed only in specific muscle tissues. However, β- and γ-actin promoters are ubiquitously active and remarkably conserved, consistent with their likely origin from a gene duplication event (Erba et al., 1988, 1986). At the same time, the levels of β- and γ-actin and their ratios in different tissues are differentially regulated during development and adulthood (Andrade, 2015; McHugh et al., 1991). In most cell types, the relative amounts of β-actin and γ-actin are within a twofold level of variation, with the exception of the stereocilia in inner hair cells and microvilli in intestinal epithelial cells, which are prevalent in γ-actin (Andrade, 2015; Khaitlina, 2001; Vandekerckhove and Weber, 1981), as well as platelets, which contain predominantly β-actin (Chen et al., 2017). The ratios of the mRNAs for the two actin isoforms are different, often much higher than that of the proteins, indicating that the relative protein abundance between these two actins is regulated at the post-transcriptional level, probably to a greater extent than by their promoters (Erba et al., 1988; Patrinostro et al., 2017). Thus, promoter-level regulation is unlikely to substantially contribute to the functional differences between β- and γ-actin in organismal survival.
Introns
The γ-actin gene contains a unique and highly conserved intron III (Lloyd and Gunning, 1993) and an alternatively spliced exon 3a (Drummond and Friderici, 2013). While the intron III of the γ-actin gene has been shown to play a role in regulating actin structures in cells, the alternatively spliced exon 3a, when included, introduces a premature stop codon, resulting in nonsense-mediated decay of γ-actin transcript. These data suggest that intron regions are likely to be important for the functionality of the actin genes, even though their role is virtually unexplored.
It is worth noting, however, that in most of the studies on β-actin disruption, the intron sequences were intact, with the exception of the work by Bunnell et al. (2011), where one intron of the β-actin gene, between exons 2 and 3, was deleted. All these strategies led to a largely similar phenotype (Shawlot et al., 1998; Shmerling et al., 2005; Strathdee et al., 2008; Tondeleir et al., 2013, 2014), suggesting that the introns are unlikely to play a specific role in the essential function of β-actin. Further experiments are needed to fully test the role of introns in actin function.
Untranslated regions of mRNA
Given the fact that the relative abundance of intracellular β- and γ-actin mRNA is not directly reflective of the ratios of their protein levels, it seems likely that some post-transcriptional control is employed to modulate the translation and accumulation of these actins in the cell (for a comprehensive review, see Simiczyjew et al., 2017). For instance, it has been shown that an alternative polyA site in β-actin mRNA increases its translation (Ghosh et al., 2008). β-actin also exhibits both 3′UTR-dependent (Ghosh et al., 2008; Lyubimova et al., 1999) and 3′UTR-independent (Lloyd et al., 1992; Schevzov et al., 1992) feedback regulation of its own expression. The exogenous expression of different actin isoforms in cells causes a differential feedback regulation that has been attributed to the 3′UTR, promoter and the protein itself (Ballestrem et al., 1998; Lloyd et al., 1992; Lyubimova et al., 1999; Schevzov et al., 1992).
The unique 3′UTR of β-actin contains the mRNA localization signal that targets it to the cell periphery (Condeelis and Singer, 2005; Kislauskis et al., 1993, 1994). This region has also been shown to mediate the epigenetic regulation of the promoter and gene expression (Strathdee et al., 2008). Indeed, transgenic insertion of the lacZ reporter into the β-actin 3′UTR downstream of the mRNA localization sequence resulted in pre-implantation lethality in mice as a result of epigenetic silencing of the modified alleles (Strathdee et al., 2008). However, the molecular mechanism for how this distal 3′UTR regulates gene expression through epigenetic silencing remains to be understood.
Coding sequence
A previous study, focused on characterizing migration defects in cells derived from β-actin knockout mice, describes a rescue experiment, where insertion of the coding sequence of human β-actin into the deletion site (between exons 2 and 3) of these mice rescues embryonic lethality (Tondeleir et al., 2012). Although the data for the detailed characterization of this rescue experiment were not presented in the manuscript, this result nevertheless suggests that the coding sequence of actin is sufficient to rescue its essential function, even if this coding sequence comes from another vertebrate species.
Comparison of the β- and γ-actin coding sequences reveals a 13% difference in their mRNAs – a much higher divergence compared with their near identity at the amino acid level (Zhang et al., 2010). These synonymous substitutions are only 40% randomized (Erba et al., 1986). Typically, genes that have diverged for more than 100 million years acquire sufficient mutations for the synonymous substitutions to be completely randomized, thus allowing them to serve as an evolutionary clock (Perler et al., 1980). The anomalously high conservation of the substitutions in the β- and γ-actin codons suggests that this conservation has an independent functional significance.
The actin code: silent substitutions in the coding sequence define actin isoform function by regulating their translation dynamics
So, how can differences in the nucleotide coding sequences through synonymous substitutions account for the divergence of actin functions? One of the major known effects of the coding sequence relates to the potential differences in codon usage and secondary structure of mRNA, which can lead to different ribosome translocation dynamics, and thus to different rates of translation. In the case of β- and γ-actin, coding sequence differences give rise to a different secondary structure. A stable loop is present in the 5′ coding region of γ-actin, but is not prominent in the corresponding region of β-actin, leading to slower ribosome translocation over γ-actin mRNA in this region (Zhang et al., 2010).
Translation dynamics for any given mRNA can be estimated from global ribosome profiling data that are publicly available, such as via the GWIPS-viz genome browser (http://gwips.ucc.ie) (Michel et al., 2014), which aggregates the results of multiple ribosome profiling studies across genomes. Our search of these databases for mouse actin isoforms revealed that the composite ribosome density (averaged from 26 independent studies) over the first 150 codons of β-actin mRNA (1351) is over a thousand times higher than that of γ-actin (∼1.3) (Vedula et al., 2017) (Fig. 3). This suggests that, in vivo, the translation dynamics of β-actin is dramatically different from that of γ-actin.
Fig. 3.

Composite ribosome density correlates with actin isoform function. Schematic representation of ribosome density on the translating mRNA for the 6 mouse actin isoforms, derived from the composite ribosome density calculated from the datasets deposited to the GWIPS-viz genome browser (listed on the left for the corresponding mouse mRNAs). The direction of arrows on the right indicates the functional compensation of the target actin isoform (at the end of the arrow) by the donor one (at the beginning of the arrow). Black arrows indicate full or near-full functional compensation. Gray arrow indicates major upregulation without apparent functional compensation. Generally, only the actins with a ribosome density closely resembling the ones they are replacing appear to be effective in compensating for those that exhibit a lower ribosome density. β-actin appears to be the most effective in compensating for multiple other actin isoforms, but cannot be compensated for, even by an upregulation of the isoforms with closest ribosome densities.
A high ribosome occupancy is normally interpreted as an indicator of a high translation rate. However, this appears unlikely to be the case for β-actin. According to a recent study that looked at translation rates of nascent chains, β-actin can be translated at a rate of three actin molecules per minute (Morisaki et al., 2016). However, β-actin mRNA is highly abundant, with some studies reporting estimates of 2500 β-actin mRNAs per cell (Condeelis and Singer, 2005; Kislauskis et al., 1997), while others find this number to be variable (Islam et al., 2011). Therefore, assuming 2500 molecules per cell, the constitutive translation of all β-actin mRNA would result in the production of 7500 actin molecules per minute, a rate that could quickly overpower the cell with an extreme abundance of actin. As β-actin is normally maintained at a high but constant intracellular level (∼1% of total protein), it is clear that some mechanisms must exist that prevent its continuous translation.
An alternative possibility, which is also consistent with higher ribosome occupancy, is that β-actin mRNA is maintained in a state that is constantly translationally repressed, but is nevertheless primed and translation ready, so that as soon as this repression is released, β-actin can accumulate by a localized ‘burst’ of de novo produced actin monomer (Strohl et al., 2017). Both active protein synthesis and β-actin mRNA localization to the cell leading edge, have been previously shown to be required for directional cell migration, indirectly suggesting that cell migration needs localized actin translation (Buxbaum et al., 2014; Katz et al., 2012). Bursts of de novo synthesized actin in localized spots at the cell leading edge have also been previously proposed based on live-imaging studies, in which ‘hot spots’ of actin were seen appearing at the cell leading edge, too fast to be explained by diffusion (Zicha et al., 2003). Furthermore, bursts of actin translation have also been directly observed in neurons by using fast-folding and fast-bleaching fluorescent tags or photo-convertible tags as translation reporters (Buxbaum et al., 2014; Strohl et al., 2017). Based on the length of the actin coding region (1125 nucleotides) and the estimated ribosome footprint (28 nucleotides), bursting translation from each β-actin mRNA, at a theoretical maximum, can produce a localized pool of ∼40 actin subunits. If multiple mRNA molecules are translated in the same place (e.g. ∼250 estimated to be present at the cell leading edge), this would result in a localized production of 10,000 actin subunits per burst, capable of forming >10 µm of actin filaments. This number is likely to be higher than the actual actin production rate, but anything in this ballpark could, in principle, induce a substantial change in the dynamics at the leading edge of the cell. Elucidating the mechanisms underlying these bursts of actin translation and the contribution of the actin-coding sequence to its regulation constitutes an exciting direction of further studies.
A similar translation-dependent regulation by coding sequences is probably also applicable to other members of the actin gene family. Indeed, the ribosome profiling data reveal a correlation between ribosome densities on different actin isoform mRNAs and their abilities to functionally compensate for each other in mouse knockout models (Vedula et al., 2017). The actin isoforms that tend to become upregulated in the different knockout models are typically those with the ribosome density that most closely resembles that of the isoform knocked out (the nearest neighbors in ribosome density, as shown in Fig. 3), suggesting that only isoforms with similar ribosome density can directly compensate for each other's function. For instance, α-skeletal actin is upregulated in the mouse knockout model of α-smooth muscle actin and vice versa, and the ribosome density of these isoforms are also the second and third highest, after that of β-actin. Moreover, α-cardiac actin is able to completely rescue the knockout of α-skeletal actin (Nowak et al., 2009), which has the closest ribosome density. In contrast, loss of α-cardiac actin cannot be substituted for by γ-enteric smooth muscle actin (Kumar et al., 1997), which has a ∼10-times lower ribosome density. Finally, none of the other actin isoforms can compensate for the loss of β-actin, which has orders of magnitude higher ribosome density. We propose that changes in ribosome density that arise from silent substitutions in the nucleotide sequence affect the translation dynamics of actin and its protein accumulation rates, which, in turn, serve as key determinants in the functional diversity of actins.
Interestingly, dramatic differences in ribosome density between actin isoforms can also be seen in other vertebrate and non-vertebrate genomes, including human, zebrafish, Drosophila melanogaster and Caenorhabditis elegans (Vedula et al., 2017). These differences suggest that ribosome density differences may indeed reflect at least one of the mechanisms regulating differences in the function of the actin isoforms.
Conclusions
Despite decades of studies, the major determinants driving the functional diversity of actin isoforms remains elusive. At the same time, it is now clear that this regulation is achieved at multiple levels that go beyond the amino acid sequence, previously believed to constitute the sole determinant of actin function. The discovery of the ‘actin code’ that drives actin isoform diversity through silent changes at the gene and mRNA level significantly expands our understanding of the functionality of actin across genomes, and may in principle enable new fundamental levels of functional modulation of actin in normal physiology and disease. It is exciting to think that this silent regulation may also be applicable to other proteins in eukaryotic genomes. Elucidation of the underlying mechanisms and the universality of the nucleotide-based actin code will constitute an exciting direction for further studies.
Acknowledgements
We thank Dr Satoshi Kurosaka for helpful discussions.
Footnotes
Competing interests
The authors declare no competing or financial interests.
Funding
Work in the authors’ lab is funded by the National Institute of General Medical Sciences (GM122505). Deposited in PMC for release after 12 months.
References
- Agrawal P. B., Strickland C. D., Midgett C., Morales A., Newburger D. E., Poulos M. A., Tomczak K. K., Ryan M. M., Iannaccone S. T., Crawford T. O. et al. (2004). Heterogeneity of nemaline myopathy cases with skeletal muscle alpha-actin gene mutations. Ann. Neurol. 56, 86-96. 10.1002/ana.20157 [DOI] [PubMed] [Google Scholar]
- Allen P. G., Shuster C. B., Käs J., Chaponnier C., Janmey P. A. and Herman I. M. (1996). Phalloidin binding and rheological differences among actin isoforms. Biochemistry 35, 14062-14069. 10.1021/bi961326g [DOI] [PubMed] [Google Scholar]
- Almuzzaini B., Sarshad A. A., Rahmanto A. S., Hansson M. L., Von Euler A., Sangfelt O., Visa N., Farrants A. K. and Percipalle P. (2016). In beta-actin knockouts, epigenetic reprogramming and rDNA transcription inactivation lead to growth and proliferation defects. FASEB J. 30, 2860-2873. 10.1096/fj.201600280R [DOI] [PubMed] [Google Scholar]
- Ampe C. and Van Troys M. (2017). Mammalian actins: isoform-specific functions and diseases. Handb. Exp. Pharmacol. 235, 1-37. 10.1007/164_2016_43 [DOI] [PubMed] [Google Scholar]
- Andrade L. R. (2015). Evidence for changes in beta- and gamma-actin proportions during inner ear hair cell life. Cytoskeleton 72, 282-291. 10.1002/cm.21227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antin P. B. and Ordahl C. P. (1991). Isolation and characterization of an avian myogenic cell line. Dev. Biol. 143, 111-121. 10.1016/0012-1606(91)90058-B [DOI] [PubMed] [Google Scholar]
- Arad M., Penas-Lado M., Monserrat L., Maron B. J., Sherrid M., Ho C. Y., Barr S., Karim A., Olson T. M., Kamisago M. et al. (2005). Gene mutations in apical hypertrophic cardiomyopathy. Circulation 112, 2805-2811. 10.1161/CIRCULATIONAHA.105.547448 [DOI] [PubMed] [Google Scholar]
- Bai F., Caster H. M., Rubenstein P. A., Dawson J. F. and Kawai M. (2014). Using baculovirus/insect cell expressed recombinant actin to study the molecular pathogenesis of HCM caused by actin mutation A331P. J. Mol. Cell. Cardiol. 74, 64-75. 10.1016/j.yjmcc.2014.04.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ballestrem C., Wehrle-Haller B. and Imhof B. A. (1998). Actin dynamics in living mammalian cells. J. Cell Sci. 111, 1649-1658. [DOI] [PubMed] [Google Scholar]
- Baranwal S., Naydenov N. G., Harris G., Dugina V., Morgan K. G., Chaponnier C. and Ivanov A. I. (2012). Nonredundant roles of cytoplasmic beta- and gamma-actin isoforms in regulation of epithelial apical junctions. Mol. Biol. Cell 23, 3542-3553. 10.1091/mbc.E12-02-0162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bassell G. J., Zhang H., Byrd A. L., Femino A. M., Singer R. H., Taneja K. L., Lifshitz L. M., Herman I. M. and Kosik K. S. (1998). Sorting of beta-actin mRNA and protein to neurites and growth cones in culture. J. Neurosci 18, 251-265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belyantseva I. A., Perrin B. J., Sonnemann K. J., Zhu M., Stepanyan R., McGee J., Frolenkov G. I., Walsh E. J., Friderici K. H., Friedman T. B. et al. (2009). Gamma-actin is required for cytoskeletal maintenance but not development. Proc. Natl. Acad. Sci. USA 106, 9703-9708. 10.1073/pnas.0900221106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergeron S. E., Zhu M., Thiem S. M., Friderici K. H. and Rubenstein P. A. (2010). Ion-dependent polymerization differences between mammalian beta- and gamma-nonmuscle actin isoforms. J. Biol. Chem. 285, 16087-16095. 10.1074/jbc.M110.110130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruser L. and Bogdan S. (2017). Molecular control of actin dynamics in vivo: insights from Drosophila. Handb. Exp. Pharmacol. 235, 285-310. 10.1007/164_2016_33 [DOI] [PubMed] [Google Scholar]
- Bryan K. E., Wen K.-K., Zhu M., Rendtorff N. D., Feldkamp M., Tranebjaerg L., Friderici K. H. and Rubenstein P. A. (2006). Effects of human deafness gamma-actin mutations (DFNA20/26) on actin function. J. Biol. Chem. 281, 20129-20139. 10.1074/jbc.M601514200 [DOI] [PubMed] [Google Scholar]
- Bunnell T. M. and Ervasti J. M. (2010). Delayed embryonic development and impaired cell growth and survival in Actg1 null mice. Cytoskeleton 67, 564-572. 10.1002/cm.20467 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bunnell T. M., Burbach B. J., Shimizu Y. and Ervasti J. M. (2011). beta-Actin specifically controls cell growth, migration, and the G-actin pool. Mol. Biol. Cell 22, 4047-4058. 10.1091/mbc.E11-06-0582 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buxbaum A. R., Wu B. and Singer R. H. (2014). Single beta-actin mRNA detection in neurons reveals a mechanism for regulating its translatability. Science 343, 419-422. 10.1126/science.1242939 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen A., Arora P. D., McCulloch C. A. and Wilde A. (2017). Cytokinesis requires localized beta-actin filament production by an actin isoform specific nucleator. Nat. Commun. 8, 1530 10.1038/s41467-017-01231-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Condeelis J. and Singer R. H. (2005). How and why does beta-actin mRNA target? Biol Cell 97, 97-110. 10.1042/BC20040063 [DOI] [PubMed] [Google Scholar]
- Cook R. K., Sheff D. R. and Rubenstein P. A. (1991). Unusual metabolism of the yeast actin amino terminus. J. Biol. Chem. 266, 16825-16833. [PubMed] [Google Scholar]
- Crawford K., Flick R., Close L., Shelly D., Paul R., Bove K., Kumar A. and Lessard J. (2002). Mice lacking skeletal muscle actin show reduced muscle strength and growth deficits and die during the neonatal period. Mol. Cell. Biol. 22, 5887-5896. 10.1128/MCB.22.16.5887-5896.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeNofrio D., Hoock T. C. and Herman I. M. (1989). Functional sorting of actin isoforms in microvascular pericytes. J. Cell Biol. 109, 191-202. 10.1083/jcb.109.1.191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drummond M. C. and Friderici K. H. (2013). A novel actin mRNA splice variant regulates ACTG1 expression. PLoS Genet. 9, e1003743 10.1371/journal.pgen.1003743 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dugina V., Zwaenepoel I., Gabbiani G., Clement S. and Chaponnier C. (2009). Beta and gamma-cytoplasmic actins display distinct distribution and functional diversity. J. Cell Sci. 122, 2980-2988. 10.1242/jcs.041970 [DOI] [PubMed] [Google Scholar]
- Dugina V., Alieva I., Khromova N., Kireev I., Gunning P. W. and Kopnin P. (2016). Interaction of microtubules with the actin cytoskeleton via cross-talk of EB1-containing +TIPs and gamma-actin in epithelial cells. Oncotarget 7, 72699-72715. 10.18632/oncotarget.12236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erba H. P., Gunning P. and Kedes L. (1986). Nucleotide sequence of the human gamma cytoskeletal actin mRNA: anomalous evolution of vertebrate non-muscle actin genes. Nucleic Acids Res. 14, 5275-5294. 10.1093/nar/14.13.5275 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erba H. P., Eddy R., Shows T., Kedes L. and Gunning P. (1988). Structure, chromosome location, and expression of the human gamma-actin gene: differential evolution, location, and expression of the cytoskeletal beta- and gamma-actin genes. Mol. Cell. Biol. 8, 1775-1789. 10.1128/MCB.8.4.1775 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falahzadeh K., Banaei-Esfahani A. and Shahhoseini M. (2015). The potential roles of actin in the nucleus. Cell J 17, 7-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fyrberg E. A., Fyrberg C. C., Biggs J. R., Saville D., Beall C. J. and Ketchum A. (1998). Functional nonequivalence of Drosophila actin isoforms. Biochem. Genet. 36, 271-287. 10.1023/A:1018785127079 [DOI] [PubMed] [Google Scholar]
- Galkin V. E., Orlova A. and Egelman E. H. (2012). Actin filaments as tension sensors. Curr. Biol. 22, R96-R101. 10.1016/j.cub.2011.12.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh T., Soni K., Scaria V., Halimani M., Bhattacharjee C. and Pillai B. (2008). MicroRNA-mediated up-regulation of an alternatively polyadenylated variant of the mouse cytoplasmic {beta}-actin gene. Nucleic Acids Res. 36, 6318-6332. 10.1093/nar/gkn624 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goebel H. H., Brockman K., Bonnemann C. G., Warlo I. A., Hanefeld F., Labeit S., Durling H. J. and Laing N. G. (2006). Patient with actin aggregate myopathy and not formerly identified ACTA1 mutation is heterozygous for the Gly15Arg mutation of ACTA1, which has previously been associated with actinopathy. J. Child Neurol. 21, 545 10.1177/08830738060210060103 [DOI] [PubMed] [Google Scholar]
- Gordon D. J., Boyer J. L. and Korn E. D. (1977). Comparative biochemistry of non-muscle actins. J. Biol. Chem. 252, 8300-8309. [PubMed] [Google Scholar]
- Gunning P., Ponte P., Okayama H., Engel J., Blau H. and Kedes L. (1983). Isolation and characterization of full-length cDNA clones for human alpha-, beta-, and gamma-actin mRNAs: skeletal but not cytoplasmic actins have an amino-terminal cysteine that is subsequently removed. Mol. Cell. Biol. 3, 787-795. 10.1128/MCB.3.5.787 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gunning P. W., Ghoshdastider U., Whitaker S., Popp D. and Robinson R. C. (2015). The evolution of compositionally and functionally distinct actin filaments. J. Cell Sci. 128, 2009-2019. 10.1242/jcs.165563 [DOI] [PubMed] [Google Scholar]
- Guo D. C., Pannu H., Tran-Fadulu V., Papke C. L., Yu R. K., Avidan N., Bourgeois S., Estrera A. L., Safi H. J., Sparks E. et al. (2007). Mutations in smooth muscle alpha-actin (ACTA2) lead to thoracic aortic aneurysms and dissections. Nat. Genet. 39, 1488-1493. 10.1038/ng.2007.6 [DOI] [PubMed] [Google Scholar]
- Gutierrez N., Eromobor I., Petrie R. J., Vedula P., Cruz L. and Rodriguez A. J. (2014). The beta-actin mRNA zipcode regulates epithelial adherens junction assembly but not maintenance. RNA 20, 689-701. 10.1261/rna.043208.113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halliburton W. D. (1887). On muscle-plasma. J. Physiol. 8, 133-202. 10.1113/jphysiol.1887.sp000252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill M. A. and Gunning P. (1993). Beta and gamma actin mRNAs are differentially located within myoblasts. J. Cell Biol. 122, 825-832. 10.1083/jcb.122.4.825 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hofmann W. A., Stojiljkovic L., Fuchsova B., Vargas G. M., Mavrommatis E., Philimonenko V., Kysela K., Goodrich J. A., Lessard J. L., Hope T. J. et al. (2004). Actin is part of pre-initiation complexes and is necessary for transcription by RNA polymerase II. Nat. Cell Biol. 6, 1094-1101. 10.1038/ncb1182 [DOI] [PubMed] [Google Scholar]
- Hu P., Wu S. and Hernandez N. (2004). A role for beta-actin in RNA polymerase III transcription. Genes Dev. 18, 3010-3015. 10.1101/gad.1250804 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Islam S., Kjallquist U., Moliner A., Zajac P., Fan J.-B., Lonnerberg P. and Linnarsson S. (2011). Characterization of the single-cell transcriptional landscape by highly multiplex RNA-seq. Genome Res. 21, 160-1167. 10.1101/gr.110882.110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaeger M. A., Sonnemann K. J., Fitzsimons D. P., Prins K. W. and Ervasti J. M. (2009). Context-dependent functional substitution of alpha-skeletal actin by gamma-cytoplasmic actin. FASEB J 23, 2205-2214. 10.1096/fj.09-129783 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Just I., Wollenberg P., Moss J. and Aktories K. (1994). Cysteine-specific ADP-ribosylation of actin. Eur. J. Biochem. 221, 1047-1054. 10.1111/j.1432-1033.1994.tb18823.x [DOI] [PubMed] [Google Scholar]
- Kaech S., Fischer M., Doll T. and Matus A. (1997). Isoform specificity in the relationship of actin to dendritic spines. J. Neurosci 17, 9565-9572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalhor H. R., Niewmierzycka A., Faull K. F., Yao X., Grade S., Clarke S. and Rubenstein P. A. (1999). A highly conserved 3-methylhistidine modification is absent in yeast actin. Arch. Biochem. Biophys. 370, 105-111. 10.1006/abbi.1999.1370 [DOI] [PubMed] [Google Scholar]
- Kapustina M., Read T. A. and Vitriol E. A. (2016). Simultaneous quantification of actin monomer and filament dynamics with modeling-assisted analysis of photoactivation. J. Cell Sci. 129, 4633-4643. 10.1242/jcs.194670 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karakozova M., Kozak M., Wong C. C., Bailey A. O., Yates J. R. III, Mogilner A., Zebroski H. and Kashina A. (2006). Arginylation of beta-actin regulates actin cytoskeleton and cell motility. Science 313, 192-196. 10.1126/science.1129344 [DOI] [PubMed] [Google Scholar]
- Katz Z. B., Wells A. L., Park H. Y., Wu B., Shenoy S. M. and Singer R. H. (2012). beta-Actin mRNA compartmentalization enhances focal adhesion stability and directs cell migration. Genes Dev. 26, 1885-1890. 10.1101/gad.190413.112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kemerley A., Sloan C., Pfeifer W., Smith R. and Drack A. (2017). A novel mutation in ACTG1 causing Baraitser-Winter syndrome with extremely variable expressivity in three generations. Ophthalmic Genet. 38, 152-156. 10.3109/13816810.2016.1164196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khaitlina S. Y. (2001). Functional specificity of actin isoforms. Int. Rev. Cytol. 202, 35-98. 10.1016/S0074-7696(01)02003-4 [DOI] [PubMed] [Google Scholar]
- Kislauskis E. H., Li Z., Singer R. H. and Taneja K. L. (1993). Isoform-specific 3'-untranslated sequences sort alpha-cardiac and beta-cytoplasmic actin messenger RNAs to different cytoplasmic compartments. J. Cell Biol. 123, 165-172. 10.1083/jcb.123.1.165 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kislauskis E. H., Zhu X. and Singer R. H. (1994). Sequences responsible for intracellular localization of beta-actin messenger RNA also affect cell phenotype. J. Cell Biol. 127, 441-451. 10.1083/jcb.127.2.441 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kislauskis E. H., Zhu X.-c. and Singer R. H. (1997). beta-Actin messenger RNA localization and protein synthesis augment cell motility. J. Cell Biol. 136, 1263-1270. 10.1083/jcb.136.6.1263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar A., Crawford K., Close L., Madison M., Lorenz J., Doetschman T., Pawlowski S., Duffy J., Neumann J., Robbins J. et al. (1997). Rescue of cardiac alpha-actin-deficient mice by enteric smooth muscle gamma-actin. Proc. Natl. Acad. Sci. USA 94, 4406-4411. 10.1073/pnas.94.9.4406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawrence J. B. and Singer R. H. (1986). Intracellular localization of messenger RNAs for cytoskeletal proteins. Cell 45, 407-415. 10.1016/0092-8674(86)90326-0 [DOI] [PubMed] [Google Scholar]
- Lehtimaki J., Hakala M. and Lappalainen P. (2017). Actin filament structures in migrating cells. Handb. Exp. Pharmacol. 235, 123-152. 10.1007/164_2016_28 [DOI] [PubMed] [Google Scholar]
- Lindberg U., Schutt C. E., Hellsten E., Tjader A. C. and Hult T. (1988). The use of poly(L-proline)-Sepharose in the isolation of profilin and profilactin complexes. Biochim. Biophys. Acta 967, 391-400. 10.1016/0304-4165(88)90102-X [DOI] [PubMed] [Google Scholar]
- Liu P., Li H., Ren X., Mao H., Zhu Q., Zhu Z., Yang R., Yuan W., Liu J., Wang Q. et al. (2008). Novel ACTG1 mutation causing autosomal dominant non-syndromic hearing impairment in a Chinese family. J Genet. Genomics 35, 553-558. 10.1016/S1673-8527(08)60075-2 [DOI] [PubMed] [Google Scholar]
- Lloyd C. and Gunning P. (1993). Noncoding regions of the gamma-actin gene influence the impact of the gene on myoblast morphology. J. Cell Biol. 121, 73-82. 10.1083/jcb.121.1.73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lloyd C., Schevzov G. and Gunning P. (1992). Transfection of nonmuscle beta- and gamma-actin genes into myoblasts elicits different feedback regulatory responses from endogenous actin genes. J. Cell Biol. 117, 787-797. 10.1083/jcb.117.4.787 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luxenburg C. and Geiger B. (2017). Multiscale view of cytoskeletal mechanoregulation of cell and tissue polarity. Handb. Exp. Pharmacol. 235, 263-284. 10.1007/164_2016_34 [DOI] [PubMed] [Google Scholar]
- Lyubimova A., Bershadsky A. D. and Ben-Ze'ev A. (1999). Autoregulation of actin synthesis requires the 3'-UTR of actin mRNA and protects cells from actin overproduction. J. Cell. Biochem. 76, 1-12. [DOI] [PubMed] [Google Scholar]
- Martin D. J. and Rubenstein P. A. (1987). Alternate pathways for removal of the class II actin initiator methionine. J. Biol. Chem. 262, 6350-6356. [PubMed] [Google Scholar]
- McHugh K. M., Crawford K. and Lessard J. L. (1991). A comprehensive analysis of the developmental and tissue-specific expression of the isoactin multigene family in the rat. Dev. Biol. 148, 442-458. 10.1016/0012-1606(91)90263-3 [DOI] [PubMed] [Google Scholar]
- McKane M., Wen K. K., Meyer A. and Rubenstein P. A. (2006). Effect of the substitution of muscle actin-specific subdomain 1 and 2 residues in yeast actin on actin function. J. Biol. Chem. 281, 29916-29928. 10.1074/jbc.M602251200 [DOI] [PubMed] [Google Scholar]
- Michel A. M., Fox G., C M. K. A., Bo D., O'Connor P. B., Heaphy S. M., Mullan J. P., Donohue C. A., Higgins D. G. and Baranov P. V. (2014). GWIPS-viz: development of a ribo-seq genome browser. Nucleic Acids Res. 42, D859-D864. 10.1093/nar/gkt1035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Micheva K. D., Vallée A., Beaulieu C., Herman I. M. and Leclerc N. (1998). beta-Actin is confined to structures having high capacity of remodelling in developing and adult rat cerebellum. Eur. J. Neurosci. 10, 3785-3798. 10.1046/j.1460-9568.1998.00391.x [DOI] [PubMed] [Google Scholar]
- Migocka-Patrzalek M., Makowiecka A., Nowak D., Mazur A. J., Hofmann W. A. and Malicka-Błaszkiewicz M. (2015). beta- and gamma-Actins in the nucleus of human melanoma A375 cells. Histochem. Cell Biol. 144, 417-428. 10.1007/s00418-015-1349-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miwa T., Manabe Y., Kurokawa K., Kamada S., Kanda N., Bruns G., Ueyama H. and Kakunaga T. (1991). Structure, chromosome location, and expression of the human smooth muscle (enteric type) gamma-actin gene: evolution of six human actin genes. Mol. Cell. Biol. 11, 3296-3306. 10.1128/MCB.11.6.3296 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mogensen J., Klausen I. C., Pedersen A. K., Egeblad H., Bross P., Kruse T. A., Gregersen N., Hansen P. S., Baandrup U. and Børglum A. D. (1999). Alpha-cardiac actin is a novel disease gene in familial hypertrophic cardiomyopathy. J. Clin. Invest. 103, R39-R43. 10.1172/JCI6460 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mogensen J., Perrot A., Andersen P. S., Havndrup O., Klausen I. C., Christiansen M., Bross P., Egeblad H., Bundgaard H., Osterziel K. J. et al. (2004). Clinical and genetic characteristics of alpha cardiac actin gene mutations in hypertrophic cardiomyopathy. J. Med. Genet. 41, e10 10.1136/jmg.2003.010447 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morisaki T., Lyon K., DeLuca K. F., DeLuca J. G., English B. P., Zhang Z., Lavis L. D., Grimm J. B., Viswanathan S., Looger L. L. et al. (2016). Real-time quantification of single RNA translation dynamics in living cells. Science 352, 1425-1429. 10.1126/science.aaf0899 [DOI] [PubMed] [Google Scholar]
- Mounier N. and Sparrow J. C. (1997). Structural comparisons of muscle and nonmuscle actins give insights into the evolution of their functional differences. J. Mol. Evol. 44, 89-97. 10.1007/PL00006125 [DOI] [PubMed] [Google Scholar]
- Mounier N., Perriard J. C., Gabbiani G. and Chaponnier C. (1997). Transfected muscle and non-muscle actins are differentially sorted by cultured smooth muscle and non-muscle cells. J. Cell Sci. 110, 839-846. [DOI] [PubMed] [Google Scholar]
- Müller M., Diensthuber R. P., Chizhov I., Claus P., Heissler S. M., Preller M., Taft M. H. and Manstein D. J. (2013). Distinct functional interactions between actin isoforms and nonsarcomeric myosins. PLoS ONE 8, e70636 10.1371/journal.pone.0070636 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nowak K. J., Ravenscroft G., Jackaman C., Filipovska A., Davies S. M., Lim E. M., Squire S. E., Potter A. C., Baker E., Clement S. et al. (2009). Rescue of skeletal muscle alpha-actin-null mice by cardiac (fetal) alpha-actin. J. Cell Biol. 185, 903-915. 10.1083/jcb.200812132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nowak K. J., Ravenscroft G. and Laing N. G. (2013). Skeletal muscle alpha-actin diseases (actinopathies): pathology and mechanisms. Acta Neuropathol. 125, 19-32. 10.1007/s00401-012-1019-z [DOI] [PubMed] [Google Scholar]
- Nunoi H., Yamazaki T., Tsuchiya H., Kato S., Malech H. L., Matsuda I. and Kanegasaki S. (1999). A heterozygous mutation of beta-actin associated with neutrophil dysfunction and recurrent infection. Proc. Natl. Acad. Sci. USA 96, 8693-8698. 10.1073/pnas.96.15.8693 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nyman T., Schuler H., Korenbaum E., Schutt C. E., Karlsson R. and Lindberg U. (2002). The role of MeH73 in actin polymerization and ATP hydrolysis. J. Mol. Biol. 317, 577-589. 10.1006/jmbi.2002.5436 [DOI] [PubMed] [Google Scholar]
- Ohshima S., Abe H. and Obinata T. (1989). Isolation of profilin from embryonic chicken skeletal muscle and evaluation of its interaction with different actin isoforms. J. Biochem. 105, 855-857. 10.1093/oxfordjournals.jbchem.a122765 [DOI] [PubMed] [Google Scholar]
- Olson T. M., Michels V. V., Thibodeau S. N., Tai Y. S. and Keating M. T. (1998). Actin mutations in dilated cardiomyopathy, a heritable form of heart failure. Science 280, 750-752. 10.1126/science.280.5364.750 [DOI] [PubMed] [Google Scholar]
- Olson T. M., Doan T. P., Kishimoto N. Y., Whitby F. G., Ackerman M. J. and Fananapazir L. (2000). Inherited and de novo mutations in the cardiac actin gene cause hypertrophic cardiomyopathy. J. Mol. Cell. Cardiol. 32, 1687-1694. 10.1006/jmcc.2000.1204 [DOI] [PubMed] [Google Scholar]
- O'Rourke A. R., Lindsay A., Tarpey M. D., Yuen S., McCourt P., Nelson D. M., Perrin B. J., Thomas D. D., Spangenburg E. E., Lowe D. A. et al. (2018). Impaired muscle relaxation and mitochondrial fission associated with genetic ablation of cytoplasmic actin isoforms. FEBS J. 285, 481-500. 10.1111/febs.14367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otey C. A., Kalnoski M. H., Lessard J. L. and Bulinski J. C. (1986). Immunolocalization of the gamma isoform of nonmuscle actin in cultured cells. J. Cell Biol. 102, 1726-1737. 10.1083/jcb.102.5.1726 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otey C. A., Kalnoski M. H. and Bulinski J. C. (1988). Immunolocalization of muscle and nonmuscle isoforms of actin in myogenic cells and adult skeletal muscle. Cell Motil. Cytoskelet. 9, 337-348. 10.1002/cm.970090406 [DOI] [PubMed] [Google Scholar]
- Pathan-Chhatbar S., Taft M. H., Reindl T., Hundt N., Latham S. L. and Manstein D. J. (2018). Three mammalian tropomyosin isoforms have different regulatory effects on nonmuscle myosin-2B and filamentous beta-actin in vitro. J. Biol. Chem. 293, 863-875. 10.1074/jbc.M117.806521 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patrinostro X., O'Rourke A. R., Chamberlain C. M., Moriarity B. S., Perrin B. J. and Ervasti J. M. (2017). Relative importance of betacyto- and gammacyto-actin in primary mouse embryonic fibroblasts. Mol. Biol. Cell 28, 771-782. 10.1091/mbc.E16-07-0503 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perler F., Efstratiadis A., Lomedico P., Gilbert W., Kolodner R. and Dodgson J. (1980). The evolution of genes: the chicken preproinsulin gene. Cell 20, 555-566. 10.1016/0092-8674(80)90641-8 [DOI] [PubMed] [Google Scholar]
- Perrin B. J. and Ervasti J. M. (2010). The actin gene family: function follows isoform. Cytoskeleton 67, 630-634. 10.1002/cm.20475 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pollard T. D. (2001). Genomics, the cytoskeleton and motility. Nature 409, 842-843. 10.1038/35057029 [DOI] [PubMed] [Google Scholar]
- Pollard T. D. (2017). What we know and do not know about actin. Handb. Exp. Pharmacol. 235, 331-347. 10.1007/164_2016_44 [DOI] [PubMed] [Google Scholar]
- Prassler J., Stocker S., Marriott G., Heidecker M., Kellermann J. and Gerisch G. (1997). Interaction of a Dictyostelium member of the plastin/fimbrin family with actin filaments and actin-myosin complexes. Mol. Biol. Cell 8, 83-95. 10.1091/mbc.8.1.83 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prins K. W., Call J. A., Lowe D. A. and Ervasti J. M. (2011). Quadriceps myopathy caused by skeletal muscle-specific ablation of beta(cyto)-actin. J. Cell Sci. 124, 951-957. 10.1242/jcs.079848 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Procaccio V., Salazar G., Ono S., Styers M. L., Gearing M., Davila A., Jimenez R., Juncos J., Gutekunst C. A., Meroni G. et al. (2006). A mutation of beta -actin that alters depolymerization dynamics is associated with autosomal dominant developmental malformations, deafness, and dystonia. Am. J. Hum. Genet. 78, 947-960. 10.1086/504271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rendtorff N. D., Zhu M., Fagerheim T., Antal T. L., Jones M., Teslovich T. M., Gillanders E. M., Barmada M., Teig E., Trent J. M. et al. (2006). A novel missense mutation in ACTG1 causes dominant deafness in a Norwegian DFNA20/26 family, but ACTG1 mutations are not frequent among families with hereditary hearing impairment. Eur. J. Hum. Genet. 14, 1097-1105. 10.1038/sj.ejhg.5201670 [DOI] [PubMed] [Google Scholar]
- Rodriguez A. J., Shenoy S. M., Singer R. H. and Condeelis J. (2006). Visualization of mRNA translation in living cells. J. Cell Biol. 175, 67-76. 10.1083/jcb.200512137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubenstein P. A. and Martin D. J. (1983). NH2-terminal processing of actin in mouse L-cells in vivo. J. Biol. Chem. 258, 3961-3966. [PubMed] [Google Scholar]
- Sanger J. W., Wang J., Fan Y., White J., Mi-Mi L., Dube D. K., Sanger J. M. and Pruyne D. (2017). Assembly and maintenance of myofibrils in striated muscle. Handb. Exp. Pharmacol. 235, 39-75. 10.1007/164_2016_53 [DOI] [PubMed] [Google Scholar]
- Schevzov G., Lloyd C. and Gunning P. (1992). High level expression of transfected beta- and gamma-actin genes differentially impacts on myoblast cytoarchitecture. J. Cell Biol. 117, 775-785. 10.1083/jcb.117.4.775 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schildmeyer L. A., Braun R., Taffet G., Debiasi M., Burns A. E., Bradley A. and Schwartz R. J. (2000). Impaired vascular contractility and blood pressure homeostasis in the smooth muscle alpha-actin null mouse. FASEB J. 14, 2213-2220. 10.1096/fj.99-0927com [DOI] [PubMed] [Google Scholar]
- Shawlot W., Deng J. M., Fohn L. E. and Behringer R. R. (1998). Restricted beta-galactosidase expression of a hygromycin-lacZ gene targeted to the beta-actin locus and embryonic lethality of beta-actin mutant mice. Transgenic Res. 7, 95-103. 10.1023/A:1008816308171 [DOI] [PubMed] [Google Scholar]
- Sheff D. R. and Rubenstein P. A. (1989). Identification of N-acetylmethionine as the product released during the NH2-terminal processing of a pseudo-class I actin. J. Biol. Chem. 264, 11491-11496. [PubMed] [Google Scholar]
- Sheterline P., Clayton J., and Sparrow J.C.. 1998. Actin, pp. 272 Oxford, New York: Oxford University Press. [Google Scholar]
- Shmerling D., Danzer C. P., Mao X., Boisclair J., Haffner M., Lemaistre M., Schuler V., Kaeslin E., Korn R., Burki K. et al. (2005). Strong and ubiquitous expression of transgenes targeted into the beta-actin locus by Cre/lox cassette replacement. Genesis 42, 229-235. 10.1002/gene.20135 [DOI] [PubMed] [Google Scholar]
- Shuster C. B. and Herman I. M. (1995). Indirect association of ezrin with F-actin: isoform specificity and calcium sensitivity. J. Cell Biol. 128, 837-848. 10.1083/jcb.128.5.837 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shuster C. B., Lin A. Y., Nayak R. and Herman I. M. (1996). Beta cap73: a novel beta actin-specific binding protein. Cell Motil. Cytoskelet. 35, 175-187. [DOI] [PubMed] [Google Scholar]
- Simiczyjew A., Pietraszek-Gremplewicz K., Mazur A. J. and Nowak D. (2017). Are non-muscle actin isoforms functionally equivalent? Histol. Histopathol. 32, 1125-1139. 10.14670/HH-11-896 [DOI] [PubMed] [Google Scholar]
- Sonnemann K. J., Fitzsimons D. P., Patel J. R., Liu Y., Schneider M. F., Moss R. L. and Ervasti J. M. (2006). Cytoplasmic gamma-actin is not required for skeletal muscle development but its absence leads to a progressive myopathy. Dev. Cell 11, 387-397. 10.1016/j.devcel.2006.07.001 [DOI] [PubMed] [Google Scholar]
- Strathdee D., Whitelaw C. B. and Clark A. J. (2008). Distal transgene insertion affects CpG island maintenance during differentiation. J. Biol. Chem. 283, 11509-11515. 10.1074/jbc.M709805200 [DOI] [PubMed] [Google Scholar]
- Straub F. B. (1942). Actin. Stud. Inst. Med. Chem. Univ. Szeged 2, 3-15. [Google Scholar]
- Strauch A. R. and Rubenstein P. A. (1984). A vascular smooth muscle alpha-isoactin biosynthetic intermediate in BC3H1 cells. Identification of acetylcysteine at the NH2 terminus. J. Biol. Chem. 259, 7224-7229. [PubMed] [Google Scholar]
- Strohl F., Lin J. Q., Laine R. F., Wong H. H., Urbancic V., Cagnetta R., Holt C. E. and Kaminski C. F. (2017). Single molecule translation imaging visualizes the dynamics of local beta-actin synthesis in retinal axons. Sci. Rep. 7, 709 10.1038/s41598-017-00695-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terman J. R. and Kashina A. (2013). Post-translational modification and regulation of actin. Curr. Opin. Cell Biol. 25, 30-38. 10.1016/j.ceb.2012.10.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tondeleir D., Vandamme D., Vandekerckhove J., Ampe C. and Lambrechts A. (2009). Actin isoform expression patterns during mammalian development and in pathology: insights from mouse models. Cell Motil. Cytoskelet. 66, 798-815. 10.1002/cm.20350 [DOI] [PubMed] [Google Scholar]
- Tondeleir D., Lambrechts A., Muller M., Jonckheere V., Doll T., Vandamme D., Bakkali K., Waterschoot D., Lemaistre M., Debeir O. et al. (2012). Cells lacking beta-actin are genetically reprogrammed and maintain conditional migratory capacity. Mol. Cell. Proteomics 11, 255-271. 10.1074/mcp.M111.015099 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tondeleir D., Drogat B., Slowicka K., Bakkali K., Bartunkova S., Goossens S., Haigh J. J. and Ampe C. (2013). Beta-actin is involved in modulating erythropoiesis during development by fine-tuning Gata2 expression levels. PLoS ONE 8, e67855 10.1371/journal.pone.0067855 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tondeleir D., Noelanders R., Bakkali K. and Ampe C. (2014). Beta-actin is required for proper mouse neural crest ontogeny. PLoS ONE 9, e85608 10.1371/journal.pone.0085608 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vandekerckhove J. and Weber K. (1978a). At least six different actins are expressed in a higher mammal: an analysis based on the amino acid sequence of the amino-terminal tryptic peptide. J. Mol. Biol. 12, 783-802. 10.1016/0022-2836(78)90020-7 [DOI] [PubMed] [Google Scholar]
- Vandekerckhove J. and Weber K. (1978b). Mammalian cytoplasmic actins are the products of at least two genes and differ in primary structure in at least 25 identified positions from skeletal muscle actins. Proc. Natl. Acad. Sci. USA 75, 1106-1110. 10.1073/pnas.75.3.1106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vandekerckhove J. and Weber K. (1981). Actin typing on total cellular extracts: a highly sensitive protein-chemical procedure able to distinguish different actins. Eur. J. Biochem. 113, 595-603. 10.1111/j.1432-1033.1981.tb05104.x [DOI] [PubMed] [Google Scholar]
- Van Driest S. L., Ellsworth E. G., Ommen S. R., Tajik A. J., Gersh B. J. and Ackerman M. J. (2003). Prevalence and spectrum of thin filament mutations in an outpatient referral population with hypertrophic cardiomyopathy. Circulation 108, 445-451. 10.1161/01.CIR.0000080896.52003.DF [DOI] [PubMed] [Google Scholar]
- van Wijk E., Krieger E., Kemperman M. H., De Leenheer E. M., Huygen P. L., Cremers C. W., Cremers F. P. and Kremer H. (2003). A mutation in the gamma actin 1 (ACTG1) gene causes autosomal dominant hearing loss (DFNA20/26). J. Med. Genet. 40, 879-884. 10.1136/jmg.40.12.879 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vedula P., Kurosaka S., Leu N. A., Wolf Y. I., Shabalina S. A., Wang J., Sterling S., Dong D. and Kashina A. (2017). Diverse functions of homologous actin isoforms are defined by their nucleotide, rather than their amino acid sequence. eLife 6 10.7554/eLife.31661 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viita T. and Vartiainen M. K. (2017). From cytoskeleton to gene expression: actin in the nucleus. Handb. Exp. Pharmacol. 235, 311-329. 10.1007/164_2016_27 [DOI] [PubMed] [Google Scholar]
- von Arx P., Bantle S., Soldati T. and Perriard J. C. (1995). Dominant negative effect of cytoplasmic actin isoproteins on cardiomyocyte cytoarchitecture and function. J. Cell Biol. 131, 1759-1773. 10.1083/jcb.131.6.1759 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner C. R., Mahowald A. P. and Miller K. G. (2002). One of the two cytoplasmic actin isoforms in Drosophila is essential. Proc. Natl. Acad. Sci. USA 99, 8037-8042. 10.1073/pnas.082235499 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallefeld W., Krause S., Nowak K. J., Dye D., Horváth R., Molnár Z., Szabó M., Hashimoto K., Reina C., De Carlos J. et al. (2006). Severe nemaline myopathy caused by mutations of the stop codon of the skeletal muscle alpha actin gene (ACTA1). Neuromuscul. Disord. 16, 541-547. 10.1016/j.nmd.2006.07.018 [DOI] [PubMed] [Google Scholar]
- Wallgren-Pettersson C., Sewry C. A., Nowak K. J. and Laing N. G. (2011). Nemaline myopathies. Semin. Pediatr. Neurol. 18, 230-238. 10.1016/j.spen.2011.10.004 [DOI] [PubMed] [Google Scholar]
- Weber A., Nachmias V. T., Pennise C. R., Pring M. and Safer D. (1992). Interaction of thymosin beta 4 with muscle and platelet actin: implications for actin sequestration in resting platelets. Biochemistry 31, 6179-6185. 10.1021/bi00142a002 [DOI] [PubMed] [Google Scholar]
- Welch A. Y., Riley K. N., D'Souza-Schorey C. and Herman I. M. (2005). Arf6 modulates the beta-actin specific capping protein, betacap73. Methods Enzymol. 404, 377-387. 10.1016/S0076-6879(05)04033-4 [DOI] [PubMed] [Google Scholar]
- Yao X., Chaponnier C., Gabbiani G. and Forte J. G. (1995). Polarized distribution of actin isoforms in gastric parietal cells. Mol. Biol. Cell 6, 541-557. 10.1091/mbc.6.5.541 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H. L., Singer R. H. and Bassell G. J. (1999). Neurotrophin regulation of beta-actin mRNA and protein localization within growth cones. J. Cell Biol. 147, 59-70. 10.1083/jcb.147.1.59 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang F., Saha S., Shabalina S. A. and Kashina A. (2010). Differential arginylation of actin isoforms is regulated by coding sequence-dependent degradation. Science 329, 1534-1537. 10.1126/science.1191701 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu M., Yang T., Wei S., DeWan A. T., Morell R. J., Elfenbein J. L., Fisher R. A., Leal S. M., Smith R. J. and Friderici K. H. (2003). Mutations in the gamma-actin gene (ACTG1) are associated with dominant progressive deafness (DFNA20/26). Am. J. Hum. Genet. 73, 1082-1091. 10.1086/379286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zicha D., Dobbie I. M., Holt M. R., Monypenny J., Soong D. Y., Gray C. and Dunn G. A. (2003). Rapid actin transport during cell protrusion. Science 300, 142-145. 10.1126/science.1082026 [DOI] [PubMed] [Google Scholar]