

SUMMARY
The hallmarks of age-related immune senescence are chronic inflammation, aberrant expansion of effector memory, and loss of naive T lymphocytes due in part to systemic activation of innate immune sensor NLRP3 inflammasome in myeloid lineage cells. The endogenous mechanisms that regulate inflammasome activation during aging are unknown. Here, we present evidence that growth hormone receptor (GH-R)-dependent downregulation of NLRP3 inflammasomein macrophages is linked to pro-longevity effects that maintain immune system homeostasis in aging. Deletion of GH-R prevented the macrophage-driven age-related activation of inflammasome in response to NLRP3 ligands and also increased the preservation of naive T cells, even in advanced age and with higher IFNγ secretion from effector cells. The mechanism of inflammasome inhibition is linked to autocrine somatotropic axis as ablation of IGF1R in macrophages lowered the NLRP3 inflammasome activation. Together, our findings show that functional somatotropic axis in macrophages controls inflammation, thus linking NLRP3-mediated innate immune signaling to health span and longevity.
In Brief
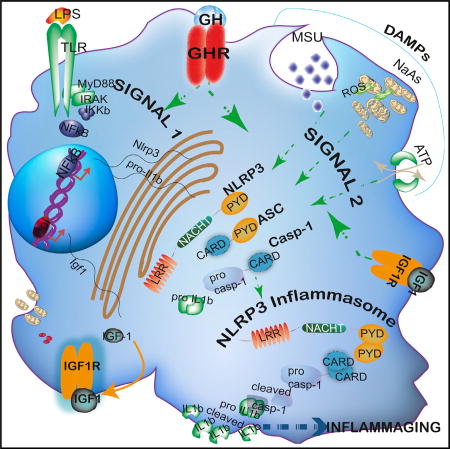
Reduction in endocrine GH and IGF1 axis extends lifespan in model organisms. In this work, Spadaro et al. show that the ablation of the somatotropic axis within macrophages dampens the NLRP3 inflammasome-mediated inflammation seen with aging and prevents against age-related loss of naive T cells.
INTRODUCTION
Systemic low-grade inflammation contributes to the development of chronic diseases and degenerative changes during aging (Ferrucci et al., 2005; Goldberg and Dixit, 2015). The increase in inflammatory cytokines in the elderly often referred to as “inflammaging” is a proposed driver of chronic diseases and shortened health span (Franceschi and Campisi, 2014). Although the cellular origin of age-related inflammation and molecular mechanism is not fully understood, emerging evidence suggests that accumulation of cellular damage, due to exhaustion of endogenous mechanisms in tissue macrophages that clear the damage-associated molecular patterns (DAMPs), may play an important role in this process (Goldberg and Dixit, 2015). These new data support Metchnikoff’s original prediction that phagocytes or macrophages drive aging-associated degenerative diseases (Metchnikoff, 1908).
It is now understood that accumulation of DAMPs, such as by-products of necrotic cells, extracellular ATP, ceramides, saturated fatty acids, uric acid, amyloid fibrils, or free cholesterol crystals, are sensed by pattern recognition receptors (PRRs) in macrophages (Medzhitov, 2008; Schroder and Tschopp, 2010) to trigger chronic low-grade inflammation seen during aging (Youm et al., 2013). The NLRP3 inflammasome is the primary sensor of structurally diverse DAMPs and initiator of a sterile inflammatory cascade (Lamkanfi and Dixit, 2014). Assembly of the NLRP3 inflammasome in response to DAMPs requires pyrin-pyrin interaction of NLRP3 with adaptor protein ASC. The active inflammasome complex is formed through CARD-CARD (caspase activation recruitment domain) interaction of ASC with procaspase-1. Once assembled, the NLRP3 inflammasome activates caspase-1, which in turn controls the secretion of IL-1β and IL-18 (Martinon et al., 2002). The ablation of NLRP3 inflammasome prevents age-related inflammation and functional decline (Youm et al., 2012, 2013). This raises the question, what are the endogenous pathways that regulate NLRP3 inflammasome during aging, and are these mechanisms relevant to longevity and maintenance of immune homeostasis?
Downregulation of somatotropic axis (GH, IGF-1, and hypothalamic factors that control GH release) is a well-established endocrine feature of the aging process that is associated with deficits in physiological processes (Bartke et al., 2013). The question whether this decline causes age-related disorders or represents a compensatory homeostatic mechanism to promote longevity has been addressed using several model organisms (Bartke et al., 2013; Kenyon, 2010). For example, the mutant mice lacking GH (growth hormone), GH-R (GH receptor), and GHRH (GH-releasing hormone) outlive their normal siblings by ~40% with better metabolic function, low risk of cancer, and severely reduced IGF-1-circulating levels (Brown-Borg et al., 1996; Coschigano et al., 2003; Sun et al., 2013). Pro-longevity intervention caloric restriction is also associated with reduction in IGF-1 in rodents. Interestingly, unlike in rodents, the recent human CALERIE-II trial, which evaluated the effect of 15% reduction in energy intake over 2 years, shows no decrease in serum-circulating IGF-1 concentration but rather increased serum IGFBP-1 and decreased IGF-1:IGFBP-1 ratio levels; this suggests an overall reduction in IGF-1 bioactivity in humans post-calorie restriction (Fontana et al., 2016). Consistent with these data, fasting and caloric-restriction-induced reduction in IGF-1 protects against age-related defects in hematopoietic stem cells and T cell progenitors (Cheng et al., 2014; Longo and Finch, 2003). Interestingly, GH-R expression increases with age in hematopoietic stem cells, but GH signaling is dispensable for normal hematopoiesis and bone marrow reconstitution post-cytoablation (Stewart et al., 2014). GH-R in macrophages is required for inflammation and glucose homeostasis in the context of diet-induced obesity (Lu et al., 2013). Given a central role of macrophage-expressed NLRP3 inflammasome in age-related inflammation, we investigated the contribution of endogenous pro-aging GH-IGF-1 pathway in controlling the innate immune-sensing machinery and immune senescence. Our findings show that downregulation of GH-R-mediated signals in macrophages represents an endogenous regulatory “break” mechanism that limits age-related inflammation by downregulation of NLRP3 inflammasome activation. These data also suggest that longevity in GH-R-deficient mice is associated with reduction in NLRP3 inflammasome.
RESULTS
Deletion of GH-R Protects against Age-Related Inflammasome Activation and T Cell Senescence
GH controls lipolysis and can likely impact the generation of metabolic DAMPs that are sensed by the NLRP3 inflammasome to induce inflammation. Given the important role of visceral adipose tissue (VAT) in age-related inflammation and insulin sensitivity (Huffman and Barzilai, 2009; Youm et al., 2013), our initial studies investigated whether GH-R-deficient long-lived mice exhibit altered regulation of inflammasome machinery during aging. Compared to young WT mice, 36-month-old mice displayed significant upregulation of IL-1β, caspase-1, Nlrp3, and the Asc mRNA in VAT that was significantly reduced in age-matched GH-R-deficient (Ghr−/−) mice (Figures 1A–1D). Ablation of GH-R prevented the age-related increase in pro-IL-1β, whereas the secreted p17 isoform could not be detected in VAT lysates (Figure 1E). Consistent with this, aged VAT shows an increased expression of inflammasome adaptor protein ASC, which was significantly reduced in age-matched Ghr−/− mice (Figure 1E). To further define the role of GH-R in downregulation of inflammasome activation in aging, we then quantified the caspase-1 cleavage into enzymatically active p20 heterodimer. Consistent with our prior data (Youm et al., 2013), aging is associated with an increase in caspase-1 activation (Figure 1F). Compared to age-matched control animals, 36-month Ghr−/− mice have reduced p20 subunit of caspase-1 (Figure 1F). These data suggest that GH-R-dependent signaling promotes age-related inflammasome activation in VAT.
Figure 1. GH-R-Deficient Mice Are Protected from Nlrp3 Inflammasome Activation in the Visceral Adipose and from T Cell Diversity Restriction in Aging.

(A–D) VAT from young (3 month) and old (30 month) WTand Ghr−/− mice was analyzed for Nlrp3, Casp-1, Asc, and Il1β gene expression by real-time PCR (data are expressed as mean ± SEM; n = 3/age group/genotype).
(E and F) The pro IL-1β and the adaptor protein ASC (E) and the active p20 Casp-1 with its proform (F) were analyzed by immunoblot analysis in the VAT of young (3 month) and old (30 month) WT and Ghr−/− mice (each band is representative of a single mouse).
(G and H) FACS analysis depicting spleen naive (CD4+/CD8+ CD62L+ CD44−) and effector memory (E/M) (CD4+/CD8+ CD62L− CD44+)-enriched T cells from 3 months and 36 months WT and Ghr−/− mice.
(I and J) Percentage of gated CD4+ and CD8+ naive T cells and effector memory T cells in the spleens of 3-month and 36-month WT and Ghr−/− mice.
(K) Luminex analysis of IFNγ secretion from CD4+ CD44+ effector-memory-sorted and activated T cells from splenocytes in 12-month WT and Ghr−/− mice siblings.
Data are expressed as mean ± SEM; n = 3–9/age group/genotype. Statistical differences were calculated by two-way ANOVA with Tukey’s test (*p < 0.05; A–D) and by two-tailed paired Student’s t test (*p < 0.05; I–K).
We previously demonstrated that ablation of NLRP3 inflammasome in aged mice prevents the homeostatic expansion of effector memory (E/M) T cells and increases naive T cell output due to delayed thymic involution (Youm et al., 2012, 2013). Therefore, we determined whether GH-R-dependent reduction in age-related inflammasome activation impacts T cell senescence. As expected, WT mice in advanced age displayed expansion of E/M cells and loss of naive T cells in both CD4 and CD8 subsets (Figures 1G–1J) with no difference in the total CD4 and CD8 T cells subsets (Figures S1A and S1B). Strikingly, the 36-month-old age-matched Ghr−/− mice had increase in naive CD4 and CD8 cells and were protected from E/M T cell expansions (Figures 1G–1J). Furthermore, ablation of GH-R in old mice increased the central memory CD8 cells (Figure 1H). To determine the functional capacity of E/M T cells, we sorted CD4 E/M T cells and stimulated them in a TCR-dependent manner to evaluate the effector cytokine production. Interestingly, compared to WT mice, the CD4 E/M T cells of middle-aged Ghr−/− mice secreted significantly higher levels of IFNγ (Figure 1K), whereas no differences in TNFα, IL-4, and IL-5 production were observed (Figures S1C–S1E). These data suggest that long-lived GH-R-deficient mice are protected from agerelated inflammasome activation and associated defects in T cell immune compartment.
Age-Dependent Increase in Macrophage Activation Is Mediated in Part by GH-R
The inflammasome activation in macrophages requires an initial priming “signal 1” via TLRs-NF-kB pathway, which leads to transcriptional upregulation of cytokines and components of the inflammasome machinery (Gross et al., 2011). To determine the mechanism of GH-R-dependent inhibition of inflammasome during aging, we investigated the potential regulation of signal 1 in macrophages. We first investigated the expression of GH-R in BMDMs of young and old mice either primed with LPS or polarized to classical M1 activation. We found that GH-R expression is upregulated with aging in activated macrophages, but not in liver and VAT (Figure 2A), suggesting a role for GH in inducing signal 1 and in the pro-inflammatory macrophage phenotype upon aging. Next, to determine whether the GH-R in macrophages is functional, BMDMs were stimulated by GH and MAPK signaling was evaluated. Consistent with a functional response, the GH-R ligation increased ERK phosphorylation (Figure 2B). To determine whether signal 1 induction in old macrophages is dependent on GH-R, the BMDMs from WT and 30-month-old long-lived GH-R-deficient mice were stimulated with LPS to investigate the NFkB and MAPK signaling. These experiments revealed that LPS-induced NFkB and ERK phosphorylation was diminished in GH-R-deficient macrophages of old mice (Figure 2C), confirming the involvement of GH signaling in the NF-kB-dependent signal 1.
Figure 2. GH Signals in BMDMs Sustaining the M1 Signature.

(A) Real-time PCR analysis of Ghr either in WT BMDMs untreated, LPS treated, and differentiated to the M1 phenotype (LPS + IFNγ) or in liver and VAT.
(B) pERK 1/2 protein expression was checked after rGH treatment in cultured WT 3-month-old BMDMs at different time points.
(C) pNF-kB and pERK signaling were evaluated in 30-month-old WT and Ghr−/− mice BMDMs either untreated or LPS primed in in vitro cell culture.
(D) Real-time gene expression analysis of the inflammasome components Nlrp3, Asc, and Casp-1 in cultured BMDMs under LPS and LPS + IFNγ (M1 phenotype) treatment. BMDMs were derived from 3-month- and 30-month-old WT and Ghr−/− mice as indicated.
(E–H) Real-time PCR analysis of Il1β, Il12β, TNFα, and Il6 in in vitro M1-differentiated BMDMs.
(I–L) Real-time PCR analysis of Ccl22, Ccl24, Arg-1, and Il4r in in vitro M2-differentiated BMDMs.
All real-time data are presented as mean ± SEM (n = 3–10/age group/ genotype); statistical differences were calculated by two-way ANOVA with Tukey’s test (*p < 0.05).
We next investigated the GH-R-dependent changes in inflammasome signal 1 and inflammatory mediators in BMDMs of young and old mice that were treated with LPS or polarized to classical M1 or alternative M2 activation. In LPS-stimulated BMDMs, ablation of GH-R prevented the age-dependent increase in inflammasome components ASC and caspase-1, whereas NLRP3 was not affected (Figures 2D and S2A). M1 macrophages demonstrated age-dependent increase in Nlrp3 (Figure 2D) and Il1β (Figure 2E) mRNA expression that was attenuated in GH-R-deficient cells with no change in ASC or caspase-1 (Figure S2A). Interestingly, no age-related increase upon LPS and M1 stimulation in BMDMs was found for NLRC4 inflammasome (Figure S2B), suggesting the NLRP3 inflammasome specifically regulates age-related inflammation in mice.
Compared to young cells, the aged BMDMs displayed significantly higher expression of pro-inflammatory cytokines Il12b, Tnfα, and Il6 that is GH-R dependent (Figures 2F–2H, S2C, and S2E). Furthermore, ablation of GH-R enhanced the expression of Ccl22, Ccl24, Arg1, and Il4r in M2 BMDMs, suggesting enhanced M2 polarization (Figures 2I–2L, S2D, and S2E). Together, these data suggest that GH-R controls transcriptional regulation of inflammasome components as well as age-related macrophage activation and inflammation.
GH-R and Macrophage-Derived IGF1 Controls the Inflammasome Assembly in Response to Diverse “Danger Signals”
After signal-1-dependent increase in transcription, the functional inflammasome is assembled when NLRP3 senses “signal 2,” which can be provided by a wide range of DAMPs (Henao-Mejia et al., 2014; Martinon et al., 2006; Wen et al., 2012). Therefore, we investigated whether GH-R expression controls NLRP3 inflammasome assembly in aged macrophages. Interestingly, compared to macrophages derived from 36-month-old WT mice, the age-matched GH-R-deficient BMDMs showed reduced caspase-1 activation and active p17 subunit response to canonical NLRP3 ligands ATP, urate crystals, and ROS damage induced by sodium arsenite (Figures 3A and 3B). Notably, ablation of GH-R attenuated the caspase-1 cleavage and IL-1β activation when NLRP3 inflammasome was primed by substituting LPS with a different TLR4 agonist lipid A (LA) or TLR2 ligands, peptidoglycan (PGN) and lipoteichoic acid (LTA), and signal-2-dependent inflammasome assembly was induced by ATP and urate (Figures 3C and 3D). Thus, the ablation of GH-R in old macrophages controls the NLRP3 inflammasome activation. Interestingly, aged GH-R-deficient macrophages expressed low levels of ASC and NLRP3, suggesting that the mechanism of reduced NLRP3 inflammasome activation is related to decrease in ASC-NLRP3 assembly availability upon activation by ATP, urate, and sodium arsenite (Figure 3E).
Figure 3. GH-R Ablation Protects against NLRP3 Inflammasome Activation, Suggesting an IGF1-IGF1R Autocrine-Paracrine Pathway in BMDMs.

(A and B) LPS-primed 36-month-old WT and 36-month-old Ghr−/− BMDMs were stimulated with multiple aging-relevant DAMPs (ATP, MSU, and NaArs), and supernatants were analyzed for the active caspase-1 p20 subunit (A) and active IL-1β p17 subunit (B) by immunoblotting. n = 3–10/group; each blot is representative of one mouse/genotype.
(C and D) Caspase-1 p20 active subunit immunoblot analysis of supernatants in LA-, PGN-, and LTA-primed BMDMs stimulated with ATP (C) or urate (D).
(E) Immunoblot analysis of ASC and NLRP3 within the protein fraction of BMDMs, LPS-primed, and stimulated ATP, MSU, or NaArs as indicated in 36-month-old WT and 36-month-old Ghr−/−.
(F and G) Real-time PCR analysis of Igf1 (F) and Igf1r (G) gene expression in liver and BMDMs from 3-month WT, LysM-Cre−, and LysM-Cre+ IGF1Rf/f (n = 5–7/group).
(H and I) LPS-primed 3-month-old LysMCre− and LysMCre+ IGF1R fl/fl BMDMs were stimulated with multiple aging-relevant DAMPs, and cells (H) and supernatants (I) were analyzed for active caspase-1 (p20) and active IL-1β (p17) by immunoblotting.
Given IGF-1 is a downstream effector of GH-R-dependent anabolic responses (Junnila et al., 2013), we next investigated the mechanism of inflammasome regulation by the somatotropic axis. Surprisingly, compared to liver, which is the predominant endocrine source of circulating IGF-1, we identified that macrophages expressed IGF-1 similar to levels measured in liver (Figure 3F). Furthermore, macrophages also highly expressed the IGF-1 receptor (Igfr), suggesting an autocrine/paracrine role of somatotrophic axis in innate immune signaling (Figure 3G). Therefore, we next specifically deleted Igf1r from macrophages by myeloid-lineage-specific LysM-Cre (Figure S3). Interestingly, deletion of Igfr in macrophages led to significant reduction in NLRP3 inflammasome-dependent caspase-1 and IL-1β activation induced by extracellular ATP, urate crystals, sodium arsenite, and ceramides (Figures 3H and 3I). Together, these data suggest that downregulation of somatotrophic axis in macrophages protects against NLRP3 inflammasome activation.
Deletion of GH-R Protects against NLRP3-Dependent Urate-Crystal-Induced Inflammation
The long-lived Ghr−/− mice show reduced expression of inflammatory markers and display significant increase in insulin sensitivity, despite an increase in adipose tissue mass (Masternak and Bartke, 2012). Given our prior data that VAT is a significant source of inflammation and displays increased NLRP3 inflammasome activation (Youm et al., 2013), we challenged the middle-aged WT and Ghr−/− mice with urate crystals and quantified myeloid cell infiltration into the VAT. The Ghr−/− displayed significant reduction in F4/80+CD11b+ adipose tissue macrophages (ATM) (Figure 4A). Importantly, compared to sham-treated mice, urate crystals induced significant increase in ATM infiltration in VAT that was significantly reduced in 12-month-old Ghr−/− animals (Figure 4B). Total number of B lymphocytes (B220+ MHCII+) shows an increase in VAT of urate-treated old WT mice, which was reduced in animals with GH-R deletion (Figure S4). Collectively, these data suggest that lack of GH-R-dependent signaling protects against NLRP3-driven inflammation in vivo.
Figure 4. GH-R-Deficient Mice Are Protected from MSU-Induced Macrophage Infiltration in Adipose Tissue.

(A) The SVF separated from subcutaneous adipose tissue was analyzed for F480+ CD11b+ cells by FACS staining in 12-month WT and 12-month Ghr−/− mice.
(B) Twelve-month WT and twelve-month Ghr−/− mice were challenged with MSU, and 4 hr later, the SVF from the VAT was analyzed for F480+ CD11b+ cells by FACS staining.
All data are presented as mean ± SEM (n = 4 or 5/genotype/treatment); statistical differences were calculated by two-tailed paired Student’s t test (*p < 0.05).
DISCUSSION
Chronic diseases such as kidney disease, atherosclerosis, gout, cancer, diabetes, Alzheimer’s disease, and immunopathology resulting from aberrant immune responses are the major contributors to death and disability in the elderly (Goldberg and Dixit, 2015; Pawelec et al., 2014). Epidemiological evidence suggests that aging is the single biggest risk factor for these chronic diseases, and mechanistically, inflammation is thought to be a common link between aging and disease. Despite increasing awareness of the health and economic impact of the growing elderly population, limited progress has been made in understanding the immunological mechanisms that control age-related inflammation and whether harnessing the endogenous regulatory pathways can enhance health span and reduce disease burden. Here, we show that downregulation of GH-R-IGF1 axis in macrophages is one such signal that links longevity to innate immune sensor NLRP3 and control of age-related inflammation.
Given a vital role of innate immune system in maintaining tissue homeostasis and regenerative responses through regulated production of growth factors and pro-inflammatory cytokines, tissue macrophages located in every organ are the prime cellular candidates for potential role in age-related inflammation and degenerative changes (Medzhitov, 2008). Thus, sustained activation of tissue macrophages by endogenous metabolic danger signals or exposure to exogenous environmental contaminants during the lifespan can propagate inflammatory damage and tissue dysfunction. Interestingly, macrophages can sense diverse DAMPs via nucleotide-binding domain leucine-rich repeat (NLR) protein such as NLRP1, NLRP3, NLRC4, NLRC5, NLRP6, or NLRP7 (Martinon et al., 2009). Among these NLRs, the NLRP3 has been demonstrated to sense the environmental danger signals such as silica and asbestos and endogenous DAMPs such as urate (Martinon et al., 2006), extracellular ATP (Mariathasan et al., 2006), cytosolic DNA (Muruve et al., 2008), mitochondrial damage (Zhou et al., 2011), ceramides, and fatty acids (Youm et al., 2012). Importantly, downregulation of the NLRP3 inflammasome complex in macrophages protects against age-related inflammation (Youm et al., 2013). Thus, the endogenous-tissue-derived mechanisms that negatively control the NLRP3-dependent inflammatory responses may be especially relevant to aging, longevity, and health span extension. Our data show that downregulation of GH-R signaling, which extends lifespan, controls inflammation in aging by inhibiting both the transcriptional signal1 as well as signal-2-dependent post-translational processing of NLRP3 inflammasome.
Importantly, T cells are not the predominant sources of inflammasome-dependent cytokines IL-1 or IL-18 and NLRP3 machinery (Guarda et al., 2011). Thus, GH-R-dependent increase in naive T cells in advanced aging could result from indirect effect of overall reduction in systemic inflammation or reduced inflammasome activation in thymic macrophages that could result in protection from thymic involution and higher T cell export. Importantly, data from long-lived GH-R-deficient mice showing higher naive T cells are intriguing as prior studies show that treatment of mice and humans with GH increases thymic naive T cell production (Dixit, 2010; Murphy et al., 1992; Napolitano et al., 2008). Future studies using aged cell-specific gene-targeted mice would be required to gain definitive mechanistic insights of potential cell-intrinsic thymic effects of GH and inflammasome interactions. The GH-R deficiency enhanced the naive T cell repertoire and reduced the compensatory effector/memory T cell expansions typically seen in aging. Functionally, the E/M T cells of aged GHRKO mice displayed enhanced IFNγ secretion upon TCR-mediated activation. These findings are consistent with recent reports on the importance of the increased fibroblast responsiveness to IFNγ in long-lived primates (Pickering et al., 2015), which imply a role for the interferon response in the biology of aging.
The importance of the immune endocrine cross-talk mediated by GH-GH-R interactions in aging adipose tissue suggests that changes in secretion of adipocytes-derived hormones adiponectin and leptin may impact the adipose inflammation (Masternak et al., 2012). GH-IGF1 axis is thought to primarily operate at the endocrine level (Junnila et al., 2013). Surprisingly, we found macrophages are a substantial source of IGF-1 and have significant IGF-1R expression. Existing transcriptional profiling data sets from Immunological Genome Project (http://www.immgen.org) further confirmed that macrophages and myeloid origin cells highly express IGF1 axis, suggesting an auto/paracrine function. Consistent with this hypothesis, ablation of IGF1R in macrophages and downregulation of somatotrophic axis demonstrated reduced inflammasome activation in response to diverse NLRP3 activators. In addition, downregulation of GHRH-GH-IGF1 axis exerts regulatory effects on the immune system by lowering experimental ocular inflammation (Qin et al., 2014). Furthermore, upon challenge with urate-crystal-induced NLRP3 inflammasome activation in vivo, the GH-R-deficient middle-aged mice are protected from VAT leukocytosis. Collectively, our data show that inhibition of NLRP3 inflammasome by ablation of GH-R and IGF1R is one of the mechanisms that links reduced inflammation to pro-longevity effects.
EXPERIMENTAL PROCEDURES
Experimental Animals
Young and long-lived Ghr−/− mice and WT have been described previously (Zhou et al., 1997). Mice were sacrificed 10 days after the quarantine period; during this time mice were multi-housed and fed ad libitum with normal chow diet (5002; LabDiet). Igf1R-flox strain can be purchased from Jackson Laboratories (B6;129-Igf1rtm2Arge). The colony was multi-housed, fed ad libitum with normal chow diet (5002; LabDiet), and kept following guidelines issued by Yale University’s Institutional Animal Care and Use Committee (IACUC).
Cell Culture
The murine bone marrow was collected from femurs in RPMI (Life Technologies) + 10% FBS (R10 Omega Scientific) + 5% antibacterial/antimycotic (Life Technologies), and red blood cells were lysed using ACK lysis buffer. Bone-marrow-derived macrophages (BMDMs) were differentiated using MCSF (10 ng/ml; R&D Systems) and L929-conditioned media for 7 days. Non-adherent BMDMs were re-plated and treated the following day (see Supplemental Experimental Procedures).
For Luminex analysis, CD4+ CD62L− CD44+ E/M T cells were sorted from splenocytes and cultured on anti-CD3-coated plates (BD Pharmigen) + CD28 soluble antibody 2 µg/ml (BD Pharmigen) added to the media (RPMI + 10% FBS + 5% antibacterial/antimycotic). After 96 hr, the supernatants were measured for IFNγ, TNFα, IL-4, and IL-5 by Bio-Plex Pro Mouse Cytokine Th1/Th2 Assay (Bio-Rad no. M60-00003J7) on Luminex xPONENT system.
Western Blot
BMDM cell lysates were prepared in RIPA buffer by vortexing samples every 10 min for 30’. Samples were then centrifuged at 14,000 g for 15 min, the supernatant was collected, and the protein concentration was determined using the DC Protein Assay (Bio-Rad). Visceral fat was homogenized in liquid nitrogen using pestle and mortar and then mixed in RIPA as described above. Immunoblot analysis was performed as described previously (Vandanmagsar et al., 2011).
Antibodies for caspase-1 (1:250; 4B4.2.1; Genentech), IL-1β (1:500; GTX74034; Genetex), Asc (1:500; ADI-905-173-100; Enzo Life Science), and β-actin (1:1,000; 4967L; Cell Signaling Technology) were used at the dilutions specified by the manufacturer. The immune complexes were visualized by incubation with horseradish-peroxidase-conjugated anti-rat (PI31470; Pierce) or anti-rabbit secondary antibody (PI31460). Immuno-reactive bands were visualized by enhanced chemiluminescence (PI32209; Pierce).
Gene Expression Analysis
Total RNA was extracted using the Trizol method and transferred to the QIAGEN RNeasy mini kit and purified according to the manufacturer’s instructions. Synthesis of cDNA and qPCR was performed as described previously (Nolan et al., 2006). The primer pairs used for real-time PCR are listed in the Table S1.
Flow Cytometry
Antibodies used were CD4, CD8, CD44, and CD62L (eBioscience Affymetrix). Cells were acquired on a BD FACSCalibur, and data were analyzed in FlowJo (Tree Star). SVF was stained for Fixable Viability Dye Aqua (Life Technologies), B220, MHCII, F4/80, and CD11b. Data were acquired on a BD LSR II and analyzed in FlowJo.
Statistical Analyses
Statistical significance of the differences between groups was calculated either by two-tailed paired Student’s t test or by two-way ANOVA using Tukey’s test, which protects the significance of all pair combinations (GraphPad Prism 6 software).
p < 0.05 was considered significant for all the analyses.
Supplementary Material
Highlights.
The long-lived GH-R-deficient mice are protected from inflammaging
Loss of GH-R deactivates NLRP3 inflammasome and increases naive T cells in aging
Ablation of macrophage IGF1-IGF1R axis inhibits the inflammasome
Macrophage somatotrophic axis regulates NLRP3 inflammasome in aging
Acknowledgments
V.D.D. lab is supported in part by NIH grants DK090556, AG043608, and AI105097. C.D.C. is supported by supplemental funding from NIHAG043608. E.L.G. is supported by postdoctoral fellowship from American Federation of Aging Research (AFAR). L.Y.S. is supported by NIH grant AG048264, and A.B. is supported by AG019899 and AG031736.
Footnotes
Supplemental Information includes Supplemental Experimental Procedures, four figures, and one table and can be found with this article online at http://dx.doi.org/10.1016/j.celrep.2016.01.044.
AUTHOR CONTRIBUTIONS
O.S. designed and conducted the majority of experiments, analyzed/interpreted the data, and participated in writing the manuscript. E.L.G. performed FACS analysis and interpreted the data, and C.D.C. performed the sorting analyses and interpreted the data. Y.-H.Y. conducted the analyses of T cell senescence, and K.Y.N. assisted in generation and validation of macrophage IGF1R mice and assisted in data analysis. J.J.K. generated the GH-R-null mice and assisted in data interpretation. A.B. and L.Y.S. aged the GH-R-deficient and control mice and participated in study design and data analysis. V.D.D. conceived and supervised the project, interpreted the data, and wrote the manuscript.
References
- Bartke A, Sun LY, Longo V. Somatotropic signaling: trade-offs between growth, reproductive development, and longevity. Physiol. Rev. 2013;93:571–598. doi: 10.1152/physrev.00006.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown-Borg HM, Borg KE, Meliska CJ, Bartke A. Dwarf mice and the ageing process. Nature. 1996;384:33. doi: 10.1038/384033a0. [DOI] [PubMed] [Google Scholar]
- Cheng CW, Adams GB, Perin L, Wei M, Zhou X, Lam BS, Da Sacco S, Mirisola M, Quinn DI, Dorff TB, et al. Prolonged fasting reduces IGF-1/PKA to promote hematopoietic-stem-cell-based regeneration and reverse immunosuppression. Cell Stem Cell. 2014;14:810–823. doi: 10.1016/j.stem.2014.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coschigano KT, Holland AN, Riders ME, List EO, Flyvbjerg A, Kopchick JJ. Deletion, but not antagonism, of the mouse growth hormone receptor results in severely decreased body weights, insulin, and insulin-like growth factor I levels and increased life span. Endocrinology. 2003;144:3799–3810. doi: 10.1210/en.2003-0374. [DOI] [PubMed] [Google Scholar]
- Dixit VD. Thymic fatness and approaches to enhance thymopoietic fitness in aging. Curr. Opin. Immunol. 2010;22:521–528. doi: 10.1016/j.coi.2010.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrucci L, Corsi A, Lauretani F, Bandinelli S, Bartali B, Taub DD, Guralnik JM, Longo DL. The origins of age-related proinflammatory state. Blood. 2005;105:2294–2299. doi: 10.1182/blood-2004-07-2599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fontana L, Villareal DT, Das SK, Smith SR, Meydani SN, Pittas AG, Klein S, Bhapkar M, Rochon J, Ravussin E, et al. Effects of 2-year calorie restriction on circulating levels of IGF-1, IGF-binding proteins and cortisol in nonobese men and women: a randomized clinical trial. Aging Cell. 2016;15:22–27. doi: 10.1111/acel.12400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franceschi C, Campisi J. Chronic inflammation (inflammaging) and its potential contribution to age-associated diseases. J. Gerontol. A Biol. Sci. Med. Sci. 2014;69(Suppl. 1):S4–S9. doi: 10.1093/gerona/glu057. [DOI] [PubMed] [Google Scholar]
- Goldberg EL, Dixit VD. Drivers of age-related inflammation and strategies for healthspan extension. Immunol. Rev. 2015;265:63–74. doi: 10.1111/imr.12295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gross O, Thomas CJ, Guarda G, Tschopp J. The inflammasome: an integrated view. Immunol. Rev. 2011;243:136–151. doi: 10.1111/j.1600-065X.2011.01046.x. [DOI] [PubMed] [Google Scholar]
- Guarda G, Zenger M, Yazdi AS, Schroder K, Ferrero I, Menu P, Tardivel A, Mattmann C, Tschopp J. Differential expression of NLRP3 among hematopoietic cells. J. Immunol. 2011;186:2529–2534. doi: 10.4049/jimmunol.1002720. [DOI] [PubMed] [Google Scholar]
- Henao-Mejia J, Elinav E, Thaiss CA, Flavell RA. Inflammasomes and metabolic disease. Annu. Rev. Physiol. 2014;76:57–78. doi: 10.1146/annurev-physiol-021113-170324. [DOI] [PubMed] [Google Scholar]
- Huffman DM, Barzilai N. Role of visceral adipose tissue in aging. Biochim. Biophys. Acta. 2009;1790:1117–1123. doi: 10.1016/j.bbagen.2009.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Junnila RK, List EO, Berryman DE, Murrey JW, Kopchick JJ. The GH/IGF-1 axis in ageing and longevity. Nat. Rev. Endocrinol. 2013;9:366–376. doi: 10.1038/nrendo.2013.67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kenyon CJ. The genetics of ageing. Nature. 2010;464:504–512. doi: 10.1038/nature08980. [DOI] [PubMed] [Google Scholar]
- Lamkanfi M, Dixit VM. Mechanisms and functions of inflammasomes. Cell. 2014;157:1013–1022. doi: 10.1016/j.cell.2014.04.007. [DOI] [PubMed] [Google Scholar]
- Longo VD, Finch CE. Evolutionary medicine: from dwarf model systems to healthy centenarians? Science. 2003;299:1342–1346. doi: 10.1126/science.1077991. [DOI] [PubMed] [Google Scholar]
- Lu C, Kumar PA, Sun J, Aggarwal A, Fan Y, Sperling MA, Lumeng CN, Menon RK. Targeted deletion of growth hormone (GH) receptor in macrophage reveals novel osteopontin-mediated effects of GH on glucose homeostasis and insulin sensitivity in diet-induced obesity. J. Biol. Chem. 2013;288:15725–15735. doi: 10.1074/jbc.M113.460212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mariathasan S, Weiss DS, Newton K, McBride J, O’Rourke K, Roose-Girma M, Lee WP, Weinrauch Y, Monack DM, Dixit VM. Cryopyrin activates the inflammasome in response to toxins and ATP. Nature. 2006;440:228–232. doi: 10.1038/nature04515. [DOI] [PubMed] [Google Scholar]
- Martinon F, Burns K, Tschopp J. The inflammasome: a molecular platform triggering activation of inflammatory caspases and processing of proIL-beta. Mol. Cell. 2002;10:417–426. doi: 10.1016/s1097-2765(02)00599-3. [DOI] [PubMed] [Google Scholar]
- Martinon F, Pétrilli V, Mayor A, Tardivel A, Tschopp J. Gout-associated uric acid crystals activate the NALP3 inflammasome. Nature. 2006;440:237–241. doi: 10.1038/nature04516. [DOI] [PubMed] [Google Scholar]
- Martinon F, Mayor A, Tschopp J. The inflammasomes: guardians of the body. Annu. Rev. Immunol. 2009;27:229–265. doi: 10.1146/annurev.immunol.021908.132715. [DOI] [PubMed] [Google Scholar]
- Masternak MM, Bartke A. Growth hormone, inflammation and aging. Pathobiol. Aging Age Relat. Dis. 2012;2 doi: 10.3402/pba.v2i0.17293. http://dx.doi.org/10.3402/pba.v2i0.17293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masternak MM, Bartke A, Wang F, Spong A, Gesing A, Fang Y, Salmon AB, Hughes LF, Liberati T, Boparai R, et al. Metabolic effects of intra-abdominal fat in GHRKO mice. Aging Cell. 2012;11:73–81. doi: 10.1111/j.1474-9726.2011.00763.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medzhitov R. Origin and physiological roles of inflammation. Nature. 2008;454:428–435. doi: 10.1038/nature07201. [DOI] [PubMed] [Google Scholar]
- Metchnikoff É. In: The Prolongation of Life: Optimistic Studies. Chalmers Mitchell P, editor. G.P. Putnam’s Sons; 1908. [Google Scholar]
- Murphy WJ, Durum SK, Longo DL. Role of neuroendocrine hormones in murine T cell development. Growth hormone exerts thymopoietic effects in vivo. J. Immunol. 1992;149:3851–3857. [PubMed] [Google Scholar]
- Muruve DA, Pétrilli V, Zaiss AK, White LR, Clark SA, Ross PJ, Parks RJ, Tschopp J. The inflammasome recognizes cytosolic microbial and host DNA and triggers an innate immune response. Nature. 2008;452:103–107. doi: 10.1038/nature06664. [DOI] [PubMed] [Google Scholar]
- Napolitano LA, Schmidt D, Gotway MB, Ameli N, Filbert EL, Ng MM, Clor JL, Epling L, Sinclair E, Baum PD, et al. Growth hormone enhances thymic function in HIV-1-infected adults. J. Clin. Invest. 2008;118:1085–1098. doi: 10.1172/JCI32830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nolan T, Hands RE, Bustin SA. Quantification of mRNA using real-time RT-PCR. Nat. Protoc. 2006;1:1559–1582. doi: 10.1038/nprot.2006.236. [DOI] [PubMed] [Google Scholar]
- Pawelec G, Goldeck D, Derhovanessian E. Inflammation, ageing and chronic disease. Curr. Opin. Immunol. 2014;29:23–28. doi: 10.1016/j.coi.2014.03.007. [DOI] [PubMed] [Google Scholar]
- Pickering AM, Lehr M, Miller RA. Lifespan of mice and primates correlates with immunoproteasome expression. J. Clin. Invest. 2015;125:2059–2068. doi: 10.1172/JCI80514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin YJ, Chan SO, Chong KK, Li BF, Ng TK, Yip YW, Chen H, Zhang M, Block NL, Cheung HS, et al. Antagonist of GH-releasing hormone receptors alleviates experimental ocular inflammation. Proc. Natl. Acad. Sci. USA. 2014;111:18303–18308. doi: 10.1073/pnas.1421815112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schroder K, Tschopp J. The inflammasomes. Cell. 2010;140:821–832. doi: 10.1016/j.cell.2010.01.040. [DOI] [PubMed] [Google Scholar]
- Stewart MH, Gutierrez-Martinez P, Beerman I, Garrison B, Gallagher EJ, LeRoith D, Rossi DJ. Growth hormone receptor signaling is dispensable for HSC function and aging. Blood. 2014;124:3076–3080. doi: 10.1182/blood-2014-05-575308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun LY, Spong A, Swindell WR, Fang Y, Hill C, Huber JA, Boehm JD, Westbrook R, Salvatori R, Bartke A. Growth hormone-releasing hormone disruption extends lifespan and regulates response to caloric restriction in mice. eLife. 2013;2:e01098. doi: 10.7554/eLife.01098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vandanmagsar B, Youm YH, Ravussin A, Galgani JE, Stadler K, Mynatt RL, Ravussin E, Stephens JM, Dixit VD. The NLRP3 inflammasome instigates obesity-induced inflammation and insulin resistance. Nat. Med. 2011;17:179–188. doi: 10.1038/nm.2279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wen H, Ting JP, O’Neill LA. A role for the NLRP3 inflammasome in metabolic diseases–did Warburg miss inflammation? Nat. Immunol. 2012;13:352–357. doi: 10.1038/ni.2228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Youm YH, Kanneganti TD, Vandanmagsar B, Zhu X, Ravussin A, Adijiang A, Owen JS, Thomas MJ, Francis J, Parks JS, Dixit VD. The Nlrp3 inflammasome promotes age-related thymic demise and immunosenescence. Cell Rep. 2012;1:56–68. doi: 10.1016/j.celrep.2011.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Youm YH, Grant RW, McCabe LR, Albarado DC, Nguyen KY, Ravussin A, Pistell P, Newman S, Carter R, Laque A, et al. Canonical Nlrp3 inflammasome links systemic low-grade inflammation to functional decline in aging. Cell Metab. 2013;18:519–532. doi: 10.1016/j.cmet.2013.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Y, Xu BC, Maheshwari HG, He L, Reed M, Lozykowski M, Okada S, Cataldo L, Coschigamo K, Wagner TE, et al. A mammalian model for Laron syndrome produced by targeted disruption of the mouse growth hormone receptor/binding protein gene (the Laron mouse) Proc. Natl. Acad. Sci. USA. 1997;94:13215–13220. doi: 10.1073/pnas.94.24.13215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou R, Yazdi AS, Menu P, Tschopp J. A role for mitochondria in NLRP3 inflammasome activation. Nature. 2011;469:221–225. doi: 10.1038/nature09663. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.