

Abstract
Translesion synthesis (TLS) is an important mechanism through which proliferating cells tolerate DNA damage during replication. The mutagenic Rev1/Polζ-dependent branch of TLS helps cancer cells survive first-line genotoxic chemotherapy and introduces mutations that can contribute to the acquired resistance so often observed with standard anti-cancer regimens. As such, inhibition of Rev1/Polζ-dependent TLS has recently emerged as a strategy to enhance the efficacy of first-line chemotherapy and reduce the acquisition of chemoresistance by decreasing tumor mutation rate. The TLS DNA polymerase Rev1 serves as an integral scaffolding protein that mediates the assembly of the active multi-protein TLS complexes. Protein-protein interactions (PPIs) between the C-terminal domain of Rev1 (Rev1-CT) and the Rev1-interacting region (RIR) of other TLS DNA polymerases play an essential role in regulating TLS activity. To probe whether disrupting the Rev1-CT/RIR PPI is a valid approach for developing a new class of targeted anti-cancer agents, we designed a fluorescence polarization-based assay that was utilized in a pilot screen for small molecule inhibitors of this PPI. Two small molecule scaffolds that disrupt this interaction were identified and secondary validation assays confirmed that compound 5 binds to Rev1-CT at the RIR interface. Finally, survival and mutagenesis assays in mouse embryonic fibroblasts and human fibrosarcoma HT1080 cells treated with cisplatin and ultraviolet light indicate that these compounds inhibit mutagenic Rev1/Polζ-dependent TLS in cells, validating the Rev1-CT/RIR PPI for future anti-cancer drug discovery and identifying the first small molecule inhibitors of TLS that target Rev1-CT.
INTRODUCTION
Genotoxic chemotherapeutics (platinating agents, alkylating agents, etc.) are standard first-line chemotherapy for many types of human cancer.1–5 Typically, cancer patients initially respond well to these agents; however, many patients rapidly develop resistance to these chemotherapeutics and experience relapse, which requires either an increase in drug dosage or a change in drug regimen to combat the relapsed cancer. In addition, the high doses of these drugs used in chemotherapy regimens can result in toxic side effects in other tissues. These disadvantages limit both the short- and long-term effectiveness of traditional genotoxic agents, while also highlighting the therapeutic potential of small molecules that could reduce the high dosage and avert acquired chemoresistance associated with these therapies.
Translesion synthesis (TLS) is an important mechanism through which proliferating cells tolerate DNA damage during replication.6–9 In humans, specialized TLS DNA polymerases, including the Y-family polymerases Rev1, Polη, Polκ, Polι and the B-family polymerase Polζ (Rev3/Rev7/PolD2/PolD3 complex10–12), carry out TLS over sites of DNA damage thereby enabling cells to replicate while leaving the DNA lesions unrepaired.6–9 The mutagenic Rev1/Polζ-dependent branch of TLS is responsible for most of the mutations caused by DNA damage9,13–16 and is also a mechanistic component of the repair of interstrand DNA crosslinks.17 TLS not only plays a central role in helping normal cells survive DNA damage, but also allows cancer cells to survive genotoxic chemotherapy.8 Furthermore, the error-prone TLS DNA polymerases increase the rate of mutation in tumors contributing to the rapid emergence of drug-resistant cells; therefore, TLS has also been implicated as an underlying mechanism responsible for the acquisition of resistance to genotoxic chemotherapeutics.18–22
Inhibition of TLS has been shown to sensitize several types of cancer cells to first-line chemotherapeutics while reducing mutagenesis.22–25 For example, suppression of Rev1/Polζ-dependent TLS sensitizes lung adenocarcinoma24 and ovarian carcinoma25 cells to the cytotoxic effect of cisplatin and decreases their mutation frequency, as well as results in improved chemotherapeutic efficacy in the KrasG12D;p53−/− mouse model of human non-small cell lung cancer.25 In the well-established Eμ-myc arf−/− mouse model of Burkitt’s lymphoma, suppression of Rev1/Polζ-dependent TLS greatly reduced the frequency of acquired drug resistance during DNA damaging chemotherapy.22 In another example, co-administration of a cisplatin prodrug with siRNAs specific for Rev1 and Rev3 (the catalytic subunit of Polζ) enhanced chemotherapy response in a murine xenograft model of prostate cancer.23 These findings provide strong evidence that TLS inhibition holds potential as a new therapeutic strategy to enhance the efficacy of first-line chemotherapy. As such, small molecule inhibitors of TLS are emerging as a promising new class of adjuvant agents that could reduce both the dose of genotoxic agents and the associated toxic side effects of first-line therapies, as well as avert chemoresistance.8
The majority of replication-blocking DNA lesions are bypassed by a combination of TLS DNA polymerases in a two-step process of Rev1/Polζ-dependent TLS,9,13–16 in which an ‘inserter’ DNA polymerase (commonly Polη, Polκ, Polι) incorporates a nucleotide across the lesion followed by extension of the primer-template by an ‘extender’ Polζ (a complex of Rev3/Rev7/PolD2/PolD3 subunits10–12). These TLS enzymes replace replicative DNA polymerases, Polδ and Polε, at sites of DNA damage in a reversible process controlled by ubiquitination of Proliferating Cell Nuclear Antigen (PCNA).26–28 The sliding clamp PCNA and the DNA polymerase Rev1 play key structural roles in Rev1/Polζ-dependent TLS by serving as scaffolds that control the fine-tuned assembly of TLS DNA polymerase complexes and mediate polymerase switching through a series of protein-protein interactions (PPIs).6,9 In humans, Rev1 PPIs with all other TLS enzymes are mediated by its C-terminal (Rev1-CT) domain that can bind Rev1-interacting regions (RIR) from Polκ, Polη, Polι and PolD3 and, at the same time, interact with the accessory Rev7 subunit of Polζ.29–35 Cells deficient in Rev1 exhibit increased sensitivity to DNA damage and a significantly reduced mutation rate.36–38 Deletion of the Rev1-CT domain confers a similar DNA damage sensitive non-mutable phenotype, suggesting that this domain is critical for the cellular function of the TLS pathway.39–40
Based on the essential role that Rev1-CT interactions with other TLS DNA polymerases play in mediating Rev1/Polζ-dependent TLS, small molecules that disrupt these PPIs hold potential as a novel class of adjuvant agents for first-line genotoxic chemotherapy. Therefore, we developed a fluorescence polarization based assay to identify small molecules that disrupt interactions between Rev1-CT and other TLS polymerases containing RIR motifs. Optimization and implementation of this assay in a pilot screen has resulted in the identification of the first small molecule scaffolds that inhibit TLS through direct binding interactions with Rev1. In addition, the ability of these compounds to enhance cisplatin and UV sensitivity and reduce cisplatin-mediated mutagenesis in mouse embryonic fibroblasts and human cancer cells validates the Rev1-CT/RIR PPI as an anti-cancer drug target.
RESULTS AND DISCUSSION
Assay Optimization and Pilot Screen
Our overall assay design focused on developing a fluorescence polarization (FP) assay to identify small molecule scaffolds that disrupt Rev1-CT/Polκ-RIR PPIs. We chose the Polκ-RIR for our screen because it exhibits greater binding affinity (Kd = 1.7 μM) for Rev1-CT than other RIR motifs, suggesting a more stable complex for assay development.29 The Rev1-CT domain (residues 1157-1251) was expressed as described elsewhere31 and purified in complex with a synthesized (GenScript) human Polκ-RIR peptide (residues 560-575) modified with an N-terminal fluorescein amidite label (FAM-Polκ-RIR). To determine the optimal concentration of the Rev1-CT/FAM-Polκ-RIR complex for our assay, we evaluated the ability of unlabeled Polκ-RIR to disrupt the complex across a wide-range of concentrations (Supplemental Figure 2). When the complex concentrations were below 10 nM, the changes in fluorescence polarization (measured in mP units) upon displacement of the FAM-labeled with the unlabeled peptide were modest. When the concentration of the complex was above 100 nM, the noise associated with the fluorescently labeled peptide was too high and a concentration-dependent response was not observed. By contrast, a robust, concentration-dependent displacement of the FAM-Polκ-RIR was observed with complex concentrations of either 10 nM (IC50 = 7.3 ± 1.4 μM) or 100 nM (IC50 = 6.6 ± 0.1 μM). In addition, the ability of the unlabeled peptide to disrupt the complex at these concentrations correlated well with the Rev1-CT/Polκ-RIR binding affinity previously determined.29–30,41 In our hands, the assay was most reproducible when the complex concentration was 100 nM; therefore, we utilized this concentration for the remainder of our assays. These optimized parameters provide a robust, reproducible assay with a Z’ score of 0.74 (Figure 1A).42
Figure 1.

Initial screening results. (A) Optimized assay parameters provide a robust, reproducible disruption of the Rev1-CT/FAM-Polκ-RIR complex (100 nM) by unlabeled Polκ-RIR. Graph shows a single representative experiment that was reproduced at least three separate times. (B) Scatter plot of fluorescence polarization values (mP) from the initial pilot screen. Each compound (4,800) was evaluated at 10 μM. Rev1-CT/FAM-Polκ-RIR concentration was 100 nM.
Utilizing the optimized FP displacement assay, we performed a pilot screen of 4,800 compounds to identify small molecules that disrupt Rev1-CT/RIR PPIs. Compounds evaluated in this screen were from the ChemBridge DIVERSet library, which was chosen because it represents a highly diverse series of structures with stringent drug-like properties that cover a broad range of molecular scaffolds. In this initial screen, 83 compounds (1.7%) completely displaced FAM-Polκ-RIR from Rev1-CT at 10 μM (Figure1B). Re-screening of the 83 hit compounds at 1 μM identified several compounds that displaced approximately 50% of the FAM-Polκ-RIR from the complex at this concentration (Figure 2). Follow-up studies for these compounds demonstrated that each of them inhibited Rev1-CT/Polκ-RIR interactions in a concentration-dependent fashion with IC50 values in the low micromolar range (Table 1 and Supplemental Figure 3). Structurally, our screen identified two general scaffolds as inhibitors of the Rev1-CT/RIR PPI (Figure 2). The first scaffold contained a central piperazine flanked by two phenyl rings (represented here by 1–3). The second scaffold contains a ‘right-side’ piperidinyl ketone linked through an amide bond to a ‘left-side’ substituted thiophene (4–5).
Figure 2.

Initial hits identified from the pilot screen.
Table 1.
IC50 and cLogP values for initial hits.
| Compound | IC50 (μM)a | Predicted cLogPb |
|---|---|---|
|
| ||
| 1 | 3.5 ± 0.6 | 1.72 |
| 2 | 4.6 ± 0.2 | 1.68 |
| 3 | 7.5 ± 1.2 | 3.99 |
| 4 | 4.1 ± 0.5 | 1.55 |
| 5 | 2.5 ± 1.2 | 1.45 |
Values represent mean ± SEM of three separate experiments performed in triplicate.
cLogP values provided by ChemBridge.
NMR Binding Studies
The results of previous structural and mutational studies for PPIs between the Rev1-CT domain and RIR motifs from multiple TLS DNA polymerases highlighted the importance of a Phe-Phe pair (Polκ, Phe567-Phe568; Polη, Phe531-Phe532; PolD3, Phe238-Phe239) within the RIR motif as ‘hot spot’ residues critical for binding Rev1-CT.29–31,34–35 The Rev1-CT domain adopts a four-helix bundle fold with the binding site for RIR motifs formed by the first two α-helices and the N-terminal β-hairpin (Figure 3).31 Upon binding, the RIR motif folds into an α-helix that positions the side-chains of the two Phe residues towards the domain surface. The side-chains of these Phe residues interact with a pre-formed binding site on Rev1-CT and account for the majority of intermolecular contacts that stabilize the Rev1-CT/RIR complex, representing a ‘hot spot’ region for this PPI in Rev1-CT (Figure 3C).
Figure 3.

Compound 5 binds directly to Rev1-CT at the RIR interface. (A) Regions of 1H-15N HSQC NMR spectrum of the Rev1-CT domain upon its titration with increasing amount of compound 5, showing gradual shifts for selected peaks toward their positions in the compound bound state. (B) Cumulative 1H and 15N NMR chemical shift changes for the backbone and Trp side-chain HN groups of the Rev1-CT domain induced by binding compound 5 mapped onto Rev1-CT structure (PDB: 2LSY; chemical shift changes Δϖ are proportional to sphere radii). (C) ‘Hot spot’ residues of the Polη-RIR motif (purple) and corresponding RIR binding pocket on Rev1-CT (PDB: 2LSK).31
In order to confirm that the hit compounds disrupt the Rev1-CT/Polκ-RIR complex through direct binding interactions with the Rev1-CT site for RIR motifs, we performed NMR binding experiments in which 50 μM 15N labeled Rev1-CT domain was gradually titrated with 1 or 5 (Figure 3). These two compounds were specifically chosen for the binding studies because they are predicted to exhibit greater aqueous solubility (as determined by their cLogP values) at the concentrations needed for our NMR binding studies (above 100–150 μM). A number of backbone and Trp side-chain NH resonances in 1H-15N HSQC spectrum of the Rev1-CT domain shifted upon addition of compound 5 (Figure 3A), suggesting direct binding (with exchange between the free and bound forms fast on the NMR time-scale). The changes in positions of amide resonances of the Rev1-CT domain were quantified, and the resulting titration profiles (1H and 15N chemical shifts vs. the domain and compound concentrations) were analyzed to extract per-residue binding-induced amide chemical shift changes Δϖ and dissociation constants Kd (see Methods and Supplemental Figure 4). The obtained binding-induced chemical shift changes were mapped onto the Rev1-CT structure, outlining the Rev1-CT binding site for compound 5 (Figure 3B). On the other hand, titration of compound 1 into 50 μM 15N labeled Rev1-CT resulted in only minimal chemical shift changes for amino acid residues at the RIR interface, possibly due to lower aqueous solubility of 1 as compared to 5 (Table 1). Overall, the assumed aqueous concentrations of 1 and 5 were at the limit of compound solubility and the actual concentrations evaluated are likely overestimated. Because of this low solubility, we were not able to saturate Rev1-CT with either compound and obtain accurate estimates of either Kd or absolute values of chemical differences between the free and bound states (Δϖ). Nonetheless, relative binding-induced NMR chemical shift changes for different NH groups provide clear evidence that compound 5 interacts with the Rev1-CT region encompassing the binding site for RIR-motifs formed by α-helices H1 and H2 and the N-terminal β-hairpin (Figure 3A and 3B). The most pronounced chemical shift changes upon binding were observed for Rev1-CT residues located in the vicinity of the known RIR binding pocket, including the backbone NH and side-chain NHε1 groups of Trp1175 and Trp1225, and the backbone amides of Arg1173 and several other residues. Overall, these results clearly demonstrate that 5 directly binds to Rev1-CT near the RIR interaction site and provides strong evidence that this binding results in disruption of the Rev1-CT/RIR PPI.
Computational Studies
In parallel to our experimental NMR-based binding analysis, we also performed a series of computational studies to explore both the overall energetics of Rev1-CT/RIR binding and the specific intermolecular interactions between Rev1-CT and the small molecule scaffolds. Initially, we performed molecular dynamics (MD) simulations (5 ns, AMBER) followed by per-residue and pairwise energy decompositions to evaluate and predict the binding free energy (ΔG) associated with the Rev1-CT/Polκ-RIR complex. It is important to note that the absolute values for binding free energies calculated through these methods cannot be directly compared to the association free energies obtained from experimentally determined dissociation constants; instead, these values are most useful for either comparing the overall binding trends between Rev1-CT and RIR motifs or to provide preliminary context for the overall activity of the compounds. These simulations supported strong binding interactions between Rev1-CT and the Polκ-RIR motif (ΔG = −54.15 kcal/mol) and demonstrated that the Phe-Phe pair contributes a majority of the binding energy to the PPI (ΔG = −19.41 kcal/mol, Supplemental Figure 5) clearly defining a binding site on Rev1-CT that can be targeted with small molecules. These results are in agreement with previous structural works and mutational analyses characterizing the essential nature of the Phe-Phe pair in the RIR motif for tight binding interactions with Rev1-CT.29–31,34–35
Following our NMR binding studies localizing interactions between Rev1-CT and 5 to the Phe-Phe binding pocket we performed a series of docking and MD simulations for compounds 1–2 and 4–5. These computational studies were designed to both predict specific binding interactions between the compounds and amino acid residues in the Rev1-CT/RIR binding site and to explore how various energy terms contribute to the overall binding energy for each compound (Table 2). While there was a wide-range of predicted binding affinities, compound 5 demonstrated the greatest binding energy through the MM/GBSA calculations (ΔG values = −13.26 kcal/mol). The van der Waals term contributed most to the binding energy for these compounds; not a surprising result considering aromatic functionalities from each of the compounds was predicted to insert into the Phe binding pocket on Rev1-CT. In fact, the results of the docking/MD simulations for the piperazine scaffold (1 and 2) did not predict any specific electrostatic or hydrogen bonding interactions between this scaffold and Rev1-CT (Supplemental Figures 6 and 7). Compound 5 was predicted to have the strongest binding due to increased electrostatic interactions with Rev1-CT that result from a hydrogen bond between the side chain of Gln1189 and the carbonyl of the central amide (Figure 4). By contrast, the amide bond of compound 4 is predicted to orient toward Leu1172 and does not form a hydrogen bond with any amino acid residues present in the RIR interacting site (Supplemental Figure 8). In addition, the entire methyl ester of 4 is solvent exposed and interacts with multiple water molecules present at the Rev1-CT surface, reducing its overall electrostatic interactions with Rev1-CT. For both 4 and 5, the thiophene is buried in the Phe binding pocket while the piperidine and terminal acetyl group are solvent exposed (Figure 4B and Supplemental Figure 8). These computational studies provide key preliminary insight into the potential binding orientations and specific interactions between each scaffold and Rev1-CT while providing a starting point for the future design of improved analogues.
Table 2.
Binding energies of Rev1-CT and compounds from docking/MD simulations.
| Compound | ΔGbinding | ΔH | TΔS | ΔEvdw | ΔEele | ΔGGB | ΔGSA |
|---|---|---|---|---|---|---|---|
|
| |||||||
| 1 | −6.51 | −17.45 | −10.94 | −30.52 | −9.39 | 26.47 | −4.01 |
| 2 | −0.09 | −19.06 | −18.97 | −31.50 | −0.43 | 16.81 | −3.94 |
| 4 | −2.59 | −21.49 | −18.90 | −26.25 | −1.44 | 11.54 | −3.48 |
| 5 | −13.26 | −24.74 | −11.48 | −30.00 | −24.46 | 33.09 | −3.36 |
Gbinding = Total binding energy in kcal/mol.
ΔEvdw, ΔEele = Total van der Waals and electrostatic energies in kcal/mol.
ΔGGB and ΔGSA = Total polar and nonpolar contributions in kcal/mol.
Figure 4.

Predicted binding of compound 5 in the Rev1-CT/RIR ‘hot spot’. (A) Three-dimensional representation showing key Rev1-CT amino acid residues. (B) Two-dimensional representation of binding showing key interactions between the scaffold and Rev1-CT. Green line, pi-pi interaction; dotted purple arrow, side chain hydrogen bond; gray sphere, solvent exposed.
Cellular Studies
Our next step with these analogues was to determine whether their ability to disrupt the Rev1-CT/RIR PPI through direct binding interactions with Rev1-CT would result in functional inhibition of TLS in human cancer cells following treatment with genotoxic agents such as cisplatin and ultraviolet (UV) light. Cisplatin and other platinating agents are standard first-line chemotherapeutics for many types of human cancer that block replication by forming inter- and intra-strand DNA crosslinks.1–2,6 The replicative bypass of 1,2-d(GpG) cisplatin intra-strand crosslinks is contingent on Rev1/Polζ-dependent TLS and requires the coordinated action of Polη or Polκ to insert nucleotides across the lesion and Polζ to extend the primer-template.13–14,43 UV light, one of the best characterized genotoxic agents, causes two major types of DNA lesion, TT cis-syn cyclobutane pyrimidine dimers (TT-CPD) and (6-4) photoproducts (6-4 PP).9 While Polη alone can accurately and efficiently copy over TT-CPDs, the replicative bypass of 6-4 PPs relies on multi-step Rev1/Polζ-dependent TLS.13–14,44–45 Furthermore, TT-CPDs in cells from xeroderma pigmentosum variant (XPV) patients deficient in functional Polη can be bypassed by a combination of Polι or Polκ and Polζ, suggesting that the Rev1/Polζ-dependent TLS provides a backup mechanism for replicating over this lesion (albeit in a mutagenic manner).13–14
Initially, we evaluated the ability of the more soluble compounds (1–2 and 4–5) to enhance cisplatin sensitivity of the HT1080 human fibrosarcoma cell line.46–47 While the IC50 values for each compound was comparable in the FP displacement assay (Table 1), only 4 and 5 enhanced the sensitivity of HT1080 cells to cisplatin at concentrations as low as 1.5 μM (Supplemental Figure 9). Compounds 1 and 2 had little effect on cell viability either alone or in combination with cisplatin. The inability of either 1 or 2 to enhance cellular sensitivity to cisplatin could be a result of reduced solubility or permeability of the scaffold. These standard cytotoxicity assays reveal that by targeting Rev1-CT/RIR PPI, 4 and 5 lower the capacity of the cells to tolerate cisplatin-induced DNA damage. Based on the promising activity of 4 and 5 in this initial sensitivity assay, we further characterized their ability to enhance cisplatin sensitivity by determining whether they could improve the ability of cisplatin to reduce clonogenic survival of HT1080 cells. Treatment of HT1080 cells with cisplatin (0.6 μM), 4 (1.5 μM), or 5 (1.5 μM) had a minimal effect on colony formation; however, co-administration of cisplatin with either compound significantly reduced the clonogenic survival of HT1080 cells (Figure 5A).
Figure 5.

Compounds 4 and 5 sensitize HT1080 cells to killing by cisplatin and reduce mutagenesis. (A) Compounds 4 and 5 enhance sensitivity to cisplatin in a colony survival assay. Cells were incubated with cisplatin (0.6 μM) for 24 hrs, followed by the addition of either 4 or 5 (1.5 μM) and colonies were allowed to form for seven days. Colonies were stained and counted with coomassie blue. *P<0.05 and ***P<0.001 were calculated with the Student t-test using comparisons as indicated. (B) Compounds 4 and 5 reduce the cisplatin-induced HPRT mutations in HT1080 cells. Data are the Ave ± SD of two separate experiments with 3 replicates each. All experiments were performed at least two separate times and results were comparable across all replicates. **P<0.01 and ***P<0.001 versus cisplatin treated cells, Student t-test.
Previous studies have shown that Rev1 is essential for cisplatin-induced mutagenesis monitored by measuring mutation frequency of the hypoxanthine phosphoribosyl transferase (HPRT) gene, the standard assay to measure TLS activity in cells.48–50 To probe the effect of Rev1-CT/RIR PPI inhibitors on mutagenesis in human cancer cells, we evaluated the ability of 4 and 5 to reduce cisplatin-induced mutagenesis of the HPRT gene in the HT1080 fibrosarcoma cell line. Cisplatin alone (0.6 μM) increases HPRT mutations approximately 1.9-fold compared to non-treated cells (Figure 5B). By contrast, neither 4 nor 5 increased the frequency of HPRT mutations, indicating that these compounds were not inherently mutagenic under these assay conditions. The addition of either 4 or 5 to cisplatin-treated HT1080 cells significantly reduced HPRT mutations to levels comparable to the non-treated control cells, demonstrating that small molecule disruption of the Rev1-CT/RIR PPI reduces cisplatin-induced mutagenesis in a cultured cellular model of human cancer. Overall, increased cisplatin sensitivity and reduced mutagenesis in HT1080 cells treated with 4 or 5 (Figure 5) suggests that small molecule compounds targeting Rev1-CT/RIR PPIs effectively inhibit Rev1/Polζ-dependent TLS across cisplatin DNA adducts.
In addition to the studies performed in the HT1080 cells, we also evaluated the ability of compounds 4 and 5 to enhance cisplatin sensitivity in wild-type (WT) and Rev1−/− mouse embryonic fibroblasts (MEFs).51 Similar to the HT1080 cells, treatment of WT MEFs with either cisplatin (0.6 μM), 4 (1.5 μM), or 5 (1.5 μM) had minimal effect on clonogenic growth (Figure 6A), but WT MEFs treated with a combination of cisplatin and either 4 or 5 demonstrated a significant reduction in colony survival. As expected, the Rev1−/− MEFs lacking functional Rev1 were significantly more sensitive to cisplatin treatment, presumably due to their inability to utilize Rev1/Polζ-dependent TLS for the bypass of cisplatin DNA adducts. Co-administration of either 4 or 5 with cisplatin in the Rev1−/− MEFs did not further enhance the ability of cisplatin to reduce colony survival, providing strong evidence that the compounds were specific to Rev1 and their anti-TLS activity is dependent on the presence of functional Rev1. Similar to our results in HT1080 cells, compounds 4 and 5 reduced the HPRT mutagenesis frequencies in WT MEF cells when combined with cisplatin, but did not significantly alter these frequencies by themselves (Figure 6B). These results suggest that 4 and 5 are potent inhibitors of the Rev1-CT/RIR interface and, subsequently, TLS activity in cultured cells.
Figure 6.

Compounds 4 and 5 sensitize MEF cells to killing by cisplatin in a REV1+/+-dependent manner and reduce mutagenesis. (A) Compounds 4 and 5 enhance sensitivity of WT MEFs to cisplatin in a colony survival assay and not Rev1−/− cells. Cells were incubated with cisplatin (0.6 μM) for 24 hrs, followed by the addition of either 4 or 5 (1.5 μM) and colonies were allowed to form for seven days. Colonies were stained and counted with coomassie blue. Data are the Ave ± SD of two separate experiments performed in triplicate. NS = Not significant. (B) Compounds 4 and 5 reduce cisplatin-induced HPRT mutations in the WT MEF cells. Data are the Ave ± SD of two separate experiments with 6 replicates each. ***P<0.001 versus cisplatin treated cells, Student t-test.
To further explore whether the ability of 4 and 5 to sensitize cells to cisplatin is a broader effect of inhibiting Rev1/Polζ-dependent TLS and identify whether these compounds can effectively bypass DNA lesions caused by other carcinogens, we evaluated the ability of 4 and 5 to sensitize the HT1080 cells to UV light and observed a similar result (Figure 7), further highlighting their anti-TLS activity. Taken together, the results of our cellular studies demonstrate that disruption of the Rev1-CT/RIR PPI in human cancer cells inhibits Rev1/Polζ-dependent TLS across DNA lesions induced by different genotoxic agents and thus provides a new strategy to enhance the efficacy of first-line genotoxic chemotherapy.
Figure 7.
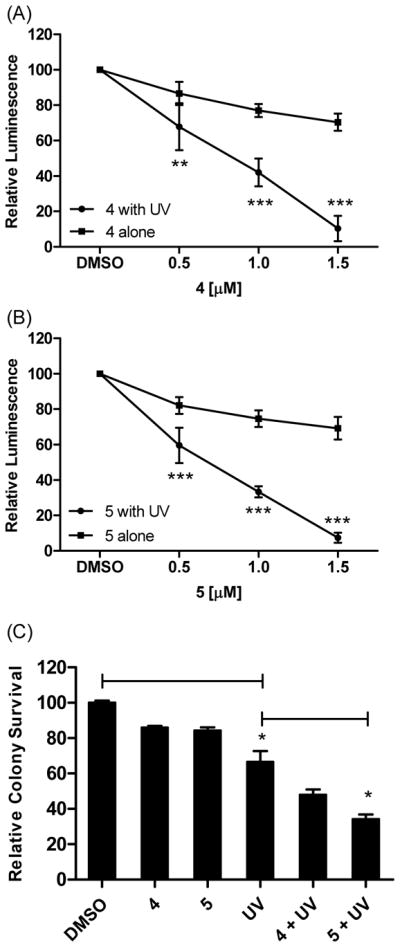
Enhanced sensitivity to UV light in HT1080 cells treated with Rev1-CT/RIR PPI inhibitors. In (A) and (B) cells were plated and exposed to UV light (5 J/m2) or left untreated. The next day, 4 or 5 was added and cell viability was measured 24 hrs later. **P<0.01 and ***P<0.001 versus cells treated with compound alone, Student t-test. (C) Cells were treated with UV light followed by dosing with 4 or 5 (1.5 μM). Cells were incubated to allow for the formation of colonies that were counted with commassie blue. *P<0.05 were calculated with the Student t-test using comparisons as indicated. Data are the Ave ± SD of two separate experiments performed in triplicate. Experiments were performed at least two separate times and results were comparable across all replicates.
The Rev1-CT domain plays an essential scaffolding role in Rev1/Polζ-dependent TLS by mediating PPIs with other TLS DNA polymerases via two independent binding interfaces.29–33 The recruitment of the ‘extender’ TLS polymerase Polζ to DNA lesions requires interaction of its accessory Rev7 subunit with a binding interface in the C-terminal part of Rev1-CT.32–33,52 On the other hand, a different binding interface in the N-terminal part of Rev1-CT (Figure 3) was shown to interact with the RIR motifs from Polη, Polκ and Polι and guide the selection of an appropriate DNA polymerase for an insertion step of Rev1/Polζ-dependent TLS.30 The recent discovery of an RIR motif in PolD3, which is a component of both the replicative DNA polymerase Polδ and TLS polymerase Polζ (Rev3/Rev7/PolD2/PolD3 complex10–12), suggested an important new role for Rev1-CT/RIR PPI in mediating the assembly of the ‘extender’ four-subunit Polζ4 complex and facilitating polymerase switching upon Rev1/Polζ-dependent TLS.29 Consistent with these findings, previous cellular studies have shown that Rev1 variants with deleted Rev1-CT or mutated Rev1-CT/Rev7 interface are unable to restore survival and mutagenesis of Rev1-deficient avian DT40 cells and mouse embryonic fibroblasts (MEFs) after treatment with cisplatin and other genotoxic agents, highlighting the importance of Rev1-CT PPIs for supporting functional TLS.39–40,52 Our work demonstrates that small-molecule inhibitors of Rev1-CT/RIR PPIs decrease survival and mutagenesis of the HT1080 fibrosarcoma cells and MEFs after treatment with the two genotoxic agents, cisplatin and UV light, that cause DNA lesions whose replicative bypass is contingent on Rev1/Polζ-dependent TLS.9,13–14,44–45 These findings provide clear evidence that blocking the RIR-binding interface of Rev1-CT with small molecules inhibits Rev1/Polζ-dependent TLS, likely through interfering with the assembly of functional Rev1/Polζ complex and/or preventing polymerase exchange events.29
Conclusion
We have identified and characterized the first small molecules that exhibit anti-TLS activity in human cancer cells through disruption of the Rev1-CT/RIR PPI. In addition, the ability of compound 5 to disrupt Rev1-CT/RIR PPI by directly binding the pocket on Rev1-CT that accommodates side-chains of Phe-Phe pair of RIR motifs clearly identifies this region as a druggable ‘hot spot’ for the Rev1-CT/RIR PPI. We have also demonstrated that compounds 4 and 5 enhance sensitivity of human cancer cells to both cisplatin and UV light, and decrease cisplatin-mediated mutagenesis. We used an isogenic Rev1−/− MEF cell line to show that this increased sensitization to killing requires that the cells be REV1+/+, thereby demonstrating that the compounds are specifically inhibiting Rev1 scaffolding function. Our MD simulations suggest several structure-based approaches to improve the binding affinity and potency of the identified compounds. Taken together, these results not only validate that disruption of the Rev1-CT/RIR interface inhibits functional TLS in human cancer cells, but they also establish the 4/5 molecular structure as a lead scaffold for further development.
METHODS
Protein Expression and Purification
The C-terminal domain from human Rev1 (Rev1-CT) was expressed in E. coli BL21(DE3) cells and purified as described previously.31 Briefly, cells were transformed with pET-28b(+) (Novagen) based plasmid encoding Rev1 residues 1158-1251 inserted after the cleavage site for TEV protease. Cells were grown at 37 °C to OD600 ~ 0.8–1.0 in either unlabeled LB medium or minimal M9 medium using 15NH4Cl as the sole nitrogen source. Protein expression (20 °C, 12 hrs) was induced by 1 mM IPTG. Cells were harvested and lysed by sonication followed by purification of recombinant His-tagged Rev1-CT from a soluble fraction on a Co2+ affinity column. The His-tag was removed by TEV cleavage at 4 °C (12 hrs, or longer, as needed) followed by protein purification on a HiLoad Superdex 75 gel-filtration column (GE Healthcare). The 15N labeled Rev1-CT domain for small molecule NMR binding studies (expressed in M9 medium) was exchanged into 50 mM NaH2PO4, 100 mM NaCl, 0.5% DMSO, 10% D2O, pH 7.0 buffer. The 1:1 Rev1-CT/Polκ-RIR complex for fluorescence polarization (FP) based screening assays was prepared by mixing the unlabeled Rev1-CT domain (expressed in LB medium) with an excess of Polκ-RIR peptide (residues 560-575) containing the N-terminal fluorescent FAM label (FAM-Polκ-RIR; custom-synthesized by GenScript) followed by gel-filtration and exchange into 50 mM NaH2PO4, 100 mM NaCl, 0.5% DMSO, pH 7.0 buffer. The Rev1-CT/Polκ-RIR interaction is the strongest among those mediated by RIR motifs (Kd = 1.7 μM measured by SPR)29–30 so that in our purifications the protein and the peptide co-eluted on a gel-filtration chromatography column (Supplemental Figure 1).
Optimized Displacement Assay Protocol
The Rev1-CT/FAM-Polκ-RIR complex diluted in phosphate buffered saline (10 μL, 0.2 μM) was added to a black 384-well plate (ProxiPlate-384 F Plus, PerkinElmer). Unlabeled Polκ-RIR (10 μL, varying concentrations) was added to give final concentrations of Polκ-RIR (0.1–1000 nM) in a total volume of 20 μL/well. The mixture was incubated at room temperature for 1 hour with gentle mixing (see Supplemental Figure 2). Fluorescence polarization was measured on a Synergy H1 Hybrid multi-mode microplate reader (Biotek, excitation 485 nM, emission 528 nM). For the pilot screen, compounds were diluted to 20 μM in PBS from a 2 mM DMSO stock solution for screening at a final concentration of 10 μM. Compounds (10 μL) and Rev1-CT/FAM-Polκ-RIR complex (10 μL, 0.2 μM) were mixed in 384-well black plates on a Microlab Nimbus (Hamilton Robotics) automated liquid handler. The mixture was incubated and analyzed as described above. All graphs and statistical analysis were carried out with GraphPad Prism 5.
NMR Binding Studies
All NMR experiments were performed at 15 ºC on Agilent VNMRS spectrometer operating at 18.8 T magnetic field (800 MHz 1H frequency) equipped with a cold probe. Direct binding of the identified compounds to the RIR-binding site on Rev1-CT was tested by NMR titration measurements. In these experiments, 50 μM 15N labeled Rev1-CT was gradually titrated with increasing amounts of compound 1 (125 μM stock solution) or compound 5 (250 μM stock solution) dissolved in the matching buffer and monitored by recording 1H-15N HSQC spectra of the domain at each step of the titrations (Figure 3; Supplemental Figure 4). Resonance frequencies for all non-overlapped amide peaks of the Rev1-CT domain were quantified, and the resulting titration profiles (cumulative 1H and 15N chemical shifts ϖ = (ϖH2+(γN/γH ϖN)2)1/2 vs protein P0 and ligand L0 concentrations, where γH and γN are 1H and 15N gyromagnetic ratios) were analyzed to extract dissociation constant Kd and binding-induced chemical shift changes Δϖ for each amide group of the Rev1-CT domain (providing NMR chemical shift mapping of the binding site). The data analysis was performed in two steps. In the first step, the total chemical shift change (sum of ϖ for all non-overlapped amide resonances) was fit to the following equation to extract dissociation constant Kd:53
| (1) |
In the second step, binding-induced chemical shift changes Δϖ for individual amide groups were obtained from fits of per-residue titration profiles with Kd fixed to the previously obtained value.
Molecular Docking
Docking studies were performed using the Glide module of Schrödinger suite 2016. The structure of Rev1-CT in complex with Polκ-RIR (PDB code 2LSI)35 was used for docking with the RIR peptide stripped out from the complex. Rev1-CT was refined and prepared for docking with the protein preparation wizard module from Schrödinger. A grid box of 40Å × 40Å × 40Å was centered on the mass of the ‘hot spot’ residues in Rev1-CT and solvated using the TIP3P water model. All ligand structures were built using Maestro module and the ionization states, stereochemistry, and ring conformations of the ligands were assigned using the Ligprep module. Glide extra-precision mode was selected for initial docking and the docking poses for each ligand were examined according to their relative total energy scores. The highest scoring docking pose was subjected to molecular dynamics.
Molecular Dynamics
The optimal docking pose of the ligand with Rev1-CT was used as the starting structure for the molecular dynamics simulations. The atomic partial charges of all the compounds were derived from semiempirical AM1 geometry optimization using the general AMBER force field (GAFF).54–55 To neutralize the net charge of the system, sodium counter ions were placed to grids with the largest negative coulombic potentials around the protein, and the whole system was immersed in the rectangular box of TIP3P water molecules. The water box extended 8 Å away from any solute atoms. In molecular minimization and molecular dynamics simulations, particle mesh Ewald (PME) was employed to treat the long-range electrostatic interactions.56 Before MD simulations, the complexes were gradually relaxed using 10,000 cycles of minimization procedure (500 cycles of steepest descent57–58 and 9,500 cycles of conjugate gradient minimization59). After minimization, MD simulations in the NPT ensemble with a target temperature of 298 K and a target pressure of 1 atm were performed. The SHAKE procedure was employed to constrain all hydrogen atoms and the time step was set to 2.0 fs.60 Before the actual MD simulations, the system was gradually heated in the NVT ensemble from 10K to 298K over 50 ps. Initial velocities were assigned from a Maxwellian distribution at the starting temperature. MD samplings at 3 ns were performed for all complexes.
MM/GBSA Energy Calculations
Protein–ligand binding free energies were calculated as the difference between the energy of the bound complex and the energies of the free protein and compound. The energy of binding in solvent is calculated by estimating the energy change associated with the solvation of the protein and the ligand and then subtracting those values from the energy of binding in vacuum and from the estimated energy change for the solvation of the complex.
| (2) |
ΔGcomplex(i), ΔGprotein(i), ΔGligand(i) are binding energies of the complex, protein, and ligand respectively. The (i) notation represents the average of the number of extracted coordinate ΔG values of the complex, protein, and ligand calculated by summing the contribution of the gas phase energy (Hgas), solvation energy (Gsolvation), and entropy (TΔS).
| (3) |
In Equation 2, ΔEmm represents the gas phase interaction energy between protein and ligand (including both van der Waals, ΔEvdw, and electrostatic interactions, ΔEele). ΔGGB and ΔGSA indicate the polar and nonpolar desolvation energies, respectively. For our studies, MM/GBSA calculations were performed using MMPBSA.py in AMBERTools14. The conformational entropy (TΔS) upon ligand binding was calculated using Normal Mode Analysis (NMA) in the gas phase. The distance-dependent dielectric constant was set to 1.0, and the convergence criteria for minimized energy gradient were set at 0.0001 with 10,000 minimization cycles per snapshot. Due to limited computational resources, we used only 50 frames, which were evenly extracted from 0 to 5 ns of the MD trajectories, for entropy calculations.
Clonogenic Survival Assay
HT1080 cells were purchased from ATCC and grown in RPMI 1640 (Gibco), supplemented with 10% FBS (HyClone) and 1% Penicillin/Streptomycin. MEF cells (kindly provided by Niels de Wind) were propagated in DMEM (Gibco), supplemented with 10% FBS (HyClone) and 1% Penicillin/Streptomycin. Both cell lines were incubated at 37°C, 5% CO2 incubator. For passaging, cells were trypsinized with 0.25% Trypsin-EDTA (Corning). Cells were seeded in 6-well plates (600 cells/well, 3 mL) and incubated overnight (37°C, 5% CO2). Cells were incubated with cisplatin (0.6 μM) or exposed to UV light (5 J/m2) and then incubated for additional 24 hrs. Fresh media was added along with compound 4 or 5 (1.5 μM) and the cells returned to the incubator for 24 hrs. Fresh media was added after a 24 hr incubation with the compounds and the cells were returned to the incubator for 7 days. To count the colonies at the end of the 7 day period, media was removed and fixative (50% methanol and 10% glacial acetic acid) was added to the cells for 10 min, followed by the addition of Coomassie Brilliant Blue R-250 in MeOH:AcOH:H2O (46.5:7:46.5; v/v/v).61 Colonies containing at least 40 cells were counted and relative survival rates were calculated as a ratio of colony counts from the treated samples and colony counts from the DMSO controls.
HPRT Mutagenesis Assay
HT1080 and WT MEF cells were grown in HAT media (complete media described above supplemented with 100 μM Hypoxanthine, 0.4 μM Aminopterin, and 16 μM Thymidine) for 14 days to remove any spontaneous HPRT mutants. Cells were exposed to cisplatin (0.6 μM) for 24 hrs followed by the addition of 4 or 5 (1.5 μM) in fresh complete media. To determine clonal efficiency, cells were trypsinized after 24 hrs, washed with PBS and plated in triplicate in 6-well plates (200 cells/well). The remaining cells were plated in complete media and incubated for 8 days to allow sufficient time for the expression of the HPRT mutation. After 8 days, colonies were counted from the 6-well plates and the recovering—treated or untreated—cells (500,000) were plated in triplicate in 10 cm plates in complete media described above supplemented with 6-thioguanine (6-TG, 1.5 mM) to allow for proliferation and colony formation of the HPRT+ cells. After additional 14–20 days of incubation, colonies were counted by fixing and staining the cells as described above. The HPRT mutation frequency was calculated as a ratio of the number of HPRT mutants arising from a given treatment and the number of surviving colonies present after drug treatment.
UV Sensitivity Assay
In order to test the UV sensitivity, HT1080 cells were trypsinized and transferred to 10 cm, UV-permeable plates and exposed to UV light (5 J/m2). Next, 10,000 cells were transferred to luminescent dye compatible 96-well plates (10,000 cells/well, 100 μL) and incubated for 24 hours at 37°C, 5% CO2. Number of viable cells were quantitated the next day using the CellTiter-Glo® Luminescent Cell Viability Assay kit per the manufacturer’s instructions (Promega). Luminescence was quantified on a Tecan Spark 10M plate reader. The relative survival of metabolically active cells was calculated by dividing the luminescence of treated samples by the luminescence of DMSO controls.
Supplementary Material
Acknowledgments
This research was supported by the Connecticut Institute for Clinical and Translational Sciences (D.M.K. and M.K.H.) the University of Connecticut Research Foundation (D.M.K. and M.K.H.), the National Science Foundation (D.M.K., 1615866), the National Institute of Environmental Health Sciences grant R01-ES015818 to G.C.W. and NIEHS grant P30 ES002109 to the Massachusetts Institute of Technology Center of Environmental Health Sciences. G.C.W. is an American Cancer Society Professor. The authors thank Yulia Pustovalova and Mariana Quezado for their kind assistance with protein sample preparation at early stages of the project and Irina Bezsonova for help with preparation of the manuscript graphics.
Footnotes
Author Contributions
V.S. performed assay optimization, screening, and follow-up biochemical studies. A.R. performed protein expression and purification and all the structural biology studies. N.C. performed all the cellular assays. Z.O. performed assay optimization and screening. R.D. performed all the modeling and computational studies. All authors contributed to preparation of the manuscript.
Notes
The authors declare no competing financial interest.
Supporting Information. The following Supporting Information is available free of charge on the ACS Publications Website at DOI:… Supporting figures and additional methods.
References
- 1.Chen X, Wu Y, Dong H, Zhang CY, Zhang Y. Platinum-based agents for individualized cancer treatment. Curr Mol Med. 2013;13:1603–1612. doi: 10.2174/1566524013666131111125515. [DOI] [PubMed] [Google Scholar]
- 2.Swift LH, Golsteyn RM. Genotoxic anti-cancer agents and their relationship to DNA damage, mitosis, and checkpoint adaptation in proliferating cancer cells. Int J Mol Sci. 2014;15:3403–3431. doi: 10.3390/ijms15033403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chen X, Wu Y, Dong H, Zhang CY, Zhang Y. Platinum-based agents for individualized cancer treatment. Curr Mol Med. 2013;13:1603–1612. doi: 10.2174/1566524013666131111125515. [DOI] [PubMed] [Google Scholar]
- 4.Kelland L. The resurgence of platinum-based cancer chemotherapy. Nat Rev Cancer. 2007;7:573–584. doi: 10.1038/nrc2167. [DOI] [PubMed] [Google Scholar]
- 5.Siddik ZH. Cisplatin: mode of cytotoxic action and molecular basis of resistance. Oncogene. 2003;22:7265–7279. doi: 10.1038/sj.onc.1206933. [DOI] [PubMed] [Google Scholar]
- 6.Sale JE, Lehmann AR, Woodgate R. Y-family DNA polymerases and their role in tolerance of cellular DNA damage. Nat Rev Mol Cell Biol. 2012;13:141–152. doi: 10.1038/nrm3289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ghosal G, Chen J. DNA damage tolerance: a double-edged sword guarding the genome. Transl Cancer Res. 2013;2:107–129. doi: 10.3978/j.issn.2218-676X.2013.04.01. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Korzhnev DM, Hadden MK. Targeting the translesion synthesis pathway for the development of anti-cancer chemotherapeutics. J Med Chem. 2016;59:9321–9336. doi: 10.1021/acs.jmedchem.6b00596. [DOI] [PubMed] [Google Scholar]
- 9.Waters LS, Minesinger BK, Wiltrout ME, D’Souza S, Woodruff RV, Walker GC. Eukaryotic translesion polymerases and their role in regulation in DNA damage tolerance. Microbiol Mol Biol Rev. 2009;73:134–154. doi: 10.1128/MMBR.00034-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Baranovskiy AG, Lada AG, Siebler HM, Zhang Y, Pavlov YI, Tahirov TH. DNA polymerase δ and ζ switch by sharing accessory subunits of DNA polymerase δ. J Biol Chem. 2012;287:17281–17287. doi: 10.1074/jbc.M112.351122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lee YS, Gregory MT, Yang W. Human pol ζ purified with accessory subunits is active in translesion DNA synthesis and complements pol η in cisplatin bypass. Proc Natl Acad Sci USA. 2014;111:2954–2959. doi: 10.1073/pnas.1324001111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Makarova AV, Burgers PM. Eukaryotic DNA polymerase zeta. DNA Rep. 2015;29:47–55. doi: 10.1016/j.dnarep.2015.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Shachar S, Ziv O, Avkin S, Adar S, Wittschieben J, Reibner T, Chaney S, Friedberg EC, Wang Z, Carell T, Geacintov N, Livneh Z. Two-polymerase mechanisms dictate error-free and error-prone translesion DNA synthesis in mammals. EMBO J. 2009;28:383–393. doi: 10.1038/emboj.2008.281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Livneh Z, Ziv O, Shachar S. Multiple two-polymerase mechanisms in mammalian translesion DNA synthesis. Cell Cycle. 2010;9:729–735. doi: 10.4161/cc.9.4.10727. [DOI] [PubMed] [Google Scholar]
- 15.Lawrence CW. Cellular functions of DNA polymerase ζ and Rev1 protein. Adv Protein Chem. 2004;69:167–203. doi: 10.1016/S0065-3233(04)69006-1. [DOI] [PubMed] [Google Scholar]
- 16.Prakash S, Prakash L. Translesion DNA synthesis in eukaryotes: A one- or two-polymerase affair. Genes Dev. 2002;16:1872–1883. doi: 10.1101/gad.1009802. [DOI] [PubMed] [Google Scholar]
- 17.Budzowska M, Graham TG, Sobeck A, Waga S, Walter JC. Regulation of the Rev1-pol ζ complex during bypass of a DNA interstrand cross-link. EMBO J. 2015;34:1971–1985. doi: 10.15252/embj.201490878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nojima K, Hochegger H, Saberi A, Fukushima T, Kikuchi K, Yoshimura M, Orelli BJ, Bishop DK, Hirano S, Ohzeki M, Ishiai M, Yamamoto K, Takata M, Arakawa H, Buerstedde JM, Yamazoe T, Araki K, Takahashi JA, Hashimoto N, Takeda S, Sonoda E. Multiple repair pathways mediate tolerance to chemotherapeutic cross-linking agents in vertebrate cells. Cancer Res. 2005;65:11704–11711. doi: 10.1158/0008-5472.CAN-05-1214. [DOI] [PubMed] [Google Scholar]
- 19.Wu F, Lin X, Okuda T, Howell SB. DNA polymerase ζ regulates cisplatin cytotoxicity, mutagenicity, and the rate of development of cisplatin resistance. Cancer Res. 2004;64:8029–8035. doi: 10.1158/0008-5472.CAN-03-3942. [DOI] [PubMed] [Google Scholar]
- 20.Lin X, Okuda T, Trang J, Howell SB. Human Rev1 modulates the cytotoxicity and mutagenicity of cisplatin in human ovarian carcinoma cells. Mol Pharm. 2006;69:1748–1754. doi: 10.1124/mol.105.020446. [DOI] [PubMed] [Google Scholar]
- 21.Lin XJ, Howell SB. DNA mismatch repair and p53 function are major determinants of the rate of development of cisplatin resistance. Mol Cancer Ther. 2006;5:1239–1247. doi: 10.1158/1535-7163.MCT-05-0491. [DOI] [PubMed] [Google Scholar]
- 22.Xie K, Doles J, Hemann MT, Walker GC. Error-prone translesion synthesis mediates acquired chemoresistance. Proc Natl Acad Sci USA. 2010;107:20792–20797. doi: 10.1073/pnas.1011412107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Xu X, Xie K, Zhang XQ, Pridgen EM, Park GY, Cui DS, Shi J, Wu J, Kantoff PW, Lippard SJ, Langer R, Walker GC, Farokhzad OC. Enhancing tumor cell response to chemotherapy through nanoparticle-mediated codelivery of siRNA and cisplatin prodrug. Proc Natl Acad Sci U S A. 2013;110:18638–18643. doi: 10.1073/pnas.1303958110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Doles J, Oliver TG, Cameron ER, Hsu G, Jacks T, Walker GC, Hemann MT. Suppression of Rev3, the catalytic subunit of polζ, sensitizes drug-resistant lung tumors to chemotherapy. Proc Natl Acad Sci U S A. 2010;107:20786–20791. doi: 10.1073/pnas.1011409107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Okuda T, Lin X, Trang J, Howell SB. Suppression of hRev1 expression reduces the rate at which human ovarian carcinoma cells acquire resistance to cisplatin. Mol Pharm. 2005;67:1852–1860. doi: 10.1124/mol.104.010579. [DOI] [PubMed] [Google Scholar]
- 26.Hoege C, Pfander B, Moldovan GL, Pyrowolakis G, Jentsch S. RAD6-dependent DNA repair is linked to modification of PCNA by ubiquitin and SUMO. Nature. 2002;419:135–141. doi: 10.1038/nature00991. [DOI] [PubMed] [Google Scholar]
- 27.Friedberg EC, Lehmann AR, Fuchs RPP. Trading places: How do DNA polymerases switch during translesion DNA synthesis? Mol Cell. 2005;18:499–505. doi: 10.1016/j.molcel.2005.03.032. [DOI] [PubMed] [Google Scholar]
- 28.Ulrich HD. Regulating post-translational modifications of the eukaryotic replication clamp PCNA. DNA Rep. 2009;8:461–469. doi: 10.1016/j.dnarep.2009.01.006. [DOI] [PubMed] [Google Scholar]
- 29.Pustovalova Y, Magalhães MTQ, D’Souza S, Rizzo AA, Korza G, Walker GC, Korzhnev DM. Interaction between the Rev1 C-termial domain and the PolD3 subunit of Polζ suggests a mechanism of polymerase exchange upon Rev1/Polζ-dependent translesion synthesis. Biochemistry. 2016;55:2043–2053. doi: 10.1021/acs.biochem.5b01282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ohashi E, Hanafusa T, Kamei K, Song I, Tomida J, Hashimoto H, Vaziri C, Ohmori H. Identification of a novel Rev1-interacting motif necessary for DNA polymerase κ function. Genes Cells. 2009;14:101–111. doi: 10.1111/j.1365-2443.2008.01255.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pozhidaeva A, Pustovalova Y, D’Souza S, Bezsonova I, Walker GC, Korzhnev DM. NMR structure and dynamics of the C-terminal domain from human Rev1 and its complex with Rev1 interacting region of DNA polymerase η. Biochemistry. 2012;51:5506–5520. doi: 10.1021/bi300566z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pustovalova Y, Bezsonova I, Korzhnev DM. The C-terminal domain of human Rev1 contains independent binding sites for DNA polymerase η and Rev7 subunit of polymerase ζ. FEBS Lett. 2012;586:3051–3056. doi: 10.1016/j.febslet.2012.07.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kikuchi S, Hara K, Shimizu T, Sato M, Hashimoto H. Structural basis of recruitment of DNA polymerase ζ by interaction between Rev1 and Rev7. J Biol Chem. 2012;287:33847–33852. doi: 10.1074/jbc.M112.396838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wojtaszek J, Liu J, D’Souza S, Wang S, Xue Y, Walker GC, Zhou P. Multifaceted recognition of vertebrate Rev1 by translesion polymerases ζ and κ. J Biol Chem. 2012;287:26400–26408. doi: 10.1074/jbc.M112.380998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Liu D, Ryu KS, Ko J, Sun D, Lim K, Lee JO, Hwang J, Lee Z-w, Choi BS. Insights into the regulation of human Rev1 for translesion synthesis polymerases is revealed by the structural studies on its polymerase-interacting domain. J Mol Cell Biol. 2013;5:204–206. doi: 10.1093/jmcb/mjs061. [DOI] [PubMed] [Google Scholar]
- 36.Lawrence CW. Cellular functions of DNA polymerase ζ and Rev1 protein. Adv Protein Chem. 2004;6(9):167–203. doi: 10.1016/S0065-3233(04)69006-1. [DOI] [PubMed] [Google Scholar]
- 37.Simpson LJ, Sale JE. Rev1 is essential for DNA damage tolerance and non-templated immunoglobulin gene mutation in a vertebrate cell line. EMBO J. 2003;22:1654–1664. doi: 10.1093/emboj/cdg161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Jansen JG, Tsaalbi-Shtylik A, Hendriks G, Gali H, Hendel A, Johansson F, Erixon K, Livneh Z, Mullenders LHF, Haracska L, de Wind N. Separate domains of Rev1 mediate two modes of DNA damage bypass in mammalian cells. Mol Cell Biol. 2009;29:3113–3123. doi: 10.1128/MCB.00071-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ross AL, Simpson LJ, Sale JE. Vertebrate DNA damage tolerance requires the C-terminus but not BRCT or transferase domains of Rev1. Nucleic Acids Res. 2005;33:1280–1289. doi: 10.1093/nar/gki279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hashimoto K, Cho Y, Yang IY, Akagi JI, Ohashi E, Tateishi S, de Wind N, Hanaoka F, Ohmori H, Moriya M. The vital role of polymerase ζ and Rev1 in mutagenic, but not correct, DNA synthesis across benzo[a]pyrene-dG and recruitment of polymerase ζ by Rev1 to replication-stalled site. J Biol Chem. 2012;287:9613–9622. doi: 10.1074/jbc.M111.331728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.London RE. The structural basis of XRCC1-mediated DNA repair. DNA Rep. 2015;30:90–103. doi: 10.1016/j.dnarep.2015.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zhang JH, Chung TD, Oldenburg KR. A simple statistical parameter for use in evaluation and validation of high throughput screening assays. J Biomol Screen. 1999;4:67–73. doi: 10.1177/108705719900400206. [DOI] [PubMed] [Google Scholar]
- 43.Lee YS, Gregory MT, Yang W. Human pol ζ purified with accessory subunits is active in translesion DNA synthesis and complements pol η in cisplatin bypass. Proc Natl Acad Sci USA. 2014;111:2954–2959. doi: 10.1073/pnas.1324001111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Acharya N, Johnson RE, Prakash S, Prakash L. Complex formation with Rev1 enhances the profeiciency of Saccaromyces cerevisiae DNA polymerase zeta for mismatch extension and for extension opposite from DNA lesions. Mol Cell Biol. 2006;26:9555–9563. doi: 10.1128/MCB.01671-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Johnson RE, Haracska L, Prakash S, Prakash L. Role of DNA polymerase eta in the bypass of a (6-4) TT photoproduct. Mol Cell Biol. 2001;21:3558–3563. doi: 10.1128/MCB.21.10.3558-3563.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Chu G, Chang E. Cisplatin-resistant cells express increased levels of a factor that recognizes damaged DNA. Proc Natl Acad Sci USA. 1990;87:3324–3327. doi: 10.1073/pnas.87.9.3324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Imle A, Polzer B, Alexander S, Klein CA, Friedl P. Genomic instability of micronucleated cells revealed by singe-cell comparative genomic hybridization. Cytometry A. 2009;75:562–568. doi: 10.1002/cyto.a.20733. [DOI] [PubMed] [Google Scholar]
- 48.Silva MJ, Costa P, Dias A, Valente M, Luoro H, Boavida MG. Comparative analysis of the mutagenic activity of oxaliplatin and cisplatin in the Hprt gene of CHO cells. Environ Mol Mutagen. 2005;46:104–115. doi: 10.1002/em.20138. [DOI] [PubMed] [Google Scholar]
- 49.Lin X, Okuda T, Trang J, Howell SB. Human REV1 modulates the cytotoxicity and mutagenicity of cisplatin in human ovarian carcinoma cells. Mol Pharmacol. 2009;69:1748–1754. doi: 10.1124/mol.105.020446. [DOI] [PubMed] [Google Scholar]
- 50.Okuda T, Lin X, Trang J, Howell SB. Suppression of hREV1 expression reduces the rate at which human ovarian carcinoma cells acquire resistance to cisplatin. Mol Pharmacol. 2005;67:1852–1860. doi: 10.1124/mol.104.010579. [DOI] [PubMed] [Google Scholar]
- 51.Jansen JG, Tsaalbi-Shtylik A, Hendriks G, Gali H, Hendel A, Johansson F, Erixon K, Livneh Z, Mullenders LHF, Haracska L, et al. Separate domains of Rev1 mediate two modes of DNA damage bypass in mammalian cells. Mol Cell Biol. 2009;29:3113–3123. doi: 10.1128/MCB.00071-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wojtaszek J, Lee CJ, D’Souza S, Minesinger B, Kim H, D’Andrea AD, Walker GC, Zhou P. Structural basis of Rev1-mediated assembly of a quaternary vertebrate translesion polymerase complex consisting of Rev1, heterodimeric polζ and polκ. J Biol Chem. 2012;287:33836–33846. doi: 10.1074/jbc.M112.394841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Furukawa A, Konuma T, Yanaka S, Sugase K. Quantitative analysis of protein-ligand interactions by NMR. Prog Nucl Magn Reson Spectrosc. 2016;96:47–57. doi: 10.1016/j.pnmrs.2016.02.002. [DOI] [PubMed] [Google Scholar]
- 54.Wang J, Wang W, Kollman PA, Case DA. Automatic atom type and bond type perception in molecular mechanical calculations. J Mol Graph Model. 2006;25:247–260. doi: 10.1016/j.jmgm.2005.12.005. [DOI] [PubMed] [Google Scholar]
- 55.Wang J, Wolf RM, Caldwell JW, Kollman PA, Case DA. Development and testing of a general amber force field. J Comput Chem. 2004;25:1157–1174. doi: 10.1002/jcc.20035. [DOI] [PubMed] [Google Scholar]
- 56.Darden T, York D, Pedersen L. Particle mesh Ewald: An N·log(N) method for Ewald sums in large systems. J Chem Phys. 1993;98:10089–10092. [Google Scholar]
- 57.Kiwiel KC, Murty K. Convergence of the steepest descent method for minimizing quasiconvex functions. J Optim Theory Appl. 1996;89:221–226. [Google Scholar]
- 58.Petrova SS, Solov’ev AD. The origin of the method of steepest descent. Historia Mathematica. 1997;24:361–375. [Google Scholar]
- 59.Hestenes MR. Methods of conjugate gradients for solving linear systems. 1952;49 [Google Scholar]
- 60.Ryckaert JP, Ciccotti G, Berendsen HJC. Numerical integration of the cartesian equations of motion of a system with constraints: molecular dynamics of n-alkanes. J Comput Phys. 1977;23:327–341. [Google Scholar]
- 61.Mochizuki Y, Furukawa K. Applications of coomassie brilliant blue staining to cultured hepatocytes. Cell Biol Int Rep. 1987;11:367–371. doi: 10.1016/0309-1651(87)90003-8. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.