

Abstract
Background
Bacterial meningitis is associated with high mortality and long-term neurological sequelae. Increasing the phagocytic activity of microglia could improve the resistance of the CNS against infections. We studied the influence of activin A, a member of the TGF-β family with known immunoregulatory and neuroprotective effects, on the functions of microglial cells in vitro.
Methods
Primary murine microglial cells were treated with activin A (0.13 ng/ml–13 μg/ml) alone or in combination with agonists of TLR2, 4, and 9. Phagocytosis of Escherichia coli K1 as well as release of TNF-α, IL-6, CXCL1, and NO was assessed.
Results
Activin A dose-dependently enhanced the phagocytosis of Escherichia coli K1 by microglial cells activated by agonists of TLR2, 4, and 9 without further increasing NO and proinflammatory cytokine release. Cell viability of microglial cells was not affected by activin A.
Conclusions
Priming of microglial cells with activin A could increase the elimination of bacteria in bacterial CNS infections. This preventive strategy could improve the resistance of the brain to infections, particularly in elderly and immunocompromised patients.
Keywords: Bacterial meningitis, Activin, Proinflammatory cytokines, TLR, Innate immune system, Nitric oxide, CNS infection, Phagocytosis, E. coli
Background
Bacterial meningitis is associated with high mortality and long-term neurological sequelae despite the use of bactericidal antibiotics [1, 2]. The incidence and the frequency of an unfavorable outcome of bacterial central nervous system (CNS) infections are increased in immunocompromised and aged persons. In these patients, the Gram-negative bacterium Escherichia coli is one of the most prevalent causative pathogens [3, 4]. The presence of the polysaccharide capsule K1 allows E. coli strains to survive in the bloodstream, to cross the blood-brain barrier by penetrating the brain microvascular endothelial cell layer and to enter the CNS [5]. In the CNS, meningeal and perivascular macrophages and microglia, the resident immune cells and the major constituents of innate immunity in the brain parenchyma, represent the first line of defense against bacteria [6]. They express toll-like receptors (TLR) that recognize pathogen-associated molecular patterns (PAMPS) [7, 8]. TLR on microglia are stimulated during the early phase of CNS infections and systemic infections [9, 10]. TLR2 is activated by bacterial lipopeptides [11], TLR4 recognizes endotoxin (LPS) [12], and TLR9 is activated by bacterial DNA [13]. In response to an inflammatory stimulus, microglia undergo changes in morphology and functions, such as production of proinflammatory cytokines, chemokines and reactive oxygen species (ROS), phagocytic activity, antigen presentation, clearance of toxic cellular debris, and promotion of tissue repair [14–16].
We previously demonstrated that the age-related decline of microglia and macrophage functions, particularly the age-related decline of their phagocytic capacity, plays an essential role for the impaired elimination of bacteria and the higher mortality after an intracerebral bacterial challenge in aged mice [17]. Thus, strategies to increase the phagocytic potential of macrophages and microglial cells appear promising for the prevention and therapy of CNS infections, especially in elderly and immunocompromised patients. On the other hand, stimulation of microglial cells bears the risk of microglia-mediated neuronal damage.
In vitro, activation of microglial cells with agonists of TLR 2, 4, and 9 increases phagocytosis and intracellular killing of E. coli K1 [18]. However, microglia activated by these TLR agonists also produce proinflammatory cytokines (e.g., TNF-α, IL-6, CXCL1) and nitric oxide (NO) [18–20] which can cause destruction of neuronal axons and somata [21–25]. Palmitoylethanolamide (PEA) enhances phagocytosis of E. coli K1 by microglial cells in vitro without inducing the release of proinflammatory cytokines and increases survival in mouse models of E. coli meningitis and sepsis [26]. The identification of other compounds which increase phagocytosis of pathogens without exerting collateral damage to the brain tissue is a promising approach for the prophylaxis and early therapy of intracerebral infections in high-risk individuals [27].
We considered activin A an ideal compound for this purpose, as it has been closely linked to bacterial CNS infections and microglial cells, and both immunoregulatory and neuroprotective effects have been described (for review, see [28, 29]). Activin A is a multifunctional member of the TGF-β-superfamily [30]. Together with its binding protein follistatin, activin A is involved in the fine-tuning of the host’s inflammatory response [28, 31]. Levels of activin A and follistatin are elevated in serum during sepsis [32] and in CSF during meningitis [33, 34]. Depending on the circumstances, activin A can be both pro- or anti-inflammatory by regulating key mediators of the inflammatory response such as cytokines and [28, 31]. Microglial cells have been shown to be a source of activin A during bacterial infections [32, 35, 36], as well as a target of activin A. Microglia express activin A receptor type II (Act-RII) and Act-RI [36, 37] by which Smad and non-Smad signaling pathways are initiated. In several experiments with murine peritoneal macrophages and macrophage cell lines, activin A modulated not only the release of cytokines and ROS but also the phagocytic activity as assessed by the uptake of chicken red blood cells (cRBC) or latex particles. Results from these studies indicate that activin A increases the phagocytic capacity of resting macrophages [38–40] and inhibits the phagocytic activity of LPS-activated macrophages [40, 41]. To our knowledge, the effect of activin A on phagocytosis of a living bacterium and on the phagocytic activity of microglia has not been examined so far.
Here, we investigated the effect of activin A on resting and activated primary murine microglial cells with a focus on their ability to phagocytose E. coli K1.
Methods
Primary murine microglia cell cultures
Primary cultures of microglial cells were prepared from brains of newborn C57BL/6 mice (1–3 days) [19]. After removal of the meninges, cells were mechanically dissociated and suspended in Dulbecco’s modified Eagle’s medium (DMEM) with Glutamax I (Gibco, Karlsruhe, Germany) supplemented with 10% fetal calf serum (FCS), 100 U/ml penicillin, and 100 μg/ml streptomycin. Cells were plated at a density of two brains per T75 culture flask (Corning Costar GmbH, Wiesbaden, Germany) and incubated at 37 °C in a humid atmosphere with 5% CO2. Culture medium was changed twice a week. After 10–14 days, microglial cells were separated from the confluent astrocyte layer by shaking 200×/min for 30 min and plated in 96-well plates at a density of 50,000 cells/well for phagocytosis experiments. For staining, microglial cells were seeded on coverslips placed in a 24-well plate at a density of 50,000 cells/well.
Stimulation of microglial cells
Two hours after plating, cells were treated with activin A (R&D Systems, Wiesbaden, Germany) for 24 h. Different concentrations of activin A were chosen for the experiments: 0.13 ng/ml (= 10 pM), 1.3 ng/ml (= 100 pM), 13 ng/ml (= 1 nM), and a high concentration of 13 μg/ml (= 1 μM). Unstimulated control cells were treated with medium only. After 24 h, microglial cells were additionally treated with tripalmitoyl-S-glycerl-cysteine (Pam3CSK4; EMC Microcollections, Tübingen, Germany) as an agonist of TLR1/2, endotoxin (lipopolysaccharide, LPS) from E. coli serotype 026:B6 (Sigma-Aldrich, Taufkirchen, Germany) as an agonist of TLR4, or CpG oligodesoxynucleotide (ODN) 1668 (TCC ATG ACG TTC CTG ATG CT) containing unmethylated cytosine-guanosine motifs (CpG; TIB Molbiol, Berlin, Germany) as an agonist of TLR9 for 24 h. Concentrations of the different TLR agonists which induced a 2- to 3-fold increase of phagocytosis of E. coli K1 in our previous experiments were used [18, 20]. Control cells were treated with medium only. Treatment with 1 μg/ml LPS was used as a positive control of stimulation based on previous experiments [18, 19]. In experiments for measurement of NO release, microglial cells were additionally stimulated with 100 U/ml interferon-γ, because a basal level of interferon-γ is essential to reach a substantial NO release of microglial cells after stimulation [15] Unstimulated cells were treated with medium containing 100 U/ml interferon-γ only. After 24 h of stimulation, supernatants were stored at − 80 °C until measurement of NO, cytokine, and chemokine levels. Cells were used for the bacterial phagocytosis assay, the cell viability assay, or were fixated in 4% formaldehyde for staining.
Bacteria
The E. coli strain K1 (serotype O18:K1:H7) originally isolated from the cerebrospinal fluid of a child with neonatal meningitis (gift of Dr. Gregor Zysk, Institute for Medical Microbiology, Düsseldorf, Germany) was used for phagocytosis experiments. Bacteria were grown over night on blood agar plates, harvested in 0.9% saline, and stored at − 80 °C. Frozen aliquots were thawed directly before the experiment and diluted with saline to the required bacterial concentration.
Phagocytosis assay
The phagocytosis assay was performed as previously described [18]. After stimulation, microglia were exposed to the encapsulated E. coli K1 for 90 min at 37 °C, 5% CO2 with a ratio of 100 bacteria per phagocyte (5 × 106 colony-forming units (CFU)/well). After incubation with bacteria, microglial cells were washed with PBS and incubated with DMEM containing gentamicin (final concentration 100 μg/ml; Sigma-Aldrich, Taufkirchen, Germany) for 60 min to kill extracellular bacteria. Thereafter, cells were washed twice with PBS and lysed with 100 μl of distilled water. The number of intracellular bacteria was determined by quantitative plating of serial 1:10 dilutions of the lysate on blood agar plates.
Cell viability test
Cell viability of microglial cells was determined using the WST-1 Cell Proliferation Reagent (Roche Applied Science, Mannheim, Germany). The assay is based on the cleavage of the tetrazolium salt WST-1 by active mitochondria producing a soluble formazan. This conversion only occurs in viable cells. Cells were incubated with WST-1 for 2 h. Then, the formazan dye formed was quantified by measuring the optical density at 490 nm using a Genios multiplate reader (Tecan, Crailsheim, Germany). The absorbance directly correlated with the number of metabolically active cells.
Cytokine and chemokine measurements
Concentrations of tumor necrosis factor alpha (TNF-α), interleukin-6 (IL-6), and the chemokine (C-X-C motif) ligand 1 (CXCL1, also called GRO α or KC) in the cell culture supernatants were measured by ELISA. TNF-α levels were determined using antibody pairs from BioLegend (Biozol, Munich, Germany), and DuoSet ELISA Development Kits (R&D Systems, Wiesbaden, Germany) were used for the measurement of IL-6 and CXCL1. The color reaction was measured at 450 nm in a microplate reader (Bio-Rad, Munich, Germany). Detection limits were 19 pg/ml for TNF-α, 38 pg/ml for IL-6, and 64 pg/ml for CXCL1.
Quantification of nitric oxide release
NO release was quantified by the measurement of nitrite, one of its stable reaction products, in the supernatant of microglial cultures using the Griess reagent. One hundred microliters of the supernatant were mixed with 100 μl Griess reagent [equal volumes of 1% sulfonilamide in 30% acetate and 0.1% N-(1-naphthyl) ethylenediamine in 60% acetate] in a 96-well plate. After 10 min, the optical density at 570 nm was measured with a Genios multiplate reader (Tecan, Crailsheim, Germany). Concentrations were calculated by comparison of absorptions with a standard curve.
Isolectin B4 staining
Isolectin B4 staining was used to assess the purity and density of microglial cultures and the morphology of microglial cells. For this purpose, microglial cells were plated on poly-l-lysine-coated cover slips. After stimulation, cells were fixated in 4% formalin. Fixated cells were permeabilized with Triton X (0.1% in PBS) for 30 min and then incubated with biotinylated isolectin B4 (5 μg/ml, diluted in PBS + 1% BSA; Sigma-Aldrich, Taufkirchen, Germany) for 90 min. Thereafter, cells were treated with avidin-biotin complex (ABC, Vector, Burlingame, CA) for 30 min, and diaminobenzidine was used for visualization (5 min) resulting in a brown staining of the somata of microglial cells. The purity of microglia in the cultures was greater than 98%.
Statistics
GraphPad Prism 5.0 Software (GraphPad Software, San Diego, CA, USA) was used to perform statistical analyses and graphical presentation. Parametric data were expressed as means ± standard deviations (SD); nonparametric data were expressed as medians (25th percentile/75th percentile). Student’s t test was performed to compare two groups of parametric data; ANOVA followed by Bonferroni’s multiple comparison test was used to compare more than two groups of parametric data. Nonparametric data were analyzed by Mann-Whitney U test or Kruskal-Wallis test followed by Dunn’s multiple comparison test. P values < 0.05 were considered statistically significant.
Results
Activin A alone did not influence phagocytosis of E. coli K1 by primary murine microglial cells
Treatment of microglial cells with activin A alone in concentrations between 0.13 and 13 ng/ml (Fig. 1) and in a high concentration of 13 μg/ml (Table 1) did not influence phagocytosis of E. coli K1.
Fig. 1.
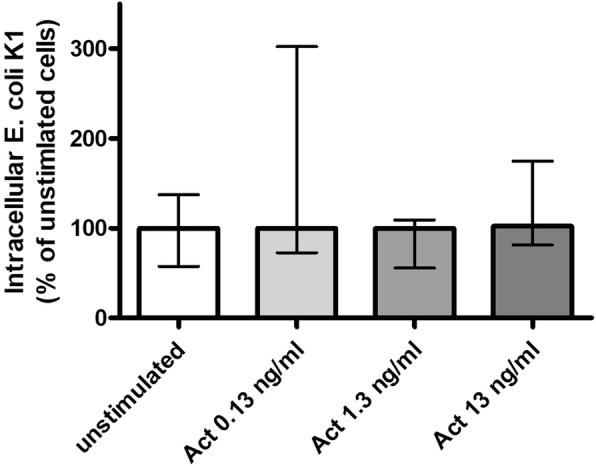
Phagocytosis of E. coli by microglial cells treated with activin A alone. Comparison of E. coli K1 phagocytosis between unstimulated microglial cells and microglial cells treated with activin A in concentrations of 0.13, 1.3, and 13 ng/ml (n = 11–30 from up to seven experiments). In each experiment, the mean number of bacteria ingested by unstimulated control cells was defined as 100%. Phagocytic rates of the stimulated groups are presented as percentages of phagocytosis of the unstimulated control group (medians with interquartile ranges). Data were analyzed by Kruskal-Wallis test followed by Dunn’s multiple comparison test
Table 1.
Phagocytosis of E. coli K1 and release of proinflammatory cytokines and NO by microglial cells treated with 13 μg/ml activin A alone and in combination with different TLR agonists
| No stim. | Act A 13 μg/ml | P3C | P3C + Act A | LPS | LPS + Act A | CpG | CpG + Act A | |
|---|---|---|---|---|---|---|---|---|
| Phagocytosis (%) | 100 (52/143) | 56 (27/96) | 337 (147/561) | 276 (219/500) | 493 (324/750) | 275 (180/949) | 493 (270/1399) | 750 (183/7914) |
| p = 0.11 | p = 0.85 | p = 0.33 | p = 0.94 | |||||
| TNF-α (pg/ml) | 125 (85/195) | 112 (33/157) | 7280 (1884/8315) | 4046 (1615/7189) | 8103 (7126/8636) | 4776 (1255/5505) | 4325 (932/4829) | 4826 (1313/7199) |
| p = 0.34 | p = 0.31 | p < 0.0003*** | p = 0.11 | |||||
| IL-6 (pg/ml) | 38 (38/95) | 38 (38/38) | 38 (38/7383) | 2785 (38/5913) | 3910 (2832/4192) | 2017 (1894/2215) | 656 (435/727) | 751 (656/941) |
| p = 0.56 | p = 0.93 | p = 0.10 | p = 0.27 | |||||
| CXCL1 (pg/ml) | 928 (352/1536) | 690 (64/1656) | 9168 (1908/19508) | 6445 (2051/14133) | 2802 (2751/3003) | 3204 (2412/4460) | 6650 (4309/8876) | 4410 (4108/8820) |
| p = 0.77 | p = 0.82 | p = 0.70 | p = 0.70 | |||||
| NO (μM) | 4.6 (4.0/5.3) | 4.0 (3.7/4.9) | 32.4 (22.0/34.7) | 40.5 (29.4/42.8) | 26.1 (18.7/28.2) | 31.6 (28.0/32.8) | 24.4 (15.0/26.5) | 25.4 (23.8/32.2) |
| p = 0.45 | p = 0.03* | p = 0.02* | p = 0.11 | |||||
Phagocytosis of E. coli K1 by primary murine microglial cells [percentages of phagocytosis of the unstimulated control group (medians with interquartile ranges); n = 9–16 from up to four experiments] as well as concentrations (medians with interquartile ranges) of TNF-α (pg/ml), IL-6 (pg/ml), CXCL1(pg/ml), and NO (μM) in the supernatants after treatment with 13 μg/ml activin A (Act A) alone and in combination with the TLR2 agonist Pam3CSK4 (P3C), the TLR4 agonist LPS, and the TLR9 agonist CpG. Data were analyzed by Mann-Whitney U test (*p < 0.05, ***p < 0.001)
Activin A dose-dependently enhanced phagocytosis of E. coli K1 by microglial cells stimulated with agonists of TLR2, 4 and 9
Stimulation with the different TLR agonists increased phagocytosis of E. coli K1 by microglial cells compared to unstimulated cells. Based on previous experiments [18], concentrations of TLR agonists evoking an approximately 3-fold increase of phagocytosis were chosen: Pam3CSK4 0.1 μg/ml: 273 (129/515) %, LPS 0.01 μg/ml: 324 (182/641) %, CpG 1 μg/ml: 255 (150/510) % (Fig. 2a–c).
Fig. 2.

Phagocytosis of E. coli by microglial cells treated with activin A and different TLR agonists. Comparison of E. coli K1 phagocytosis between microglial cells treated with activin A in concentrations of 0.13, 1.3, and 13 ng/ml in combination with the TLR2 agonist Pam3CSK4 (P3C; 0.1 μg/ml, a), the TLR4 agonist LPS (0.01 μg/ml, b), and the TLR9 agonist CpG (1 μg/ml, c), and microglial cells treated with the respective TLR agonist alone (n = 14–30 from up to seven experiments). Data were analyzed by Kruskal-Wallis test followed by Dunn’s multiple comparison test (*p < 0.05; **p < 0.01, ***p < 0.001). In each experiment, the mean number of bacteria ingested by unstimulated control cells was defined as 100%. Phagocytic rates of the stimulated groups are presented as percentages of phagocytosis of an unstimulated control group (medians with interquartile ranges). Please mind the interrupted y-axis
Additional treatment of microglial cells with activin A in concentrations of 1.3 and 13 ng/ml starting 24 h before stimulation with TLR agonists further increased phagocytosis of E. coli K1 compared to treatment with the respective TLR agonists alone (Fig. 2a–c). Compared to treatment with Pam3CSK4 alone [273 (129/515) %], additional treatment with activin A 1.3 ng/ml increased the phagocytic rate 3.4-fold [927 (403/1503) %, p < 0.01] and activin 13 ng/ml increased the phagocytic rate 2.8-fold [750 (440/1490) %, p < 0.001; Fig. 2a]. Compared to treatment with LPS alone [324 (182/641) %], additional treatment with activin 13 ng/ml increased the phagocytic rate 1.7-fold [540 (364/900) %, p < 0.05; Fig. 2b]. Compared to treatment with CpG alone [255 (150/510) %], additional treatment with activin 13 ng/ml increased the phagocytic rate 2.7-fold [676 (393/1337) %, p < 0.01; Fig. 2c].
Additional treatment of microglial cells with activin A in the high concentration of 13 μg/ml starting 24 h before stimulation with TLR agonists did not significantly influence phagocytosis of E. coli K1 compared to treatment with the respective TLR agonists alone (Table 1).
In concentrations enhancing phagocytosis, activin A did not increase the release of proinflammatory cytokines and nitric oxide by microglial cells
Treatment of microglial cells with activin A alone in concentrations between 0.13 and 13 ng/ml did not significantly influence the release of TNF-α (Fig. 3a), IL-6 (Fig. 3b), and CXCL1 (Fig. 3c) and slightly decreased the release of NO (activin A 0.13 and 13 ng/ml: p < 0.05; Fig. 3d).
Fig. 3.

Release of proinflammatory cytokines and NO by microglial cells treated with activin A alone. Comparison of concentrations of TNF-α (pg/ml) (a), IL-6 (pg/ml) (b), CXCL1 (pg/ml) (c), and NO (μM) (d) in the supernatants of unstimulated primary murine microglial cells and microglial cells treated with activin A in concentrations of 0.13, 1.3, and 13 ng/ml [a: n = 8–27 from up to six experiments; b, c: n = 8–18 from up to four experiments; d: n = 3 from one single experiment / b: almost all values were below the detection limit of 38 pg/ml IL-6; d: for activin A 0.13 and 13 ng/ml, all values were below the detection limit of 3 μM nitrite]. Data are presented as medians with interquartile ranges and were analyzed by Kruskal-Wallis test followed by Dunn’s multiple comparison test (*p < 0.05)
As known from previous studies [18–20], the TLR agonists Pam3CSK4, LPS, and CpG enhanced the release of TNF-α, IL-6, CXCL1, and NO by primary murine microglial cells. Additional treatment of microglial cells with activin A in concentrations of 0.13, 1.3, and 13 ng/ml starting 24 h before stimulation with TLR agonists did not significantly influence the release of TNF-α (Fig. 4a–c), IL-6 (Fig. 4d–f), CXCL1 (Fig. 4g–i), and NO (Fig. 4j–l) compared to treatment with the respective TLR agonists alone.
Fig. 4.

Release of proinflammatory cytokines and NO by microglial cells treated with phagocytosis-enhancing concentrations of activin A and different TLR agonists. Comparison of concentrations of TNF-α (pg/ml) (a–c), IL-6 (pg/ml) (d–f), CXCL1 (pg/ml) (g–i), and NO (μM) (j–l) in the supernatants of microglial cells after treatment with activin A in concentrations of 0.13, 1.3, and 13 ng/ml in combination with the TLR2 agonist Pam3CSK4 (P3C; 0.1 μg/ml; a, d, g, j), the TLR4 agonist LPS (0.01 μg/ml; b, e, h / 0.003 μg/ml; k) and the TLR9 agonist CpG (1 μg/ml; c, f, i / 0.1 μg/ml; l) and of microglial cells treated with the respective TLR agonist alone [a–c: n = 8–28 from up to 6 experiments; d–i: n = 8–17 from up to four experiments; j–l: n = 3 from one single experiment]. Data are presented as medians with interquartile ranges and were analyzed by Kruskal-Wallis test followed by Dunn’s multiple comparison test. No significant differences were detected. Please mind the interrupted y-axis in d and g–i
In a higher concentration, activin A influenced the release of TNF-α and NO by microglia activated with TLR agonists
Treatment of microglial cells with activin A alone in a high concentration of 13 μg/ml did not significantly influence the release of TNF-α, IL-6, and CXCL1, and NO (Table 1). Additional treatment with 13 μg/ml activin A decreased TNF-α release of LPS-stimulated microglial cells (p < 0.0003) and increased NO release of microglial cells stimulated with Pam3CSK4 (p = 0.03) and LPS (p = 0.02). No further significant influences of 13 μg/ml activin A on cytokine and NO release were observed (Table 1).
Activin A did not influence the viability of primary murine microglial cells
Viability of microglial cells was not influenced by treatment with high doses of activin A (13 μg/ml) as assessed by the WST-1 assay [unstimulated cells (n = 9): 100 ± 11%, activin A 13 μg/ml (n = 9): 96.9 ± 11.5%, p = 0.56; Fig. 5a]. Activin A in lower doses and in combination with the TLR agonists Pam3CSK4, LPS, and CpG also did not reduce WST-1 values (n = 3 per group; p > 0.05). Isolectin B4 staining did not reveal changes of morphology or density of microglial cells after treatment with activin A (Fig. 5b).
Fig. 5.
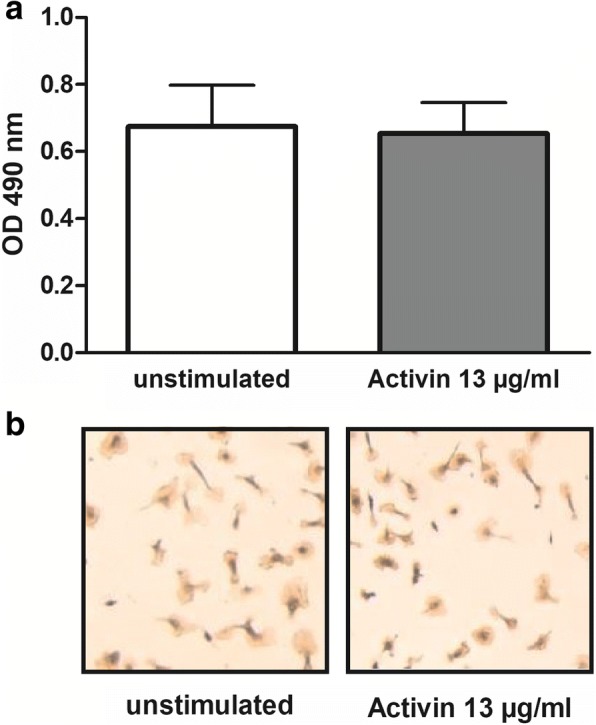
Viability of primary murine microglial cells after treatment with a high concentration of activin A. a Comparison of WST-1 assay results of unstimulated primary murine microglial cells and microglial cells after treatment with 13 μg/ml activin A for 48 h (p = 0.69). Data are presented as means + SD of the optical density (OD) at 490 nm (n = 9 from three experiments). b Representative isolectin B4 stainings of unstimulated primary murine microglial cells and microglial cells after treatment with 13 μg/ml activin A for 48 h. By light microscopy, no differences in morphology and density of microglial cells were detected after treatment with activin A
Discussion
In search of compounds that increase phagocytosis of pathogens without exerting collateral damage to the brain tissue with the aim to improve the resistance of the brain to infections, we investigated the effect of activin A on primary murine microglial cells. Activin A appears particularly interesting for modulation of brain immune cell functions as neuroprotective effects have been described in vitro [42] and in vivo. Intracerebroventricular (i.c.v.) infusion of activin A in mice had anti-inflammatory and neuroprotective effects in a stroke model [43], enhanced neurogenesis after excitotoxic neurodegeneration [37], and only recently has been shown to protect midbrain neurons in mouse models of Parkinson’s disease (MPTP and LPS model) [44].
We used different concentrations of activin A in our study. The lower concentrations from 0.13 to 13 ng/ml covered the range of concentrations used in the majority of in vitro experiments with different types of immune cells (for review, see [28]) and also the concentrations of activin A measured in human cerebrospinal fluid (CSF) of persons with an age over 60 years without any neurological disease (median 0.14 ng/ml, maximum value 0.33 ng/ml [34]) and mainly younger patients with meningitis (median 0.13 ng/ml [34]) or traumatic brain injury (0.2 ng/ml [45]). To exclude direct toxic effects, we investigated the effect of a 1000-fold higher concentration of activin A (13 μg/ml) which is unphysiolgically high and higher than the estimated CSF concentrations (based on a mouse CSF volume of 35 μl [46]) achieved in mouse experiments by intracerebroventricular (i.c.v.) injection (15 μg/kg bodyweight [43]) or continuous infusion (295 ng/day [44]).
By light microscopic evaluation of isolectin B4 stainings and the WST-1 cell viability assay, we did not detect effects of activin A on viability and morphology of primary microglial cells and could exclude cytotoxic effects of even high activin A concentrations. Similarly, activin A in concentrations from 0.8 to 10 ng/ml did not affect viability and proliferation of cells from a murine macrophage cell line and of primary murine macrophages as assessed by quantitating viable cells with the MTT test which is based on the same principles as the WST-1 test used in our study. Other studies documented an influence of activin A on the density of microglial cells in vitro and in vivo that might be explained by the different experimental settings. In rat microglial cells in vitro, treatment with 10 ng/ml activin A for 24 h enhanced proliferation [36], whereas in primary murine microglial cells, treatment with 100 ng/ml and 1 μg/ml activin A reduced cell numbers alone and in combination with LPS [37]. It also inhibited LPS-induced morphological changes, but did not influence morphology/phenotype of microglial cells when given alone [37]. In mouse models in vivo, i.c.v. administration of activin A alone did not affect numbers of microglial cells; however, continuous administration of activin A for 7 or 15 days after intracerebral challenge with LPS reduced LPS-induced proliferation of microglial cells in specific brain regions [37, 44]. Shorter periods of activin A exposure are more relevant for acute intracerebral infections and might not influence microglial proliferation.
The effect of activin A on phagocytic activity of different phagocytes, mainly macrophages, monocytes [28, 40], and neutrophils [47], has been addressed before; however, to our knowledge, this is the first study investigating the effect of activin A on phagocytic activity of microglial cells. Other groups examined phagocytosis of chicken red blood cells (cRBC), latex particles, and microspheres or pinocytosis of natural red, whereas we investigated phagocytosis of living bacteria. Activin A alone did not significantly affect phagocytosis of E. coli K1 by primary murine microglial cells. Differently to these results in resting microglia, in resting murine macrophages (peritoneal macrophages and a macrophage cells line) activin A increased the capacity to phagocytose cRBC and microspheres [38–40]. As in our previous in vitro experiments [18], activation of microglial cells with agonists of TLR 2, 4, and 9 (Pam3CSK4, LPS, and CpG) increased phagocytosis of E. coli K1. Concentrations of TLR agonists which increased the phagocytosis rate approximately 2- to 3-fold were chosen. Treatment with activin A in concentrations of 1.3 and 13 ng/ml starting 24 h before TLR activation, further increased phagocytosis of E. coli K1 up to 3.4-fold compared to treatment with the respective TLR agonist alone. Thus, microglial cells treated with activin A in these concentrations followed by activation with one TLR agonist ingested up to 9-fold more bacteria than unstimulated microglial cells. The higher concentration of 13 μg/ml activin A did not influence phagocytosis. In preliminary experiments, we had tested phagocytosis of E. coli by microglial cells after treatment with a broader range of activin A concentrations (0.0013 pg/ml to 1.3 μg/ml in intervals of factor ten) in combination with Pam3CSK4: Bacterial uptake was increased after treatment with Pam3CSK4 in combination with activin A 1.3 and 13 ng/ml; higher or lower concentrations of activin A neither increased nor decreased bacterial uptake (n = 3 per group, data not shown). This shows that the effect of activin A on phagocytic activity is dose dependent. The phagocytosis-inducing effect of activin A differed only slightly between the different TLR agonists and was strongest in case of co-treatment with the TLR2 agonist Pam3CSK4, followed by CpG and LPS (approximately 2-fold increase compared to LPS alone). In contrast, in mouse macrophages, activin A inhibited the phagocytic activity of LPS-activated macrophages [40, 41, 48]. Reasons for these differences might be the different cells types studied and the different timing of activin A treatment: In our experiment, activin A treatment of microglial cells started 24 h before administration of the different TLR agonists, whereas macrophages in the other studies were simultaneously treated with activin A and LPS. Furthermore, the viable E. coli K1 used in our phagocytosis experiments can induce a strong immune reaction itself involving signaling pathways that are not activated by non-bacterial particles used in the other studies. Activin A in combination with the different TLR agonists increased phagocytosis of E. coli K1 by primary murine microglial cells up to 9-fold and thus to a similar extent as pretreatment with PEA which subsequently has been shown to reduce bacterial titers and mortality in our mouse model of E. coli meningitis [26].
The release of the proinflammatory cytokines TNF-α, IL-6, and CXCL1 from primary murine microglial cells was not increased after treatment with activin A concentrations that enhanced phagocytosis (0.13, 1.3, and 13 ng/ml) or a much higher concentration of activin A (13 μg/ml). Activin A also did not further enhance cytokine release induced by Pam3CSK4, LPS, and CpG. TNF-α even showed a tendency to be decreased upon activin A treatment. These results are in line with data from previous studies in which activin A downregulated the expression of proinflammatory cytokines in murine primary microglial cells [49] and decreased the LPS-induced release of TNF-α, IL-6, and IL-1 in primary microglial cells of mice and rats [36, 37]. However, in resting mouse macrophages, activin A increased the release of IL-1 and IL-6 [38, 39].
NO production upon microglial activation contributes to microglia-mediated neuronal injury [21–25]. NO release from primary mouse microglial cells was not increased after treatment with activin A alone. Activin A in phagocytosis-enhancing concentrations of 0.13, 1.3, and 13 ng/ml also did not increase NO release induced by Pam3CSK4, LPS, and CpG. However, in a high concentration of 13 μg/ml, activin A slightly increased NO release induced by Pam3CSK4 and LPS. In previous studies, activin A has been shown to decrease iNOS mRNA expression in LPS-activated rat microglia [36] and NO release of LPS-activated mouse peritoneal macrophages [41]; however, it enhanced NO release of resting macrophages [38, 39, 50, 51].
Our results indicate that despite multiple similarities of microglial cells and macrophages, there are considerable differences between these cell types concerning the modulation of their functions by activin A. Furthermore, concentration and timing of activin A treatment seem to be crucial parameters for activin A effects. Concentrations of activin A that increased phagocytosis of TLR-activated microglia in vitro are higher than CSF concentrations of activin A that have been measured in patients with meningitis [34]. Thus, endogenous activin A released during bacterial infections probably is not sufficient to influence phagocytic activity of microglial cells. Additional activin A administration is required to achieve an effect on phagocytosis. As described above, i.c.v. administration of activin A has already been shown to be beneficial in mouse models of stroke, excitotoxic neurodegeneration, and Parkinson’s disease [37, 43, 44]. Concentrations that increase NO release under infectious conditions are unlikely to be achieved in vivo.
In mouse macrophages, activin A influenced the expression of TLR, and LPS upregulated the mRNA of Act-RII and Smad 2/3 indicating a mutual regulation of signaling pathways [40]. Timing of activin A administration—before, simultaneously or after an inflammatory stimulus—appears to be critical for activin A effects. In our experimental setting, activin A was administered 24 h before microglial activation by agonists of TLR2, 4, and 9 and 48 h before addition of E. coli resembling a preventive approach. TLR2, 4, and 9 are the principal TLR involved in the pathogenesis of bacterial infections which are activated in microglia during the early phase of bacterial CNS infection [10]. Thus, we assessed microglial functions under conditions mimicking a patient at high risk or in an early phase of an already ongoing bacterial CNS infection. In a mouse model of stroke, both prophylactic i.c.v. administration of activin A 1 h before as well as administration 6 h after acute cerebral ischemia had anti-inflammatory and neuroprotective effects [43].
In summary, we demonstrated that activin A dose-dependently enhances phagocytosis of E. coli K1 by primary microglial cells activated by different TLR agonists. In phagocytosis-enhancing concentrations, activin A does not increase the release of different proinflammatory cytokines and NO. This suggests that there is no risk of aggravating neuronal injury by the prophylactic or therapeutic administration of activin A. Higher phagocytic activity of microglia without exerting collateral damage to the brain might lead to faster bacterial clearance and prevention or a better outcome of bacterial CNS infections.
Conclusions
Because of its known neuroprotective effects, activin A appears to be particularly promising for modulation of brain immune cell functions. Our present cell culture results suggest that priming of microglial cells with activin A could increase the elimination of bacteria in bacterial CNS infections. This strategy could improve the resistance of the brain to infections, particularly in elderly and immunocompromised patients. The potential effect of prophylactic activin A administration needs to be investigated in models of bacterial CNS infections in vivo.
Acknowledgments
Funding
The study was supported by a grant of the Robert Bosch Stiftung, Stuttgart – Forschungskolleg Geriatrie (to SS).
Availability of data and materials
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
Abbreviations
- ABC
Avidin-biotin complex
- Act-RI
Activin A receptor type I
- Act-RII
Activin A receptor type II
- ANOVA
Analysis of variance
- BSA
Bovine serum albumin
- CFU
Colony-forming units
- CNS
Central nervous system
- CpG
Cytosine-guanosine motifs
- cRBC
Chicken red blood cells
- CSF
Cerebrospinal fluid
- CXCL1
Chemokine (C-X-C-motif) ligand 1
- DMEM
Dulbecco’s modified Eagle’s medium
- DNA
Deoxyribonucleic acid
- ELISA
Enzyme-linked immunosorbent assay
- E. coli
Escherichia coli
- FCS
Fetal calf serum
- GRO α
Growth-related oncogene alpha
- i.c.v.
Intracerebroventricular
- IL-1
Interleukin-1
- IL-6
Interleukin-6
- iNOS
Inducible nitric oxide synthase
- KC
Keratinocyte chemotractant
- LPS
Lipopolysaccharide
- MPTP
1-Phenyl-4-methyl-1,2,3,6-tetrahydropyridine
- mRNA
Messenger ribonucleic acid
- NO
Nitric oxide
- OD
Optical density
- ODN
Oligodesoxynucleotide
- Pam3CSK4
Tripalmitoyl-S-glycerl-cysteine
- PAMPS
Pathogen-associated molecular patterns
- PBS
Phosphate-buffered saline
- PEA
Palmitoylethanolamide
- ROS
Reactive oxygen species
- SD
Standard deviation
- TGF-β
Transforming growth factor beta
- TLR
Toll-like receptor
- TNF-α
Tumor necrosis factor alpha
- WST
Water-soluble tetrazolium salt
Authors’ contributions
CD performed the experiments, analyzed the results, and wrote the manuscript. SRi, UM, and RN planned the experiments and discussed the results. AK and SRe performed the phagocytosis assays. SB and UKH analyzed the cytokine release. JS performed the experiments for the revision of the paper. SS planned and designed the study, analyzed the results, and wrote the manuscript. All authors read and approved the final version of the manuscript, with exception of UKH who unfortunately died before the completion of the manuscript.
Ethics approval and consent to participate
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Nau R, Djukic M, Spreer A, Ribes S, Eiffert H. Bacterial meningitis: an update of new treatment options. Expert Rev Anti-Infect Ther. 2015;13:1401–1423. doi: 10.1586/14787210.2015.1077700. [DOI] [PubMed] [Google Scholar]
- 2.van de Beek D, Brouwer M, Hasbun R, Koedel U, Whitney CG, Wijdicks E. Community-acquired bacterial meningitis. Nat Rev Dis Primers. 2016;2:16074. doi: 10.1038/nrdp.2016.74. [DOI] [PubMed] [Google Scholar]
- 3.Choi C. Bacterial meningitis in aging adults. Clin Infect Dis. 2001;33:1380–1385. doi: 10.1086/322688. [DOI] [PubMed] [Google Scholar]
- 4.Cabellos C, Verdaguer R, Olmo M, Fernández-Sabé N, Cisnal M, Ariza J, et al. Community-acquired bacterial meningitis in elderly patients: experience over 30 years. Medicine (Balitmore) 2009;88:115–119. doi: 10.1097/MD.0b013e31819d50ef. [DOI] [PubMed] [Google Scholar]
- 5.Kim KS. Strategy of Escherichia coli for crossing the blood-brain barrier. J Infect Dis. 2002;186:220–224. doi: 10.1086/344284. [DOI] [PubMed] [Google Scholar]
- 6.Aloisi F. Immune function of microglia. Glia. 2001;36:165–179. doi: 10.1002/glia.1106. [DOI] [PubMed] [Google Scholar]
- 7.Akira S, Takeda K, Kaisho T. Toll-like receptors: critical proteins linking innate and acquired immunity. Nat Immunol. 2000;2:675–680. doi: 10.1038/90609. [DOI] [PubMed] [Google Scholar]
- 8.Bsibsi M, Ravid R, Gveric D, van Noort JM. Broad expression of toll-like receptors in the human central nervous system. J Neuropathol Exp Neurol. 2002;61:1013–1021. doi: 10.1093/jnen/61.11.1013. [DOI] [PubMed] [Google Scholar]
- 9.Perry VH, Newman TA, Cunningham C. The impact of systemic infection on the progression of neurodegenerative disease. Nat Rev Neurosci. 2003;4:103–112. doi: 10.1038/nrn1032. [DOI] [PubMed] [Google Scholar]
- 10.van Rossum D, Hanisch UK. Microglia. Metab Brain Dis. 2004;19:393–411. doi: 10.1023/B:MEBR.0000043984.73063.d8. [DOI] [PubMed] [Google Scholar]
- 11.Takeda K, Kaisho T, Akira S. Toll-like receptors. Ann Rev Immunol. 2003;21:335–376. doi: 10.1146/annurev.immunol.21.120601.141126. [DOI] [PubMed] [Google Scholar]
- 12.Poltorak A, He X, Smirnova I, Liu MY, Van Huffel C, Du X, et al. Defective LPS signaling in C3H/HeJ and C57BL/10ScCr mice: mutations in Tlr4 gene. Science. 1998;282:2085–2088. doi: 10.1126/science.282.5396.2085. [DOI] [PubMed] [Google Scholar]
- 13.Hemmi H, Takeuchi O, Kawai T, Kaisho T, Sato S, Sanjo H, et al. A toll-like receptor recognizes bacterial DNA. Nature. 2000;408:740–745. doi: 10.1038/35047123. [DOI] [PubMed] [Google Scholar]
- 14.Smith ME, van der Maesen K, Somera FP. Macrophage and microglial responses to cytokines in vitro: phagocytic activity, proteolytic enzyme release, and free radical production. J Neurosci Res. 1998;54:68–78. doi: 10.1002/(SICI)1097-4547(19981001)54:1<68::AID-JNR8>3.0.CO;2-F. [DOI] [PubMed] [Google Scholar]
- 15.Häusler KG, Prinz M, Nolte C, Weber JR, Schumann RR, Kettenmann H, et al. Interferon-gamma differentially modulates the release of cytokines and chemokines in lipopolysaccharide- and pneumococcal cell wall-stimulated mouse microglia and macrophages. Eur J Neurosci. 2002;16:2113–2122. doi: 10.1046/j.1460-9568.2002.02287.x. [DOI] [PubMed] [Google Scholar]
- 16.Hanisch UK, Kettenmann H. Microglia: active sensor and versatile effector cells in the normal and pathologic brain. Nat Neurosci. 2007;10:1387–1394. doi: 10.1038/nn1997. [DOI] [PubMed] [Google Scholar]
- 17.Schütze S, Ribes S, Kaufmann A, Manig A, Scheffel J, Redlich S, et al. Higher mortality and impaired elimination of bacteria in aged mice after intracerebral infection with E. coli are associated with an age-related decline of microglia and macrophage functions. Oncotarget. 2014;5:12573–12592. doi: 10.18632/oncotarget.2709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ribes S, Ebert S, Czesnik D, Regen T, Zeug A, Bukowski S, et al. Toll-like receptor prestimulation increases phagocytosis of Escherichia coli DH5alpha and Escherichia coli K1 strains by murine microglial cells. Infect Immun. 2009;77:557–564. doi: 10.1128/IAI.00903-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ebert S, Gerber J, Bader S, Mühlhauser F, Brechtel K, Mitchell TJ, et al. Dose-dependent activation of microglial cells by toll-like receptor agonists alone and in combination. J Neuroimmunol. 2005;159:87–96. doi: 10.1016/j.jneuroim.2004.10.005. [DOI] [PubMed] [Google Scholar]
- 20.Redlich S, Ribes S, Schütze S, Eiffert H, Nau R. Toll-like receptor stimulation increases phagocytosis of Cryptococcus neoformans by microglial cells. J Neuroinflamm. 2013;10:71. doi: 10.1186/1742-2094-10-71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chao CC, Hu S, Close K, Choi CS, Molitor TW, Novick WJ, et al. Cytokine release from microglia: differential inhibition by pentoxifylline and dexamethasone. J Infect Dis. 1992;166:847–853. doi: 10.1093/infdis/166.4.847. [DOI] [PubMed] [Google Scholar]
- 22.Dawson VL, Brahmbhatt HP, Mong JA, Dawson TM. Expression of inducible nitric oxide synthase causes delayed neurotoxicity in primary mixed neuronal-glial cortical cultures. Neuropharmacology. 1994;33:1425–1430. doi: 10.1016/0028-3908(94)90045-0. [DOI] [PubMed] [Google Scholar]
- 23.Lehnardt S, Massillon L, Follett P, Jensen FE, Ratan R, Rosenberg PA, et al. Activation of innate immunity in the CNS triggers neurodegeneration through a toll-like receptor 4-dependent pathway. Proc Natl Acad Sci U S A. 2003;100:8514–8519. doi: 10.1073/pnas.1432609100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Iliev AI, Stringaris AK, Nau R, Neumann H. Neuronal injury mediated via stimulation of microglial toll-like receptor-9 (TLR9) FASEB J. 2004;18:412–414. doi: 10.1096/fj.03-0670fje. [DOI] [PubMed] [Google Scholar]
- 25.Schütze S, Loleit T, Zeretzke M, Bunkowski S, Brück W, Ribes S, et al. Additive microglia-mediated neuronal injury caused by amyloid-β and bacterial TLR agonists in murine neuron-microglia co-cultures quantified by an automated image analysis using cognition network technology. J Alzheimers Dis. 2012;31:651–657. doi: 10.3233/JAD-2012-120856. [DOI] [PubMed] [Google Scholar]
- 26.Redlich S, Ribes S, Schütze S, Nau R. Palmitoylethanolamide stimulates phagocytosis of Escherichia coli K1 by macrophages and increases the resistance of mice against infections. J Neuroinflamm. 2014;11:108. doi: 10.1186/1742-2094-11-108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Nau R, Ribes S, Djukic M, Eiffert H. Strategies to increase the activity of microglia as efficient protectors of the brain against infections. Front Cell Neurosci. 2014;8:138. doi: 10.3389/fncel.2014.00138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ebert S, Nau R, Michel U. Role of activin in bacterial infection: a potential target for immunointervention? Immunotherapy. 2010;2:673–684. doi: 10.2217/imt.10.64. [DOI] [PubMed] [Google Scholar]
- 29.Chen W, Ten Dijke P. Immunoregulation by members of the TGFβ superfamily. Nat Rev Immunol. 2016;16:723–740. doi: 10.1038/nri.2016.112. [DOI] [PubMed] [Google Scholar]
- 30.Vale W, Rivier J, Vaughan J, McClintock R, Corrigan A, Woo W, et al. Purification and characterization of an FSH releasing protein from porcine ovarian follicular fluid. Nature. 1986;321:776–779. doi: 10.1038/321776a0. [DOI] [PubMed] [Google Scholar]
- 31.Phillips DJ, de Kretser DM, Hedger MP. Activin and related proteins in inflammation: not just interested bystanders. Cytokine Growth Factor Rev. 2009;20:153–164. doi: 10.1016/j.cytogfr.2009.02.007. [DOI] [PubMed] [Google Scholar]
- 32.Michel U, Ebert S, Phillips D, Nau R. Serum concentrations of activin and follistatin are elevated and run parallel in patients with septicemia. Eur J Endocrinol. 2003;148:559–564. doi: 10.1530/eje.0.1480559. [DOI] [PubMed] [Google Scholar]
- 33.Michel U, Ebert S, Schneider O, Shintani Y, Bunkowski S, Smirnov A, et al. Follistatin (FS) in human cerebrospinal fluid and regulation of FS expression in a mouse model of meningitis. Eur J Endocrinol. 2000;143:809–816. doi: 10.1530/eje.0.1430809. [DOI] [PubMed] [Google Scholar]
- 34.Ebert S, Phillips DJ, Jenzewski P, Nau R, O’Connor AE, Michel U. Activin A concentrations in human cerebrospinal fluid are age-dependent and elevated in meningitis. J Neurol Sci. 2006;250:50–57. doi: 10.1016/j.jns.2006.06.026. [DOI] [PubMed] [Google Scholar]
- 35.Ebert S, Zeretzke M, Nau R, Michel U. Microglial cells and peritoneal macrophages release activin A upon stimulation with toll-like receptor agonists. Neurosci Lett. 2007;413:241–244. doi: 10.1016/j.neulet.2006.11.065. [DOI] [PubMed] [Google Scholar]
- 36.Wilms H, Schwark T, Brandenburg L, Sievers J, Dengler R, Deuschl G, et al. Regulation of activin A synthesis in microglial cells: pathophysiological implications for bacterial meningitis. J Neurosci Res. 2010;88:16–23. doi: 10.1002/jnr.22185. [DOI] [PubMed] [Google Scholar]
- 37.Abdipranoto-Cowley A, Park JS, Croucher D, Daniel J, Henshall S, Galbraith S, Mervin K, Vissel B. Activin A is essential for neurogenesis following neurodegeneration. Stem Cells. 2009;27:1330–1346. doi: 10.1002/stem.80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ge J, Wang Y, Feng Y, Liu H, Cui X, Chen F, et al. Direct effects of activin A on the activation of mouse macrophages RAW264.7 cells. Cell Mol Immunol. 2009;6:129–133. doi: 10.1038/cmi.2009.18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wang Y, Cui X, Tai G, Ge J, Li N, Chen F, et al. A critical role of activin A in maturation of mouse peritoneal macrophages in vitro and in vivo. Cell Mol Immunol. 2009;6:387–389. doi: 10.1038/cmi.2009.50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Li N, Cui X, Ge J, Li J, Niu L, Liu H, et al. Activin A inhibits activities of lipopolysaccharide-activated macrophages via TLR4, not of TLR 2. Biochem Biophys Res Commun. 2013;435:222–228. doi: 10.1016/j.bbrc.2013.04.077. [DOI] [PubMed] [Google Scholar]
- 41.Zhou J, Tai G, Liu H, Ge J, Feng Y, Chen F, et al. Activin A down-regulates the phagocytosis of lipopolysaccharide-activated mouse peritoneal macrophages in vitro and in vivo. Cell Immunol. 2009;255:69–75. doi: 10.1016/j.cellimm.2008.11.001. [DOI] [PubMed] [Google Scholar]
- 42.Park SE, Lee J, Chang EH, Kim JH, Sung J-H, Na DL, et al. Activin A secreted by human mesenchymal stem cells induces neuronal development and neurite outgrowth in an in vitro model of Alzheimer’s disease: neurogenesis induced by MSCs via activin A. Arch Pharm Res. 2016;39:1171–1179. doi: 10.1007/s12272-016-0799-4. [DOI] [PubMed] [Google Scholar]
- 43.Mukerji S, Rainey RN, Rhodes J, Hall A. Delayed activin A administration attenuates tissue death after transient focal cerebral ischemia and is associated with decreased stress-responsive kinase activation. J Neurochem. 2009;111:1138–1148. doi: 10.1111/j.1471-4159.2009.06406.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Stayte S, Rentsch P, Tröscher AR, Bamberger M, Li KM, Vissel B. Activin A inhibits MPTP and LPS-induced increases in inflammatory cell populations and loss of dopamine neurons in the mouse midbrain in vivo. PLoS One. 2017;12:e0167211. doi: 10.1371/journal.pone.0167211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Phillips DJ, Nguyen P, Adamides AA, Bye N, Rosenfeld JV, Kossmann T, et al. Activin A release into cerebrospinal fluid in a subset of patients with severe traumatic brain injury. J Neurotrauma. 2006;23:1283–1294. doi: 10.1089/neu.2006.23.1283. [DOI] [PubMed] [Google Scholar]
- 46.Pardridge WM. Advances in cell biology of blood-brain barrier transport. Semin Cell Biol. 1991;2:419–426. [PubMed] [Google Scholar]
- 47.Qi Y, Ge J, Ma C, Wu N, Cui X, Liu Z. Activin A regulates activation of mouse neutrophils by Smad3 signalling. Open Biology. 2017;7:160342. doi: 10.1098/rsob.160342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wang S, Tai G, Zhang P, Mu D, Zhang X, Liu Z. Inhibitory effect of activin A on activation of lipopolysaccharide-stimulated mouse macrophage RAW264.7 cells. Cytokine. 2008;42:85–91. doi: 10.1016/j.cyto.2008.01.010. [DOI] [PubMed] [Google Scholar]
- 49.Mitchell K, Shah JP, Tsytsikova LV, Campbell AM, Affram K, Symes AJ. LPS antagonism of TGF-β signaling results in prolonged survival and activation of rat primary microliga. J Neurochem. 2014;129:155–168. doi: 10.1111/jnc.12612. [DOI] [PubMed] [Google Scholar]
- 50.Nüsing RM, Barsig J. Induction of prostanoid, nitric oxide, and cytokine formation in rat bone marrow derived macrophages by activin A. Br J Pharmacol. 1999;127:919–926. doi: 10.1038/sj.bjp.0702626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zhang XJ, Li Y, Tai GX, Xu GY, Zhang PY, Yang Y, et al. Effects of activin A on the activities of the mouse peritoneal macrophages. Cell Mol Immunol. 2005;2:63–67. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.