

Abstract
Posttranslational modifications of DNA repair proteins have been linked to their function. However, it is not clear if posttranslational acetylation affects subcellular localization of these enzymes. Here, we show that the human DNA glycosylase NEIL1, which is involved in repair of both endo- and exogenously generated oxidized bases via the base excision repair (BER) pathway, is acetylated by histone acetyltransferase p300. Acetylation occurs predominantly at Lys residues 296, 297 and 298 located in NEIL1’s disordered C-terminal domain. NEIL1 mutant having the substitution of Lys 296–298 with neutral Ala loses nuclear localization, whereas Lys>Arg substitution (in 3KR mutant) at the same sites does not affect NEIL1’s nuclear localization or chromatin binding, presumably due to retention of the positive charge. Although non-acetylated NEIL1 can bind to chromatin, acetylated NEIL1 is exclusively chromatin-bound. NEIL1 acetylation while dispensable for its glycosylase activity enhances it due to increased product release. The acetylation-defective 3KR mutant forms less stable complexes with various chromatin proteins, including histone chaperones and BER/single-strand break repair partners, than the wild-type (WT) NEIL1. We also showed that the repair complex with WT NEIL1 has significantly higher BER activity than the 3KR mutant complex. This is consistent with reduced resistance of non-acetylable mutant NEIL1 expressing cells to oxidative stress relative to cells expressing acetylable WT enzyme. We thus conclude that the major role of acetylable Lys residues in NEIL1 is to stabilize the formation of chromatin-bound repair complexes which protect cells from oxidative stress.
Keywords: base excision repair (BER), NEIL1, chromatin, acetylation, oxidative stress, repair complex
1. Introduction
In concert with epigenetic modifications of histones and DNA in chromatin which control cellular functions [1, 2], reversible, covalent modifications of non-histone proteins also occur during various DNA transactions [3–5]. The acetyltransferases p300, and its close homolog CBP, initially characterized for their histone acetyltransferase (HAT) activity, were named transcriptional co-activators, based on their regulation of a wide variety of genes via interaction with the cognate transcription factors [6–8]. Along with the discovery of acetylation of PCNA, FEN-1, Dna2 and their linkage to DNA replication in mammalian cells [9–12], we discovered acetylation of APE1, a key enzyme in the base excision repair (BER) pathway [13]. Acetylation of other BER proteins including DNA Pol β [14], and TDG [15], a DNA glycosylase (DG) which preferentially excises T(U) from G/T and G/U mismatches in the CpG context, were also discovered. We subsequently characterized acetylation of NEIL2 [16] and OGG1 [17], the oxidized base-specific DGs, and mapped their acetyl-accepting Lys residues. These findings implicated acetylation in the BER process. For almost all of the proteins undergoing acetylation, p300 was identified as the major acetyltransferase, while class I histone deacetylases (HDACs) were mainly found to be involved in controlling the acetylation level in cells. However, SIRT1 (a member of class III HDACs), together with HDAC1 deacetylates acetylated APE1 [18, 19].
The effects of acetylation on the enzymatic activity of BER enzymes are variable, presumably because acetylation affects their interaction with the partner proteins and DNA. Acetylation of APE1 increases its AP-endonuclease activity thereby stimulating BER [19–21], and also its transcriptional co-regulatory activity [22–24]. Acetylation also affects APE1’s role in RNA metabolism [25]. Acetylation of OGG1 enhances its activity [17], while NEIL2’s activity is inhibited by acetylation [16]. Acetylation of the Werner syndrome helicase (WRN), a member of the human RECQ helicase family, stimulates its catalytic activities and affects BER [26]. However, whether acetylation impacts subcellular localization of repair enzymes was not investigated earlier. Here we characterized acetylation of NEIL1, a key DG for oxidized base repair [27–32], and showed that acetylation is required for the formation of NEIL1’s repair competent complex in the chromatin. Based on the studies of intracellular distribution of wild-type (WT) and acetylation defective NEIL1 mutants, we postulate that acetylation is required neither for NEIL1’s chromatin binding, nor its enzymatic activity, but is required for constitutive binding to histones (along with other chromatin factors), and BER/single-strand break repair (SSBR) partner proteins to form active repair complexes in chromatin, thus regulating oxidized base repair.
2. Materials and Methods
2.1. In vitro acetylation of NEIL1
Recombinant full-length WT, K296R/K297R/K298R (3KR) mutant, truncated N1-349, N1-288 NEIL1 polypeptides were purified from E.coli as described previously [27, 29, 30, 32, 33]. Five μg of the proteins were individually incubated with 0.2 μg recombinant human p300 HAT domain in the presence of 1 mM acetyl CoA (AcCoA; Sigma) or 1 μCi of [3H] AcCoA (200 mCi/mM; NEN) in 50 μl HAT buffer (50 mM Tris-HCl pH 8.0, 0.1 mM EDTA, 10% glycerol, 1 mM DTT, 10 mM sodium butyrate) at 30°C for 2 h. Acetylation was confirmed either by SDS-polyacrylamide gel electrophoresis (PAGE) followed by Western analysis with antibodies specific for NEIL1 or acetylated NEIL1 (AcNEIL1), or fluorography for 3H incorporation with enhancing solution (Amplify; Amersham).
2.2. Identification of acetyl-accepting Lys residues in NEIL1
A peptide corresponding to aa residues 287-305 (LAPKGRKSRKKKSKATQLS) in NEIL1 was chemically synthesized and HPLC-purified at the University of Texas Medical Branch (UTMB) Protein Chemistry core facility, and then incubated with 0.2 μg p300 HAT and 1 mM AcCoA at 30°C for 2 h. After desalting, the peptide was analyzed by MALDI-TOF (Applied Biosystems) and N-terminal Sequencing (Edman degradation) at UTMB’s Biomolecular Resource Facility.
2.3. Analyses of AcNEIL1 activity and kinetic parameters
NEIL1’s DG activity was analyzed using a 5′-32P-labeled 5-hydroxyuracil (5-OHU)-containing duplex oligonucleotide substrate, as previously described [28–30, 32, 33]. Briefly, the 5-OHU-containing strand was 5′ end labeled with [γ-32P] ATP using T4 polynucleotide kinase, and annealed to the complementary strand with a G opposite 5-OHU lesion. The duplex oligonucleotide was purified on a gel filtration column (Chroma Spin TE 10, Clontech). For measuring NEIL1’s DG activity, the labeled oligonucleotide was incubated with 10 or 20 nM unmodified NEIL1 or AcNEIL1 for 10 min at 37°C in a 10 μl reaction mixture that contained 50 mM Tris-HCl pH 8.5, 50 mM KCl, 1 mM MgCl2, 1 mM DTT, 0.1 mM EDTA, and 100 μg/ml bovine serum albumin. The reaction was stopped with 10 μl of 80% formamide + 40 mM NaOH containing 0.05% xylene cyanol dye, followed by heating at 95°C and ran in 20% polyacrylamide denaturing gel, and imaged in a PhosphorImager. The kinetic parameters Km and kcat were calculated from the enzyme activity, after incubating 20 nM unmodified NEIL1 or AcNEIL1 with 20 fM of labeled substrate together with increasing concentration (1–160 nM) of unlabeled substrate in a steady state reaction for 5 min at 37°C. Km, Vmax, and kcat were calculated from the linear range of the reaction by regression analysis using Michaelis-Menten equation and Line weaver-Burk plot [30, 33].
2.4. Cell lines, plasmids, siRNAs, transfection and treatments
The human embryonic kidney epithelial HEK293 (ATCC # CRL-1573) cell line was maintained in DMEM-high glucose medium (Hyclone) containing 10% fetal calf serum (FCS; Sigma) and antibiotic mixture of 100 IU penicillin and 100 μg/ml streptomycin (Corning) at 37°C under 5% CO2 and 95% relative humidity. The human colorectal adenocarcinoma HCT116 (ATCC # CCL-247) and HT29 (ATCC # HTB-38) cell lines were grown in MCoy’s 5A medium (Gibco/Life Technologies), supplemented with FCS and antibiotic mixture. PCMV5.1 recombinant plasmid encoding FLAG-tagged human WT [28, 29, 33], K296A/K297A/K298A (3KA) mutant [34], 3KR mutant (generated using Stratagene’s Site Directed Mutagenesis Kit following manufacturer’s protocol), truncated N1-311 mutant [32] NEIL1 were used for ectopic expression of NEIL1 by Lipofectamine 2000 mediated transfection of exponentially growing cells following manufacturer’s protocol (plasmid DNA: 1 μg/10 cm plate; Lipofectamine 2000: 3 μl/μg plasmid DNA). Downregulation of endogenous NEIL1, p300, CBP, PCAF, or HDAC1 was achieved by transiently transfecting cells with specific siRNAs by Lipofectamine RNAimax following manufacturer’s protocol (siRNA: 80 nM; Lipofectamine RNAimax: 10 μl/transfection in 5ml medium). NEIL1 siRNA [29, 33], targeting 3′UTR of the NEIL1 gene was obtained from Sigma; p300 (# L-003486-00) and HDAC1 (# L-003493-00) siRNAs were obtained from Dharmacon; CBP siRNA (# sc-29952) was obtained from Santa Cruz Biotechnology; PCAF siRNA (# 4390824) was obtained from Ambion; universal negative control siRNA (# SIC001) was obtained from Sigma. Cells were harvested 48 h post transfection to check the expression of ectopic proteins or downregulated endogenous proteins by Western analysis.
Oxidative stress in cells was induced by glucose oxidase (GOx; 2–100 ng/ml) treatment for 1 h followed by washing in phosphate buffered saline (PBS), incubating in fresh medium and harvesting immediately or after various times according to the experiments. HDAC inhibition was achieved by treating cells with TSA (100 ng/ml; Calbiochem) or NAM (2 mM; Sigma).
2.5. Antibodies (α)
α-H3 (# 4499; 1:1000), α-FLAG (# 2368; 1:1000), and α-ASF1A (# 2990; 1:500) were purchased from Cell Signaling; α-HDAC1 (# 05-100-1; 1:500) was obtained from Millipore; α-p300 (# sc-584; 1:250) was obtained Santa Cruz Biotechnology; α-CBP (# GTX101249; 1:500) and α-PCAF (# GTX109666; 1:500) were obtained from GeneTex; α-CAF-1 p48 (# ab47456; 1:500), α-CHAF1B (# ab72520; 1:500), α-CHAF1A (# ab126625; 1:1000) and α-XRCC1 (# 134056; 1:1000) were obtained from Abcam; α-DNA Pol β (# 18003-1-AP; 1:1000), α-βactin (# 66009-1-Ig; 1:3000), α-GAPDH (# 60004-1-Ig; 1:3000) were obtained from Proteintech. α-PNKP [35] (1:500) was a kind gift from Dr. Michael Weinfeld (Cross Cancer Institute, University of Alberta, Canada) and α-NEIL1 [27] (1:500) was custom-generated. α-AcNEIL1 (1:500) was custom generated through EZBiolab using a chemically synthesized NEIL1 peptide (288APKGRKSRK*K*K*SKA301) containing acetylated Lys (*) as a hapten to elicit IgG antibody in rabbit and purified the IgG from the sera. All antibodies were diluted in 5% non-fat dry milk in tris-buffered saline (TBS) containing 0.05% Tween20.
2.6. Subcellular fractionation
Cell fractionation for isolating soluble nuclear and chromatin extracts was performed following a published protocol [36]. Briefly, eighty per cent confluent cells grown in 10 cm plates were washed twice in PBS and then lysed in cytoplasmic lysis buffer (10 mM Tris-HCl pH 7.9, 0.34 M sucrose, 3 mM CaCl2, 2 mM MgCl2, 0.1 mM EDTA, 1 mM DTT, 0.1% NP-40, and protease inhibitor mixture (Roche Applied Science); 500 μl lysis buffer per 10 cm plate). After pelleting the nuclei by centrifugation at 3500 g for 15 min at 4°C, they were lysed in nuclear lysis buffer (20 mM HEPES pH 7.9, 1.5 mM MgCl2, 3 mM EDTA, 150 mM K-acetate, 10% glycerol, 0.5% NP-40, and protease inhibitor mixture; 100 μl lysis buffer per 10 cm plate), vortexed for 15 min at 4°C, followed by centrifugation at 14000 rpm for 30 min at 4°C to pellet the chromatin. The supernatant was labeled as the soluble nuclear extract. The chromatin pellet was dissolved in chromatin lysis buffer (150 mM HEPES pH 7.9, 1.5 mM MgCl2, 150 mM K-acetate, 10% glycerol, and protease inhibitor mixture; 100 μl lysis buffer per 10 cm plate), and incubated with 0.15 unit/μl of Benzonase (Novagen) at 37°C for 30 min, followed by centrifugation at 14000 rpm for 30 min at 4°C. The supernatant was called the chromatin extract. Cell extract was isolated by harvesting cells in cell lysis buffer (50 mM Tris-HCl pH 7.5, 150 mM NaCl, 1 mM EDTA, 1% Triton X-100 and protease inhibitor mixture; 700 μl lysis buffer per 10 cm plate), followed by vortexing for 15 min at 4°C, and centrifugation at 14000 rpm for 15 min at 4°C. The supernatant is the cell extract. To isolate whole nuclear extract containing chromatin fraction, the nuclear pellet after removing the cytoplasmic fraction was lysed in cell lysis buffer (500 μl lysis buffer per 10 cm plate) and followed accordingly.
2.7. Co-Immunoprecipitation (Co-IP) analysis
For Co-IP analysis, total nuclear extracts containing the chromatin fractions isolated from HEK293 cells expressing FLAG-tagged WT NEIL1 or the 3KR mutant were immunoprecipitated for 3 h at 4°C with α-FLAG M2 conjugated agarose beads (Sigma; # A2220), as described earlier [17]. Approximately, 50 μl of TBS-washed beads were incubated with whole nuclear extracts containing 1 mg total proteins in a total volume of 1 ml with constant rotation. The beads were washed 3–5 times with 1 ml of cold TBS containing 1% Triton X-100, eluted in 40 μl Laemmli buffer. The eluate was fractionated by SDS-PAGE for Western analysis with the appropriate antibodies.
2.8. In vitro pulldown assay
For His-affinity pulldown assay, His-tagged CHAF1A (1 μg) was bound to 25 μl suspension of Ni-NTA magnetic agarose beads (Qiagen; # 36111), and then mixed with 0.5 μg of either non-tagged WT or 3KR mutant NEIL1 proteins in 0.5 ml TBS, and incubated for 1 h with constant rotation at 4°C. After washing the beads 5 times with cold 0.5 ml TBS containing 0.5% Tween20, the bound proteins were eluted with Laemmli buffer and fractionated by SDS PAGE followed by Western analysis.
2.9. NEIL1 activity and BER assay using NEIL1 IP
FLAG IP from FLAG-tagged WT or 3KR mutant NEIL1-expressing cells was eluted using FLAG peptide (Sigma). The eluates were normalized for the FLAG (NEIL1) level by Western analysis before the assay. The DG activity was measured as described before (section 2.3). NEIL1 initiated BER assay in the FLAG IP eluates using a 5-OHU-containing labeled duplex oligonucleotide substrate (containing a labeled 21-mer control non-cleavable duplex oligonucleotide) was performed as described previously [28, 29, 33, 34]. The IP eluates were incubated in 20 μl of the repair reaction mixture containing 1 mM ATP, 2 μCi [α-32P] dCTP and 25 μM unlabeled dCTP for 30 min at 37°C. The products were then analyzed by electrophoresis in 20% polyacrylamide denaturing gel.
2.10. Clonogenic cell survival assay
Endogenous NEIL1 was downregulated in HEK293 or HT29 cells using NEIL1’s 3′UTR specific siRNA [29, 33], followed by ectopic expression of WT, 3KA or 3KR mutant NEIL1. The siRNA did not affect WT, 3KA or 3KR mutant NEIL1 proteins expressed from PCMV5.1 constructs. At 48 h after transfection, 400 cells were plated on 35 mm dishes and treated with various doses of GOx for 1 h, then washed with PBS and allowed to grow in fresh medium for 10–14 days until visible colonies appear. The colonies were stained with crystal violet, counted and plotted as surviving fraction with respect to untreated cells.
3. Results
3.1. Acetylation of human NEIL1 by p300 and identification of acetyl-accepting Lys residues
Following up on our earlier characterization of acetylation of human BER enzymes APE1 [13], NEIL2 [16] and OGG1 [17], we tested whether NEIL1 could also be acetylated by the HAT domain of p300. We observed incorporation of 3H acetyl group in full-length recombinant NEIL1 after incubation with 3H AcCoA and p300 HAT (Fig. 1A). We had shown earlier that the intrinsically disordered C-terminal domain (CTD) of NEIL1 is involved in its interaction with partner proteins [28, 31, 32]. This situation is analogous to that of APE1 where its N-terminal disordered region containing the acetylation sites is essential for its interaction with partner proteins [24, 37–40]. Hence, we expected that the Lys residues in NEIL1’s CTD are the acetylation targets. We compared acetylation of the full-length (389 aa residues) NEIL1, C-terminal truncated mutants (1-349 aa residues and 1-288 aa residues) by 3H AcCoA, and found 3H incorporation in WT, as well as in the 1-349, but not in the 1-288 truncated mutant (Fig. 1A). It was thus evident that the acetyl-accepting Lys residues are predominantly localized in the region containing 289-349 aa residues. From mass spectrometric analysis of a synthetic peptide corresponding to 287-305 aa residues in NEIL1, followed by N-terminal sequencing (data not shown), we observed that major acetylation sites were at Lys residues 296, 297 and 298 (Fig. 1B). We thus concluded that NEIL1 is predominantly tri-acetylated.
Fig. 1. Acetylation of human NEIL1 by p300 and identification of acetyl-accepting residues.
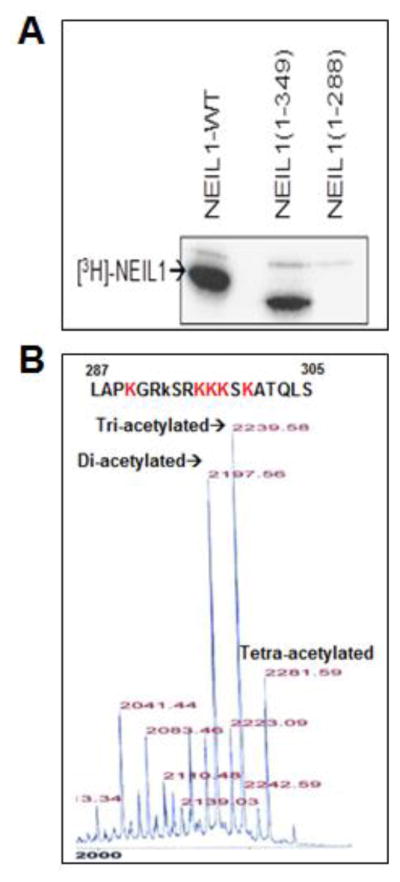
(A) Recombinant full-length WT, truncated N1-349 or N1-288 NEIL1 were acetylated in vitro with p300 HAT and 1 μCi of [3H] AcCoA (200 mCi/mM) in HAT buffer (details in Materials and Methods), followed by SDS-PAGE and fluorography.
(B) Mass spectrometric analysis of a synthetic NEIL1 peptide (aa 287-305) after in vitro acetylation with p300 HAT and AcCoA, which identified tri-acetylated NEIL1 as the predominant acetylated form with K296, K297 and K298 as the major acetyl-accepting residues.
3.2. NEIL1 acetylation enhances its DG activity by increasing enzymatic turnover
We tested the effect of acetylation on NEIL1’s DG activity in vitro. Recombinant NEIL1 was in vitro acetylated by incubating with p300 HAT domain in the presence or absence of AcCoA and analyzed the DG activity using a 5-OHU-containing duplex oligonucleotide substrate (Fig. 2A). Acetylation was confirmed by Western analysis using α-AcNEIL1 as shown in Fig. 3A. Acetylation enhanced NEIL1’s DG activity (Fig. 2A). To further understand the kinetic mechanism of acetylation mediated enhancement of activity, we analyzed the effect of acetylation on NEIL1’s substrate affinity (Km) as well as catalytic turnover (kcat) using a steady state reaction as described in the Materials and Methods. Acetylation increased kcat by ~5-fold, while there was moderate decrease in the Km (Fig. 2B and C). Thus ~8-fold higher kcat/Km ratio for AcNEIL1 relative to the non-acetylated form indicates that acetylation increases NEIL1’s enzymatic activity, mostly due to reduced product affinity.
Fig. 2. Acetylation enhances NEIL1’s DG activity.

(A) Incision of the 5-OHU-containing 51-mer oligonucleotide (top) by NEIL1 (lane 2, 10 nM and lane 4, 20 nM) and in vitro acetylated NEIL1 (AcNEIL1; lane 3, 10 nM and lane 5, 20 nM). Histogram (bottom) represents quantitation of activity. S: substrate, P: product.
(B) Analysis of kinetic parameters of NEIL1 vs. AcNEIL1. Increasing amounts of unlabeled plus constant amount (20 fM) of radio-labeled 5-OHU-containing substrate was incubated with 20 nM of NEIL1 or AcNEIL1 for 5 min at 37°C and the products were analyzed by denaturing gels. The Km, Vmax and kcat values were calculated from the linear range of the reaction by regression analysis using Lineweaver-Burk equation.
(C) Tabulation of kinetic parameters (average ± SE). Other details are provided in Materials and Methods.
Fig. 3. Characterization of α-AcNEIL1 and detection of AcNEIL1 in cells.

(A) 25-200 ng of unmodified (−Ac) or in vitro acetylated (+Ac) recombinant (Rec) WT NEIL1 was subjected to SDS PAGE for Western analysis with α-AcNEIL1 and α-NEIL1.
(B) Western analysis of cell extracts from control vs. siRNA-mediated NEIL1 downregulated HT29 cells for AcNEIL1, total NEIL1 and GAPDH levels.
(C and D) Western analysis of (C) soluble nuclear extracts and chromatin extracts of HEK293, and (D) chromatin extracts of HCT116 cells after GOx treatment (25 ng/ml) for the indicated times for AcNEIL1, total NEIL1 and H3 levels; quantitation of AcNEIL1 level (ImageJ software) shown in the associated histograms.
3.3. NEIL1 acetylation is detectable only in the chromatin fraction and oxidative stress moderately increases its level
We used a custom generated α-AcNEIL1 (described in Materials and Methods). This antibody is highly specific for AcNEIL1 (≈ 200-fold preference for AcNEIL1 relative to non-acetylated NEIL1), and could detect as little as 25 ng AcNEIL1 in Western analysis (Fig. 3A). The specificity of this antibody was further confirmed in endogenous NEIL1 downregulated cells, which hardly detected any signal compared to the control cells (Fig. 3B). We used this antibody to characterize the parameters that regulate acetylation of NEIL1 at Lys 296-298 in cells, although it could not detect acetylation at other sites in NEIL1.
In our earlier studies on acetylation of OGG1 and APE1, we observed oxidative stress dependent enhancement of acetylation of these enzymes [17, 24]. In the present study, AcNEIL1 could be detected appreciably even in unstressed cells (Fig. 3C and D). However, oxidative stress induced by GOx treatment that generates reactive oxygen species (ROS), transiently led to modest increase in AcNEIL1 level (Fig. 3C and 3D). Moreover, AcNEIL1 could only be detected in chromatin and not in the soluble nuclear extract (Fig. 3C).
3.4. NEIL1 acetylation and deacetylation in cells are mediated by p300/CBP/PCAF and HDACs, respectively
We showed earlier that acetylation of both APE1 and OGG1 is carried out by p300 [13, 17], while HDAC1 and NAD+-dependent SIRT1 catalyze APE1’s deacetylation [18, 19]. We confirmed involvement of p300 in acetylation of NEIL1 via siRNA mediated downregulation of endogenous p300 and analyzing the level of AcNEIL1 in the downregulated cells (Fig. 4A). This result strongly suggests that p300, and probably also CBP belonging to the same HAT family [6, 41, 42], are primarily responsible for acetylating NEIL1. However, our result showing modest reduction in AcNEIL1 level after downregulation of endogenous CBP (Fig. 4B), indicates that CBP is not a major contributor to NEIL1 acetylation. We also tested another HAT family member namely, PCAF (p300/CBP associated factor) [43, 44] for its contribution to acetylating NEIL1. Downregulation of PCAF also reduced AcNEIL1 level (Fig. 4C). The increased level of AcNEIL1 in cells (Fig. 4D and E) observed after treatment with trichostatin A (TSA), inhibitor of Class I and II HDACs [45], and nicotinamide (NAM), inhibitor of class III HDACs [46], indicate that multiple HDACs deacetylate AcNEIL1; class III HDACs may have a marginal effect. However, HDAC1 is not involved in deacetylating AcNEIL1 (Fig. 4F).
Fig. 4. Regulation of AcNEIL1 level in cells.

(A–C and F) Endogenous (A) p300, (B) CBP, (C) PCAF, or (F) HDAC1 were downregulated in HCT116 cells by specific siRNAs, and whole nuclear extracts were subjected to SDS PAGE followed by Western analysis with the indicated antibodies.
(D and E) Western analysis of GOx (25 ng/ml; 1–3 h), TSA (100 ng/ml; 1–6 h) or NAM (2 mM; 4 h) treated cells’ whole nuclear extracts with the indicated antibodies.
3.5. Acetylable Lys residues in NEIL1 are required for interaction with chromatin and BER factors
We used site-directed mutagenesis to generate two types of acetylation-negative NEIL1 mutants in PCMV5.1 mammalian vectors expressing FLAG-tagged WT NEIL1. In one mutant (NEIL1 3KA), Lys 296-298 were substituted with Ala resulting in the loss of positive charges present in WT enzyme. In the second mutant (NEIL1 3KR), these residues were substituted with Arg. The 3KR mutant retains the positive charge cluster but is non-acetylable, and did bind to the chromatin, like the WT enzyme (Fig. 5A). Interestingly, the 3KA mutant was undetectable either in the soluble nuclear, or chromatin extracts (Fig. 5B). It appears likely that the 3KA mutant lost NEIL1’s nuclear localization sequence (NLS) which possibly overlaps with the acetylation sites, and could not translocate to the nucleus. This is consistent with our earlier finding that the N1-311 NEIL1 mutant, while retaining the acetylable Lys residues could not bind to the chromatin, in spite of retaining the putative NLS [32].
Fig. 5. Chromatin association of WT vs. non-acetylable mutant NEIL1 and analysis of their IP complexes.

(A and B) Western analysis of different extracts of HEK293 cells expressing FLAG-tagged WT vs. (A) 3KR, or (B) 3KA, N1-311 mutant NEIL1 for FLAG (NEIL1) level.
(C) FLAG Co-IP analysis on the whole nuclear extracts containing chromatin fractions of FLAG-tagged WT vs. 3KR mutant NEIL1 expressing cells for different chromatin and BER/SSBR proteins.
While the 3KR mutant binds to the chromatin like the WT enzyme, Co-IP analysis showed that the immunoprecipitation (IP) complex of the mutant contained significantly lower levels of chromatin factors (histone H3, histone chaperones CHAF1A, CHAF1B and ASF1A), compared to that of the WT protein (Fig. 5C). The levels of BER/SSBR proteins, except for DNA Pol β, were also significantly lower in the IP of 3KR mutant relative to that of the WT enzyme (Fig. 5C).
Because, the 3KA mutant was unable to translocate to the nucleus where all DNA transactions occur, we excluded it from the interaction and repair activity studies.
3.6. Acetylation is dispensable for NEIL1’s DG activity but is required for NEIL1-initiated repair of oxidized bases
To examine the role of NEIL1’s acetylable Lys residues in the repair activity of its complexes, we first analyzed the DG activity of FLAG-tagged WT vs. 3KR mutant enzyme present in the FLAG IP complexes. We used a 32P labeled duplex oligonucleotide substrate containing 5-OHU [29, 32] and the same amount of WT vs. mutant enzymes in the assay by normalizing the FLAG immunoblotting signal, for comparing their enzymatic activity. Surprisingly, the WT NEIL1 IP exhibited significantly lower activity than the 3KR mutant (Fig. 6A and B). This is probably because the 3KR mutant IP had lesser amount of the inhibitory histone chaperone (CHAF1A) than the WT IP (Fig. 5C). We showed recently that CHAF1A sequesters and inhibits the BER initiating DGs including NEIL1 and OGG1 through direct interaction [34]. To examine the effect of purified CHAF1A on the DG activity of WT vs. 3KR mutant proteins, we first showed by His-tag pulldown assay, that unlike WT NEIL1, the 3KR mutant did not bind to His-tagged CHAF1A (Fig. 6C). The DG activity of purified WT enzyme was higher than that of the 3KR mutant (Fig. 6D; lanes 3 vs. 5). However, the DG activity of WT, but not of the 3KR mutant was inhibited by the added CHAF1A (Fig. 6D; lanes 2 and 3 vs. 4 and 5), probably due to the absence of direct interaction between CHAF1A and the 3KR mutant (Fig. 6C). We subsequently analyzed complete repair of 5-OHU present in a linear duplex oligonucleotide [29, 34] using these FLAG IP complexes. Fig. 6E and F show that the 3KR NEIL1 repair complex was much less active in complete repair than the repair complex of acetylable WT enzyme. This is presumably due to weaker association of the mutant with the downstream BER partner proteins (Fig. 5C).
Fig. 6. Estimation of DG activity and complete BER in WT vs. non-acetylable mutant NEIL1 IP complexes.

(A and B) DG activity assay in the eluates of FLAG IP from FLAG-tagged WT vs. 3KR mutant NEIL1 expressing HEK293 cells; level of FLAG (NEIL1) shown in A. A 32P-labeled 5-OHU lesion-containing oligonucleotide substrate as shown in B was used with the IP eluates containing equivalent amount of FLAG (NEIL1) in the assay. Bottom panel shows relative quantification of activity measured as the fraction of the substrate remaining.
(C) His-pull down assay showing interaction of Ni column-bound His-CHAF1A with WT NEIL1, but not with 3KR NEIL1 proteins. Western analysis of WT and 3KR mutant NEIL1 proteins shown in the side panel.
(D) DG activity assay with purified WT vs. 3KR mutant NEIL1 proteins in the presence or absence of added CHAF1A. Side panel shows relative quantification of activity measured as the fraction of the substrate remaining.
(E and F) BER assay in FLAG IP eluates from the same cells as in A and B; FLAG (NEIL1) level shown in E. Schematic representation of the 51-mer substrate oligonucleotide indicating incorporation of radiolabeled [α-32P] dCMP reflecting single nucleotide BER shown. Relative quantification of repair product is shown in the side panel. Details are described in Materials and Methods.
3.7. Acetylation of NEIL1 increases resistance to ROS toxicity
To test if NEIL1 acetylation confers protection of the cell genome from ROS toxicity, we performed clonogenic cell survival assay after treatment of cells with GOx and compared the effect of siRNA mediated downregulation of endogenous NEIL1, and subsequent complementation with ectopic WT or the acetylation defective mutants. The level of endogenous and ectopic NEIL1 in these cells are shown in Fig. 7A and C. The percent surviving fraction of each cell type was calculated relative to the untreated control. GOx induced cell death in a dose dependent manner (Fig. 7B and D). Downregulation of endogenous NEIL1 caused higher level of cell death compared to the control cells (Fig. 7B and D). Ectopic expression of the 3KA mutant (which has weaker nuclear localization) showed negligible protective effect unlike the ectopic WT NEIL1 from ROS-induced killing of the NEIL1 depleted cells (Fig. 7B). In contrast, the 3KR mutant (that translocates to the nucleus and remains bound to chromatin like the WT protein, but is not acetylated and is deficient in forming stable repair complexes) could partially rescue the effect of NEIL1 deficiency (Fig. 7D). Thus, cells expressing WT NEIL1 with acetylable Lys residues are best protected from induced oxidative stress.
Fig. 7. Sensitivity of WT vs. non-acetylable mutant NEIL1-expressing cells to ROS.

(A and C) Western analysis of HEK293 and HT29 cell extracts for ectopic expression of (A) WT and 3KA, and (C) WT and 3KR mutant NEIL1, respectively after downregulation of endogenous NEIL1.
(B and D) Clonogenic cell survival assay was performed in these cells after treating with various doses of glucose oxidase. Details are described in Materials and Methods. * denotes p value < 0.05 (unpaired T Test; online Graphpad software).
4. Discussion
This report documents characterization of NEIL1 acetylation, and its impact on repair of oxidized bases in human cells. We showed earlier that NEIL1 initiated BER is regulated by NEIL1’s interaction with the downstream repair proteins [28, 32, 47], as well as the recently identified chromatin factors [34]. We discovered that while the unmodified NEIL1 can be present in the nucleoplasm, AcNEIL1 is detected only in the chromatin. The interesting observation that the Lys>Ala (3KA) mutant loses ability for nuclear translocation, suggests a critical role of these acetylable, positively charged Lys residues within the putative NLS. The NLS in NEIL1 has not been characterized. A recent study reported that the NEIL1 variant Q282Stop does not translocate to the nucleus, which the authors attributed to the absence of a putative NLS encompassing aa 359-378 in this variant [48]. Our recent study showing that nuclear localization of the truncated N1-311 NEIL1 mutant was comparable to that of the WT enzyme [32], suggests retention of the NLS in the truncated protein, presumably localized in the positive charge patch containing the acetylable Lys residues. This would be consistent with the essentiality of these Lys residues for nuclear translocation which subsequently undergo acetylation. The Lys>Arg (3KR) mutant, retaining the positive charge localizes to the nucleus and binds to the chromatin but cannot be acetylated. It is thus likely that acetylation occurs after NEIL1’s chromatin binding. One possible scenario is that the positive charge cluster is involved in non-specific DNA binding as we had proposed earlier [31]. Once bound to chromatin, it undergoes acetylation. While there may be additional, unidentified acetyl-accepting Lys residues in NEIL1, this study attests to the critical role of Lys 296-298 residues in NEIL1’s nuclear localization and subsequent acetylation after chromatin binding.
We have analyzed by now the effect of acetylation on several oxidized base-specific DGs [16, 17] and on APE1 [13, 19, 21]. Interestingly, the enzymatic activities of NEIL1 (present study), OGG1 and APE1 in vitro are strongly enhanced by acetylation, primarily due to increased product release. In contrast, acetylation of NEIL2 at Lys 49 inhibited its activity, while Lys 153 acetylation had no effect [16]. Similarly, acetylation inhibits TDG’s activity [49]. Taken together, these results show that acetylation has diverse functions for the BER enzymes, even though these are all engaged in early steps of repair. The unique role of acetylation in the case of NEIL1 is in stabilizing its interaction with the partner proteins in forming repair competent BER complexes, together with the nucleosome remodeling histone chaperones CAF-1 and ASF1A (Fig. 5C), and probably acetylated histones H3 and H4 as well. These chaperones are involved in the loading of H3/H4 in chromatin [50]. While the positive charge cluster at aa 296-298 in NEIL1 may be involved in electrostatic binding to DNA in chromatin which is retained in the 3KR mutant, hydrophobic interaction of NEIL1 after neutralization of the charge via acetylation probably stabilizes its complexes with nucleosome components and other downstream proteins, identified in this study.
We recently showed the inhibitory role of CHAF1A (the largest subunit of CAF-1) on DGs, specifically by affecting NEIL1-initiated single nucleotide BER, where the chaperone temporally dissociates from BER complexes after oxidative stress [34]. In the present study, we showed that the acetylation negative NEIL1 3KR mutant was not inhibited by CHAF1A (Fig. 6D), probably due to the absence of direct interaction (Fig. 6C), unlike the WT enzyme. However, the 3KR IP complex exhibited significantly more DG activity than the WT complex (Fig. 6A and B), most likely due to the reduced level of the inhibitory chromatin components, e.g. CHAF1A, ASF1A and H3 in the 3KR complex (Fig. 5C). On the other hand, NEIL1 initiated complete BER carried out by NEIL1 IP was more proficient with the WT than the mutant DG (Fig. 6E and F), because of deficiency of BER partners in the mutant IP complex. Consequently, cells expressing the WT protein are less sensitive to oxidative stress than the non-acetylable mutants (Fig. 7).
Several studies have shown multiple signal-dependent posttranslational modifications (phosphorylation, acetylation, etc.) of a protein to occur sequentially, with one modification influencing the subsequent ones [51]. Recently, NEIL1 was reported to be phosphorylated at Ser 61, 207, 306 residues [52]. Further studies on possible cross talks between phosphorylation and acetylation to regulate its repair function is worth pursuing. Analogous to the situation in APE1 and OGG1, the acetyl-accepting Lys residues located in NEIL1’s CTD are dispensable for the DG activity, but are involved in interaction with other repair proteins and chromatin factors. We had extensively characterized NEIL1’s CTD for its intrinsic disordered conformation which acts as a hub for interaction with more than dozen proteins [28, 31, 32, 53]. We postulated that CTD’s inherent lack of secondary structure allows its stable interaction with various partners via inducing conformational changes which could be modulated by acetylation. This possibility warrants in-depth investigation.
5. Conclusion
This study documents how posttranslational modification of an oxidized base-specific DG affects its activity and cellular survival. We have shown here that acetylation of NEIL1 enhances formation of its repair complex in chromatin, thereby increasing the repair efficiency of oxidized bases. This contributes to cellular protection from oxidative stress.
Highlights.
Human NEIL1 is acetylated by p300 at Lys 296-298 residues located in its disordered C-terminal domain.
Although dispensable for NEIL1’s activity, acetylation enhances the DG activity via enhanced product release.
Acetylated NEIL1 (AcNEIL1) forms active repair complexes in chromatin, and cross talks with different chromatin factors including histone chaperones.
Acetylation stabilizes NEIL1 repair complexes in chromatin, thereby protecting cells from oxidative stress.
Acknowledgments
This work was supported by NIH R01 CA158910 (SM), R01 GM105090 (SM), P01 CA92584 (SM), R01 NS088645 (MLH) and R01 CA148941 (KKB).
Footnotes
Conflict of interest statement
The authors declare that there are no conflicts of interest associated with this study.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Portela A, Esteller M. Epigenetic modifications and human disease. Nature biotechnology. 2010;28:1057–1068. doi: 10.1038/nbt.1685. [DOI] [PubMed] [Google Scholar]
- 2.Reik W, Dean W, Walter J. Epigenetic reprogramming in mammalian development. Science. 2001;293:1089–1093. doi: 10.1126/science.1063443. [DOI] [PubMed] [Google Scholar]
- 3.Choudhary C, Kumar C, Gnad F, Nielsen ML, Rehman M, Walther TC, Olsen JV, Mann M. Lysine acetylation targets protein complexes and co-regulates major cellular functions. Science. 2009;325:834–840. doi: 10.1126/science.1175371. [DOI] [PubMed] [Google Scholar]
- 4.Polevoda B, Sherman F. The diversity of acetylated proteins. Genome biology. 2002;3:reviews0006. doi: 10.1186/gb-2002-3-5-reviews0006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Spange S, Wagner T, Heinzel T, Kramer OH. Acetylation of non-histone proteins modulates cellular signalling at multiple levels. The international journal of biochemistry & cell biology. 2009;41:185–198. doi: 10.1016/j.biocel.2008.08.027. [DOI] [PubMed] [Google Scholar]
- 6.Bannister AJ, Kouzarides T. The CBP co-activator is a histone acetyltransferase. Nature. 1996;384:641–643. doi: 10.1038/384641a0. [DOI] [PubMed] [Google Scholar]
- 7.Ogryzko VV, Schiltz RL, Russanova V, Howard BH, Nakatani Y. The transcriptional coactivators p300 and CBP are histone acetyltransferases. Cell. 1996;87:953–959. doi: 10.1016/s0092-8674(00)82001-2. [DOI] [PubMed] [Google Scholar]
- 8.Snowden AW, Perkins ND. Cell cycle regulation of the transcriptional coactivators p300 and CREB binding protein. Biochemical pharmacology. 1998;55:1947–1954. doi: 10.1016/s0006-2952(98)00020-3. [DOI] [PubMed] [Google Scholar]
- 9.Balakrishnan L, Stewart J, Polaczek P, Campbell JL, Bambara RA. Acetylation of Dna2 endonuclease/helicase and flap endonuclease 1 by p300 promotes DNA stability by creating long flap intermediates. J Biol Chem. 2010;285:4398–4404. doi: 10.1074/jbc.M109.086397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hasan S, Hassa PO, Imhof R, Hottiger MO. Transcription coactivator p300 binds PCNA and may have a role in DNA repair synthesis. Nature. 2001;410:387–391. doi: 10.1038/35066610. [DOI] [PubMed] [Google Scholar]
- 11.Hasan S, Stucki M, Hassa PO, Imhof R, Gehrig P, Hunziker P, Hubscher U, Hottiger MO. Regulation of human flap endonuclease-1 activity by acetylation through the transcriptional coactivator p300. Mol Cell. 2001;7:1221–1231. doi: 10.1016/s1097-2765(01)00272-6. [DOI] [PubMed] [Google Scholar]
- 12.Naryzhny SN, Lee H. The post-translational modifications of proliferating cell nuclear antigen: acetylation, not phosphorylation, plays an important role in the regulation of its function. J Biol Chem. 2004;279:20194–20199. doi: 10.1074/jbc.M312850200. [DOI] [PubMed] [Google Scholar]
- 13.Bhakat KK, Izumi T, Yang SH, Hazra TK, Mitra S. Role of acetylated human AP-endonuclease (APE1/Ref-1) in regulation of the parathyroid hormone gene. Embo J. 2003;22:6299–6309. doi: 10.1093/emboj/cdg595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hasan S, El-Andaloussi N, Hardeland U, Hassa PO, Burki C, Imhof R, Schar P, Hottiger MO. Acetylation regulates the DNA end-trimming activity of DNA polymerase beta. Mol Cell. 2002;10:1213–1222. doi: 10.1016/s1097-2765(02)00745-1. [DOI] [PubMed] [Google Scholar]
- 15.Tini M, Benecke A, Um SJ, Torchia J, Evans RM, Chambon P. Association of CBP/p300 acetylase and thymine DNA glycosylase links DNA repair and transcription. Mol Cell. 2002;9:265–277. doi: 10.1016/s1097-2765(02)00453-7. [DOI] [PubMed] [Google Scholar]
- 16.Bhakat KK, Hazra TK, Mitra S. Acetylation of the human DNA glycosylase NEIL2 and inhibition of its activity. Nucleic Acids Res. 2004;32:3033–3039. doi: 10.1093/nar/gkh632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bhakat KK, Mokkapati SK, Boldogh I, Hazra TK, Mitra S. Acetylation of human 8-oxoguanine-DNA glycosylase by p300 and its role in 8-oxoguanine repair in vivo. Mol Cell Biol. 2006;26:1654–1665. doi: 10.1128/MCB.26.5.1654-1665.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yamamori T, DeRicco J, Naqvi A, Hoffman TA, Mattagajasingh I, Kasuno K, Jung SB, Kim CS, Irani K. SIRT1 deacetylates APE1 and regulates cellular base excision repair. Nucleic Acids Res. 2010;38:832–845. doi: 10.1093/nar/gkp1039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sengupta S, Mantha AK, Song H, Roychoudhury S, Nath S, Ray S, Bhakat KK. Elevated level of acetylation of APE1 in tumor cells modulates DNA damage repair. Oncotarget. 2016;7:75197–75209. doi: 10.18632/oncotarget.12113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lirussi L, Antoniali G, Vascotto C, D’Ambrosio C, Poletto M, Romanello M, Marasco D, Leone M, Quadrifoglio F, Bhakat KK, Scaloni A, Tell G. Nucleolar accumulation of APE1 depends on charged lysine residues that undergo acetylation upon genotoxic stress and modulate its BER activity in cells. Mol Biol Cell. 2012;23:4079–4096. doi: 10.1091/mbc.E12-04-0299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Roychoudhury S, Nath S, Song H, Hegde ML, Bellot LJ, Mantha AK, Sengupta S, Ray S, Natarajan A, Bhakat KK. Human Apurinic/Apyrimidinic Endonuclease (APE1) Is Acetylated at DNA Damage Sites in Chromatin, and Acetylation Modulates Its DNA Repair Activity. Mol Cell Biol. 2017;37:16. doi: 10.1128/MCB.00401-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chattopadhyay R, Das S, Maiti AK, Boldogh I, Xie J, Hazra TK, Kohno K, Mitra S, Bhakat KK. Regulatory role of human AP-endonuclease (APE1/Ref-1) in YB-1-mediated activation of the multidrug resistance gene MDR1. Mol Cell Biol. 2008;28:7066–7080. doi: 10.1128/MCB.00244-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fantini D, Vascotto C, Deganuto M, Bivi N, Gustincich S, Marcon G, Quadrifoglio F, Damante G, Bhakat KK, Mitra S, Tell G. APE1/Ref-1 regulates PTEN expression mediated by Egr-1. Free radical research. 2008;42:20–29. doi: 10.1080/10715760701765616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sengupta S, Mantha AK, Mitra S, Bhakat KK. Human AP endonuclease (APE1/Ref-1) and its acetylation regulate YB-1-p300 recruitment and RNA polymerase II loading in the drug-induced activation of multidrug resistance gene MDR1. Oncogene. 2011;30:482–493. doi: 10.1038/onc.2010.435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fantini D, Vascotto C, Marasco D, D’Ambrosio C, Romanello M, Vitagliano L, Pedone C, Poletto M, Cesaratto L, Quadrifoglio F, Scaloni A, Radicella JP, Tell G. Critical lysine residues within the overlooked N-terminal domain of human APE1 regulate its biological functions. Nucleic Acids Res. 2010;38:8239–8256. doi: 10.1093/nar/gkq691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Muftuoglu M, Kusumoto R, Speina E, Beck G, Cheng WH, Bohr VA. Acetylation regulates WRN catalytic activities and affects base excision DNA repair. PLoS One. 2008;3:e1918. doi: 10.1371/journal.pone.0001918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hazra TK, Izumi T, Boldogh I, Imhoff B, Kow YW, Jaruga P, Dizdaroglu M, Mitra S. Identification and characterization of a human DNA glycosylase for repair of modified bases in oxidatively damaged DNA. Proc Natl Acad Sci U S A. 2002;99:3523–3528. doi: 10.1073/pnas.062053799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hegde ML, Hegde PM, Arijit D, Boldogh I, Mitra S. Human DNA Glycosylase NEIL1’s Interactions with Downstream Repair Proteins Is Critical for Efficient Repair of Oxidized DNA Base Damage and Enhanced Cell Survival. Biomolecules. 2012;2:564–578. doi: 10.3390/biom2040564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hegde ML, Hegde PM, Bellot LJ, Mandal SM, Hazra TK, Li GM, Boldogh I, Tomkinson AE, Mitra S. Prereplicative repair of oxidized bases in the human genome is mediated by NEIL1 DNA glycosylase together with replication proteins. Proc Natl Acad Sci U S A. 2013;110:E3090–3099. doi: 10.1073/pnas.1304231110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hegde ML, Theriot CA, Das A, Hegde PM, Guo Z, Gary RK, Hazra TK, Shen B, Mitra S. Physical and functional interaction between human oxidized base-specific DNA glycosylase NEIL1 and flap endonuclease 1. The Journal of biological chemistry. 2008;283:27028–27037. doi: 10.1074/jbc.M802712200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hegde ML, Tsutakawa SE, Hegde PM, Holthauzen LM, Li J, Oezguen N, Hilser VJ, Tainer JA, Mitra S. The disordered C-terminal domain of human DNA glycosylase NEIL1 contributes to its stability via intramolecular interactions. J Mol Biol. 2013;425:2359–2371. doi: 10.1016/j.jmb.2013.03.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hegde PM, Dutta A, Sengupta S, Mitra J, Adhikari S, Tomkinson AE, Li GM, Boldogh I, Hazra TK, Mitra S, Hegde ML. The C-terminal Domain (CTD) of Human DNA Glycosylase NEIL1 Is Required for Forming BERosome Repair Complex with DNA Replication Proteins at the Replicating Genome: DOMINANT NEGATIVE FUNCTION OF THE CTD. J Biol Chem. 2015;290:20919–20933. doi: 10.1074/jbc.M115.642918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hegde ML, Banerjee S, Hegde PM, Bellot LJ, Hazra TK, Boldogh I, Mitra S. Enhancement of NEIL1 protein-initiated oxidized DNA base excision repair by heterogeneous nuclear ribonucleoprotein U (hnRNP-U) via direct interaction. J Biol Chem. 2012;287:34202–34211. doi: 10.1074/jbc.M112.384032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yang C, Sengupta S, Hegde PM, Mitra J, Jiang S, Holey B, Sarker AH, Tsai MS, Hegde ML, Mitra S. Regulation of oxidized base damage repair by chromatin assembly factor 1 subunit A. Nucleic Acids Res. 2017;45:739–748. doi: 10.1093/nar/gkw1024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Fanta M, Zhang H, Bernstein N, Glover M, Karimi-Busheri F, Weinfeld M. Production, characterization, and epitope mapping of monoclonal antibodies against human polydeoxyribonucleotide kinase. Hybridoma. 2001;20:237–242. doi: 10.1089/027245701753179811. [DOI] [PubMed] [Google Scholar]
- 36.Aygun O, Svejstrup J, Liu Y. A RECQ5-RNA polymerase II association identified by targeted proteomic analysis of human chromatin. Proc Natl Acad Sci U S A. 2008;105:8580–8584. doi: 10.1073/pnas.0804424105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bhakat KK, Mantha AK, Mitra S. Transcriptional regulatory functions of mammalian AP-endonuclease (APE1/Ref-1), an essential multifunctional protein. Antioxidants & redox signaling. 2009;11:621–638. doi: 10.1089/ars.2008.2198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ray S, Lee C, Hou T, Bhakat KK, Brasier AR. Regulation of signal transducer and activator of transcription 3 enhanceosome formation by apurinic/apyrimidinic endonuclease 1 in hepatic acute phase response. Molecular endocrinology (Baltimore, Md. 2010;24:391–401. doi: 10.1210/me.2009-0319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sengupta S, Chattopadhyay R, Mantha AK, Mitra S, Bhakat KK. Regulation of mouse-renin gene by apurinic/apyrimidinic-endonuclease 1 (APE1/Ref-1) via recruitment of histone deacetylase 1 corepressor complex. J Hypertens. 2012;30:917–925. doi: 10.1097/HJH.0b013e3283525124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sengupta S, Mitra S, Bhakat KK. Dual regulatory roles of human AP-endonuclease (APE1/Ref-1) in CDKN1A/p21 expression. PloS one. 2013;8:e68467. doi: 10.1371/journal.pone.0068467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chan HM, La Thangue NB. p300/CBP proteins: HATs for transcriptional bridges and scaffolds. J Cell Sci. 2001;114:2363–2373. doi: 10.1242/jcs.114.13.2363. [DOI] [PubMed] [Google Scholar]
- 42.Dancy BM, Cole PA. Protein lysine acetylation by p300/CBP. Chem Rev. 2015;115:2419–2452. doi: 10.1021/cr500452k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Marmorstein R. Structure and function of histone acetyltransferases. Cell Mol Life Sci. 2001;58:693–703. doi: 10.1007/PL00000893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Nagy Z, Tora L. Distinct GCN5/PCAF-containing complexes function as co-activators and are involved in transcription factor and global histone acetylation. Oncogene. 2007;26:5341–5357. doi: 10.1038/sj.onc.1210604. [DOI] [PubMed] [Google Scholar]
- 45.Yoshida M, Kijima M, Akita M, Beppu T. Potent and specific inhibition of mammalian histone deacetylase both in vivo and in vitro by trichostatin A. J Biol Chem. 1990;265:17174–17179. [PubMed] [Google Scholar]
- 46.Porcu M, Chiarugi A. The emerging therapeutic potential of sirtuin-interacting drugs: from cell death to lifespan extension. Trends in pharmacological sciences. 2005;26:94–103. doi: 10.1016/j.tips.2004.12.009. [DOI] [PubMed] [Google Scholar]
- 47.Hegde ML, Hazra TK, Mitra S. Functions of disordered regions in mammalian early base excision repair proteins. Cell Mol Life Sci. 2010;67:3573–3587. doi: 10.1007/s00018-010-0485-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Shinmura K, Kato H, Kawanishi Y, Goto M, Tao H, Inoue Y, Nakamura S, Sugimura H. NEIL1 p.Gln282Stop variant is predominantly localized in the cytoplasm and exhibits reduced activity in suppressing mutations. Gene. 2015;571:33–42. doi: 10.1016/j.gene.2015.06.043. [DOI] [PubMed] [Google Scholar]
- 49.Mohan RD, Litchfield DW, Torchia J, Tini M. Opposing regulatory roles of phosphorylation and acetylation in DNA mispair processing by thymine DNA glycosylase. Nucleic Acids Res. 2010;38:1135–1148. doi: 10.1093/nar/gkp1097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Verreault A, Kaufman PD, Kobayashi R, Stillman B. Nucleosome assembly by a complex of CAF-1 and acetylated histones H3/H4. Cell. 1996;87:95–104. doi: 10.1016/s0092-8674(00)81326-4. [DOI] [PubMed] [Google Scholar]
- 51.Brooks CL, Gu W. Ubiquitination, phosphorylation and acetylation: the molecular basis for p53 regulation. Current opinion in cell biology. 2003;15:164–171. doi: 10.1016/s0955-0674(03)00003-6. [DOI] [PubMed] [Google Scholar]
- 52.Prakash A, Cao VB, Doublie S. Phosphorylation Sites Identified in the NEIL1 DNA Glycosylase Are Potential Targets for the JNK1 Kinase. PLoS One. 2016;11:e0157860. doi: 10.1371/journal.pone.0157860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hegde ML, Izumi T, Mitra S. Oxidized base damage and single-strand break repair in mammalian genomes: role of disordered regions and posttranslational modifications in early enzymes. Prog Mol Biol Transl Sci. 2012;110:123–153. doi: 10.1016/B978-0-12-387665-2.00006-7. [DOI] [PMC free article] [PubMed] [Google Scholar]