

Abstract
The diarylisoxazole molecular scaffold is found in several NSAIDs, especially those with high selectivity for COX-1. Here, we have determined the structural basis for COX-1 binding to two diarylisoxazoles: mofezolac, which is polar and ionizable, and 3-(5-chlorofuran-2-yl)-5-methyl-4-phenylisoxazole (P6) that has very low polarity. X-ray analysis of the crystal structures of COX-1 bound to mofezolac and 3-(5-chlorofuran-2-yl)-5-methyl-4-phenylisoxazole allowed the identification of specific binding determinants within the enzyme active site, relevant to generate structure/activity relationships for diarylisoxazole NSAIDs.
Keywords: Cyclooxygenase-1 inhibition, mofezolac, P6, diarylisoxazole, X-ray crystallography, molecular modelling
Graphical abstract
INTRODUCTION
Prostaglandin endoperoxide H synthase-1 and -2 (PGHS -1 and -2) also known as cyclooxygenases (COXs) - are bifunctional, membrane-bound and heme-containing enzymes that catalyze the conversion of free arachidonic acid (AA) into Prostaglandin H2 (PGH2), which represents the committed step of prostanoid biosynthesis [1–3]. COX isoenzymes are inhibited through the action of non-steroidal anti-inflammatory drugs (NSAIDs), which bind the COX active site, preventing AA binding.
NSAIDs are a broad and heterogeneous group of drugs that belong to different chemical classes. NSAIDs can be classified on the basis of their mechanisms of COX inhibition in three groups: (a) rapid and reversible competitive inhibitors (e.g. ibuprofen and naproxen); (b) rapid, lower affinity, reversible inhibitors followed by time-dependent, higher affinity, slowly reversible binding (e.g. indomethacin and flurbiprofen); (c) rapid, reversible inhibitors followed by irreversible, covalent modification of the enzyme (e.g. acetylation by aspirin) [4]. Another recurring classification of COX inhibitors is based on their relative inhibitory potency for COX isoforms, quantitatively expressed as IC50 and selectivity index (SI = COX-2 IC50 /COX-1 IC50). In this context, COX inhibitors can be divided into five main groups: (a) compounds capable of producing full inhibition of both COX-1 and COX-2 with poor selectivity; (b) compounds capable of producing full inhibition of COX-1 and COX-2 with preference toward COX-2; (c) compounds that strongly inhibit COX-2 with only weak activity against COX-1; (d) compounds that strongly inhibit COX-1 with only weak activity against COX-2; and (e) compounds that are weak inhibitors of both COX-1 and COX-2 [4–5]. Finally, from a chemical point of view, NSAIDs are grouped into three main classes: (a) carboxylic acids (salicylic acid and its esters, acetic acids, propionic acids, fenamic acids), (b) phenazones (pyrazolones, oxicams) and (c) non-acidic compounds [5].
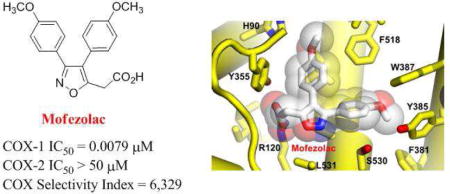
At the core of COX-1 catalytic activity is the ability to trap AA between residues R120/Y355 and the catalytic Y385. Similar to the substrate AA, the majority of NSAIDs containing a carboxylic acid interacts with COXs forming a salt bridge with the guanidinium group of the conserved R120, located at the entrance of the hydrophobic active site channel. This residue orients the aromatic portion of NSAIDs toward the catalytic Y385, located at the top of the COX active site. A tyrosyl radical at Y385, generated upon transfer of an electron to the heme, removes a hydrogen atom from the carbon-13 of AA: the AA-radical intermediate is then converted into prostaglandine G2 (PGG2) and prostaglandine H2 (PGH2) by COX cyclooxygenase- and peroxidase-activities, respectively [1–3]. Aspirin, the only irreversible COXs inhibitor, is recognized by R120 through its benzoic acid moiety and covalently modifies both COX-1 and COX-2 through acetylation of S530. Acetylation renders COXs completely inactive [6], preventing AA access to the catalytic site. Unlike aspirin, other NSAIDs reversibly inhibit COX activity with different selectivity for the two isoforms.
Over the last two decades, limited effort has been devoted to developing selective COX-1 inhibitors [7–9], possibly due to the scarce knowledge of COX-1 biology and its involvement in human diseases [10]. In contrast, many COX-2 inhibitors (COXIBs) have been identified and characterized at the molecular level, and found in most cases to contain a diarylheterocycle bearing either a sulfonamide or a methylsulfamoyl group. The development of COXIBs solved in part the selectivity problem in favor of COX-2 [4–5]. This effort was undertaken to reduce the gastrointestinal (GI) adverse effects associated with the poor COXs selectivity of most traditional NSAIDs, which was mistakenly associated with selective COX-1 inhibition [11–13]. Our discovery of 3-(5-chlorofuran-2-yl)-5-methyl-4-phenylisoxazole (P6) [7] (Fig. 1) as a highly selective COX-1 inhibitor lacking GI toxicity [11] prompted us to attempt in-depth structural modifications of this inhibitor, in order to understand how replacement of chemical moieties affects COX-1 selectivity [5,14–16]. Overall, the selectivity of NSAIDs for COXs remains an open target, mainly due to the similarity between COX isoforms and the complex chemistry of NSAIDs binding to the active site [4,5].
Figure 1.
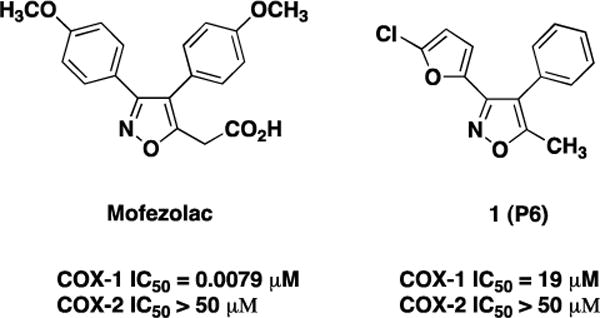
Chemical structures of mofezolac and 1 (P6), and their IC50 for COX-1 and COX-2 obtained using a colorimetric inhibitor screening assay [21].
Crystallographic studies on COX-1 and COX-2 have elucidated the detailed architecture and active site organization of these vital enzymes. COXs contain a long hydrophobic channel that spans ~25 Å from the membrane binding domain to the active site [4,5]. The entry of this channel contains a ‘lobby’ that narrows down into a constriction formed by three crucial residues: R120, Y355 and E524. This constriction opens up to allow substrates or inhibitors to entry into the channel, which is mainly hydrophobic. Crystal structures of COX-1 and COX-2 in complex with AA found that as many as nineteen amino acids in this channel make fifty close contacts with the substrate [1–4], underscoring a remarkably complex chemistry of substrate-binding. As of 2017, twenty-one of the twenty-seven crystal structures of COX-1 deposited in the RCSB database include complexes with NSAIDs. In all these structures, which were determined between 2.0 and ~3.4 Å resolution, the carboxylate moiety of acidic NSAIDs is always found to interact with the guanidinium group of R120. Consistently, binding of AA and NSAIDs containing a carboxylic moiety to COX-1 is greatly perturbed when R120 is mutated to a smaller uncharged residue [17–18]. Although most classical NSAIDs are non-selective inhibitors, in general, they seem to bind more tightly to COX-1 than COX-2, possibly due to the strength of the ionic interaction between the inhibitor carboxylate anion and the guanidinium cation of COX-1 R120 [3]. In this paper, we report an optimized synthetic procedure for mofezolac and 1 (P6), and the identification of their interactions with COX-1 active site through X-ray crystallographic analysis and molecular modeling. This work paves the way to decipher the structural basis for selective inhibition of COX-1 by diarylisoxazole-moiety containing compounds.
RESULTS AND DISCUSSION
Two diarylisoxazoles were selected for this study (Fig. 1): mofezolac, a polar and ionizable NSAID, and 1 (P6) that has very low overall polarity. In mofezolac, the isoxazole group is linked to two mildly polar 4-methoxyphenyl groups at position C3 and C4 and to a highly polar acetic moiety at C5.
In contrast, 1 (P6) isoxazole is linked to a moderately polar 5-chlorofuranyl group at position C3 and two apolar groups, a phenyl at C4 and a methyl at C5. Mofezolac is clinically used as an analgesic drug in Japan, and preferentially inhibits COX-1 [19,20]. It functions like a time-dependent/slowly reversible inhibitor, similar to indomethacin [8] and its IC50 values are 0.0079 and >50 μM for COX-1 and COX-2, respectively [21]. In contrast, 1 (P6) is a weaker, time-independent/competitive reversible inhibitor similar to ibuprofen [8], for which we measured IC50 values of 19 and >50 μM for COX-1 and COX-2, respectively [21].
Docking studies of selected diarylisoxazole NSAIDs with COX-1 revealed several equally plausible binding poses [5,10]. This ambiguity, which is a direct consequence of the large number of bonding groups in the COX-1 channel, hinders optimization and rational design of new diarylisoxazole NSAIDs with enhanced therapeutic properties. To gain insights into the chemistry of binding and inhibition of COX-1, we have determined crystal structures of the ovine COX-1 (oCOX-1) in complex with mofezolac and 1 (P6). oCOX-1:mofezolac and oCOX-1:1 (P6) complexes were solved by molecular replacement and refined to an Rwork/free of 19.5/22.8% and 20.0/23.4%, at 2.75 Å and 2.93 Å resolution, respectively (Table S1). Unlike previous structures of oCOX-1 [22–24], the current crystals do not suffer from twinning, allowing for an accurate structural analysis of mofezolac and 1 (P6) binding to oCOX-1 even at moderate resolution.
The overall architecture of oCOX-1 in our crystallographic complexes (Fig. 2A and 2B) is similar to previously reported structures of the enzyme [22]. oCOX-1 crystallizes as a homodimer in the asymmetric unit, though the two subunits are thought to function as a heterodimer in vivo [22]. oCOX-1 consists of two ∼72 kDa subunits tightly packed against each other via an extensive binding interface spanning ~2,500 Å2. Each COX-1 protomer contains an epidermal growth factor-like domain, a membrane binding domain (MBD), and a large catalytic core bound to a Fe3+-protoprophyrin IX ring (heme group) that harbors both COX and peroxidase (POX) enzymatic activities. The COX active site lies on the opposite side of the POX active site, which activates the heme necessary for the cyclooxygenase reaction. The two active sites are connected via a ~25 Å long L-shaped hydrophobic channel that originates in the MBD and is partially accessible to NSAIDs. Mofezolac and 1 (P6) were identified in unbiased Fo-Fc OMIT maps at the entry of the COX-1 hydrophobic channel in the proximity of R120 and Y355, where NSAIDs are known to interact with COXs [23,24]. Fig. 2A shows a representative Fo-Fc OMIT electron density map for mofezolac contoured at 4σ(purple) and 2σ (cyan) above background and overlaid to the final refined model of mofezolac that has an overall B-factor of ~70 Å2, comparable to that of oCOX-1 atoms. The orientation of mofezolac inside oCOX-1 active site was unambiguously determined due to the excellent electron density and asymmetric shape of this compound that has a carboxyl group above the diarylisoxazole (Fig. 2A).
Figure 2.
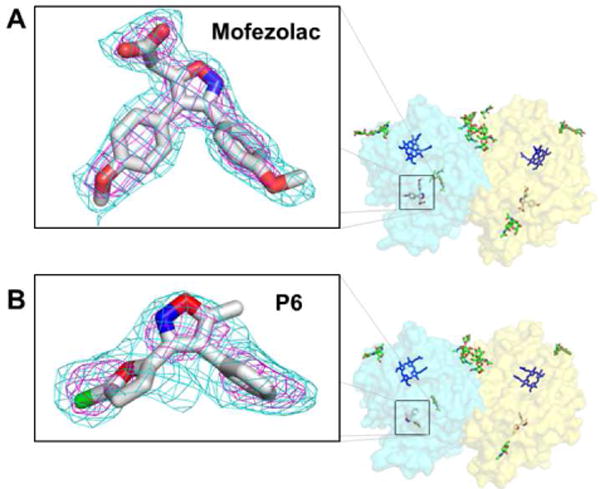
Crystallographic analysis of mofezolac and 1 (P6) bound to oCOX-1. (A, B) Right panel: surface representation oCOX-1 with the two chains colored in cyan and yellow. Fe3+-protoprophyrin IX (blue), carbohydrates moieties and βOG (green) are also shown. Left panel: Fo-Fc polder OMIT map for mofezolac (A) and 1 (P6) (B) contoured at 2σ (cyan) and 4σ (purple) above background. The OMIT maps were calculated using all reflections between 15 - 2.75 Å resolution for mofezolac and 15 - 2.93 Å for 1 (P6) and are overlaid to the final refined atomic models.
In contrast, the electron density for 1 (P6) was less continuous and could be unambiguously identified only after excluding the bulk solvent around the omitted region (Fig. 2B). An Fo-Fc polder OMIT map countered at 2σ above background revealed a ‘V-shaped’ density consistent with the expected quasi-2-fold symmetric structure of 1 (P6) (Fig. 1). At higher contour, the density breaks off into three peaks (colored in purple in Fig. 2B), two of which are connected (right hand side in Fig. 2B) and one that is more spherical (left hand side in Fig. 2B) and visible up to 6σ We assigned the central density to the isoxazole group of 1 (P6), and the globular peak on the left that lacks continuity with the isoxazole group to the more electron-dense chlorine atom of the 5-chlorofuranyl group at position C3. In turn, the density peak continuous to the isoxazole was assigned to the phenyl ring at position C4 that has delocalized π-electrons. No density was observed for the methyl group at position C5 of the isoxazole. The refined B-factor of 1 (P6) (~115 Å2) is much higher than oCOX-1 atoms and mofezolac, underscoring the high isotropic displacement of 1 (P6) atoms inside the active site channel. 1 (P6) atoms move dynamically around the positions defined by the atomic model and thus the electron density in Fig. 2B represents the resultant of different conformations averaged over all COX-1:1 (P6) complexes in the crystallographic lattice.
The crystal structure of COX-1 bound to mofezolac, refined at 2.75 Å resolution, reveals the drug binds the enzyme active site in a planar conformation, with one methoxyphenyl group inserted deep inside the active site channel facing Y385 and the other methoxyphenyl group sandwiched between Y355 and F518 (Fig. 3A). The carboxyl moiety at position 5 of the isoxazole group faces the active site channel entry point, occupied by an n-octyl-β-D-glucoside (βOG) in our structure. Hence, mofezolac makes two sets of interactions with COX-1 residues lining the active site channel. First, the anionic carboxylate makes a salt bridge with the cationic guanidinium group of R120. This salt bridge is the combination of an electrostatic contact between opposite charges (e.g. both mofezolac and guanidinium are charged at the pH of crystallization) and three close-distance (e.g. 2.5-2.8 Å) hydrogen bonds (H-bonds), namely two H-bonds between mofezolac carboxylate and R120 ε- and η-nitrogen atoms and one H-bond with Y355 hydroxyl group (Fig. 3C). Second, mofezolac makes 83 non-bonded, mainly van der Waals and hydrophobic contacts with 17 residues in the COX-1 channel in a distance range between 3.5-4.5 Å (Fig. 3A). Notably, the two methoxyphenyl groups see different chemical environments. The methoxyphenyl at C3 is surrounded by almost exclusively hydrophobic residues (Y385, W387, F381, L384 and G526), including the catalytic Y385, while the methoxyphenyl group at C4 makes van der Waals interactions with more polar residues such as Q192, S353, H90 and Y355, as well as hydrophobic contacts with I523, F518 and L352. Overall, the combination of electrostatic, H-bonds, hydrophobic and van der Waals contacts results in a remarkable surface complementarity that cements mofezolac inside the COX-1 active site channel, explaining its low IC50 (Fig. 1).
Figure 3.
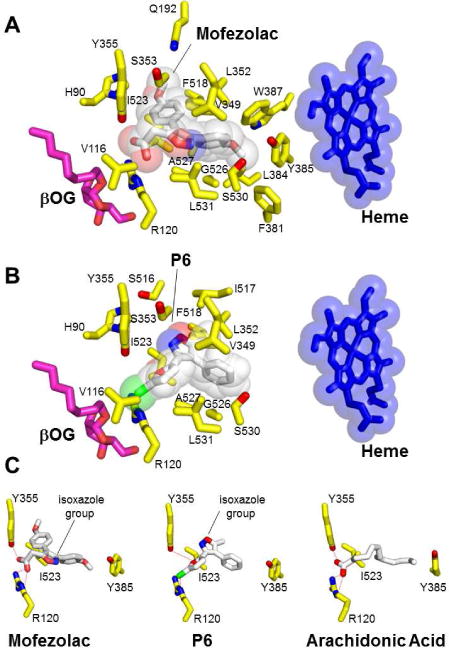
Structural determinants for mofezolac and 1 (P6) binding to oCOX-1 active site. Residues in oCOX-1 active site within 2.5 - 4.5 Å bonding distance for (A) mofezolac and (B) 1 (P6). The semi-transparent spheres around mofezolac, 1 (P6) and heme represent van der Waals radii. An n-octyl-β-D-glucoside (βOG) molecule located at the entrance of the channel is shown in magenta. (C) Comparing the position of mofezolac and 1 (P6) isoxazole group with AA bound to the COX-1 active site.
In the crystal structure of COX-1 bound to 1 (P6), the chlorofuranyl group of 1 (P6) faces down toward the active site channel entry point (Fig. 3B) at a position occupied by the bulkier carboxyl group in the COX-1:mofezolac complex (Fig. 3A). The 1 (P6) chlorine atom is coordinated by Y355 and R120, similar to the free chlorine atom found in the active site of the RNA phosphatase PIR1, which is also coordinated by a Y/R pair [25]. Analysis of the chemical interactions between 1 (P6) and COX-1 in a distance range 2.5 - 4.5 Å reveals that the drug is stabilized by two H-bonds and 56 non-bonded contacts with 9 residues of COX-1 (Fig. 3B). The H-bonds involve the ε-nitrogen of COX-1 R120 and 1 (P6) chlorine atom (2.4 Å distance) and the hydroxyl group of Y355 with the furanyl oxygen atom of 1 (P6) (Fig. 3C). 1 (P6) isoxazole group makes van der Waals and hydrophobic contacts with S353, L352, V349, I517 and I523 (Fig. 3B) and the phenyl ring engages in hydrophobic interactions with I517, L352, V349 and F518. However, COX-1 aromatic residues F381, Y385 and W387, which are important binding determinants for mofezolac, are located more than 5 Å away from 1 (P6), and thus unlikely to significantly contribute to the overall energetics of interaction. The binding free energies (∆G) of mofezolac and 1 (P6) for oCOX-1 calculated from atomic coordinates are −10.2 and −6.9 kcal/mol, respectively (Table S2). A free energy difference of ~3 kcal/mol is in good agreement with the structural data, supporting the notion that mofezolac binds oCOX-1 with much higher affinity than 1 (P6).
Both mofezolac and 1 (P6) are isoxazole-derivatives, but the position of the isoxazole-group differs greatly in the two complexes. 1 (P6)-isoxazole is rotated by 180° as compared to mofezolac, with the phenyl ring occupying a position almost superimposable to the methoxyphenyl group of mofezolac (Fig. 3C). In both drugs, this hydrophobic moiety mimics the aliphatic chain of AA, which, however, is longer and inserts itself deeper inside the COX-1 hydrophobic channel (Fig. 3C). Thus, 1 (P6) weak inhibition of COX-121 as compared to mofezolac (IC50 ~19 vs 0.0079 μM, respectively) can be explained by the lack of an ionic interaction with R120, at the mouth of the COX-1 tunnel, and fewer hydrophobic contacts with COX-1 channel residues facing the heme, especially F381, Y385 and W387. This imperfect complementarity may cause 1 (P6) to ‘wobble’ or even partially rotate inside the COX-1 substrate/inhibitor-binding channel, explaining the high B-factor of 1 (P6) atoms observed in our structure. Despite these differences, mofezolac and 1 (P6) do not make water-mediated contacts with COX-1 residues [26] and in both structures the conformation of Leu531 is consistent with the closed conformation of the enzyme [27].
The IC50 values of mofezolac and 1 (P6) for COX-1 are over two orders of magnitude lower than for COX-2 (mofezolac and 1 (P6) SI is ~ 3 and 6,300, respectively) (Fig. 1). Comparing the structure of human COX-2 [29] with the crystallographic complexes of oCOX-1 described in this paper provides clues to decipher the structural basis for this selectivity. COX-2 has a 20-25% larger and more accessible substrate/inhibitor-binding channel than COX-1 [3], and contains an additional hydrophilic side-pocket in the proximity of F518 (Fig. 4A,B). Access to this pocket is restricted in COX-1 by three aminoacid substitutions: I523 and I434, both replaced by valines in COX-2, and H513 (R513 in COX-2) that fills the pocket. 1 (P6) poor IC50 for COX-2 (>50 μM) can be explained by the larger substrate/inhibitor-binding channel of this isoform and the more hydrophilic nature of the drug-binding site, which provides an energetically unfavorable chemical environment for a poorly polar molecule like 1 (P6) (Fig. 4B). Though more polar and ionizable, mofezolac inserts one of the two methoxyphenyl group toward F518, making a stacking interaction with I523 (Fig. 4A). This contact is likely weaker in COX-2 where I523 is replaced by the smaller and less hydrophobic V523. In summary, mofezolac and 1 (P6) selectivity for COX-1 are a direct consequence of the snugger fit between these diarylisoxazole NSAIDs and the smaller active site channel of this isoform.
Figure 4.
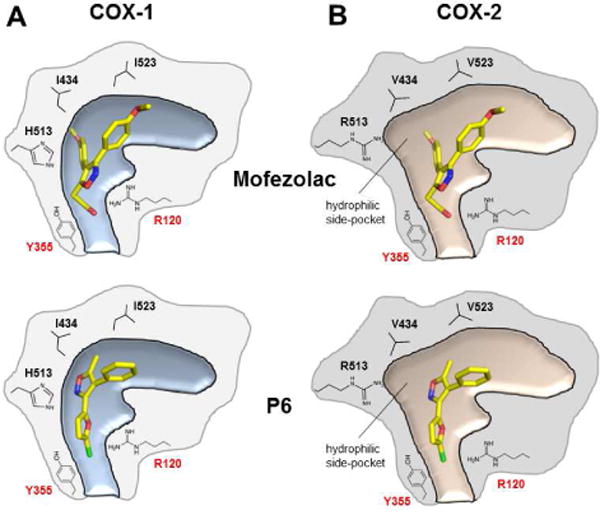
Schematic representation of the structural differences between mofezolac and 1 (P6) bound to the substrate/inhibitor-binding channels of (A) COX-1 and docked inside (B) COX-2. Adapted from reference [28].
To investigate how mofezolac and 1 (P6) affect the overall structure of oCOX-1, we performed molecular modelling studies using the software FLAP (Finger Print for Ligands and Proteins) [30]. We removed all ligands from the atomic coordinates of the oCOX-1:mofezolac and oCOX-1:1 (P6) complexes and calculated all binding cavities in oCOX-1 for each structure (Fig. 5). Interestingly, we identified eleven internal cavities in the structure of oCOX-1 bound to mofezolac and only nine in the complex with 1 (P6). As the two complexes were crystallized under identical conditions, have equal crystal contacts and are crystallographically isomorphous, the difference in internal cavities is presumably caused by bound inhibitors and may reflect the way different NSAIDs affect the enzyme breathing motion. The larger, more polar and potent mofezolac fits tightly inside the oCOX-1 channel, bridging the Y355/R120 pair at the channel entry with the catalytic Y385 (Fig. 4A), thereby reducing the enzyme breathing motion and freezing its internal cavities. On the contrary, 1 (P6) is smaller, lacks contacts with the aromatic residues next to the heme (Fig. 3B) and moves dynamically inside COX-1 substrate channel allowing the enzyme to breathe more dynamically. This is consistent with the lower resolution of oCOX-1:1 (P6) crystals and smaller number of internal cavities identified by FLAP.
Figure 5.
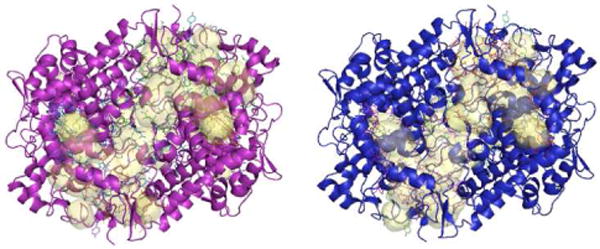
Difference in cavities (yellow) in COX-1 bound to mofezolac (purple) versus 1 (P6) (blue).
CONCLUSION
In recent years, there has been a growing interest in developing selective inhibitors of COX-1. This isoform is proven to play a critical role in the inflammatory processes that lead to many neurodegenerative diseases and cancers, mainly ovarian cancer. In addition, low dose aspirin, a commonly used antithrombotic drug and a selective and irreversible inhibitor of platelet COX-1, is a poor drug that fails to prevent as many as 80% of non-fatal and fatal cardiovascular events [31]. Incomplete suppression of platelet thromboxane A2 (TXA2) biosynthesis by aspirin has also been implicated in aspirin resistance [32]. Thus, the development of novel and potent COX-1 inhibitors is a major focus of modern pharmaceutical research. A number of highly selective COX-1 inhibitors share a diarylisoxazole scaffold [7,8]. This study describes the structural basis for selective inhibition of COX-1 by diarylisoxazoles mofezolac and 1 (P6). Our structural data suggest the low IC50 of mofezolac for COX-1 depends on the snug fit of this drug with the enzyme active site, whereas 1 (P6) lower potency correlates with the smaller size of this NSAID and its wobble inside the active site channel. In turn, the selectivity of both diarylisoxazoles for COX-1 appears to be a direct consequence of the smaller substrate/inhibitor-binding channel of this isoform that has greater van der Waals complementarity than COX-2. We validate the importance of a carboxylic group in mofezolac, or a halogen in 1 (P6), essential to make contacts with Arg120/Try355 at the entry of the active site channel, while the bulkiness and chemical features of substituents linked to the central heterocycle appears to control the avidity for the COX-1 channel [5]. In summary, this work paves the way for the development of novel COX-1 inhibitors with enhanced potency, greater selectivity and reduced toxicity.
EXPERIMENTAL SECTION
Reagents and procedures
AA was purchased from Cayman Chemical Co (Ann Arbor, MI). Hemin (heme) was purchased from Frontier Scientific (Logan, UT). N-octyl β-D-glucopyranoside (β-OG) and C10E6 were purchased from Anatrace (Maumee, OH). BCA protein reagent was purchased from Pierce (Thermo Scientific). EDTA free protease inhibitor was purchased from Roche Applied Science. Nickel-NTA Agarose beads were purchased from Gold Biotechnology. All other chemicals (reagents and solvents) were purchased from Sigma Life Science. Mofezolac was prepared by us following a known procedure [33] with a slight modification. Briefly, desoxyanisoin oxime was prepared by reacting desoxyanisoin (10 g, 39 mmol) and NH2OH•HCl (76 mmol) in methanol/water (60 and 50 mL, respectively). NaOH (0.12 mol) was, then, slowly added. The reaction mixture was stirred for 15 minutes and then heated to 70°C. After 1h, methanol was added to the hot mixture until dissolution was almost complete. The mixture was filtered and the methanol distilled under reduced pressure. The residue was cooled by adding ice-water, filtered and the resulting solid dissolved in ethyl acetate (EtOAc) and treated with brine. The organic layer was dried over anhydrous Na2SO4, filtered and concentrated to obtain the oxime as a yellow solid (9 g, 85% yield). It was used without any further purification to prepare the 3,4-di(4-methoxyphenyl)-5-methylisoxazole direct precursor of mofezolac. Hence, 2.4 N n-BuLi (3.1 mL, 7.4 mmol) was slowly added to a solution of the oxime (1 g, 3.7 mmol) in tetrahydrofuran (THF) (40 mL) kept at −15 °C and under argon atmosphere. The reaction mixture was stirred at 0°C for 30 minutes, and then ethyl acetate (0.15 g, 2 mmol) in THF (15 mL) was added. After 15 minutes, 6 N HCl (100 mL) was added and the reaction mixture refluxed with stirring by an oil-bath for 18h, cooled and the layers separated. The aqueous layer was extracted with EtOAc (3 × 100 mL). The combined organic layers were dried with Na2SO4, filtered and the solvent removed under reduced pressure. Methanol was added to the reaction crude, cooled and the crystalline desoxyanisoin removed by filtration. The filtrate was concentrated and then treated with warm ethanol (5 mL). On cooling in a freezer overnight the 3,4-di(4-methoxyphenyl)-5-methylisoxazole was obtained as a colorless solid. Mp 95-98 °C after recrystallization from ethanol (307 mg, 50% yield). 1H-NMR (300 MHz, CDCl3, δ): 7.37 (d, 2H, J = 9.0 Hz); 7.09 (d, 2H, J = 9.0 Hz); 6.90 (d, 2H, J = 9.0 Hz); 6.83 (d, 2H, J = 9.0 Hz); 3.80 (s, 3H); 3.83 (s, 3H); 2.40 (s, 3H). 3,4-Di(4-methoxyphenyl)isoxazol-5-acetic acid (mofezolac) was prepared by dropwise adding 1.6 N n-BuLi (5 mL) to a stirred cold (dry ice– acetone bath) solution of 3,4-di(4-methoxyphenyl)-5-methylisoxazole (2 g, 7 mmol) in THF (30 mL) under an argon atmosphere. After stirring for 1h at −75 °C, anhydrous gaseous CO2 was flushed into the stirred red colored reaction mixture till the disappearance of the colour. Then, the stirred reaction mixture was allowed to warm to room temperature, concentrated, and the residue dissolved in water. The resulting solution was twice extracted with EtOAc. The organic phase was cooled and acidified with concentrated HCl. The layers were separated and the aqueous phase extracted with ethyl acetate. The combined organic extracts were dried over anhydrous Na2SO4, filtered and the solvent removed under reduced pressure. The obtained sticky foam residue was recrystallized from toluene to give mofezolac as a colorless solid (1.68 g, 71% yield). Mp 142-143 °C. 1H-NMR (300 MHz, CDCl3, δ): 9.55 (bs, 1H: exchanges with D2O); 7.40-7.38 (d, 2H, J = 8.8 Hz, aromatic protons); 7.15-7.13 (d, 2H, J = 8.8 Hz, aromatic protons); 6.93-6.91 (d, 2H, J = 8.8 Hz, aromatic protons); 6.84-6.82 (d, 2H, J = 8.8 Hz, aromatic protons); 3.83 (s, 3H); 3.81 (s, 2H); 3.79 (s, 3H).
3-(5-Chlorofuran-2-yl)-5-methyl-4-phenylisoxazole was synthesized starting from 2-furancarbaldehydoxime, in turn prepared from the reaction of the 2-furancarbaldehyde and NH2OH•HCl in aqueous/EtOH (1:1) in the presence of NaOH. 2-Furancarbaldehydoxime (0.5 g, 4.5 mmol) dissolved in anhydrous dimethyl formamide (DMF) (5 mL), contained in a round-bottom flask equipped with magnetic stirrer, was cooled to 0°C. N-Chlorosuccinimide (NCS) (1.2 g, 9.0 mmol) was slowly added, and the obtained suspension was stirred for 5 h to room temperature. Then, ethyl ether was added and the solution was washed three times with water to remove DMF. The combined organic extracts were dried over anhydrous Na2SO4, and then the solvent was evaporated under vacuum. The residue was dissolved in EtOAc. A pale yellow solid of 5-chloro-2-furancarbohydroximoyl chloride formed (75% yield) by slow addition of petroleum ether (ethyl acetate/petroleum ether = 1:1). The obtained 5-chloro-2-furancarbohydroximoyl chloride (3.87 mmol) was then converted into the nitrile oxide by NEt3 (3.87 mmol), that after NEt3•HCl removal by filtration, was added dropwise to a yellow suspension of NaH (95% w/w, 4.26 mmol) and phenylacetone (0.518 mL, 3.87 mmol) in THF (20 mL) at 0 °C for 1h under nitrogen atmosphere, using a nitrogen-flushed, three-necked flask equipped with a magnetic stirrer, a nitrogen inlet, and two dropping funnels. The reaction mixture was allowed to reach room temperature and stirred overnight. The reaction was quenched by adding aqueous NH4Cl solution. The reaction product was extracted three times with ethyl acetate. The combined organic phases were dried over anhydrous Na2SO4 and then the solvent distilled under vacuum affording 1 (P6) in 60% yield. Mp 71-73 °C (yellow crystals). FT-IR (KBr): 3147, 3051, 2927, 2848, 1633, 1520, 1435, 1412, 1236, 1204, 1134, 1020, 985, 940, 926, 897, 796, 775 cm-1. 1H-NMR (300 MHz, CDCl3, δ): 7.40-7.47 (m, 3H, aromatic protons); 7.25-7.30 (m, 2H, aromatic protons); 6.25-6.27 (d, 1H, J = 3.6 Hz); 6.11-6.12 (d, 1H, J = 3.6 Hz); 2.36 (s, 3H). 13C-NMR (75 MHz, CDCl3, δ): 11.41, 108.14, 113.87, 114.99, 128.58, 129.01, 129.63, 130.20, 138.59, 143.76, 152.42, 167.10. GC-MS (70 eV) m/z (rel int): 261 [M(37Cl)+, 5], 259 [M(35Cl)+, 15], 219 (11), 217 (36), 154 (17), 127 (10), 89 (14), 77 (9), 63 (10), 51 (12), 43 (100). If 3-(5-chlorofuran-2-yl)-5-hydroxy-5-methyl-4-phenyl-2-isoxazoline [direct 1 (P6) precursor] is present in the ethyl acetate extracts (TLC analysis) obtained after quenching the reaction mixture with NH4Cl, it can be separated by column chromatography (silica gel, petroleum ether/ethyl acetate = 15/1) of the reaction crude and converted into 1 (P6) by Na2CO3/methanol under reflux for 2h.
Protein expression and purification
The gene encoding oCOX-1 was cloned in a modified pFastBac vector (Invitrogen) engineered with an N-terminal 8X-his tag and a TEV protease cleavage. Generation of recombinant baculovirus, expression of recombinant his-tagged oCOX-1, and purification of untagged oCOX-1 were carried out as previously described [22]. O2 consumption (see functional assay paragraph) was measured at each step of purification and increased from 136,165 to 210,998 nmol/min: the final purified oCOX-1 had specific activity of 40,000 unit/mg ± 0.49. Purified oCOX-1was concentrated using a Millipore Ultrafree-15 spin concentrator to 5-6 mg/ml (as assessed by BCA protein assay, Pierce, Rockford, IL) in HEPES pH = 7.0, 40 mM NaCl and 0.4% ß-OG and used for crystallization.
Crystallographic methods
oCOX-1 was reconstituted with a 2-fold molar excess of heme (Fe3+-protoprophyrin IX) and 2-fold molar excess of mofezolac [or 1 (P6) ]and allowed to incubate at room temperature for 10 min before setting up crystallization trays. Crystallization trials were set up at 25°C using the sitting-drop vapor diffusion method. 1 μL of protein was mixed with 1 μL of drop solution consisting of 0.5-0.9 M LiCl, 0.7 M sodium citrate pH 6.5, 1 mM sodium azide and 0.3 %(w/v) β-OG and was equilibrated within a reservoir containing 0.6-0.85 M LiCl, 0.7 and 0.85 M sodium citrate pH 6.5 and 1mM sodium azide. Crystals appeared within 2-3 weeks. Prior to data collection, crystals were harvested, briefly soaked in a solution containing 1M sodium citrate, 1M LiCl, 0.15% β-OG, and 1mM sodium malonate as a cryo-protectant and flash-frozen in liquid nitrogen. Diffraction data were collected at beam line 21-ID-F, Life Science-Cat (Argonne National Laboratory, Argonne, IL) on a MARMOSAIC 225 CCD detector and processed using HKL2000 [34]. The structure was solved by molecular replacement (MR) using the program PHASER [35] and oCOX-1 (PDB 3KK6) as a search model. For both crystal structures, the hexagonal asymmetric unit contains a dimer of oCOX-1 that was subjected to iterative cycles of positional and B-factor refinement using distinct TLS groups using phenix.refine [36]. Ligands and water molecules in the structure were identified in Fo-Fc electron density difference maps. Mofezolac and 1 (P6) were identified in Fo-Fc polder maps, as implemented in phenix.polder[37]. Visualization and model building were done using Coot [38]. The final models consist of residues 32-584, Fe3+-protoprophyrin IX, carbohydrates moieties linked to N68, N144 and N410, four βOGs and mofezolac or 1 (P6) bound in the oCOX-1 active site of each monomer and a few water molecules (Table S1). Figures were generated using PYMOL [39].
Functional analysis of oCOX-1
COX activity was monitored by O2 consumption using a Clark-type O2 sensitive electrode (YSI 5300 A, Yelow Springs, Ohio). A typical assay consisting of 100 mM Tris-HCl (pH 8.0) containing 1 mM phenol, 1.5 μL of 1 mM heme, 50 μL of 100 μM AA, 50 μL of protein (3 mL final volume). Mofezolac and 1 (P6) inhibition of COXs was measured using a colorimetric inhibitor screening assay, as described in reference [21].
Computational Methods
The computational tools employed in this work are mainly part of FLAP package [30]. FLAP was employed in structure-based mode using the crystallographic structures of COX-1:mofezolac and COX-1:1 (P6) as templates. Binding free energies were computed from atomic coordinates as described in reference [40].
Supplementary Material
Highlights.
X-ray analysis of the crystal structure of COX-1 bound to mofezolac
X-ray analysis of the crystal structure of COX-1 bound to P6
Binding determinants for highly selective COX-1 inhibition
Selective diarylisoxazole scaffold inhibitors as tools to study COX-1 active site
Acknowledgments
We thank Dr. Sidhu at Cayman Chemical Company for help throughout the project. Coordinates and structure factors have been deposited in the protein data bank (PDB id 5WBE and 5U6X). Authors will release the atomic coordinates and experimental data upon article publication.
Funding Sources
This work was supported by research funds through the University of Bari; MIUR (Rome - Italy) for Progetti di Ricerca Industriale nell’ambito del Programma Operativo Nazionale R&C 2007-2013 e Project “Research, Application, Innovation, Services in Bioimaging (R.A.I.SE.)” code PON01_03054; First AIRC Grant-MFAG2015 (Project Id. 17566). GC was supported in part by a DTSA intramural award. Research in this publication includes work carried out at the SKCC X-ray Crystallography and Molecular Interaction Facility at Thomas Jefferson University, which is supported in part by NCI Cancer Center Support Grant P30 CA56036 and S10 OD017987.
ABBREVIATIONS
- COXIB
selective cyclooxygenase-2 inhibitor
- FLAP
Finger Print for Ligands and Proteins
- MBD
membrane binding domain
- PGHS -1 and -2
Prostaglandin endoperoxide H synthase-1 and -2
- PGG2
prostaglandin G2
- PGH2
prostaglandin H2
- POX
peroxidase
- SI
selectivity index
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Smith WL, Garavito RM, DeWitt DL. Prostaglandin endoperoxide H synthases (cyclooxygenases) -1 and -2. J Biol Chem. 1996;271:33157–33160. doi: 10.1074/jbc.271.52.33157. [DOI] [PubMed] [Google Scholar]
- 2.Marnett LJ, Rowlinson SW, Goodwin DC, Kalgurtkar AS, Lanzo CA. Arachidonic acid oxygenation by COX-1 and COX-2 – Mechanism of catalysis and inhibition. J Biol Chem. 1999;274:22903–22906. doi: 10.1074/jbc.274.33.22903. [DOI] [PubMed] [Google Scholar]
- 3.Smith WL, DeWitt DL, Garavito RM. Cyclooxygenase: structural, cellular, and molecular biology. Annual Rev Biochem. 2000;69:149–182. doi: 10.1146/annurev.biochem.69.1.145. [DOI] [PubMed] [Google Scholar]
- 4.Perrone MG, Scilimati A, Simone L, Vitale P. Selective COX-1 inhibition: a therapeutic target to be reconsidered. Curr Med Chem. 2010;17:3769–3805. doi: 10.2174/092986710793205408. [DOI] [PubMed] [Google Scholar]
- 5.Vitale P, Scilimati A, Perrone MG. Update on SAR studies toward new COX-1 selective inhibitors. Curr Med Chem. 2015;22:4271–4292. doi: 10.2174/0929867322666151029104717. [DOI] [PubMed] [Google Scholar]
- 6.Vane JR, Botting RM. The mechanism of action of aspirin. Thromb Res. 2003;110:255–258. doi: 10.1016/s0049-3848(03)00379-7. [DOI] [PubMed] [Google Scholar]
- 7.Di Nunno L, Vitale P, Scilimati A, Tacconelli S, Patrignani P. Novel synthesis of 3,4-diarylisoxazole analogues of valdecoxib: reversal cyclooxygenase-2 selectivity by sulfonamide group removal. J Med Chem. 2004;47:4881–4890. doi: 10.1021/jm040782x. [DOI] [PubMed] [Google Scholar]
- 8.Vitale P, Tacconelli S, Perrone MG, Malerba P, Simone L, Scilimati A, Lavecchia A, Dovizio M, Marcantoni E, Bruno A, Patrignani P. Synthesis, pharmacological characterization, and docking analysis of a novel family of diarylisoxazoles as highly selective cyclooxygenase-1 (COX-1) inhibitors. J Med Chem. 2013;56:4277–4299. doi: 10.1021/jm301905a. [DOI] [PubMed] [Google Scholar]
- 9.Uddin MJ, Elleman AV, Ghebreselasie K, Daniel CK, Crews BC, Nance KD, Huda T, Marnett LJ. Design of fluorine-containing 3,4-diarylfuran-2(5H)-ones as selective COX-1 inhibitors. ACS Med Chem Lett. 2014;5:1254–1258. doi: 10.1021/ml500344j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Perrone MG, Vitale P, Panella A, Scilimati A. COX-1 inhibitors: beyond structure towards therapy. Med Rev Res. 2016;36:641–671. doi: 10.1002/med.21389. [DOI] [PubMed] [Google Scholar]
- 11.Perrone MG, Lofrumento DD, Vitale P, De Nuccio F, La Pesa V, Panella A, Calvello R, Cianciulli A, Panaro MA, Scilimati A. Selective cyclooxygenase-1 inhibition by P6 and gastrotoxicity: preliminary investigation. Pharmacol. 2015;95:22–28. doi: 10.1159/000369826. [DOI] [PubMed] [Google Scholar]
- 12.Kakuta H, Zheng X, Oda H, Harada S, Sugimoto Y, Sasaki K, Tai A. Cyclooxygenase-1-selective inhibitors are attractive candidates for analgesics that do not cause gastric damage. Design and in vitro/in vivo evaluation of a benzamide-type cyclooxygenase-1 selective inhibitor. J Med Chem. 2008;51:2400–2411. doi: 10.1021/jm701191z. [DOI] [PubMed] [Google Scholar]
- 13.Kakuta H, Fukai R, Xiaoxia Z, Ohsawa F, Bamba T, Hirata K. A Tai Identification of urine metabolites of TFAP, a cyclooxygenase-1 inhibitor. Bioorg Med Chem Lett. 2010;20:840–843. doi: 10.1016/j.bmcl.2010.01.161. [DOI] [PubMed] [Google Scholar]
- 14.Perrone MG, Vitale P, Panella P, Fortuna CG, Scilimati A. General role of the amino and methylsulfamoyl groups in selective cyclooxygenase(COX)-1 inhibition by 1,4-diaryl-1,2,3-triazoles and validation of a predictive pharmacometric PLS model. Eur J Med Chem. 2015;94:252–264. doi: 10.1016/j.ejmech.2015.02.049. [DOI] [PubMed] [Google Scholar]
- 15.Vitale P, Perrone MG, Malerba P, Lavecchia A, Scilimati A. Selective COX-1 inhibition as a target of theranostic novel diarylisoxazoles. Eur J Med Chem. 2014;74:1–13. doi: 10.1016/j.ejmech.2013.12.023. [DOI] [PubMed] [Google Scholar]
- 16.Perrone MG, Vitale P, Panella A, Ferorelli S, Contino M, Lavecchia A, Scilimati A. Isoxazole-based scaffold inhibitors targeting cyclooxygenase(COX)s. Chem Med Chem. 2016;11:1172–1187. doi: 10.1002/cmdc.201500439. [DOI] [PubMed] [Google Scholar]
- 17.Picot D, Loll PJ, Garavito RM. The X-ray crystal structure of the membrane protein prostaglandin H2 synthase-1. Nature. 1994;367:243–249. doi: 10.1038/367243a0. [DOI] [PubMed] [Google Scholar]
- 18.Mancini JA, Riendeau D, Falgueyret JP, Vickers PJ, O’Neill GP. Arginine 120 of prostaglandin G/H synthase-1 is required for the inhibition by nonsteroidal anti-inflammatory drugs containing a carboxylic acid moiety. J Biol Chem. 1995;270:29372–29377. doi: 10.1074/jbc.270.49.29372. [DOI] [PubMed] [Google Scholar]
- 19.Goto K, Ochi H, Yasunaga Y, Matsuyuki H, Imayoshi T, Kusuhara H, Okumoto T. Analgesic effect of mofezolac, a non-steroidal anti-inflammatory drug, against phenylquinone-induced acute pain in mice - A new antiinflammatory agent, selectively inhibits prostaglandin G/H synthase/cyclooxygenase (COX-2) activity in vitro. Prostaglandins & Other Lipid Mediator. 1998;56:245–254. doi: 10.1016/s0090-6980(98)00054-9. [DOI] [PubMed] [Google Scholar]
- 20.Kitamura T, Kawamori T, Uchiya N, Itoh M, Noda T, Matsuura M, Sugimura T, Wakabayashi K. Inhibitory effects of mofezolac, a cyclooxygenase-1 selective inhibitor, on intestinal carcinogenesis. Carcinogenesis. 2002;23:1463–1466. doi: 10.1093/carcin/23.9.1463. [DOI] [PubMed] [Google Scholar]
- 21.Perrone MG, Vitale P, Panella A, Ferorelli S, Contino M, Lavecchia A, Scilimati A. Isoxazole-Based-Scaffold Inhibitors Targeting Cyclooxygenases (COXs) ChemMedChem. 2016;11:1172–1187. doi: 10.1002/cmdc.201500439. [DOI] [PubMed] [Google Scholar]
- 22.Sidhu RS, Lee JY, Yuan C, Smith WL. Comparison of cyclooxygenase-1 crystal structures: cross-talk between monomers comprising cyclooxygenase-1 homodimers. Biochemistry. 2010;49:7069–7079. doi: 10.1021/bi1003298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gupta K, Selinsky BS, Kaub CJ, Katz AK, Loll PJ. The 2.0 A resolution crystal structure of prostaglandin H2 synthase-1: structural insights into an unusual peroxidase. J Mol Biol. 2004;335:503–518. doi: 10.1016/j.jmb.2003.10.073. [DOI] [PubMed] [Google Scholar]
- 24.Xu S, Hermanson DJ, Banerjee S, Ghebreselasie K, Clayton GM, Garavito RM, Marnett LJ. Oxicams bind in a novel mode to the cyclooxygenase active site via a two-water-mediated H-bonding Network. J Biol Chem. 2014;289:6799–6808. doi: 10.1074/jbc.M113.517987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sankhala SR, Lokareddy KR, Cingolani G. Structure of human PIR1, an atypical dual specificity phosphatase. Biochemistry. 2014;53:862–871. doi: 10.1021/bi401240x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Xu S, Hermanson DJ, Banerjee S, Ghebreselasie K, Clayton GM, Garavito RM, Marnett LJ. Oxicams bind in a novel mode to the cyclooxygenase active site via a two-water-mediated H-bonding Network. J Biol Chem. 2014;289:6799–6808. doi: 10.1074/jbc.M113.517987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Khan YS, Kazemi M, Gutiérrez-de-Terán H, Åqvist J. Origin of the enigmatic stepwise tight-binding inhibition of cyclooxygenase-1. Biochemistry. 2015;54:7283–7291. doi: 10.1021/acs.biochem.5b01024. [DOI] [PubMed] [Google Scholar]
- 28.Grosser T, Fries S, FitzGerald GA. Biological basis for the cardiovascular consequences of COX-2 inhibition: therapeutic challenges and opportunities. J Clin Invest. 2006;116:4–15. doi: 10.1172/JCI27291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Luong C, Miller A, Barnett J, Chow J, Ramesha C, Browner MF. Flexibility of the NSAID binding site in the structure of human cyclooxygenase-2. Nat Struct Biol. 1996;3:927–933. doi: 10.1038/nsb1196-927. [DOI] [PubMed] [Google Scholar]
- 30.Baroni M, Cruciani G, Sciabola S, Perruccio F, Mason JS. A common reference framework for analyzing/comparing proteins and ligands. Fingerprints for ligands and proteins (FLAP): theory and application. J Chem Inf Model. 2007;47:279–294. doi: 10.1021/ci600253e. [DOI] [PubMed] [Google Scholar]
- 31.Antithrombotic Trialists Collaboration. Collaborative meta-analysis of randomized trials of antiplatelet therapy for prevention of death, myocardial infarction, and stroke in high risk patients. Brit Med J. 2002;324:71–86. doi: 10.1136/bmj.324.7329.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Patrignani P. Aspirin insensitive eicosanoid biosynthesis in cardiovascular disease. Thromb Res. 2003;110:281–286. doi: 10.1016/s0049-3848(03)00382-7. [DOI] [PubMed] [Google Scholar]
- 33.Micetich RG, Shaw CC, Rastogi RB. Eur Patent. 1981;26928 [Google Scholar]
- 34.Otwinowski Z, Minor W. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]; Carter CW Jr, Sweet RM, editors. Macromolecular Crystallography. Academic Press; part A. [Google Scholar]
- 35.McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ. Phaser crystallographic software. J Appl Crystallogr. 2007;40:658–674. doi: 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Adams PD, Grosse-Kunstleve RW, Hung LW, Ioerger TR, McCoy AJ, Moriarty NW, Read RJ, Sacchettini JC, Sauter NK, Terwilliger TC. PHENIX: building new software for automated crystallographic structure determination. Acta Crystallogr D Biol Crystallogr. 2002;58:1948–1954. doi: 10.1107/s0907444902016657. [DOI] [PubMed] [Google Scholar]
- 37.Liebschner D, Afonine PV, Moriarty NW, Poon BK, Sobolev OV, Terwilliger TC, Adams PD. Polder map: improving OMIT maps by excluding bulk solvent. Acta Crystallogr D Struct Biol. 2017;73(Pt 2):148–157. doi: 10.1107/S2059798316018210. 2017 Feb 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 39.DeLano WL. World Wide Web; 2002. The PyMOL Molecular Graphics System. http://www.pymol.org. [Google Scholar]
- 40.Rahaman O, Estrada TP, Doren DJ, Taufer M, Brooks CL, 3rd, Armen RS. Evaluation of several two-step scoring functions based on linear interaction energy, effective ligand size, and empirical pair potentials for prediction of protein-ligand binding geometry and free energy. J Chem Inf Model. 2011;51:2047–2065. doi: 10.1021/ci1003009. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.