

Summary
Succinylation is a post-translational protein acylation modification that converts the cationic lysine side chain to an anion with large potential impacts on protein structure and function. Here we characterize the epigenome-wide distribution of succinyllysine marks in chromatin using chromatin immunoprecipitation sequencing (ChIP-seq). We estimate that more than one-third of all nucleosomes contain lysine succinylation marks and demonstrate a potential role of chromatin succinylation in modulating gene expression. We further demonstrate that defective tricarboxylic acid (TCA) cycle metabolism perturbs the succinyllysine distribution in chromatin, correlating with transcriptional responses. This is consistent with previous observations linking nucleosome succinylation with enhanced in vitro transcription. We additionally demonstrate that defective TCA cycle metabolism results in a DNA repair defect and sensitivity to genotoxic agents, consistent with previously reported chromatin hypersuccinylation effects observed in the context of SIRT7 depletion. Chromatin succinylation may thus represent a mechanism by which metabolism modulates both genome-wide transcription and DNA repair activities.
Subject Areas: Cellular Physiology, Molecular Mechanism of Gene Regulation, Proteomics
Graphical Abstract
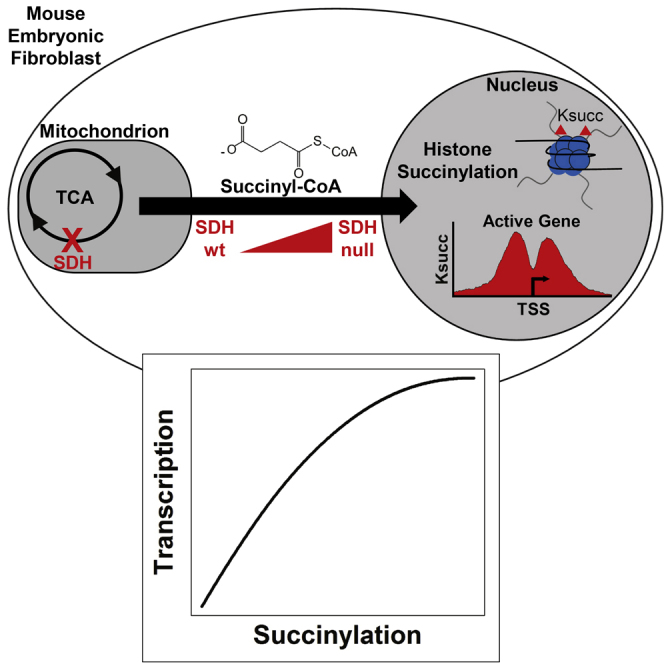
Highlights
-
•
SDH loss TCA cycle defect results in succinyl-CoA increase and hypersuccinylation
-
•
Succinyllysine modification of chromatin correlates with active gene expression
-
•
Chromatin succinyllysine change in SDH loss correlates with transcriptional change
Cellular Physiology; Molecular Mechanism of Gene Regulation; Proteomics
Introduction
Lysine succinylation (Figure 1A [Zhang et al., 2011]) has emerged as a novel post-translational modification (PTM) that directly couples tricarboxylic acid (TCA) cycle metabolism, via succinyl-CoA, to alterations in the charges, structures, and activities of proteins involved in diverse cellular processes. Global proteomic approaches have revealed that the lysine succinylome spans multiple biological compartments, including the nucleus, but with concentrated effects in the mitochondria (Park et al., 2013). Succinylation is one of many known acyl modifications capable of decorating the lysine residues in proteins, with others including acetylation, propionylation, malonylation, crotonylation, butyrylation, glutarylation, 2-hydroxyisobutyrylation, and β-hydroxybutyrylation (Chen et al., 2007, Choudhary et al., 2009, Dai et al., 2014, Du et al., 2011, Garrity et al., 2007, Tan et al., 2011, Tan et al., 2014, Xie et al., 2016).
Figure 1.

Global Lysine Succinylation Affects Multiple Cellular Compartments and Is Perturbed in the Context of SDH Loss
(A) Reaction of the ɛ-amino group of lysine with succinyl-CoA to form succinyllysine.
(B) TCA cycle metabolism and succinyl-CoA as a protein succinylating agent. Also shown are TCA cycle metabolites quantified for Sdhc fl/fl (experimental) and Sdhc fl/wt (control) cells 16 days after induction of Sdhc gene rearrangement with the doxycycline-inducible tetOcre system. Error bars indicate measurement standard deviations. Asterisks indicate comparisons that were statistically significant (p value < 0.05) by heteroscedastic 1-tailed t test.
(C) Pan-succinyllysine immunostaining of experimental and control cell lines.
(D) Quantification of mean cytoplasmic and nuclear pan-succinyllysine immunostaining intensity. Asterisks indicate degree of statistical significance by heteroscedastic 1-tailed t test (*p value < 2.2 × 10−16).
(E) Schematic of experimental design to assess differential protein succinylation in the context of SDHC loss. TetOcre-mediated Sdhc gene rearrangement is induced on day 0 by doxycycline. On the same day, Sdhc fl/fl (experimental) cells are transitioned to SILAC light media and Sdhc fl/+ (control) cells are transitioned to SILAC heavy media containing isotopically labeled lysine and arginine.
(F) Overlap of succinylated sites quantified between two biological replicates.
(G) Distribution of SILAC log2 ratios of heavy and light peptides (averaged between replicate experiments).
(H) Spearman correlation analysis of site succinylation values from two biological replicates.
(I) Analysis of correlation between protein succinylation change (y axis) and protein abundance change (x axis). Subset of data points for which succinylation log2 ratio and abundance log2 ratio differed by more than 0.2 is highlighted in blue.
(J) Analysis of gene ontologies and KEGG pathways preferentially affected by differential protein succinylation in the context of SDHC loss. Terms are grouped according to ontology (MF, molecular function; CC, cellular component; BP, biological process) or KEGG pathway. Color reflects the average succinylation log2(fold-change) for identified proteins mapping to a given term [green, negative heavy/light log2(fold-change) and increased succinylation in SDHC loss]. See also Figure S1.
(K) Box plot showing succinylation stoichiometries (fraction of a given protein site that is succinylated) for proteins quantified in experimental and control cell lines, separated according to intracellular compartment. See also Figure S1.
(L) Succinylation stoichiometries for histone and non-histone chromatin proteins quantified in experimental (green) and control (gray) cell lines.
(M) Relative percent increase in histone protein succinylation quantified between experimental and control cell lines.
Significant discussion persists within the field regarding whether the attachment of the various acyl PTMs to lysine residues is enzyme mediated. Acetylation is the oldest known and best studied acyl PTM, and many acetyltransferases and deacetylases catalyzing the attachment and removal of acetyl marks, respectively, have been identified (Ito et al., 2000, McManus and Hendzel, 2000, Michishita et al., 2008, Ravindra et al., 2009, Tafrova and Tafrov, 2014, Warrener et al., 2010, Xiong et al., 2010, Yang et al., 2011; Zhang et al., 1998). Several acetyltransferases have also been identified that promiscuously attach propionyl and butyryl groups to lysine residues (Leemhuis et al., 2008, Simithy et al., 2017). It remains less clear whether other acyl PTMs are attached to lysine residues in an enzyme-mediated manner. Recent work has suggested that several of the larger acyl modifications such as succinylation, malonylation, crotonylation, glutarylation, and β-hydroxybutyrylation may predominantly functionalize proteins via a non-enzymatic mechanism (Simithy et al., 2017, Wagner et al., 2017). This suggests that the rates of attachment for the various acyl-CoA species to lysine residues may be primarily governed by the concentrations of the involved reactants.
In contrast to the current paradigm of non-enzymatic succinylation, the process of desuccinylation may be mediated by specific enzymes. SIRT5 has been identified as a lysine desuccinylase that catalyzes the removal of succinyl marks from lysine residues in the mitochondria and cytoplasm (Du et al., 2011). Attenuation of SIRT5 alters the distribution of lysine succinylation marks (Park et al., 2013, Rardin et al., 2013). Importantly, depletion of SIRT5 has been shown to impair the function of complex II (succinate dehydrogenase [SDH]) and fatty acid β-oxidation, suggesting that succinylation inhibits mitochondrial enzymes (Park et al., 2013, Rardin et al., 2013). Recent work has extended these observations by identifying SIRT5 as an important regulator of cardiomyocyte biology via modulation of hydroxyacyl-CoA dehydrogenase trifunctional multienzyme complex subunit alpha (HADHA/ECHA) activity, a protein involved in fatty acid β-oxidation (Sadhukhan et al., 2016). These findings imply that lysine succinylation modulates enzyme activity in a previously unappreciated layer of biological regulation.
Initial reports of succinylation PTMs also included lysine succinylation of histones (Xie et al., 2012). These early studies revealed specific succinylation sites and characterized loss of yeast viability resulting from mutation of histone residues that are normally highly succinylated. These results suggested that histone succinylation is important for cell viability. Although provocative, the basis for these findings has remained unclear, and the relevance of histone succinylation for biological regulation has been uncertain. More recent work identified SIRT7 as a nuclear-localizing histone desuccinylase that functionally links the poly (ADP-ribose) polymerase (PARP) 1-dependent DNA damage response and chromatin compaction essential for cell survival following genotoxic stress (Li et al., 2016). A high priority for the field is to characterize other functions of histone lysine succinylation in regulating nuclear processes such as transcription.
We have been interested in the possibility that disease-causing TCA cycle defects may affect the lysine succinylome and contribute to pathologic processes. In the present work, we characterize succinylation in an inducible cell culture model of SDH loss, which results in accumulation of succinate and succinyl-CoA. This allowed us to characterize perturbation of the lysine succinylome relevant to several human malignancies including paraganglioma/pheochromocytoma (PPGL), gastrointestinal stromal tumor (GIST), renal cell carcinoma (RCC), thyroid cancer, and colon cancer (Agaimy, 2016, Astuti et al., 2001, Baysal et al., 2000, Gill et al., 2013, Nannini et al., 2014, Ni et al., 2015, Niemann and Müller, 2000, Pantaleo et al., 2015, Wang et al., 2016).
Our analyses reveal succinylation effects in several subcellular compartments, including histone and non-histone chromatin components in the nucleus. We characterize for the first time the epigenome-wide distribution of succinyllysine marks in chromatin using chromatin immunoprecipitation sequencing (ChIP-seq), revealing a potential role of chromatin succinylation in modulating gene expression. In addition, we analyze epigenome-wide patterns of chromatin succinylation perturbation in the context of SDH loss and document correlations with altered gene expression. These results are consistent with previous observations linking nucleosome succinylation with enhanced in vitro transcription. We also demonstrate that defective TCA cycle metabolism results in a DNA repair defect and sensitivity to genotoxic agents, consistent with previously reported chromatin hypersuccinylation effects observed in the context of SIRT7 depletion. Chromatin succinylation may thus represent a mechanism by which metabolism modulates both genome-wide transcription and DNA repair activities.
Results
SDH Loss Leads to Global Lysine Hypersuccinylation in Multiple Subcellular Compartments, Especially Mitochondria
Biallelic loss of genes encoding subunits of the SDH complex in the TCA cycle results in accumulation of succinate and succinyl-CoA (Figure 1B). Previous work has shown that these metabolic effects are only observed in the context of homozygous SDH subunit depletion, with no evidence for metabolic abnormalities in hemizygous lines (Smestad et al., 2017). We therefore hypothesized that a global hypersuccinylation effect might be observed in the context of SDH loss due to accumulation of succinyl-CoA, the presumptive succinylating agent. We therefore performed immunostaining using an anti-succinyllysine antibody (PTM-401) to assess whether SDH loss results in an observable bulk protein hypersuccinylation effect (Figure 1C). Intriguingly, hypersuccinylation effects were observable both in the cytoplasm and the nucleus of SDH loss cells, but with most effects observable in the cytoplasm (Figure 1D). This inspired us to perform a more granular analysis of protein-specific hypersuccinylation effects. We therefore performed stable isotope labeling with amino acids in cell culture (SILAC) labeling of SDH knockout (SILAC light) and control (SILAC heavy) immortalized mouse embryonic fibroblast (iMEF) cell lines using 13C-labeled lysine and arginine followed by global proteomic analysis of succinylated peptides purified by immunoprecipitation (Figure 1E). Specificity of the anti-succinyllysine antibody (PTM-401) was confirmed by analysis of the prevalence of succinyllysine and the various other acyl modifications in the antibody-enriched peptide pool (Figure S1A). Analysis of biological replicate experiments identified 2,776 unique succinylation sites, yielding quantifiable SILAC heavy/light ratios between replicates (Figure 1F, Table S1). Following succinylation site identification and quantification, distributions of median-normalized SILAC heavy/light ratios were log2-transformed, revealing a non-Gaussian distribution of SILAC log2 ratios with hypersuccinylation in the SDH loss cell line as hypothesized (Figure 1G). Analysis of the correlation between log2-transformed SILAC heavy/light ratios in biological replicates (Figure 1H) confirmed reproducibility (Spearman correlation coefficient: 0.82; p value < 2.2 × 10−16).
As SDH loss may alter both protein abundance and protein lysine succinylation, we identified proteins whose change in lysine succinylation was disproportionate to their change in abundance upon SDH loss (Figure 1I). This analysis identified 78 such cases (Table S2). Matching gene ontologies and KEGG pathways associated with this set of proteins demonstrated a hypersuccinylation effect concentrated in the mitochondria, but that is detectable in several other cellular compartments (Figure 1J). Analysis of mitochondrial proteins in this differentially succinylated subset revealed dramatic effects for proteins involved in glycolysis/TCA cycle, fatty acid catabolism, ketone body metabolism, heat shock response, solute transport, ATP synthesis, amino acid synthesis, and the electron transport chain (Figure S1B). Protein hypersuccinylation affecting multiple subcellular compartments in the context of SDH loss had not previously been reported. It is possible that hypersuccinylation may contribute to suppression of mitochondrial enzymatic activities in the context of SDH loss, similar to what has been previously described in the context of SIRT5 desuccinylase knockout (Park et al., 2013).
We next assessed the degree to which particular lysine residues are succinylated. A global proteomic analysis of peptide abundance was leveraged to determine stoichiometries of succinylated and unmodified peptides (Olsen et al., 2010). A sample of 363 sites was identified with adequate abundance for this calculation, yielding succinylation stoichiometries for both SDH loss and control cells (Table S3). Mean succinylation stoichiometries were 0.13 and 0.18, respectively, for control and experimental lines (Figures S1C–S1E), demonstrating succinylation to be a high-abundance PTM and confirming the bulk increase in lysine succinylation in the context of SDH loss (Figure S1E). Intriguingly, there is a strong correlation between the extent of succinylation in the control cell line and hypersuccinylation upon SDH loss (Figure S1F). Classification of proteins based upon observed subcellular localization patterns using data from the Cell Atlas (Thul et al., 2017) revealed that both normal succinylation and hypersuccinylation induced by SDH loss are detected in most subcellular compartments (Figure 1K). We were particularly intrigued that these effects were apparent in the nucleus, as was also seen in our anti-succinyllysine immunostaining analysis (Figures 1C and 1D). We further pursued this observation.
Lysine Succinylation of Chromatin Proteins Is Common and Increases upon SDH Loss
We extracted succinylation data for all histone and non-histone chromatin components and examined the absolute impact of SDH loss on succinylation stoichiometry (Figure 1L) and relative succinylation ratio (Figure 1M). Intriguingly, several sites displayed high baseline succinylation in control cells, with increased succinylation upon SDH loss. Importantly, 17% of histone H4K80 residues were succinylated in control cells. Since two copies of histone H4 are present in each nucleosome, this suggests that perhaps one-third of all nucleosomes contain at least one succinyl mark at H4K80. In the context of SDH loss, the fraction of H4K80 residues bearing succinylation marks increases to 0.24, indicating that this already highly abundant mark increases by a further 41%. This striking prevalence of histone succinylation suggests that succinylation of chromatin components may be important for regulating nuclear functions, including chromatin dynamics and transcription.
Genome-wide Chromatin Succinylation Correlates with Euchromatic Histone Marks
We hypothesized that succinylation of histones and other chromatin components encodes regulatory information and therefore that succinyllysine marks would be non-uniformly distributed across the genome. To test this hypothesis, we mapped the genome-wide distribution of succinyllysine marks in chromatin from control and SDH-loss iMEF cell lines using a ChIP-seq approach with a pan-succinyllysine antibody, comparing the pattern of DNA sequence enrichment to patterns observed for chromatin marks commonly found in euchromatic (H3K4me3) or heterochromatic (H3K27me3) regions. Following MNase digestion and sonication, gel quantification of input chromatin fragments showed the chromatin to have an approximately mononucleosomal size distribution (Figure S2A), as did our analysis of mapped read lengths from paired-end read alignment following chromatin immunoprecipitation (ChIP) experiments involving formaldehyde cross-linking (Figure S2B). Quantified yields from ChIP experiments ranged between 0.4% and 4% (Figure S2C). Comparison of mapped reads from ChIP-seq experiments with input controls using an unbiased peak-calling algorithm identified several thousand peaks for each antibody (Figure 2A), with an average succinyllysine ChIP peak width less than 500 bp (Figure S2D). Analysis of the genome-wide distributions of identified ChIP-seq peaks revealed that the most common pattern of localization is in the intergenic regions (56%), followed by the introns (25%) and gene promoters (15% within 3 kbp of a transcription start site [TSS], 11.4% within 1 kbp of a TSS) (Figures 2B and 2C). Compared to a distribution of randomly selected genomic coordinates, this represents significant enrichment for promoter-localizing peaks (6% within 3 kbp of a TSS, 2% within 1 kbp of a TSS) as opposed to what chance would have predicted (Figure 2B), suggesting that succinyllysine marks in chromatin may play a role in transcriptional regulation.
Figure 2.

Succinyllysine ChIP-Seq Peaks Correlate with Euchromatic Histone Marks
(A) Numbers of peaks for each ChIP-seq target identified via the MACS peak-calling algorithm. See also Figures S2.
(B) Genomic feature localization for identified peaks. x Axis indicates percentage of identified peaks for each immunoprecipitation mapping to a given feature type.
(C) Detailed analysis of succinyllysine peak localization. Dot plot indicates genomic feature peak localization pattern, with a solid line joining two or more dots indicating instances of peaks spanning one or more genomic feature type. Bar plot above dot plot indicates number of peaks represented by a given pattern.
(D) Correlation between input-subtracted succinyllysine peak intensities quantified between ChIP-seq biological replicates.
(E) Correlation between succinyllysine signal intensity and intensities of known histone marks (H3K4me3 and H3K27me3) at positions of identified succinyllysine peaks.
(F) Representative genomic view of mouse chromosome 3 showing several identified peaks for each ChIP-seq experiment. Shown are genomic annotations for genes, mRNAs, coding DNA sequences (CDS), identified ChIP-seq peaks, and wiggle plot representations of mapped reads from the various ChIP-seq experiments. For each experiment type, mapped reads for two biological replicates are shown.
Analysis of succinyllysine peaks between two biological replicate experiments showed high reproducibility (Pearson correlation coefficient: 0.88; p value < 2.2 × 10−16; Figure 2D), suggesting that the positions and intensities of chromatin succinylation are biologically stable. We compared succinyllysine locations to euchromatic histone mark H3K4me3 and heterochromatic mark H3K27me3. Intriguingly, positions of succinyllysine marks correlate positively with euchromatic H3K4me3 marks (Spearman correlation coefficient: 0.23; p value < 2.2 × 10−16) and correlate negatively with H3K27me3 heterochromatic marks (Spearman correlation coefficient: −0.30; p value < 2.2 × 10−16) (Figure 2E). This is the first evidence that chromatin lysine succinylation is a euchromatic mark (see also Figure 2F).
Chromatin Succinyllysine Marks Form a Distinctive Pattern Near Gene Promoters
We next assessed the pattern of promoter chromatin succinylation to discern any potential role in regulating gene transcription. We generated aligned probe plots (Figure 3A) and average probe plots (Figure 3B) displaying the abundance of identified peaks as a function of distance from the annotated TSS. Intriguingly, succinyllysine modification displays a distinctive pattern of enrichment peaking 600 bp upstream and downstream of the TSS, with relative depletion at the TSS and beyond 600 bp from the TSS. This pattern is clearly distinct from those observed for H4K3me3 and H3K27me3, suggesting that promoter succinylation encodes additional gene regulatory signals.
Figure 3.

Chromatin Succinylation Exhibits Distinct Pattern of Localization Near Gene Promoters and Correlates with Gene Expression
(A) Plots of aligned probes showing chromatin mark enrichment over a 6-kbp window centered on the TSS. Individual lines on the plots correspond to specific TSS loci, with the shading of the plot representing the presence of ChIP-seq peaks. Genomic regions in the three plots are ordered independently according to the mean intensity.
(B) Probe trend plots covering a 6-kbp window centered on the TSS. Lines demonstrate relative peak frequency as a function of position, averaged across all annotated TSS.
(C) Analysis of correlation between succinyllysine signal intensity and intensities of known histone marks (H3K4me3 and H3K27me3) at TSS (4-kbp window centered on TSS).
(D) Analysis of correlation between chromatin ChIP-seq mark TSS abundance and gene expression (log2-transformed fragments per kilobase million (FPKM) values, then scaled in range of 0–1).
(E) Analysis of expression in subsets of genes containing ChIP-seq peaks within 3 kbp of the TSS for H3K27me3, H3K4me3, or succinyllysine. y Axis shows log2-transformed gene expression FPKM values.
(F) Detailed analysis of gene expression in subset of genes containing TSS-localizing ChIP-seq peaks for H3K4me3 and/or succinyllysine.
Chromatin Succinylation in Promoters Correlates with Activating Histone Marks
We assessed whether succinylation in gene promoters (4-kbp window centered on the TSS) correlates with histone marks associated with actively transcribed (H3K4me3) or epigenetically repressed (H3K27me3) genes. Intriguingly, succinyllysine marks mapped to gene promoters were found to correlate positively with H3K4me3 marks (Pearson correlation coefficient: 0.38; p value < 2.2 × 10−16) and correlate negatively with H3K27me3 marks (Pearson correlation coefficient: −0.12; p value < 2.2 × 10−16; Figure 3C). This coincidence of succinyl marks in chromatin with H3K4me3, but not with H3K27me3, suggests that succinylation of chromatin at active gene promoters is functionally meaningful.
Chromatin Succinylation Is a Feature of Highly Expressed Genes
We directly tested the correlation between gene expression and bulk succinylation of chromatin at gene promoters using previously published transcriptomic data for our iMEF cell lines. We identified a meaningful correlation between gene promoter chromatin succinylation and gene expression (Pearson correlation coefficient: 0.32; p value < 2.2 × 10−16; Figure 3D), comparable to the relationship observed for H3K4me3. This is a highly provocative observation suggesting that chromatin succinylation may play a functional role in transcriptional regulation. Intriguingly, previous in vitro studies have shown that bulk and non-specific succinylation of nucleosome core particles with succinic anhydride enhances transcription, possibly through electrostatic effects (Piñeiro et al., 1992). The findings of these early studies ultimately support our in vivo work analyzing genome-wide patterns of succinylation in chromatin and assessing correlations with gene expression. When taken together, these data support a claim that chromatin succinylation in gene promoters is transcription activating. Unlike acetylation, which neutralizes potential favorable interactions between cationic lysine side chains and anionic DNA or histone sites, succinylation converts the cationic lysine side chain to an anion with much greater potential for electrostatic disruption. This possible electrostatic mechanism for promoting an active chromatin state remains to be explored.
To assess whether chromatin succinylation patterns merely reflect the H3K4me3 status of a given promoter, or whether chromatin succinylation encodes unique information, we next assessed gene expression as a function of mapped ChIP-seq peaks for the various immunoprecipitation experiments. Comparing gene expression with ChIP-seq peaks for H3K27me3, H4K4me3, and succinyllysine, we observe that genes whose promoter chromatin is enriched in H3K27me3 marks are poorly expressed, whereas genes whose promoter chromatin is enriched in H3K4me3 or succinyllysine marks are strongly expressed (Figure 3E). We further classified promoters as having H3K4me3 marks, succinyllysine marks, or both. This analysis showed that even for promoters enriched in succinyllysine marks without H3K4me3 marks, gene expression levels were among the highest in the cell (Figure 3F). This result suggests that chromatin succinylation represents a novel information-carrying mark of gene activation that is non-redundant with H3K4me3.
SDH Loss Perturbs Genome-wide Chromatin Succinylation
We next assessed how SDH loss affects genome-wide chromatin succinylation. Our previous analysis of bulk protein succinylation after SDH loss demonstrated increased protein succinylation, including several chromatin components (Figure 1J). This analysis did not reveal how the distribution of succinyllysine chromatin marks changes genome-wide upon SDH loss. We therefore mapped succinyllysine chromatin marks in SDH knockout and control cell lines. Interestingly, this analysis revealed a small net decrease in mapped read abundance at positions of succinyllysine peaks in the SDH knockout line relative to control, but with a particular subset of loci displaying dramatic perturbation (Figure 4A). This result was reproducible, as indicated by a high degree of correlation between the results of replicate experiments (Pearson correlation coefficient: 0.57; p value < 2.2 × 10−16; Figure 4B). Analysis of the correlation between succinyllysine change and changes in histone marks at succinyllysine peaks revealed that change in succinyllysine marks is positively correlated with perturbation of H3K4me3 (Figure 4C) and negatively correlated with H3K27me3 (Figure 4D) marks. An observed positive correlation between succinylation and H3K4me3 changes is particularly intriguing, given the potential for succinylation and methylation at a given histone lysine position to be mutually exclusive and competing PTMs.
Figure 4.

SDH Loss Results in Reproducible Perturbation of Chromatin Succinylation that Preferentially Affects the Promoters of Genes Involved in Transcriptional Regulation
(A) Distribution of observed differences in succinylation peak integrated areas between experimental (SDHC loss) and control cell lines. Vertical red lines indicate the cutoff positions for 0.05 and 0.95 quantiles.
(B) Correlation analysis of observed succinylation peak integrated intensity differences (experimental minus control) between replicate experiments (see also Figure S3).
(C) Correlation analysis of changes (experimental minus control) in succinyllysine and H3K4me3 integrated intensities measured at the positions of succinyllysine ChIP-seq peaks.
(D) Correlation analysis of changes (experimental minus control) in succinyllysine and H3K27me3 integrated intensities measured at the positions of succinyllysine ChIP-seq peaks.
(E) Analysis of genomic feature localization for succinyllysine peaks showing dramatic integrated intensity differences resulting from SDHC loss. 0.05 and 0.95 quantiles, as shown in panel (A). x Axis indicates percentage of identified peaks mapping to a given feature type. See also Figure S3.
(F) Average profile plot showing TSS relative position for succinyllysine peaks showing dramatic integrated intensity differences resulting from SDHC loss. See also Figure S3.
(G) Analysis of correlation between promoter succinyllysine integrated intensity change and gene expression change [experimental minus control log2(FPKM)]; see also Figure S3.
(H) DAVID functional enrichment analysis of gene ontology impact for top 0.95 quantile of gene TSS (ranked according to absolute value of difference) affected by differential succinylation. x Axis value indicates degree of statistical significance for term enrichment relative to random gene set of the same size. Colors indicate mean succinyllysine difference (experimental minus control integrated TSS values) for differentially succinylated genes mapping to a given term (green, increased succinylation in experimental line; red, decreased succinylation in the experimental line).
(I) Representative genomic view of mouse chromosome 3 showing a succinyllysine peak dramatically affected by SDHC loss. Shown are genomic annotations for genes, mRNA, CDS, identified ChIP-seq peaks, and wiggle plot representations of mapped reads from the various ChIP-seq experiments; see also Figure S4.
SDH Loss Selectively Perturbs Chromatin Succinylation in Promoters
We assessed which genomic regions undergo the greatest change in chromatin succinylation upon SDH loss. For this analysis, we selected the top 0.05 and bottom 0.95 quantiles of ChIP-seq peaks, ranked by observed succinylation difference (SDH knockout minus control). Thus these peaks correspond to the subset of peaks that display extreme hypersuccinylation and hyposuccinylation, respectively, upon SDH loss. We mapped these highly affected peaks relative to the population of all chromatin succinyllysine peaks. Intriguingly, the alteration preferentially affects gene promoters (Figures 4E and 4F), with 45% of hypersuccinylated peaks and 30% of hyposuccinylated peaks mapping to promoters, compared with 15% of peaks belonging to the parental distribution. The fact that succinylation change in chromatin following SDH loss is highly enriched in gene promoters over other types of genomic regions further underscores the potential transcriptional regulatory role of this mark. Subsequent analysis of the perturbation H3K4me3 and H3K27me3 marks upon SDH loss yielded the similar finding that epigenetic impacts are concentrated in promoter regions (Figures S3A–S3H and S4A–S4C). Although the mechanism remains unknown, the observation that TCA cycle dysfunction due to SDH loss has concerted effects on chromatin succinylation in gene promoters emphasizes the apparent regulatory specificity of this novel mark.
Chromatin Succinylation Changes upon SDH Loss Correlate with Gene Expression Changes
We assessed whether changes in promoter chromatin succinylation upon SDH loss correlate with induced alterations in gene expression. Our previous analysis of the correlation between promoter succinylation and gene expression in normal cells showed that succinylation is a feature of highly expressed genes (Figures 3D–3F). We therefore hypothesized that changes in chromatin succinylation upon SDH loss would directly correlate with observed gene expression changes. This hypothesis is indeed supported by the data (Spearman correlation coefficient: 0.23; p value < 2.2 × 10−16; Figure 4G). This result suggests that either perturbation of chromatin succinylation causes altered gene expression or that the succinylation status of specific genomic loci dynamically changes to reflect perturbed gene expression patterns. Under either model, this observation constitutes a quantifiable transcriptional phenotype that is correlated with alterations in genome-wide succinylation of gene promoters.
Changes in Chromatin Succinylation upon SDH Loss Selectively Affect Promoters of Genes Involved in Transcriptional Regulation
We assessed the extent to which chromatin succinylation plays a role in regulating the expression of genes controlling specific processes or pathways. We used the DAVID functional annotation database (Huang da et al., 2009) to perform functional annotation enrichment analysis on the top 0.05 quantile of differentially succinylated promoters for known gene ontologies, ranked according to the absolute value of succinylation change. The results (Figure 4H) reveal a genome-wide phenotype of selective hypersuccinylation impact on genes involved in transcriptional regulation and a specific hyposuccinylation effect upon genes involved in RNA processing. These trends provide strong evidence that chromatin succinylation cues specific transcriptional programs within cells. A representative genomic view displaying altered chromatin succinylation in the context of SDH loss is shown in Figure 4I.
Collectively, these observations support the hypothesis that chromatin succinylation is a link between the TCA cycle status and epigenetic transcriptional control. This result has implications both for normal cell growth and homeostasis, and for the mechanism of oncometabolites, such as succinate, that accumulate due to mutations affecting TCA cycle enzymes.
SDH Loss Results in a DNA Repair Defect Consistent with Chromatin Hypersuccinylation Effects
We next assessed whether SDH loss results in a DNA repair defect that is consistent with hypersuccinylation effects, similar to that observed in the context of SIRT7 depletion (Li et al., 2016). In the context of SIRT7 desuccinylase depletion, a defect in PARP1-dependent DNA repair becomes evident that is linked to failure in histone desuccinylation and chromatin compaction essential for repairing genotoxic damage. Upon genotoxic insult, SIRT7 −/− cells display abnormally elevated levels of gamma H2A.X, a marker for DNA damage. We therefore investigated whether hypersuccinylation of chromatin in the context of SDH phenocopies SIRT7 by similarly exhibiting a DNA repair defect. Strikingly, we observe that SDH loss results in heightened levels of DNA damage marker gamma H2A.X (phospho 139) (Figures 5A and 5B), consistent with a global DNA repair defect attributable to hypersuccinylation effects. Remarkably, this elevation in gamma H2A.X (phospho 139) occurs in the absence of an external genotoxic insult, suggesting that SDH loss results in a DNA repair defect that allows for accumulation of normal replication-induced errors to abnormally high levels. We additionally show that this intrinsic DNA repair defect predisposes SDH loss cells to be sensitive to genotoxic drugs such as gemcitabine (Figure 5C) and etoposide (Figure 5D), further validating the claim of a DNA repair defect in the setting of SDH loss. This enhanced sensitivity to genotoxic compounds such as these is particularly remarkable because these compounds mediate toxicity during DNA replication and SDH loss results in slowed population doubling time (Smestad et al., 2017). Collectively, these experiments suggest that chromatin hypersuccinylation may generally impede DNA repair activities and form tantalizing conceptual links to recent work connecting SIRT7 desuccinylase activity with chromatin compaction essential for repair of genotoxic DNA damage.
Figure 5.

SDH Loss Results in a DNA Repair Defect Consistent with Chromatin Hypersuccinylation Effects
(A) Immunostaining for gamma H2A.X (phospho 139), with 4',6-diamidino-2-phenylindole (DAPI) counterstain. Shown are representative images for two stable experimental (SDH loss) and control cell lines.
(B) Quantification of mean nuclear gamma H2A.X (phospho 139) immunostain intensity using automated image analysis.
(C) Analysis of differential cell line viability as a function of gemcitabine concentration. Viability quantification is in reference to untreated condition, with error bars corresponding to standard deviations from triplicate assay replicates.
(D) Analysis of differential cell line viability as a function of etoposide concentration. Viability quantification is in reference to untreated condition, with error bars corresponding to standard deviations from triplicate assay replicates.
Discussion
This work demonstrates for the first time that an SDH loss TCA cycle defect relevant to several human malignancies results in increased concentrations of succinyl-CoA and bulk protein hypersuccinylation affecting multiple subcellular compartments. Hypersuccinylation affects both histone and non-histone nuclear proteins. We estimate that more than one-third of all nucleosomes contain lysine succinylation marks and that this fraction is increased in the context of SDH loss. By investigating the genomic localization of succinyllysine modifications in chromatin via ChIP-seq, we show that succinyllysine ChIP-seq peaks are enriched in gene promoters and identify a bimodal pattern of peak enrichment flanking transcription start sites genome-wide that is suggestive of a role in transcriptional regulation. We also directly test the correlation between succinyllysine ChIP signal and gene expression. We find that there is indeed a strong correlation between gene expression and succinylation near the transcription start site and further show that promoter succinylation is strongly predictive of high gene expression. We further assess how succinyllysine marks in chromatin are perturbed in the context of SDH loss TCA cycle defect and show that chromatin succinylation changes are concentrated in gene promoters relative to other genomic regions. We show that promoter succinylation changes induced by SDH loss are correlated with observed transcriptional responses genome-wide, further underscoring the fact that promoter chromatin succinylation and gene expression are correlated.
Beyond in vivo correlative studies, the next level of evidence needed to establish that histone succinylation activates transcription could come from an in vitro assay to test this hypothesis under tightly controlled conditions. Conveniently, such assays have already been reported (Piñeiro et al., 1992). These early experiments involved treating histone octamers with succinic anhydride, reconstituting with transcriptional templates, and measuring the effects of nucleosome succinylation on transcription. Remarkably, the authors found that succinylated nucleosomes have potent transcription-activating properties. These studies, performed years before it was appreciated that succinylation is a relevant histone lysine PTM that occurs in living cells, strongly support our in vivo work analyzing genome-wide patterns of succinylation in chromatin and assessing correlations with gene expression. When taken together, these data strongly support a claim that chromatin succinylation in gene promoters activates transcription. It remains to be determined if the basis for this effect is primarily electrostatic, or if a family of regulatory succinyllysine “reader” proteins are also involved.
Finally, we demonstrate that SDH loss results in a DNA repair defect that may be linked to chromatin hypersuccinylation effects. Previous work performed by Li et al. has demonstrated that depletion of SIRT7 desuccinylase results in histone hypersuccinylation and a resultant defect in DNA double-strand break repair (Li et al., 2016). We were intrigued that SDH loss, another cause of histone hypersuccinylation, appears to phenocopy the DNA repair defect observed upon SIRT7 depletion. It is highly remarkable that the two currently known genetic causes of histone hypersuccinylation both result in DNA repair defects. Whether these observed DNA repair defects are related by a common mechanism remains to be determined.
Methods
All methods can be found in the accompanying Transparent Methods supplemental file.
Acknowledgments
This work was facilitated by the Mayo Clinic Metabolomics Resource Core Facility (NIH grant U24DK100469), the Mayo Clinic Medical Genome Facility Sequencing Core, the Mayo Clinic Epigenetics Development Laboratory, and the Mayo Clinic Research Computing Facility. Financial support for this work was from the Mayo Clinic; NIH grants R01CA166025 (L.J.M.), T32GM065841 (Mayo Clinic Medical Scientist Training Program), and F30CA220660 (J.S.); and generous support from the Paradifference Foundation (L.J.M.).
Author Contributions
Conceptualization, L.J.M., J.S., and Y.C.; methodology, L.J.M., J.S., L.E., and Y.C.; validation, J.S. and L.E.; formal analysis, J.S., L.E., and Y.C.; investigation, J.S. and L.E.; resources, L.J.M. and J.S.; data curation, J.S.; writing: original draft, J.S. and L.E.; writing: review and editing, L.J.M., J.S., Y.C., and L.E.; visualization, J.S.; supervision, L.J.M. and Y.C.; project administration, L.J.M. and Y.C.; funding acquisition, L.J.M.
Declaration of Interests
The authors declare no competing interests.
Published: April 27, 2018
Footnotes
Supplemental Information includes Transparent Methods, four figures, and three tables and can be found with this article online at https://doi.org/10.1016/j.isci.2018.03.012.
Supplemental Information
References
- Agaimy A. Succinate dehydrogenase (SDH)-deficient renal cell carcinoma. Pathologe. 2016;37:144–152. doi: 10.1007/s00292-016-0158-8. [DOI] [PubMed] [Google Scholar]
- Astuti D., Latif F., Dallol A., Dahia P.L., Douglas F., George E., Sköldberg F., Husebye E.S., Eng C., Maher E.R. Gene mutations in the succinate dehydrogenase subunit SDHB cause susceptibility to familial pheochromocytoma and to familial paraganglioma. Am. J. Hum. Genet. 2001;69:49–54. doi: 10.1086/321282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baysal B.E., Ferrell R.E., Willett-Brozick J.E., Lawrence E.C., Myssiorek D., Bosch A., van der Mey A., Taschner P.E., Rubinstein W.S., Myers E.N. Mutations in SDHD, a mitochondrial complex II gene, in hereditary paraganglioma. Science. 2000;287:848–851. doi: 10.1126/science.287.5454.848. [DOI] [PubMed] [Google Scholar]
- Chen Y., Sprung R., Tang Y., Ball H., Sangras B., Kim S.C., Falck J.R., Peng J., Gu W., Zhao Y. Lysine propionylation and butyrylation are novel post-translational modifications in histones. Mol. Cell. Proteomics. 2007;6:812–819. doi: 10.1074/mcp.M700021-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choudhary C., Kumar C., Gnad F., Nielsen M.L., Rehman M., Walther T.C., Olsen J.V., Mann M. Lysine acetylation targets protein complexes and co-regulates major cellular functions. Science. 2009;325:834–840. doi: 10.1126/science.1175371. [DOI] [PubMed] [Google Scholar]
- Dai L., Peng C., Montellier E., Lu Z., Chen Y., Ishii H., Debernardi A., Buchou T., Rousseaux S., Jin F.L. Lysine 2-hydroxyisobutyrylation is a widely distributed active histone mark. Nat. Chem. Biol. 2014;10:365–U373. doi: 10.1038/nchembio.1497. [DOI] [PubMed] [Google Scholar]
- Du J.T., Zhou Y.Y., Su X.Y., Yu J.J., Khan S., Jiang H., Kim J., Woo J., Kim J.H., Choi B.H. Sirt5 is a NAD-dependent protein lysine demalonylase and desuccinylase. Science. 2011;334:806–809. doi: 10.1126/science.1207861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garrity J., Gardner J.G., Hawse W., Wolberger C., Escalante-Semerena J.C. N-Lysine propionylation controls the activity of propionyl-CoA synthetase. J. Biol. Chem. 2007;282:30239–30245. doi: 10.1074/jbc.M704409200. [DOI] [PubMed] [Google Scholar]
- Gill A.J., Lipton L., Taylor J., Benn D.E., Richardson A.L., Frydenberg M., Shapiro J., Clifton-Bligh R.J., Chow C.W., Bogwitz M. Germline SDHC mutation presenting as recurrent SDH deficient GIST and renal carcinoma. Pathology. 2013;45:689–691. doi: 10.1097/PAT.0000000000000018. [DOI] [PubMed] [Google Scholar]
- Huang da W., Sherman B.T., Lempicki R.A. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 2009;4:44–57. doi: 10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]
- Ito K., Barnes P.J., Adcock I.M. Glucocorticoid receptor recruitment of histone deacetylase 2 inhibits interleukin-1 beta-induced histone H4 acetylation on lysines 8 and 12. Mol. Cell. Biol. 2000;20:6891–6903. doi: 10.1128/mcb.20.18.6891-6903.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leemhuis H., Packman L.C., Nightingale K.P., Hollfelder F. The human histone acetyltransferase P/CAF is a promiscuous histone propionyltransferase. Chembiochem. 2008;9:499–503. doi: 10.1002/cbic.200700556. [DOI] [PubMed] [Google Scholar]
- Li L., Shi L., Yang S., Yan R., Zhang D., Yang J., He L., Li W., Yi X., Sun L. SIRT7 is a histone desuccinylase that functionally links to chromatin compaction and genome stability. Nat. Commun. 2016;7:12235. doi: 10.1038/ncomms12235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McManus K.J., Hendzel M.J. Specificity determination of CREB binding protein's (CBP) acetyltransferase activity for various histone lysine residues. Mol. Biol. Cell. 2000;11:307a. [Google Scholar]
- Michishita E., McCord R.A., Berber E., Kioi M., Padilla-Nash H., Damian M., Cheung P., Kusumoto R., Kawahara T.L., Barrett J.C. SIRT6 is a histone H3 lysine 9 deacetylase that modulates telomeric chromatin. Nature. 2008;452:492–496. doi: 10.1038/nature06736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nannini M., Astolfi A., Urbini M., Indio V., Santini D., Heinrich M.C., Corless C.L., Ceccarelli C., Saponara M., Mandrioli A. Integrated genomic study of quadruple-WT GIST (KIT/PDGFRA/SDH/RAS pathway wild-type GIST) BMC Cancer. 2014;14:685. doi: 10.1186/1471-2407-14-685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ni Y., Seballos S., Ganapathi S., Gurin D., Fletcher B., Ngeow J., Nagy R., Kloos R.T., Ringel M.D., LaFramboise T. Germline and somatic SDHx alterations in apparently sporadic differentiated thyroid cancer. Endocr. Relat. Cancer. 2015;22:121–130. doi: 10.1530/ERC-14-0537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niemann S., Müller U. Mutations in SDHC cause autosomal dominant paraganglioma, type 3. Nat. Genet. 2000;26:268–270. doi: 10.1038/81551. [DOI] [PubMed] [Google Scholar]
- Olsen J.V., Vermeulen M., Santamaria A., Kumar C., Miller M.L., Jensen L.J., Gnad F., Cox J., Jensen T.S., Nigg E.A. Quantitative phosphoproteomics reveals widespread full phosphorylation site occupancy during mitosis. Sci. Signal. 2010;3:ra3. doi: 10.1126/scisignal.2000475. [DOI] [PubMed] [Google Scholar]
- Pantaleo M.A., Nannini M., Corless C.L., Heinrich M.C. Quadruple wild-type (WT) GIST: defining the subset of GIST that lacks abnormalities of KIT, PDGFRA, SDH, or RAS signaling pathways. Cancer Med. 2015;4:101–103. doi: 10.1002/cam4.325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park J., Chen Y., Tishkoff D.X., Peng C., Tan M., Dai L., Xie Z., Zhang Y., Zwaans B.M., Skinner M.E. SIRT5-mediated lysine desuccinylation impacts diverse metabolic pathways. Mol. Cell. 2013;50:919–930. doi: 10.1016/j.molcel.2013.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piñeiro M., Hernández F., Palacián E. Succinylation of histone amino-groups facilitates transcription of nucleosomal cores. Biochim. Biophys. Acta. 1992;1129:183–187. doi: 10.1016/0167-4781(92)90485-i. [DOI] [PubMed] [Google Scholar]
- Rardin M.J., He W., Nishida Y., Newman J.C., Carrico C., Danielson S.R., Guo A., Gut P., Sahu A.K., Li B. SIRT5 regulates the mitochondrial lysine succinylome and metabolic networks. Cell Metab. 2013;18:920–933. doi: 10.1016/j.cmet.2013.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ravindra K.C., Selvi B.R., Arif M., Reddy B.A.A., Thanuja G.R., Agrawal S., Pradhan S.K., Nagashayana N., Dasgupta D., Kundu T.K. Inhibition of lysine acetyltransferase KAT3B/p300 activity by a naturally occurring hydroxynaphthoquinone, plumbagin. J. Biol. Chem. 2009;284:24453–24464. doi: 10.1074/jbc.M109.023861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sadhukhan S., Liu X.J., Ryu D., Nelson O.D., Stupinski J.A., Li Z., Chen W., Zhang S., Weiss R.S., Locasale J.W. Metabolomics-assisted proteomics identifies succinylation and SIRT5 as important regulators of cardiac function. Proc. Natl. Acad. Sci. USA. 2016;113:4320–4325. doi: 10.1073/pnas.1519858113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simithy J., Sidoli S., Yuan Z.F., Coradin M., Bhanu N.V., Marchione D.M., Klein B.J., Bazilevsky G.A., McCullough C.E., Magin R.S. Characterization of histone acylations links chromatin modifications with metabolism. Nat. Commun. 2017;8:1141. doi: 10.1038/s41467-017-01384-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smestad J., Hamidi O., Wang L., Holte M.N., Khazal F.A., Erber L., Chen Y., Maher L.J., 3rd Characterization and metabolic synthetic lethal testing in a new model of SDH-loss familial pheochromocytoma and paraganglioma. Oncotarget. 2017;9:6109–6127. doi: 10.18632/oncotarget.23639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tafrova J.I., Tafrov S.T. Human histone acetyltransferase 1 (Hat1) acetylates lysine 5 of histone H2A in vivo. Mol. Cell. Biochem. 2014;392:259–272. doi: 10.1007/s11010-014-2036-0. [DOI] [PubMed] [Google Scholar]
- Tan M., Luo H., Lee S., Jin F., Yang J.S., Montellier E., Buchou T., Cheng Z., Rousseaux S., Rajagopal N. Identification of 67 histone marks and histone lysine crotonylation as a new type of histone modification. Cell. 2011;146:1016–1028. doi: 10.1016/j.cell.2011.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan M.J., Peng C., Anderson K.A., Chhoy P., Xie Z., Dai L., Park J., Chen Y., Huang H., Zhang Y. Lysine glutarylation is a protein posttranslational modification regulated by SIRT5. Cell Metab. 2014;19:605–617. doi: 10.1016/j.cmet.2014.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thul P.J., Åkesson L., Wiking M., Mahdessian D., Geladaki A., Ait Blal H., Alm T., Asplund A., Bjork L., Breckels L.M. A subcellular map of the human proteome. Science. 2017;356 doi: 10.1126/science.aal3321. eaal3321. [DOI] [PubMed] [Google Scholar]
- Wagner G.R., Bhatt D.P., O'Connell T.M., Thompson J.W., Dubois L.G., Backos D.S., Yang H., Mitchell G.A., Ilkayeva O.R., Stevens R.D. A class of reactive Acyl-CoA species reveals the non-enzymatic origins of protein acylation. Cell Metab. 2017;25:823–837.e8. doi: 10.1016/j.cmet.2017.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H., Chen Y., Wu G. SDHB deficiency promotes TGFβ-mediated invasion and metastasis of colorectal cancer through transcriptional repression complex SNAIL1-SMAD3/4. Transl. Oncol. 2016;9:512–520. doi: 10.1016/j.tranon.2016.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warrener R., Chia K., Warren W.D., Brooks K., Gabrielli B. Inhibition of histone deacetylase 3 produces mitotic defects independent of alterations in histone H3 lysine 9 acetylation and methylation. Mol. Pharmacol. 2010;78:384–393. doi: 10.1124/mol.109.062976. [DOI] [PubMed] [Google Scholar]
- Xie Z., Dai J., Dai L., Tan M., Cheng Z., Wu Y., Boeke J.D., Zhao Y. Lysine succinylation and lysine malonylation in histones. Mol. Cell. Proteomics. 2012;11:100–107. doi: 10.1074/mcp.M111.015875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie Z., Zhang D., Chung D., Tang Z., Huang H., Dai L., Qi S., Li J., Colak G., Chen Y. Metabolic regulation of gene expression by histone lysine beta-hydroxybutyrylation. Mol. Cell. 2016;62:194–206. doi: 10.1016/j.molcel.2016.03.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiong B., Lu S.A., Gerton J.L. Hos1 is a lysine deacetylase for the Smc3 subunit of cohesin. Curr. Biol. 2010;20:1660–1665. doi: 10.1016/j.cub.2010.08.019. [DOI] [PubMed] [Google Scholar]
- Yang Y.Y., Grammel M., Hang H.C. Identification of lysine acetyltransferase p300 substrates using 4-pentynoyl-coenzyme A and bioorthogonal proteomics. Bioorg. Med. Chem. Lett. 2011;21:4976–4979. doi: 10.1016/j.bmcl.2011.05.060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang W., Bone J.R., Edmondson D.G., Turner B.M., Roth S.Y. Essential and redundant functions of histone acetylation revealed by mutation of target lysines and loss of the Gcn5p acetyltransferase. EMBO J. 1998;17:3155–3167. doi: 10.1093/emboj/17.11.3155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z., Tan M., Xie Z., Dai L., Chen Y., Zhao Y. Identification of lysine succinylation as a new post-translational modification. Nat. Chem. Biol. 2011;7:58–63. doi: 10.1038/nchembio.495. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.