

Abstract
Maternal obesity is associated with increased oxidative stress but decreased placental mitochondrial respiration and expression of mitochondrial electron transport chain (ETC) complexes I to V. Melatonin acts as an antioxidant and prevents oxidative stress-induced changes in cytotrophoblasts. Placentas were collected at term by cesarean delivery from obese (first trimester body mass index [BMI] ≥30, n = 10) or lean (BMI < 25, n = 6) women. Cytotrophoblasts were isolated and allowed to syncytialize for 72 hours with or without melatonin (0.1-100 µM) for the last 24 hours. Mitochondrial respiratory parameters were measured in a Seahorse XF24. Expression of ETC complexes I to V and antioxidant enzymes was measured by Western blot. Maternal clinical characteristics of patients were similar except for BMI. No significant improvement in mitochondrial respiration occurred with addition of melatonin to trophoblasts of lean women. However, in trophoblasts from obese women, melatonin (10 and 100 µmol/L) significantly increased maximal respiration (P = .01 and P = .009, respectively) and spare capacity (P = .02 and P = .003, respectively) compared to the untreated control. No differences were detected in the expression of ETC complexes and superoxide dismutase 1 or 2 in trophoblasts treated with melatonin. The expression of glutathione peroxidase, which was significantly greater in trophoblast of obese compared to lean women (P < .05), was decreased back to the level seen in trophoblast of lean women with addition of melatonin (P = .02). Improved spare respiratory capacity, the cellular reserve, could impart a protective effect to the placenta and fetus in an adverse intrauterine environment or in response to additional stressors.
Keywords: obesity, placenta, trophoblast, mitochondrial respiration, melatonin
Introduction
The World Health Organization recognizes obesity as a global pandemic.1 The number of reproductive-age women who are overweight or obese is increasing with 31.9% of reproductive-aged women (20-39 years) in the United States being obese (body mass index [BMI] ≥30 kg/m2.2,3 Obesity in pregnant women can lead to immediate complications for both mother and infant during pregnancy and may also have adverse long-term health consequences for the child. Overweight and obese women are at increased risk of gestational diabetes, hypertension, preeclampsia, and cesarean delivery.4,5 Fetal complications of maternal obesity include prematurity, stillbirth, congenital anomalies, macrosomia, and subsequent childhood and adolescent obesity.6-8
Pregnancy is a state of oxidative stress,9 which becomes exaggerated with pathologic conditions including obesity.10 Oxidative stress is described as an imbalance in the production of reactive oxygen species (ROS) and the ability of the antioxidant defenses to scavenge them. The capacity of the placental antioxidant defenses to counteract the effects of highly reactive and potentially damaging free radicals is critical for healthy placental function and optimal growth and development of the fetus.11 Placental mitochondria are a major site for ROS production and themselves become a target for ROS-induced damage and can be severely compromised by prolonged oxidative stress.12-14 Mitochondrial respiration can be measured in intact syncytiotrophoblast from human term placentas and provides a physiologic readout of function.15 Endogenous homeostatic ROS levels are tightly regulated by major antioxidant enzymes,16 which are present in the human placenta.10,17 These include the primary antioxidant enzyme superoxide dismutase (SOD) which converts superoxide to hydrogen peroxide and is found in both cytosol (SOD1) and mitochondria (SOD2) together with glutathione peroxidases (GPxs) found in both the mitochondrial matrix and the cytoplasm as part of a secondary defense mechanism catalyzing the conversion of hydrogen peroxide to water.18 When the balance between prooxidant and antioxidant activities is lost, deleterious outcomes such as syncytiotrophoblast apoptosis occurs.19
Placental mitochondrial function is compromised in pregnancies complicated by obesity. Syncytiotrophoblasts from placentas of obese women have significantly reduced mitochondrial respiration, decreased adenosine triphosphate (ATP) production, decreased expression of mitochondrial electron transport chain (ETC) complexes I to V, decreased activity of mitochondrial ETC complex I, and abnormal metabolic flexibility.20,21 In obesity, there are increased placental oxidative stress markers including increased levels of reduced glutathione and increased expression of catalase and SOD22 as well as a 14-fold increase in ROS production.20
Melatonin, an indoleamine that is rhythmically secreted in regulation of circadian rhythm, is a potent antioxidant that participates in the physiologic regulation of mitochondrial homeostasis. Melatonin improves mitochondrial function, reduces oxidative stress, and increases activity of the respiratory chain23,24 and acts directly by scavenging ROS and also indirectly by inducing gene expression of antioxidant enzymes including GPx, glutathione reductase, SOD, and catalase.16,25-27 Extrapineal sources of melatonin include local synthesis of melatonin in human trophoblasts where its receptors are also expressed.28 In vitro treatment of human cytotrophoblast with melatonin has been reported to reverse hypoxia/reoxygenation (H/R)-induced damage.29 Fetal tissues, especially the brain, are vulnerable to the effects of ROS.30 In an ovine model, maternal antenatal melatonin administration has been shown to improve fetal outcomes as evidenced by decreased newborn neurodevelopmental deficits and brain injury caused by intrauterine growth restriction (IUGR).31 There are currently phase I trials underway using antenatal maternal melatonin administration to establish whether melatonin will clinically benefit women and pregnancies with early-onset preeclampsia and IUGR with pregnancy prolongation and to determine the oxidative stress response in maternal, placental, and fetal circulation.32,33
We hypothesized that addition of melatonin to syncytiotrophoblasts isolated from placentas of obese women would lead to (1) improved mitochondrial respiration, (2) increased expression of mitochondrial ETC complexes, and (3) alterations in the expression of the antioxidant enzymes SOD1, SOD2, and GPx4.
Materials and Methods
Study Participants
We obtained placentas immediately following cesarean delivery at term without labor from otherwise uncomplicated pregnancies in women who were either lean (BMI < 25, n = 6) or obese (BMI ≥ 30, n = 10) during early pregnancy (<17 weeks’ gestational age). Deidentified relevant medical information was also collected under a protocol approved by the University of Texas Health Science Center at San Antonio institutional review board, to which pregnant women were recruited following written informed consent.
Isolation and Culture of Primary Cytotrophoblasts
Cytotrophoblasts were isolated from placental villous tissue using trypsin/DNAse digestion and percoll purification as described previously.15,20 Cytotrophoblasts were plated at a density of ∼3.5 × 106 cells on 6-well plates or ∼8 × 105 cells in Seahorse XF24 plates (Agilent, Santa Clara, California) and incubated for 72 hours to allow fusion and differentiation into syncytiotrophoblast. Cells were cultured at 37°C in 5% CO2 in air and media (Dulbecco’s modified Eagle medium/Hams F-12, supplemented with l-glutamine, penicillin, streptomycin, gentamicin, and 10% fetal bovine serum) was changed daily.15,20,34 For the last 24 hours (48-72 hours in culture), cells were cultured with or without melatonin 0.1, 1.0, 10, and 100 µmol/L. Stock melatonin was dissolved in dimethyl sulfoxide (M5250; Sigma-Aldrich, Billerica, MA). Melatonin has been previously shown to have no effect on human chorionic gonadotropin (hCG) production (a biochemical measure of villous cytotrophoblast differentiation rate) of in vitro cultured primary cytotrophoblasts.29 The melatonin concentrations were based on data from prior studies29,35 as well the calculated human physiologic dose to a level that would be well below the maternal no-adverse-effect-level (NOAEL) at 100 mg/kg/d and fetal NOAEL established at ≥200 mg/kg/d.36,37
Assessment of Mitochondrial Respiration
Mitochondrial respiration of the cultured syncytiotrophoblasts was measured using a Seahorse XF24 analyzer (Seahorse Bioscience, Agilent, Santa Clara, CA) as we have described previously.15 Data are expressed as oxygen consumption rate (OCR) in pmol/min. Total cellular protein was measured following each experiment by the Bradford method to normalize the mitochondrial OCR. The OCR was measured under basal conditions followed by the sequential addition of oligomycin (1 μmol/L), carbonyl cyanide p-trifluoromethoxy-phenylhydrazone (1 µmol/L), and a mixture of rotenone (3 μmol/L) and antimycin A (1.5 μmol/L) to determine the ATP-coupled respiration, maximal respiration, spare capacity, and nonmitochondrial respiration (Mito stress kit).15,20,34
Western Blots
In addition, separate syncytiotrophoblast cultures treated with and without melatonin for the last 24 hours were collected with buffer-containing protease and phosphatase inhibitors (Sigma-Aldrich, Billerica, MA). Total protein concentration of each sample was determined using Bradford reagent (Bio-Rad, Hercules, CA). Proteins were separated on 4% to 20% gradient precast gels (Bio-Rad), transferred onto nitrocellulose membranes, and blocked with 5% milk in 0.1% Tween, 20 mmol/L Tris (pH7.5)-buffered saline (TTBS; w/v) for 1 hour. Blots were probed with primary antibody in 1% nonfat milk powder/TTBS overnight at 4°C and were detected using appropriate peroxidase-conjugated secondary antibody in 5% milk/TTBS for 1 hour. Mitochondrial ETC complexes antibody cocktail (Abcam ab110411, Cambridge, MA) was used at 1:5000 dilution, anti-SOD1 antibody was used at 1:2000 dilution (Abcam ab16831, Cambridge, MA), anti-SOD2 was used at 1:10 000 dilution (EMD Millipore 06-984); and anti-GPx4 was used at 1:5000 (Abcam ab125066, Cambridge, MA). Protein bands were visualized by enhanced chemiluminescence (Millipore, Billerica, Massachusetts, USA) and band intensity was measured by densitometry using Gene Tools software (Syngene, Frederick, Mary land) and ImageJ software (National Institutes of Health). All gels were run at the same time, in the same buffers, and incubated with the same primary and secondary antibodies. The density of each band was normalized to β-actin.
Statistical Analysis
There were a total of 16 study participants across the range of maternal BMIs. Summary data are presented as means ± standard error of the mean (SEM). Because our data did not significantly deviate from normality for most variables (as tested using F-test or Brown-Forsythe test), comparisons between 2 groups was performed using a Student t-test and comparison of more than 2 groups was performed using 1- or 2-way analysis of variance (ANOVA) and Dunnett post hoc test. Mann-Whitney U test or Kruskal-Wallis test was utilized for nonparametric data. Given that there may be sexual dimorphism in the adaptation of the placenta,38 we also separated the samples by placental sex and evaluated for male and female differences. A P value of <.05 was considered significant.
Results
Demographic Data and Birth Outcome
Placentas were obtained from 6 lean (BMI < 25) and 10 obese (BMI ≥ 30) women at term at the time of scheduled repeat cesarean delivery. The demographics of the participants involved in this study are summarized in Table 1. As per study design, obese women had a significant higher maternal BMI (36.9 ± 2.0, mean ± SEM) at their initial prenatal visit when compared to lean pregnant women (21.6 ± 1.0, P < .001). Lean and obese women were otherwise matched for maternal age, gravidity, and gestational age at delivery. All of the women had an early entry to prenatal care including similar gestational age at initial visit. The mean weight gain during pregnancy of each group, although not statistically different, followed the Institute of Medicine recommendations.6 There were no differences in placental or infant weight. All of the pregnancies were otherwise uncomplicated, and there were no cases of gestational diabetes, preeclampsia, macrosomia, IUGR, or poor immediate fetal or neonatal outcomes.
Table 1.
Maternal and Pregnancy Clinical Characteristics.a
| Descriptions | Lean (BMI < 25) | Obese (BMI ≥ 30) | P Value |
|---|---|---|---|
| n | 6 | 10 | |
| Maternal age, years | 26.5 ± 2.0 | 27 ± 1.2 | .15 |
| Gravidity | 3.0 ± 0.2 | 3.2 ± 0.3 | .64 |
| Parity | 1.5 ± 0.2 | 1.7 ± 0.2 | .57 |
| Ethnicity (Hispanic/non-Hispanic) | 6/0 | 10/0 | |
| Early pregnancy BMI, kg/m2 | 21.6 ± 1.0 | 36.9 ± 2.0 | <.001 |
| Gestational age at initial prenatal visit, weeks | 14.5 ± 2.7 | 12.3 ± 1.8 | .47 |
| Gestational age at delivery, weeks | 39.2 ± 0.1 | 39.3 ± 0.1 | .48 |
| Gestational weight gain, lb | 24.9 ± 2.8 | 19.3 ± 3.1 | .15 |
| Placental weight, g | 565 ± 32 | 693 ± 67 | .09 |
| Infant weight, g | 3234 ± 62 | 3484 ± 91 | .06 |
| Infant sex, M/F | 4/2 | 5/5 |
Abbreviations: BMI, body mass index; M, male; F, female.
aValues are mean ± SEM.
The Effect of Melatonin on Placental Mitochondrial Respiration in Trophoblasts of Obese Women
We have previously shown that mitochondrial respiration is reduced in trophoblasts of obese versus lean women.20 Representative tracings of mitochondrial respiration of the cultured syncytiotrophoblasts from an obese and a lean woman, as measured by the Seahorse XF24 analyzer and showing the effect of addition of increasing concentrations of melatonin, are shown in Figure 1. Addition of melatonin to trophoblasts of lean women did not have a significant effect with either improvement or detrimental decrease in basal respiration, ATP-coupled respiration, nonmitochondrial respiration, maximal respiration, and spare capacity (Figure 2A-D). Melatonin also had no effect on basal respiration or ATP-coupled respiration in trophoblasts of obese women (Figure 3A and B). However, in trophoblasts from obese women, which we have previously shown to have significantly lower maximal respiration compared to trophoblasts of lean women,20 there is a significant concentration-dependent increase in maximal respiration upon addition of melatonin as shown by 1-way ANOVA (P = .008) which reached significance at melatonin concentrations of 10 and 100 µmol/L (P = .01 and P = .009; Figure 3C). Correspondingly, evaluation of spare capacity in trophoblasts from obese women demonstrates a significant concentration-dependent increase in spare capacity upon addition of melatonin (one-way ANOVA, P = .003), which reached significance at 10 and 100 µmol/L melatonin (P = .02 and P = .003; Figure 3D). Indeed, maximal respiration and spare capacity of trophoblasts from obese women is increased by melatonin to a level similar to that found in trophoblast of lean patients (Figure 4A and B).
Figure 1.

Effect of melatonin on mitochondrial oxygen consumption rate: representative traces of mitochondrial respiration of the cultured synctiotrophoblasts from (A) an obese woman and (B) a lean woman with or without increasing melatonin concentrations (0.1-100 µmol/L). Each data point is a specific oxygen consumption rates (OCR) measurement in time representing the parameters of basal respiration, ATP-coupled respiration, maximal respiration, and nonmitochondrial respiration. ATP indicates adenosine triphosphate.
Figure 2.

Effect of melatonin on respiratory parameters in trophoblasts of lean women: mitochondrial respiratory parameters of the syncytiotrophoblasts from lean women (n = 5), with and without melatonin (0.1-100 µmol/L) were measured in SeahorseXF24. Measurements in the presence of melatonin were expressed as a ratio normalized to the values in the untreated control, scatter plot with mean ± SEM. Basal respiration (A), ATP-coupled respiration (B), maximal respiration (C), and spare capacity (D). ATP indicates adenosine triphosphate; SEM, standard error of the mean.
Figure 3.

Effect of melatonin on respiratory parameters in trophoblasts of obese women: mitochondrial respiratory parameters of the synctiotrophoblasts from obese women (n = 9), with and without melatonin (0.1-100 µmol/L) were measured in SeahorseXF24. Measurements in the presence of melatonin were expressed as a ratio normalized to the values in the untreated control, scatter plot with mean ± SEM. Basal respiration (A), ATP-coupled respiration (B), maximal respiration (C), and spare capacity (D). C, Obese maximal respiration melatonin versus control: 10 µmol/L (**P = .01) and 100 µmol/L (*P = .009). D, Obese spare capacity melatonin versus control: 10 µmol/L (**P = .02) and 100 µmol/L (*P = .003). ATP indicates adenosine triphosphate; SEM, standard error of the mean.
Figure 4.
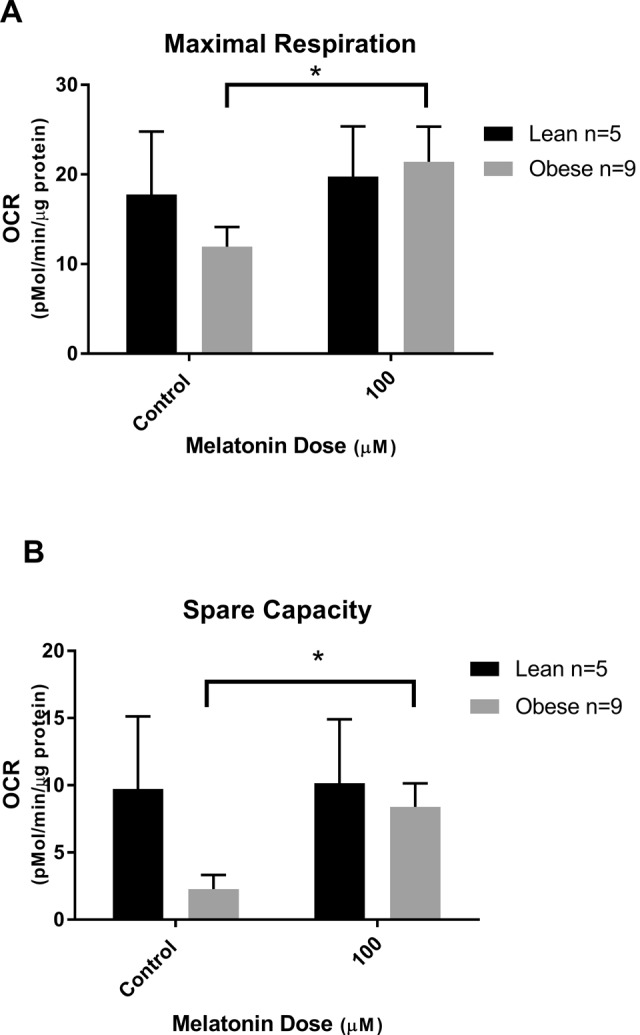
Improvement in maximal respiration and spare capacity by melatonin: oxygen consumption rate (OCR) in trophoblasts from lean and obese women with or without addition of 100 μmol/L melatonin. (A) Maximal respiration 100 µmol/L melatonin versus control (*P = .009) and (B) spare capacity 100 µmol/L melatonin versus control (*P = .003). Mean ± SEM. SEM indicates standard error of the mean.
When stratifying the data by placental sex, there were also no sex-specific differences detected in OCR in either lean or obese placentas with the addition of melatonin (data not shown).
Expression of Mitochondrial ETC Complexes and Antioxidant Enzymes
To determine the expression of ETC complexes and antioxidant enzymes, trophoblasts were cultured from placentas of lean (n = 4) and obese (n = 7) women and treated with or without melatonin 100 µmol/L for 24 hours. Unfortunately, cellular homogenates were not available from all samples to study. Western blotting using an antibody cocktail recognizing epitopes of purified selected subunits of complexes I to V on syncytiotrophoblasts from lean and obese placentas was performed (Figure 5A). We were unable to visualize complex II on the Western blot, and there were no differences detected in the expression of the mitochondrial, ETC complexes I, III, IV, and V in trophoblasts from lean or obese women treated with melatonin (Figure 5B to E, lean not shown). To determine whether melatonin’s effect on OCR is mediated by alteration in the expression of antioxidant enzymes, Western blotting of SOD1, SOD2, and GPx4 was performed (Figure 6A and B). In trophoblasts of both lean and obese women, addition of melatonin resulted in no significant alteration in the expression of SOD1 or SOD2 (Figure 6C and D, lean not shown). While expression of GPx4, which has a high preference for lipid hydroperoxides, was significantly greater in untreated trophoblast of obese compared to lean women (P < .05), the addition of melatonin significantly reduced (P = .02) GPx4 expression to a level not significantly different from that in lean women (Figure 6E). There was no effect of melatonin on GPx4 expression in trophoblast of lean women.
Figure 5.

Expression of mitochondrial electron transport chain complexes: (A) representative Western blots of complexes I, III, IV, and V in trophoblast of lean (n = 4) and obese (n = 7) women treated with and without melatonin 100 µmol/L for 24 hours. Relative protein expression of complex I (B), complex III (C), complex IV (D) or complex V (E) in trophoblasts of obese women. Represented in box and whiskers plot with median, minimum, and maximum values shown due to nonparametric distribution of data.
Figure 6.

Expression of antioxidant enzymes: (A) representative Western blots of SOD1 and SOD2 and (B) GPx4 in trophoblast of lean (n = 4) and obese (n = 7) women with and without the addition of melatonin (100 μmol/L) for 24 hours. Relative protein expression of SOD1 (C) and SOD2 (D) in trophoblasts of obese women. E, GPx4 protein expression was greater in trophoblast of obese compared to lean women (#P < .05) and the addition of melatonin significantly reduced (*P = .02) GPx4 expression to a level not significantly different from that in lean women. Represented in box and whiskers plot with median, minimum, and maximum values shown due to nonparametric distribution of data. SOD indicates superoxide dismutase; GPx, glutathione peroxidase.
Discussion
We investigated the effect of a potent antioxidant, melatonin, on the mitochondrial respiratory function of trophoblasts from obese women in vitro. We have shown that certain respiratory parameters in syncytiotrophoblasts from obese women were significantly improved with the addition of melatonin. Maximal respiration is, as the name suggests, the maximal mitochondrial oxygen consumption a cell can achieve, and was significantly improved in trophoblasts of obese placentas upon addition of melatonin. Spare capacity, or reserve respiratory capacity, is the difference between ATP produced by oxidative phosphorylation at the basal resting state and at maximal activity39 and represents the ability of cells to increased work and to respond to stress.15 Many cells operate at a basal level that only requires a part of their total bioenergetics capability. It is perhaps not surprising then that there is no change in basal OCR of trophoblast from obese women or from lean women upon addition of melatonin. Similarly, Lanoix et al found that melatonin had no effect on cytotrophoblasts under normoxic conditions but did lead to improved oxidative stress and apoptosis-induced changes in trophoblasts that had been stressed via H/R.29 Exhaustion of spare capacity has been correlated with variety of pathologies including heart disease,40 neurodegenerative disorders,41,42 and cell death in smooth muscle.43 If the energy requirement supersedes what the spare capacity can provide, the cell and affected tissue risk cell death.39 Improved spare respiratory capacity could impart a protective effect to placenta and fetus in response to additional stressors, such as the medical comorbidities and complications that occur in conjunction with obesity including preeclampsia, chronic hypertension or other vasculopathies, IUGR, or macrosomia.
There also appears to be different overall mitochondrial OCR response to the addition of melatonin in syncytiotrophoblasts from lean versus obese women. In syncytiotrophoblasts of obese women, a significant concentration-dependent increase in OCR from 0.1 to 100 μmol/L melatonin is seen. However, in trophoblasts of lean women, there is no significant effect with an increasing concentration of melatonin. There is considerable variability in response in trophoblasts of lean women, and we acknowledge that an increase number of samples may have reduced this variability. We have previously shown decreased respiratory parameters in placental mitochondria from obese compared to lean women,20 and the overall aim of this study was to determine whether the depressed respiratory function in obesity could be improved with addition of melatonin. The ROS induces pathology as part of oxidative stress, but they also serve as signaling molecules to regulate biological and physiological processes.44 In rat vascular smooth muscle, ligand binding of growth factors stimulates a burst of ROS, and inhibition of the ROS rise was shown to block normal signaling.45,46 Therefore, alterations in this homeostasis, in a cell without dysfunctional oxidative stress, could lead to detrimental cellular effects. The lack of effect of melatonin in trophoblasts from lean women perhaps reflects their low level of oxidative stress but also that there are no detrimental effects of melatonin. Similarly, in Lanoix et al’s study, the addition of melatonin to cytotrophoblasts in vitro showed no effect on hCG production and synctialization.29
The enzymes synthesizing melatonin and melatonin receptors are expressed in the human placenta throughout pregnancy, and melatonin promotes syncytiotrophoblast formation.47 Hence, melatonin likely plays an essential role in the normal function of the placenta and therefore may be a viable treatment option to protect against oxidative stress generated in the setting of maternal obesity. Maternal melatonin administration has been shown to improve outcomes in animal models in conditions of increased oxidative stress and ameliorates placental insufficiency and the detrimental newborn neurodevelopmental effects of IUGR.31,48 Administration of melatonin in rats with undernourished pregnancies, a model known to promote oxidative stress, leads to improved placental efficiency and restored birth weight.31 Antenatal maternal melatonin treatment normalized myelination and rescued axonopathy within IUGR lamb brains and gave functional improvements with reduced time to suckle at birth.48
We found that acute melatonin treatment for 24 hours had no significant effect on the expression of mitochondrial respiratory chain complexes. The effect of melatonin may therefore be via reduction in oxidative stress or increased respiratory chain activity rather than increased expression of the complexes in this time period. In vitro administration of melatonin to mouse liver cells has been shown to increase the activity of respiratory complexes I, III, and IV.35 Melatonin administration has been reported to prevent the alteration of antioxidant enzymes induced by H/R including prevention of the H/R-induced reduction in SOD1, SOD2, GPx mRNA and protein expression, and enzymatic activity in cytotrophoblasts.29 We find no effect of melatonin on either the cytosolic or the mitochondrial isoforms (SOD1 and 2) of SOD in trophoblasts from lean or obese women. However, GPx4 was significantly increased in trophoblasts of obese compared to lean women, probably as a response to the increased oxidative stress but was reduced, back to the levels seen in the lean placentas, with the addition of melatonin. This would imply that melatonin decreased oxidative stress and the need for increased antioxidant enzymes. We have recently (Evans and Myatt) shown decreased SOD1 activity and protein expression in placental homogenates from obese women compared to lean women, which is significant when the fetus is male. In contrast, placental GPx activity was increased with obesity and significantly so if the fetus is male, which we suggest is a compensatory response to attempt to decrease oxidative stress.49 In the present study, GPx4 expression is increased in the placenta of obese compared to lean women which agrees with this concept and is further reinforced by the overall antioxidant effect of melatonin, reducing the compensatory increase of GPx4. The GPxs are selenoenzymes thought to play an important role in protecting trophoblast mitochondria from oxidative stress. Indeed prior incubation of trophoblasts with Na selenite and selenomethionine mitigated a significant dose-dependent decrease in GPx activity after induced oxidative stress with rotenone and antimycin.50 The primary effect of melatonin may not be via induction of antioxidant enzyme expression per se, but by another mechanism of reducing oxidative stress to which GPx4, which specifically targets lipid hydroperoxides, appears to be secondarily responsive.
Strengths of the study include the use of tissue collected at cesarean delivery to avoid labor-induced oxidative stress, the ethnically homogeneous population, the absence of confounding medical conditions other than obesity, and the absence of adverse pregnancy outcomes. Limitations of this study include an ethnically homogenous population that may not represent effects seen in other ethnicities, and the small sample size, which also limits stratification by placental sex. The participants have an ethnic background which is representative of the patient population in our region. However, the effect of maternal obesity on the placenta may show ethnic variances. For example, in a cohort of predominantly African American women, maternal obesity was reported to be associated with placental macrophage accumulation and inflammation, whereas a similar study in caucasian women failed to find infiltration of immune cells in the placentas of obese women.51,52 In the setting of poor clinical outcome, more progressive placental dysfunction and abnormalities may be seen and is an area that should be expanded in future research. Indeed, we have already described further decrements in trophoblast respiration in placentas of women with GDM compared to their obese BMI-matched controls.34 In a recent meta-analysis, even modest increases in maternal BMI were associated with increased risk of fetal death, stillbirth, and neonatal, perinatal, and infant death, but the greatest risk was observed in the category of severely obese women—women with a BMI of 40 had an approximate 2- to 3-fold increase in the relative risk (RR) of these outcomes versus those with a BMI of 20, with absolute risks in the range of 0.69% to 2.7% for BMI of 40 versus 0.20% to 0.76% for BMI of 20.8 Placental oxidative stress and mitochondrial dysfunction may play a role in this pathophysiology. Although the clinical outcomes were normal, the mitochondrial function of placentas from obese women was decreased and could be rescued with melatonin administration in vitro.
Acknowledgments
The authors thank women who donated their placentas for this study. KM would like to thank Sribalasubashini Muralimanoharan, Calais Prince, and LaShauna Evans for their technical support in the laboratory.
Authors’ Note: This study was presented at the 63rd Annual Scientific Meeting, Society for Reproductive Investigation, Montreal, Quebec, Canada, March 2016.
Declaration of Conflicting Interests: The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding: The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This study was supported by NIH HD076259 (AM and LM).
References
- 1. World Health Organization. Global prevalence and secular trends in obesity In: Obesity: Preventing and Managing the Global Epidemic: Report of a WHO Consultation. Vol 894 Geneva, Switzerland: World Health Organization; 2000. [PubMed] [Google Scholar]
- 2. Flegal KM, Carroll MD, Kit BK, Ogden CL. Prevalence of obesity and trends in the distribution of body mass index among us adults, 1999–2010. JAMA. 2012;307(5):491–497. [DOI] [PubMed] [Google Scholar]
- 3. Ogden C, Kit B, Flegal K. Prevalence of obesity among adults: United States, 2011–2012. NCHS Data Brief. 2013;(131):1–8. [PubMed] [Google Scholar]
- 4. American College of Obstetricians and Gynecologists. ACOG committee opinion no. 549: obesity in pregnancy. Obstet Gynecol. 2013;121(1):213–217. [DOI] [PubMed] [Google Scholar]
- 5. Lu GC, Rouse DJ, DuBard M, et al. The effect of the increasing prevalence of maternal obesity on perinatal morbidity. Am J Obstet Gynecol. 2001;185(4):845–849. [DOI] [PubMed] [Google Scholar]
- 6. American College of Obstetricians and Gynecologists. ACOG committee opinion no. 548: weight gain during pregnancy. Obstet Gynecol. 2013;121(1):210–212. [DOI] [PubMed] [Google Scholar]
- 7. Scott-Pillai R, Spence D, Cardwell CR, Hunter A, Holmes VA. The impact of body mass index on maternal and neonatal outcomes: a retrospective study in a UK obstetric population, 2004–2011. BJOG. 2013;120(8):932–939. [DOI] [PubMed] [Google Scholar]
- 8. Aune D, Saugstad O, Henriksen T, Tonstad S. Maternal body mass index and the risk of fetal death, stillbirth, and infant death: a systematic review and meta-analysis. JAMA. 2014;311(15):1536–1546. [DOI] [PubMed] [Google Scholar]
- 9. Wisdom S, Wilson R, McKillop JH, Walker JJ. Antioxidant systems in normal pregnancy and in pregnancy-induced hypertension. Am J Obstet Gynecol. 1991;165(6 pt 1):1701–1754. [DOI] [PubMed] [Google Scholar]
- 10. Myatt L, Cui X. Oxidative stress in the placenta. Histochem Cell Biol. 2004;122(4):369–382. [DOI] [PubMed] [Google Scholar]
- 11. Longtine MS, Nelson DM. Placental dysfunction and fetal programming: the importance of placental size, shape, histopathology, and molecular composition. Semin Reprod Med. 2011;29(3):187–196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Ishihara N, Matsuo H, Murakoshi H, et al. Increased apoptosis in the syncytiotrophoblast in human term placentas complicated by either preeclampsia or intrauterine growth retardation. Am J Obstet Gynecol. 2002;186(1):158–166. [DOI] [PubMed] [Google Scholar]
- 13. Burton GJ, Jauniaux E. Oxidative stress. Best Pract Res Clin Obstet Gynaecol. 2011;25(3):287–299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Lanoix D, Guérin P, Vaillancourt C. Placental melatonin production and melatonin receptor expression are altered in preeclampsia: new insights into the role of this hormone in pregnancy. J Pineal Res. 2012;53(4):417–425. [DOI] [PubMed] [Google Scholar]
- 15. Maloyan A, Mele J, Muralimanohara S, Myatt L. Measurement of mitochondrial respiration in trophoblast culture. Placenta. 2012;33(5):456–458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Rodriguez C, Mayo JC, Sainz RM, et al. Regulation of antioxidant enzymes: a significant role for melatonin. J Pineal Res. 2004;36(1):1–9. [DOI] [PubMed] [Google Scholar]
- 17. Hempstock J, Bao YP, Bar-Issac M, et al. Intralobular differences in antioxidant enzyme expression and activity reflect the pattern of maternal arterial bloodflow within the human placenta. Placenta. 2003;24(5):517–523. [DOI] [PubMed] [Google Scholar]
- 18. Fridovich I. Fundamental aspects of reactive oxygen species, or what’s the matter with oxygen? Ann N Y Acad Sci. 1999;893(1):13–18. [DOI] [PubMed] [Google Scholar]
- 19. Cindrova-Davies T, Yung HW, Johns J, et al. Oxidative stress, gene expression, and protein changes induced in the human placenta during labor. Am J Pathol. 2007;171(4):1168–1179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Mele J, Muralimanoharan S, Maloyan A, Myatt L. Impaired mitochondrial function in human placenta with increased maternal adiposity. Am J Physiol Endocrinol Metab. 2014;307(5):E419–E425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Hastie R, Lappas M. The effect of pre-existing maternal obesity and diabetes on placental mitochondrial content and electron transport chain activity. Placenta. 2014;35(9):673–683. [DOI] [PubMed] [Google Scholar]
- 22. Malti N, Merzouk H, Merzouk SA, et al. Oxidative stress and maternal obesity: feto-placental unit interaction. Placenta. 2014;35(6):411–416. [DOI] [PubMed] [Google Scholar]
- 23. Acuna-Castroviejo D, Escames G, Rodriguez MI, Lopez LC. Melatonin role in the mitochondrial function. Front Biosci. 2007;12:947–963. [DOI] [PubMed] [Google Scholar]
- 24. Acuña-Castroviejo D, Escames G, López LC, Hitos AB, León J. Melatonin and nitric oxide: two required antagonists for mitochondrial homeostasis. Endocrine. 2005;27(2):159–168. [DOI] [PubMed] [Google Scholar]
- 25. García JJ, López-Pingarrón L, Almeida-Souza P, et al. Protective effects of melatonin in reducing oxidative stress and in preserving the fluidity of biological membranes: a review. J Pineal Res. 2014;56(3):225–237. [DOI] [PubMed] [Google Scholar]
- 26. Mayo JC, Sainz RM, Antolín I, et al. Melatonin regulation of antioxidant enzyme gene expression. Cell Mol Life Sci. 2002;59(10):1706–1713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Reiter RJ, Tan D-X. Melatonin: a novel protective agent against oxidative injury of the ischemic/reperfused heart. Cardiovasc Res. 2003;58(1):10–19. [DOI] [PubMed] [Google Scholar]
- 28. Lanoix D, Beghdadi H, Lafond J, Vaillancourt C. Human placental trophoblasts synthesize melatonin and express its receptors. J Pineal Res. 2008;45(1):50–60. [DOI] [PubMed] [Google Scholar]
- 29. Lanoix D, Lacasse AA, Reiter RJ, Vaillancourt C. Melatonin: the watchdog of villous trophoblast homeostasis against hypoxia/reoxygenation-induced oxidative stress and apoptosis. Mol Cell Endocrinol. 2013;381(1-2):35–45. [DOI] [PubMed] [Google Scholar]
- 30. Miller SL, Wallace EM, Walker DW. Antioxidant therapies: a potential role in perinatal medicine. Neuroendocrinology. 2012;96(1):13–23. [DOI] [PubMed] [Google Scholar]
- 31. Miller SL, Yawno T, Alers NO, et al. Antenatal antioxidant treatment with melatonin to decrease newborn neurodevelopmental deficits and brain injury caused by fetal growth restriction. J Pineal Res. 2014;56(3):283–294. [DOI] [PubMed] [Google Scholar]
- 32. Hobson SR, Lim R, Gardiner EE, Alers NO, Wallace EM. Phase I pilot clinical trial of antenatal maternally administered melatonin to decrease the level of oxidative stress in human pregnancies affected by pre-eclampsia (PAMPR): study protocol. BMJ Open. 2013;3(9):e003788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Alers NO, Jenkin G, Miller SL, Wallace EM. Antenatal melatonin as an antioxidant in human pregnancies complicated by fetal growth restriction—a phase I pilot clinical trial: study protocol. BMJ Open. 2013;3(12):e004141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Muralimanoharan S, Maloyan A, Myatt L. Mitochondrial function and glucose metabolism in the placenta with gestational diabetes mellitus: role of miR-143. Clin Sci (Lond). 2016;130(11):931–941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. López A, García JA, Escames G, et al. Melatonin protects the mitochondria from oxidative damage reducing oxygen consumption, membrane potential, and superoxide anion production. J Pineal Res. 2009;46(2):188–198. [DOI] [PubMed] [Google Scholar]
- 36. Okatani Y, Okamoto K, Hayashi K, et al. Maternal–fetal transfer of melatonin in pregnant women near term. J Pineal Res. 1998;25(3):129–134. [DOI] [PubMed] [Google Scholar]
- 37. Jahnke G, Marr M, Myers C, et al. Maternal and developmental toxicity evaluation of melatonin administered orally to pregnant Sprague-Dawley rats. Toxicol Sci. 1999;50(2):271–279. [DOI] [PubMed] [Google Scholar]
- 38. Muralimanoharan S, Maloyan A, Myatt L. Evidence of sexual dimorphism in the placental function with severe preeclampsia. Placenta. 2013;34(12):1183–1189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Desler C, Hansen TL, Frederiksen JB, Marcker ML, Singh KK, Juel Rasmussen L. Is there a link between mitochondrial reserve respiratory capacity and aging? J Aging Res. 2012;2012:192503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Sansbury BE, Jones SP, Riggs DW, Darley-Usmar VM, Hill BG. Bioenergetic function in cardiovascular cells: the importance of the reserve capacity and its biological regulation. Chem Biol Interact. 2011;191(1-3):288–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Yadava N, Nicholls DG. Spare respiratory capacity rather than oxidative stress regulates glutamate excitotoxicity after partial respiratory inhibition of mitochondrial complex I with rotenone. J Neurosci. 2007;27(27):7310–7317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Nicholls DG. Oxidative stress and energy crises in neuronal dysfunction. Ann N Y Acad Sci. 2008;1147(1):53–60. [DOI] [PubMed] [Google Scholar]
- 43. Hill BG, Higdon AN, Dranka BP, Darley-Usmar VM. Regulation of vascular smooth muscle cell bioenergetic function by protein glutathiolation. Biochim Biophys Acta. 2010;1797(2):285–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Schieber M, Chandel Navdeep S. ROS function in redox signaling and oxidative stress. Curr Biol. 2014;24(10):R453–R462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Bae YS, Kang SW, Seo MS, et al. Epidermal growth factor (EGF)-induced generation of hydrogen peroxide: role in EGF receptor-mediated tyrosine phosphorylation. J Biol Chem. 1997;272(1):217–221. [PubMed] [Google Scholar]
- 46. Sundaresan M, Yu ZX, Ferrans VJ, Irani K, Finkel T. Requirement for generation of H2O2 for platelet-derived growth factor signal transduction. Science. 1995;270(5234):296–299. [DOI] [PubMed] [Google Scholar]
- 47. Soliman A, Lacasse AA, Lanoix D, et al. Placental melatonin system is present throughout pregnancy and regulates villous trophoblast differentiation. J Pineal Res. 2015;59(1):38–46. [DOI] [PubMed] [Google Scholar]
- 48. Richter HG, Hansell JA, Raut S, Giussani DA. Melatonin improves placental efficiency and birth weight and increases the placental expression of antioxidant enzymes in undernourished pregnancy. J Pineal Res. 2009;46(4):357–364. [DOI] [PubMed] [Google Scholar]
- 49. Evans L, Myatt L. Sexual dimorphism in the effect of maternal obesity on antioxidant defense mechanisms in the human placenta. Placenta. 2017;51:64–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Khera A, Vanderlelie JJ, Perkins AV. Selenium supplementation protects trophoblast cells from mitochondrial oxidative stress. Placenta. 2013;34(7):594–598. [DOI] [PubMed] [Google Scholar]
- 51. Challier JC, Basu S, Bintein T, et al. Obesity in pregnancy stimulates macrophage accumulation and inflammation in the placenta. Placenta. 2008;29(3):274–281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Roberts KA, Riley SC, Reynolds RM, et al. Placental structure and inflammation in pregnancies associated with obesity. Placenta. 2011;32(3):247–254. [DOI] [PubMed] [Google Scholar]