

Abstract
Precision medicine has shed new light on the treatment of heterogeneous cancer patients. However, intratumor heterogeneity strongly constrains the clinical benefit of precision medicine. Thus, rethinking therapeutic strategies from a different facet within the precision medicine framework will not only diversify clinical interventions, but also provide an avenue for precision medicine. Here, we explore the current approaches for targeting intratumor heterogeneity and their limitations. Furthermore, we propose a theoretical strategy with a “homogenization” feature based on iatrogenic evolutionary selection to target intratumor heterogeneity.
Keywords: Acquired drug resistance, Homogenization, Intratumor heterogeneity, Plasticity, Precision medicine
Background
Tumor heterogeneity includes intertumor and intratumor heterogeneities. Genetic and phenotypic variations are observed among different tumor patients [1, 2]. Extremely high genetic diversity makes each patient unique and distinct. However, within a tumor, both genomic instability and the tumor subclone architecture vary over time [3–8]. As the tumor evolves, the parental subclone acquires an increasing number of genetic and epigenetic alterations, resulting in a tumor with different subclone phenotypes.
Intratumor heterogeneity is characterized by its dynamic changes. Tumor initiation and progression are generated from stochastic to sequential mutations that contribute to subsequent clonal expansion and intratumor heterogeneity [9]. Therefore, a single biopsy is unlikely to capture the complete genomic landscape of a patient’s tumor, considering the spatiotemporal changes in tumor heterogeneity [10]. Consequently, even if the subclone harboring the detected molecular phenotype has been targeted effectively, other subclones of the tumor may still grow. Moreover, the sensitive subclone may become resistant to therapy, causing further disease deterioration.
Tumor homogeneity refers to the cellular populations bearing the same or similar genetic or epigenetic characters within the same lesion or in different lesions of the same patient. Here, we propose an ideal situation in which the tumor becomes a homogeneous cell population, i.e., tumor cells that acquire common molecular properties. Once tumor heterogeneity is drastically confined in this manner, the cells are susceptible to a single intervention that targets this particular feature. Recent technological advances in both molecular diagnostics and targeted drugs have led to the theory of “acquiring tumor homogeneity”.
This review summarizes the recent understanding and clinical practice of precision therapy, and illustrates the current strategies and limitations for targeting intratumor heterogeneity. Then, we discuss the possibility and implementation of “homogenization” therapy for precision medicine.
Intratumor heterogeneity challenges precision medicine
Precision therapy exerts profound effects on cancer patients. Sequencing technology and genomic analyses are driving the progress of precision therapy. In the clinic, molecular diagnosis has been applied to biopsies of tumor tissues to guide the selection of precision therapy [11, 12]. However, the outcomes of clinical trials regarding the assessment of precision therapy are discouraging. For example, the SHIVA trial showed no significant difference in progression-free survival (PFS) between targeted and conventional therapies [13]. The Princess Margaret IMPACT-COMPACT study reported a non-randomized comparison, which indicated an objective tumor response rate of 20% in the matched group (between genotype and targeted therapy) versus 11% in the unmatched group [14].
Intratumor heterogeneity and plasticity
Precision therapy has been hindered by multiple factors, resulting in the limited success of clinical therapy. Tannock et al. reviewed the problem and concluded that Darwinian evolution leading to intratumor heterogeneity may weaken the effect of precision therapy [15]. Genetic alterations, including aneuploid rearrangements and point mutations, generate extensive clonal diversity [16]. A recent study [17] showed that more than 100 million coding region mutations exist in a single tumor, and such high genetic variation subverts the effect of targeted therapy. Moreover, genetically [18, 19] and epigenetically unstable tumorigenic cells contribute to tumor plasticity. Evidence from both cell line [20–23] and animal model [24, 25] indicates that tumorigenic cells display cellular plasticity that allows them to transit between different states. Overall, intratumor heterogeneity and plasticity co-exist within a tumor, and the unification of which comprehensively illustrates the difficulty of precision therapy.
Based on the current knowledge of intratumor heterogeneity and plasticity, two strategies have been proposed to partly solve the problem (Fig. 1). First, targeting a shared pathway may be practical when parallel mutations leading to pathway convergence are detected [26]. For example, different molecular subtypes of breast cancer share pathways including Notch [27], Wnt [28], Her-2 [29], and STAT3-NF-kB [30]. In renal cancer, constraints in activation of the PI3K/mTOR pathway, which manifests as the shared pathway of mutations in PTEN, PIK3CA, TSC1, or mTOR, might be exploitable for therapeutic benefit [31]. Therefore, despite the diversity of numerous mutations, these mutations affect the same pathways, and agents that target these pathways may maximize the benefit of precision therapy [32–34].
Fig. 1.
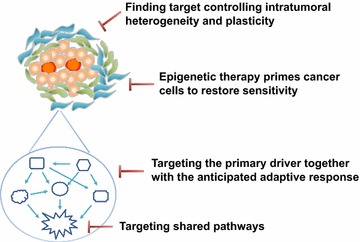
Current strategies targeting intratumor heterogeneity. Four strategies to solve this problem are as follows. First, targeting a shared pathway. Second, targeting the primary mutation together with the anticipated mutation. Third, finding a target controlling intratumor heterogeneity and plasticity. Fourth, epigenetic therapy that primes cancer to restore sensitivity
The second strategy is to block cellular plasticity by preventing the transition between cellular states, which may increase therapeutic efficacy. This strategy includes inhibitors of c-Met [35] and TGF-β [36]. Moreover, two recent studies [24, 25] showed that mutant PIK3CA in breast cancer induces multipotency in lineage-committed basal and luminal cells, which drives plasticity and intratumor heterogeneity. Likewise, in pancreatic cancer, PI3K/PDK1 signaling pathway mediates cellular plasticity and acts as a key effector of oncogenic Kras [37]. An interesting recent study [38] suggested that p53 is essential for DNA methylation homeostasis in embryonic stem cells, and the loss of which promotes clonal heterogeneity. Taken together, identifying the mechanisms contributing to intratumor heterogeneity is imperative to identify suitable targets for therapeutic interventions.
Intratumor heterogeneity and acquired drug resistance
Therapeutic intervention can induce a drug-tolerant phenotype in the absence of a pre-existing resistant clone [39, 40]. A recent study demonstrated that application of a drug initiates cellular reprogramming, revealing a mechanism of acquired drug resistance [41]. Consequently, continued targeted therapy in the presence of resistant subclones might accelerate tumor progression [42]. For example, continued BRAF inhibitor treatment results in tumor metastasis of RAS- and BRAF-mutant melanoma cells [43] and paradoxical activation of the RAS-ERK pathway in multiple myeloma clones [44]. Therefore, blind or persistent use of targeted therapy for a drug-resistant tumor is inappropriate. Accurate and timely monitoring of the evolving molecular landscape of a tumor is critical but difficult.
Currently, substantial efforts have attempted to solve the problem of intratumor heterogeneity and acquired drug resistance (Fig. 1). The first approach is to target the primary driver and simultaneously block the anticipated adaptive response [32, 45]. For example, in breast cancer, MAPK can be activated in response to PI3K inhibition. Thus, the combination of MEK and PI3K inhibitors shows great potential [46–50]. Likewise, in BRAF-mutant melanoma, resistance to BRAF inhibitors is mediated by reactivation of MAPK [43, 51] and PI3K-PTEN-AKT [51] pathways. Thus, preemptive inhibition of the MEK pathway [52, 53] or both MAPK and PI3K pathways [51] can prolong PFS. However, because epigenetics play an important role in cellular plasticity and drug resistance [54–57], epigenetic therapy has gradually gained popularity to sensitize tumor cells for therapy. For example, DNA methylation and histone deacetylase inhibitors are thought to prime cancer cells to restore sensitivity to previously ineffective drugs [58, 59].
Exploring the “homogenization” strategy for precision medicine
Limitations exist in all approaches mentioned above regarding targeting intratumor heterogeneity. Considering the complex signaling pathways, identifying the driver gene contributing to intratumor heterogeneity is difficult. Evolution from a tumor cell to an advanced cancer is a long process, and a large number of driver genes are involved in further deterioration (Fig. 2). Therefore, we propose an alternative approach to target intratumor heterogeneity. The “homogenization” strategy introduces a consistent genotype or creates a specific environment to drive all tumor cells exhibit the same phenotype. This approach weakens intratumor heterogeneity and results in a less diverse cell population. Thereafter, administration of a sensitive drug that targets these cells would eliminate the tumor population (Fig. 3).
Fig. 2.
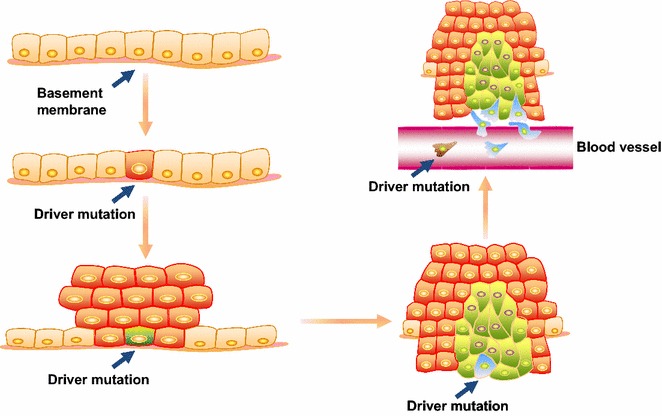
Driver mutations in tumor evolution. A cell develops a specific driver mutation that gives rise to a benign tumor. The cell then develops an additional driver mutation to invade surrounding tissues. Subsequently, the cell develops an additional driver mutation that enables the tumor to engage in hematogenous metastasis. Thereafter, the tumor may acquire additional driver mutations to propagate and sustain intratumor heterogeneity
Fig. 3.
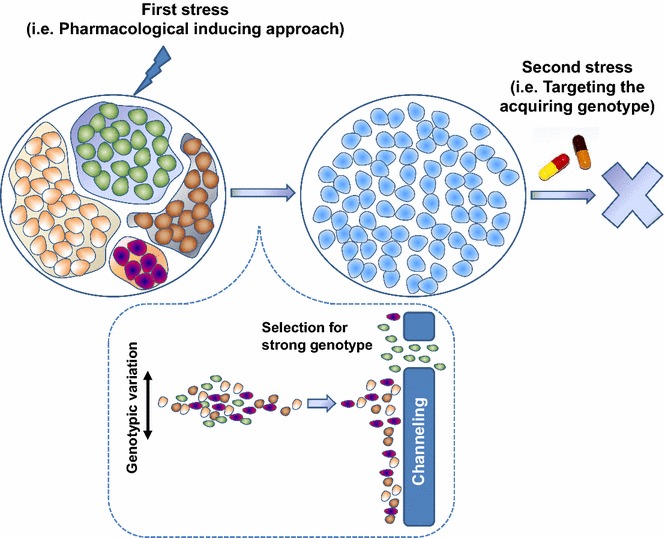
Schematic depiction of “homogenization” therapy. First, a homogeneous cell population is induced, followed by administration of a drug to which the cells are sensitive to eradicate the tumor population
Principles of the homogenization strategy
Because the tumor cell population is highly heterogeneous and unstable, evolutionary dynamics can be capitalized to select a homogenous cell population under stress. As the tumor evolves, unadapted subclones are completely lost, whereas fit subclones become dominant, and less fit minor subclones persist by forming reservoirs from which evolution can continue [60]. Consequently, therapeutic interventions may eliminate specific clones and inadvertently exert selective pressure on the propagation of resistant clones [39, 40]. Thus, homogenization imposes selective pressure on tumor cells with genetic diversity by eliminating the therapy-sensitive cell population, leading to a therapy-resistant cell population with a highly adaptive potential. Genetically, this process strongly accelerates and exacerbates a particular mutation deficiency, which emerges as an adaptive variant. Thus, surviving cells have a common and predominant genotypic variation.
To drive tumor cells into a homologous population, a selective condition must to be identified for homogenization. A general pharmacological selection approach to gain a cell population with shared adaptability and fitness from a heterogeneous cell population is outlined here. In this case, exploiting iatrogenic evolutionary selection pressure is tractable and collateral sensitivity emerges in general, i.e., sensitivity to another drug at the expense of resistance acquired to one drug has been observed [61, 62]. Such evolutionary constraints have been exploited in a murine model of acute lymphoid leukemia [63]. Specifically, dasatinib treatment results in selection of the acquired resistance BCR-ABL1 V299L mutation and renders cells sensitive to non-classical BCR-ABL inhibitors such as cabozantinib and vandetanib.
Techniques and resources facilitate implementation of a second stress
After selection of a homogenous tumor cell population via adaptation, identification of the selected genotype is required. Based on our understanding, three possibilities can be considered. First, a target gene likely exists, and genetic variation of the original drug target of the targeted gene is possible. Second, a particular genotype is functionally associated with activation of the downstream signaling pathway of the original target gene, which provides a possibility to bypass the first stress. Third, the emerging genotype of the certain gene probably locates near the original target gene in the genome. Therefore, the genotype induced by the first stress can be determined by targeted deep sequencing of these three possibilities.
With the use of targeted deep sequencing, oncologists can profile the spectrum of genomic changes in a tumor sample. For example, a typical study revealed the molecular taxonomy of prostate cancer by omics analysis as well as potentially actionable targets [64]. Moreover, research initiatives, such as The Cancer Genome Atlas (TCGA) and International Cancer Genome Consortium (ICGC), capitalize the available information of large numbers of tumor samples to identify genes and pathways important for cancer progression [65–69]. Targeted drugs, which are designed to specifically suppress certain oncogenic signaling pathways, are widely applied in clinical practice. Thus, precision therapy is appealing and changing the pattern of clinical practice for tumor patients. Notably, a multicenter and prospective study called tracking cancer evolution through therapy (TRACERx) [70–73] provided an applicable model to track tumor evolution depending on multiregion and longitudinal sampling and genetic analysis. Going forward, such large scale genomic studies may contribute to clinical practices.
Subsequently, there is a need to determine a drug that specifically targets the newly acquired genotype through drug screening or designing a targeted drug. To this end, cancer systems biology may provide a more holistic view of cancer [74, 75]. Specifically, this approach can bridge molecular characteristics with pharmacogenomics to deliver targeted therapy, which will significantly improve the specificity and efficacy of targeted therapy. Moreover, some datasets [76] are rich resources to identify therapeutic options for selected targets.
Dynamic monitoring of the homogenization genotype
The implementation of dual drug stresses theoretically leads to the extinction of the tumor cell population. Nevertheless, the theory of evolutionary dynamics proposes that the homogenous cell population can gain the ability to escape the second stress via continued phenotypic changes. To solve this problem, the genotype induced by the first stress should be monitored to avoid the emergence of resistance to the second drug. When a reduction is detected, the first drug should be used again to select and enrich the particular genotype, which makes the selected cell population more vulnerable to the second drug. Furthermore, when an increase is detected, therapy with the second drug should be resumed. The opposing selective effect of these two drugs imposes an adaptive dilemma for the selected homogenous genotype, and an analogous strategy has been demonstrated by many studies [77–81].
Current techniques that facilitate dynamic monitoring of the homogenization genotype include liquid biopsies [82–85] such as circulating tumor cells (CTCs) and circulating free DNA (cfDNA), which allows assessment of repeated samples and longitudinal measurement of clonal evolution in tumor patients. Conceivably, combining omics sequencing with liquid biopsies is required for dynamic monitoring.
Future perspectives
Homogenization therapy is based on inducing a common and druggable characteristic trait of a heterogeneous cell population. The genetic representation of clonal evolution from the primary tumor to relapse reveals convergent evolution, suggesting that this strategy is possible [86]. In addition, a proof-of-principle study in yeast has demonstrated the feasibility of this strategy [87]. Specifically, an “evolutionary trap” has been devised by reducing karyotypic heterogeneity to a defined predictable state via initial drug exposure, and then a secondary drug was subsequently applied. This study illustrated that evolutionary dynamics can be exploited for homogenization therapy, and evidence is emerging to support the efficacy of this approach. A case study [81] has reported that a patient with ALK-rearranged non-small-cell lung carcinoma, which harbored a subclonal C1156Y mutation, acquired drug resistance to crizotinib and responded to the third-generation ALK inhibitor lorlatinib. Following treatment with lorlatinib, the tumor acquired an L1198F mutation, but this mutation promoted resensitization to crizotinib, which improved the patient’s prognosis. Ultimately, other options and interventions to achieve homogeneity should be explored further. Moreover, exploration of tumor homogeneity requires the design of more clinical practices. Taken together, homogenization therapy sheds new light on our understanding of intratumor heterogeneity and provides a novel strategy to solve problems associated with tumor treatments.
Authors’ contributions
MT analyzed the data and wrote the manuscript; ZD, XZ, BH, and MY revised the manuscript; WC and QL jointly oversaw and directed the review. All authors read and approved the final manuscript.
Competing interests
The authors declare that they have no competing interests.
Availability of data and materials
Not applicable.
Consent for publication
Not applicable.
Ethics approval and consent to participate
Not applicable.
Funding
This work was supported by Innovative Research Team in University of Ministry of Education of China (No. IRT_17R15), National Natural Science Foundation of China (No. 81630005 to QL, No. 81573025 to QL), Dalian high-level talent innovation program (2016RD12 to QL) and International scientific and technological cooperation of Dalian (2015F11GH095 to QL).
Contributor Information
Mengying Tong, Email: mengyingtong@163.com.
Ziqian Deng, Email: dengziqian163@163.com.
Xiaolong Zhang, Email: xiaolongzhang2015@163.com.
Bin He, Email: hebin@sysucc.org.cn.
Mengying Yang, Email: 602227985@qq.com.
Wei Cheng, Email: chengwei2009@mail.dlut.edu.cn.
Quentin Liu, Email: liuq9@mail.sysu.edu.cn.
References
- 1.Ye XS, Yu C, Aggarwal A, et al. Genomic alterations and molecular subtypes of gastric cancers in asians. Chin J Cancer. 2016;35:42. doi: 10.1186/s40880-016-0106-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cavalli FMG, Remke M, Rampasek L, et al. Intertumoral heterogeneity within medulloblastoma subgroups. Cancer cell. 2017;31(6):737–754. doi: 10.1016/j.ccell.2017.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bolli N, Avet-Loiseau H, Wedge DC, et al. Heterogeneity of genomic evolution and mutational profiles in multiple myeloma. Nat Commun. 2014;5:2997. doi: 10.1038/ncomms3997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Schwarz RF, Ng CK, Cooke SL, et al. Spatial and temporal heterogeneity in high-grade serous ovarian cancer: a phylogenetic analysis. PLoS Med. 2015;12(2):e1001789. doi: 10.1371/journal.pmed.1001789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Izumchenko E, Chang X, Brait M, et al. Targeted sequencing reveals clonal genetic changes in the progression of early lung neoplasms and paired circulating DNA. Nat Commun. 2015;6:8258. doi: 10.1038/ncomms9258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ross-Innes CS, Becq J, Warren A, et al. Whole-genome sequencing provides new insights into the clonal architecture of barrett’s esophagus and esophageal adenocarcinoma. Nat Genet. 2015;47(9):1038–1046. doi: 10.1038/ng.3357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kim KT, Lee HW, Lee HO, et al. Single-cell mrna sequencing identifies subclonal heterogeneity in anti-cancer drug responses of lung adenocarcinoma cells. Genome Biol. 2015;16:127. doi: 10.1186/s13059-015-0692-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kim H, Zheng S, Amini SS, et al. Whole-genome and multisector exome sequencing of primary and post-treatment glioblastoma reveals patterns of tumor evolution. Genome Res. 2015;25(3):316–327. doi: 10.1101/gr.180612.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nowell PC. The clonal evolution of tumor cell populations. Science. 1976;194(4260):23–28. doi: 10.1126/science.959840. [DOI] [PubMed] [Google Scholar]
- 10.Morrissy AS, Cavalli FMG, Remke M, et al. Spatial heterogeneity in medulloblastoma. Nat Genet. 2017;49(5):780–788. doi: 10.1038/ng.3838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Moss TJ, Qi Y, Xi L, et al. Comprehensive genomic characterization of upper tract urothelial carcinoma. Eur Urol. 2017;72:641–649. doi: 10.1016/j.eururo.2017.05.048. [DOI] [PubMed] [Google Scholar]
- 12.Sun C, Fang Y, Yin J, et al. Rational combination therapy with parp and mek inhibitors capitalizes on therapeutic liabilities in ras mutant cancers. Sci Transl Med. 2017;9:392. doi: 10.1126/scitranslmed.aal5148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Le Tourneau C, Delord JP, Goncalves A, et al. Molecularly targeted therapy based on tumour molecular profiling versus conventional therapy for advanced cancer (shiva): a multicentre, open-label, proof-of-concept, randomised, controlled phase 2 trial. Lancet Oncol. 2015;16(13):1324–1334. doi: 10.1016/S1470-2045(15)00188-6. [DOI] [PubMed] [Google Scholar]
- 14.Stockley TL, Oza AM, Berman HK, et al. Molecular profiling of advanced solid tumors and patient outcomes with genotype-matched clinical trials: the princess margaret impact/compact trial. Genome Med. 2016;8(1):109. doi: 10.1186/s13073-016-0364-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tannock IF, Hickman JA. Limits to personalized cancer medicine. N Engl J Med. 2016;375(13):1289–1294. doi: 10.1056/NEJMsb1607705. [DOI] [PubMed] [Google Scholar]
- 16.Yu C, Yu J, Yao X, et al. Discovery of biclonal origin and a novel oncogene slc12a5 in colon cancer by single-cell sequencing. Cell Res. 2014;24(6):701–712. doi: 10.1038/cr.2014.43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ling S, Hu Z, Yang Z, et al. Extremely high genetic diversity in a single tumor points to prevalence of non-darwinian cell evolution. Proc Natl Acad Sci USA. 2015;47:6496–6505. doi: 10.1073/pnas.1519556112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yang Z, Li C, Fan Z, et al. Single-cell sequencing reveals variants in ARID1A, GPRC5A and MLL2 driving self-renewal of human bladder cancer stem cells. Eur Urol. 2017;71(1):8–12. doi: 10.1016/j.eururo.2016.06.025. [DOI] [PubMed] [Google Scholar]
- 19.Li C, Wu S, Yang Z, et al. Single-cell exome sequencing identifies mutations in kcp, loc440040, and loc440563 as drivers in renal cell carcinoma stem cells. Cell Res. 2017;27(4):590–593. doi: 10.1038/cr.2016.150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Liu S, Cong Y, Wang D, et al. Breast cancer stem cells transition between epithelial and mesenchymal states reflective of their normal counterparts. Stem Cell Rep. 2014;2(1):78–91. doi: 10.1016/j.stemcr.2013.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gupta PB, Fillmore CM, Jiang G, et al. Stochastic state transitions give rise to phenotypic equilibrium in populations of cancer cells. Cell. 2011;146(4):633–644. doi: 10.1016/j.cell.2011.07.026. [DOI] [PubMed] [Google Scholar]
- 22.Lim E, Vaillant F, Wu D, et al. Aberrant luminal progenitors as the candidate target population for basal tumor development in brca1 mutation carriers. Nat Med. 2009;15(8):907–913. doi: 10.1038/nm.2000. [DOI] [PubMed] [Google Scholar]
- 23.Mani SA, Guo W, Liao MJ, et al. The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell. 2008;133(4):704–715. doi: 10.1016/j.cell.2008.03.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Van Keymeulen A, Lee MY, Ousset M, et al. Reactivation of multipotency by oncogenic PIK3CA induces breast tumour heterogeneity. Nature. 2015;525(7567):119–123. doi: 10.1038/nature14665. [DOI] [PubMed] [Google Scholar]
- 25.Koren S, Reavie L, Couto JP, et al. PIK3CA(H1047R) induces multipotency and multi-lineage mammary tumours. Nature. 2015;525(7567):114–118. doi: 10.1038/nature14669. [DOI] [PubMed] [Google Scholar]
- 26.McGranahan N, Swanton C. Clonal heterogeneity and tumor evolution: past, present, and the future. Cell. 2017;168(4):613–628. doi: 10.1016/j.cell.2017.01.018. [DOI] [PubMed] [Google Scholar]
- 27.Al-Hussaini H, Subramanyam D, Reedijk M, et al. Notch signaling pathway as a therapeutic target in breast cancer. Mol Cancer Ther. 2011;10(1):9–15. doi: 10.1158/1535-7163.MCT-10-0677. [DOI] [PubMed] [Google Scholar]
- 28.King TD, Suto MJ, Li Y. The wnt/beta-catenin signaling pathway: a potential therapeutic target in the treatment of triple negative breast cancer. J Cell Biochem. 2012;113(1):13–18. doi: 10.1002/jcb.23350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Korkaya H, Paulson A, Iovino F, et al. Her2 regulates the mammary stem/progenitor cell population driving tumorigenesis and invasion. Oncogene. 2008;27(47):6120–6130. doi: 10.1038/onc.2008.207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Iliopoulos D, Hirsch HA, Struhl K. An epigenetic switch involving nf-kappab, lin28, let-7 microrna, and il6 links inflammation to cell transformation. Cell. 2009;139(4):693–706. doi: 10.1016/j.cell.2009.10.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Voss MH, Hakimi AA, Pham CG, et al. Tumor genetic analyses of patients with metastatic renal cell carcinoma and extended benefit from mTOR inhibitor therapy. Clin Cancer Res. 2014;20(7):1955–1964. doi: 10.1158/1078-0432.CCR-13-2345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zardavas D, Irrthum A, Swanton C, et al. Clinical management of breast cancer heterogeneity. Nat Rev Clin Oncol. 2015;12(7):381–394. doi: 10.1038/nrclinonc.2015.73. [DOI] [PubMed] [Google Scholar]
- 33.Shah SP, Roth A, Goya R, et al. The clonal and mutational evolution spectrum of primary triple-negative breast cancers. Nature. 2012;486(7403):395–399. doi: 10.1038/nature10933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Curtis C, Shah SP, Chin SF, et al. The genomic and transcriptomic architecture of 2000 breast tumours reveals novel subgroups. Nature. 2012;486(7403):346–352. doi: 10.1038/nature10983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Meric-Bernstam F, Brusco L, Shaw K, et al. Feasibility of large-scale genomic testing to facilitate enrollment onto genomically matched clinical trials. J Clin Oncol. 2015;33(25):2753–2762. doi: 10.1200/JCO.2014.60.4165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bhola NE, Balko JM, Dugger TC, et al. Tgf-beta inhibition enhances chemotherapy action against triple-negative breast cancer. J Clin Invest. 2013;123(3):1348–1358. doi: 10.1172/JCI65416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Eser S, Reiff N, Messer M, et al. Selective requirement of PI3K/PDK1 signaling for kras oncogene-driven pancreatic cell plasticity and cancer. Cancer Cell. 2013;23(3):406–420. doi: 10.1016/j.ccr.2013.01.023. [DOI] [PubMed] [Google Scholar]
- 38.Tovy A, Spiro A, McCarthy R, et al. P53 is essential for DNA methylation homeostasis in naive embryonic stem cells, and its loss promotes clonal heterogeneity. Genes Dev. 2017;10:959–972. doi: 10.1101/gad.299198.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hata AN, Niederst MJ, Archibald HL, et al. Tumor cells can follow distinct evolutionary paths to become resistant to epidermal growth factor receptor inhibition. Nat Med. 2016;22(3):262–269. doi: 10.1038/nm.4040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Oxnard GR. The cellular origins of drug resistance in cancer. Nat Med. 2016;22(3):232–234. doi: 10.1038/nm.4058. [DOI] [PubMed] [Google Scholar]
- 41.Shaffer SM, Dunagin MC, Torborg SR, et al. Rare cell variability and drug-induced reprogramming as a mode of cancer drug resistance. Nature. 2017;546(7658):431–435. doi: 10.1038/nature22794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.McGranahan N, Swanton C. Biological and therapeutic impact of intratumor heterogeneity in cancer evolution. Cancer Cell. 2015;27(1):15–26. doi: 10.1016/j.ccell.2014.12.001. [DOI] [PubMed] [Google Scholar]
- 43.Sanchez-Laorden B, Viros A, Girotti MR, et al. BRAF inhibitors induce metastasis in ras mutant or inhibitor-resistant melanoma cells by reactivating MEK and ERK signaling. Sci Signal. 2014;7(318):ra30. doi: 10.1126/scisignal.2004815. [DOI] [PubMed] [Google Scholar]
- 44.Lohr JG, Stojanov P, Carter SL, et al. Widespread genetic heterogeneity in multiple myeloma: implications for targeted therapy. Cancer Cell. 2014;25(1):91–101. doi: 10.1016/j.ccr.2013.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Duncan JS, Whittle MC, Nakamura K, et al. Dynamic reprogramming of the kinome in response to targeted MEK inhibition in triple-negative breast cancer. Cell. 2012;149(2):307–321. doi: 10.1016/j.cell.2012.02.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Klempner SJ, Myers AP, Cantley LC. What a tangled web we weave: emerging resistance mechanisms to inhibition of the phosphoinositide 3-kinase pathway. Cancer Discov. 2013;3(12):1345–1354. doi: 10.1158/2159-8290.CD-13-0063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Shimizu T, Tolcher AW, Papadopoulos KP, et al. The clinical effect of the dual-targeting strategy involving PI3K/AKT/mTOR and RAS/MEK/ERK pathways in patients with advanced cancer. Clin Cancer Res. 2012;18(8):2316–2325. doi: 10.1158/1078-0432.CCR-11-2381. [DOI] [PubMed] [Google Scholar]
- 48.Mirzoeva OK, Das D, Heiser LM, et al. Basal subtype and MAPK/ERK kinase (MEK)-phosphoinositide 3-kinase feedback signaling determine susceptibility of breast cancer cells to mek inhibition. Cancer Res. 2009;69(2):565–572. doi: 10.1158/0008-5472.CAN-08-3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hoeflich KP, O’Brien C, Boyd Z, et al. In vivo antitumor activity of mek and phosphatidylinositol 3-kinase inhibitors in basal-like breast cancer models. Clin Cancer Res. 2009;15(14):4649–4664. doi: 10.1158/1078-0432.CCR-09-0317. [DOI] [PubMed] [Google Scholar]
- 50.Engelman JA, Chen L, Tan X, et al. Effective use of PI3K and MEK inhibitors to treat mutant Kras G12D and PIK3CA H1047R murine lung cancers. Nat Med. 2008;14(12):1351–1356. doi: 10.1038/nm.1890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Shi H, Hugo W, Kong X, et al. Acquired resistance and clonal evolution in melanoma during BRAF inhibitor therapy. Cancer Discov. 2014;4(1):80–93. doi: 10.1158/2159-8290.CD-13-0642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Larkin J, Ascierto PA, Dreno B, et al. Combined vemurafenib and cobimetinib in BRAF-mutated melanoma. N Engl J Med. 2014;371(20):1867–1876. doi: 10.1056/NEJMoa1408868. [DOI] [PubMed] [Google Scholar]
- 53.Flaherty KT, Infante JR, Daud A, et al. Combined BRAF and MEK inhibition in melanoma with BRAF v600 mutations. N Engl J Med. 2012;367(18):1694–1703. doi: 10.1056/NEJMoa1210093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Easwaran H, Tsai HC, Baylin SB. Cancer epigenetics: tumor heterogeneity, plasticity of stem-like states, and drug resistance. Mol Cell. 2014;54(5):716–727. doi: 10.1016/j.molcel.2014.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Easwaran H, Johnstone SE, Van Neste L, et al. A DNA hypermethylation module for the stem/progenitor cell signature of cancer. Genome Res. 2012;22(5):837–849. doi: 10.1101/gr.131169.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Bernstein BE, Mikkelsen TS, Xie X, et al. A bivalent chromatin structure marks key developmental genes in embryonic stem cells. Cell. 2006;125(2):315–326. doi: 10.1016/j.cell.2006.02.041. [DOI] [PubMed] [Google Scholar]
- 57.Azuara V, Perry P, Sauer S, et al. Chromatin signatures of pluripotent cell lines. Nat Cell Biol. 2006;8(5):532–538. doi: 10.1038/ncb1403. [DOI] [PubMed] [Google Scholar]
- 58.Azad N, Zahnow CA, Rudin CM, et al. The future of epigenetic therapy in solid tumours–lessons from the past. Nat Rev Clin Oncol. 2013;10(5):256–266. doi: 10.1038/nrclinonc.2013.42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Baylin SB, Jones PA. A decade of exploring the cancer epigenome—biological and translational implications. Nat Rev Cancer. 2011;11(10):726–734. doi: 10.1038/nrc3130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kreso A, Dick JE. Evolution of the cancer stem cell model. Cell Stem Cell. 2014;14(3):275–291. doi: 10.1016/j.stem.2014.02.006. [DOI] [PubMed] [Google Scholar]
- 61.Jensen PB, Holm B, Sorensen M, et al. In vitro cross-resistance and collateral sensitivity in seven resistant small-cell lung cancer cell lines: preclinical identification of suitable drug partners to taxotere, taxol, topotecan and gemcitabin. Br J Cancer. 1997;75(6):869–877. doi: 10.1038/bjc.1997.154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Hill BT. Potential of continuous tumour cell lines for establishing patterns of cross-resistance and collateral sensitivity in vitro. Drugs Exp Clin Res. 1986;12(1–3):293–298. [PubMed] [Google Scholar]
- 63.Zhao B, Sedlak JC, Srinivas R, et al. Exploiting temporal collateral sensitivity in tumor clonal evolution. Cell. 2016;165(1):234–246. doi: 10.1016/j.cell.2016.01.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Cancer Genome Atlas Research N The molecular taxonomy of primary prostate cancer. Cell. 2015;163(4):1011–1025. doi: 10.1016/j.cell.2015.10.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Varghese AM, Berger MF. Advancing clinical oncology through genome biology and technology. Genome Biol. 2014;15(8):427. doi: 10.1186/s13059-014-0427-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Cancer Genome Atlas Research N Comprehensive molecular characterization of clear cell renal cell carcinoma. Nature. 2013;499(7456):43–49. doi: 10.1038/nature12222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Cancer Genome Atlas Research N Comprehensive molecular characterization of urothelial bladder carcinoma. Nature. 2014;507(7492):315–322. doi: 10.1038/nature12965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Cancer Genome Atlas Research N Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. New Eng J Med. 2013;368(22):2059–2074. doi: 10.1056/NEJMoa1301689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Cancer Genome Atlas Research N. Kandoth C, Schultz N, et al. Integrated genomic characterization of endometrial carcinoma. Nature. 2013;497(7447):67–73. doi: 10.1038/nature12113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Jamal-Hanjani M, Hackshaw A, Ngai Y, et al. Tracking genomic cancer evolution for precision medicine: the lung tracerx study. PLoS Biol. 2014;12(7):e1001906. doi: 10.1371/journal.pbio.1001906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Jamal-Hanjani M, Wilson GA, Horswell S, et al. Detection of ubiquitous and heterogeneous mutations in cell-free DNA from patients with early-stage non-small-cell lung cancer. Ann Oncol. 2016;27(5):862–867. doi: 10.1093/annonc/mdw037. [DOI] [PubMed] [Google Scholar]
- 72.Gerlinger M, Rowan AJ, Horswell S, et al. Intratumor heterogeneity and branched evolution revealed by multiregion sequencing. N Engl J Med. 2012;366(10):883–892. doi: 10.1056/NEJMoa1113205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Jamal-Hanjani M, Wilson GA, McGranahan N, et al. Tracking the evolution of non-small-cell lung cancer. N Engl J Med. 2017;376(22):2109–2121. doi: 10.1056/NEJMoa1616288. [DOI] [PubMed] [Google Scholar]
- 74.Werner HM, Mills GB, Ram PT. Cancer systems biology: a peek into the future of patient care? Nat Rev Clin Oncol. 2014;11(3):167–176. doi: 10.1038/nrclinonc.2014.6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Du W, Elemento O. Cancer systems biology: embracing complexity to develop better anticancer therapeutic strategies. Oncogene. 2015;34(25):3215–3225. doi: 10.1038/onc.2014.291. [DOI] [PubMed] [Google Scholar]
- 76.Iorio F, Knijnenburg TA, Vis DJ, et al. A landscape of pharmacogenomic interactions in cancer. Cell. 2016;166(3):740–754. doi: 10.1016/j.cell.2016.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Bhang HE, Ruddy DA, Krishnamurthy Radhakrishna V, et al. Studying clonal dynamics in response to cancer therapy using high-complexity barcoding. Nat Med. 2015;21(5):440–448. doi: 10.1038/nm.3841. [DOI] [PubMed] [Google Scholar]
- 78.Enriquez-Navas PM, Kam Y, Das T, et al. Exploiting evolutionary principles to prolong tumor control in preclinical models of breast cancer. Sci Transl Med. 2016;8(327):327ra324. doi: 10.1126/scitranslmed.aad7842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.McGranahan N, Furness AJ, Rosenthal R, et al. Clonal neoantigens elicit t cell immunoreactivity and sensitivity to immune checkpoint blockade. Science. 2016;351(6280):1463–1469. doi: 10.1126/science.aaf1490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Misale S, Bozic I, Tong J, et al. Vertical suppression of the EGFR pathway prevents onset of resistance in colorectal cancers. Nat Commun. 2015;6:8305. doi: 10.1038/ncomms9305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Shaw AT, Friboulet L, Leshchiner I, et al. Resensitization to crizotinib by the lorlatinib ALK resistance mutation L1198f. N Engl J Med. 2016;374(1):54–61. doi: 10.1056/NEJMoa1508887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Amirouchene-Angelozzi N, Swanton C, Bardelli A. Tumor evolution as a therapeutic target. Cancer Discov. 2017;7:805–817. doi: 10.1158/2159-8290.CD-17-0343. [DOI] [PubMed] [Google Scholar]
- 83.Qin Z, Ljubimov VA, Zhou C, et al. Cell-free circulating tumor DNA in cancer. Chin J Cancer. 2016;35:36. doi: 10.1186/s40880-016-0092-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Siravegna G, Bardelli A. Genotyping cell-free tumor DNA in the blood to detect residual disease and drug resistance. Genome Biol. 2014;15(8):449. doi: 10.1186/s13059-014-0449-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Mateo J, Gerlinger M, Rodrigues DN, et al. The promise of circulating tumor cell analysis in cancer management. Genome Biol. 2014;15(8):448. doi: 10.1186/s13059-014-0448-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Ding L, Ley TJ, Larson DE, et al. Clonal evolution in relapsed acute myeloid leukaemia revealed by whole-genome sequencing. Nature. 2012;481(7382):506–510. doi: 10.1038/nature10738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Chen G, Mulla WA, Kucharavy A, et al. Targeting the adaptability of heterogeneous aneuploids. Cell. 2015;160(4):771–784. doi: 10.1016/j.cell.2015.01.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Not applicable.