

Summary
In the clinical setting, analgesic tolerance is a primary driver of diminished pain control and opioid dose escalations. Integral to this process are primary afferent sensory neurons, the first-order components of nociceptive sensation. Here, we characterize the factors modulating morphine action and tolerance in mouse small diameter dorsal root ganglia (DRG) neurons. We demonstrate that acute morphine inactivates tetrodotoxin-resistant (TTX-R) Na+ channels in these cells. Chronic exposure resulted in tolerance to this effect, which was prevented by treatment with oral vancomycin. Using colonic supernatants, we further show that mediators in the gut microenvironment of mice with chronic morphine exposure can induce tolerance and hyperexcitability in naive DRG neurons. Tolerance (but not hyperexcitability) in this paradigm was mitigated by oral vancomycin treatment. These findings collectively suggest that gastrointestinal microbiota modulate the development of morphine tolerance (but not hyperexcitability) in nociceptive primary afferent neurons, through a mechanism involving TTX-R Na+ channels.
Subject Areas: Neuroscience, Cellular Neuroscience, Microbiome
Graphical Abstract
Highlights
-
•
Gram-positive gut bacteria depletion prevents morphine antinociceptive tolerance
-
•
Tolerance develops to morphine inhibition of TTX-R Na+ channels in DRG neurons
-
•
Tolerance in isolated DRG neurons is mitigated by Gram-positive bacteria depletion
-
•
Morphine-induced gut mediators produce tolerance in DRG neurons
Neuroscience; Cellular Neuroscience; Microbiome
Introduction
The current prescription opioid epidemic has prompted a demand for improvements in clinical pain management (Dowell et al., 2016). Over the last decade, opioid prescription rates have risen dramatically, with concurrent increases in opioid-related emergency department visits and overdose mortality. Although this is evidently concerning, opioid analgesics remain an essential component of the modern health care systems and the gold standard of therapy for moderate to severe pain. In addition to opioid-induced hypernociception (OIH), analgesic tolerance is a primary driver of diminished pain control and dose escalations in the clinical setting (Collett, 1998, Angst and Clark, 2006). Primary afferent sensory neurons are the first-order components of nociceptive sensation and integral to the process of analgesic tolerance development (Corder et al., 2017). We recently demonstrated that chronic morphine exposure in mice results in cellular level tolerance of small diameter primary afferent neurons isolated from L5-S1 dorsal root ganglia (DRG; Kang et al., 2017). Neurons from tolerant mice demonstrated resistance to morphine-mediated reductions of excitability (i.e., increases in action potential threshold). This raises critical questions about the physiological basis of morphine action and tolerance in small diameter DRG neurons, particularly in regard to modulation of ionic conductances.
Numerous distinct voltage-gated sodium (Nav) channels have been described in DRG neurons (Matsuda et al., 1978, Fukuda and Kameyama, 1980, Roy and Narahashi, 1992, Elliott and Elliott, 1993, Ogata and Tatebayashi, 1993, Rizzo et al., 1994). Only two of these (Nav1.8 and Nav1.9) demonstrate resistance to the puffer fish guanidinium toxin tetrodotoxin (TTX) in the micromolar range. These unusual Na+ channels are almost exclusively expressed by small nociceptive primary afferents (Amaya et al., 2000), with some nociceptors expressing only tetrodotoxin-resistant (TTX-R) Na+ currents (Ogata and Tatebayashi, 1992). We and others have demonstrated that chronic (5–7 days) morphine exposure can alter the voltage dependence of activation and steady-state inactivation of TTX-R Na+ channels (Chen et al., 2012, Ross et al., 2012). Specifically, Ross et al. noted hyperpolarization of the potential of half-maximum (V1/2) activation in mouse DRG neurons. Chen et al. independently reproduced this finding in rat neurons, additionally observing hyperpolarization of the action potential threshold (Vt) and acceleration of inactivation kinetics. Both of these studies utilized a chronic morphine exposure paradigm that demonstrated neuronal hyperexcitability associated with OIH. However, there is evidence that even acute morphine challenge in vitro can modulate TTX-R Na+ channel availability. In an investigation of steady-state inactivation by Smith et al. in mouse enteric neurons a nearly 50% reduction in the number of TTX-R Na+ channels available for activation at hyperpolarized potentials with acute morphine challenge was noted (3 μM; Smith et al., 2012). These findings collectively suggest that acute morphine challenge in vitro may operate on the same cellular machinery that is modified with chronic morphine exposure, that is, the voltage dependence and/or kinetics of TTX-R Na+ channel activation and inactivation in DRG neurons.
There is growing awareness for the role of gastrointestinal microbiota in numerous physiological and pathophysiological processes. Recent investigations have documented the impact of opioids in inducing dysbiosis of gastrointestinal microbiota in both mice and humans (Xu et al., 2017). In this context, dysbiosis is defined as a disruption in the composition and/or localization of the gut microflora. We and others have demonstrated that chronic morphine exposure in mice results in such dysbiosis, marked by alterations of microbial composition and compromise of gut epithelial tight junction integrity (Meng et al., 2013, Kang et al., 2017). The latter results in gut “leakiness,” which allows translocation of luminal bacteria to the gut wall and circulation. Secondary release of bacterial products and pro-inflammatory cytokines correlates with the production of antinociceptive tolerance. Indeed, we noted that concurrent treatment with antibiotics was sufficient to prevent both behavioral antinociceptive tolerance and cellular level tolerance in small diameter DRG neurons. Oral vancomycin alone was sufficient to reproduce the behavioral findings, suggesting that it may also be sufficient to reproduce the cellular level findings; namely, vancomycin may preserve the depolarizing shifts in action potential threshold with acute morphine challenge (3 μM) in vitro, which are mitigated in cells from tolerant subjects. The ionic basis of such an effect may also involve TTX-R Na+ channels.
Finally, it is as of yet unclear whether the observed impacts of gastrointestinal microbiota on morphine tolerance are a gut-localized or a more general systemic phenomenon. To address this question, we have employed a novel technique that utilizes colon tissue supernatants to grossly assess how mediators in the gut microenvironment affect naive DRG neurons. This innovative approach has arisen as a powerful paradigm for investigating the mechanisms of peripheral nociception in both mice and human patients (Barbara et al., 2004, Cenac et al., 2007, Akbar et al., 2008). The effects of supernatant exposure can be characterized in vitro; then the causal constituents may be later identified and tested in basic or whole-animal systems. To this end, colon tissue supernatants from mice with chronic morphine exposure may be applied to isolated DRG neurons from naive mice to assess the impact of gut-localized mediators (e.g., bacterial products and pro-inflammatory cytokines) on neuronal excitability and morphine tolerance. The importance of the gastrointestinal microbiome in these findings can be additionally assessed using supernatants from mice with concurrent oral vancomycin exposure.
Results
Antinociceptive Morphine Tolerance Prevention by Oral Vancomycin Is Dependent on the Duration of Treatment
We recently demonstrated that oral vancomycin treatment in mice prevents the development of antinociceptive morphine tolerance in the tail immersion assay (Kang et al., 2017). The virtual 0% bioavailability of oral vancomycin suggests a role for gastrointestinal microbiota in this process. Quantification of the total bacterial 16S rRNA gene copies in fecal samples from mice receiving a mixed antibiotic cocktail indicated a temporally dependent reduction of colonic bacterial colonization throughout the 10-day treatment period. We thereby hypothesized that varying the duration of treatment would modulate the efficacy of oral vancomycin in preventing antinociceptive tolerance.
To this end, antinociceptive tolerance development following implantation of a 75-mg morphine pellet was monitored in mice receiving 5, 10, or 15 days of vancomycin (10 mg/kg VAN) treatment by oral gavage (Figure 1 and Table S1). Control subjects received 10 days of saline (SAL) by oral gavage. Latency to tail flick in the tail immersion assay was recorded daily for each treatment cohort. Mice from all cohorts demonstrated a robust antinociceptive response on day 1 after pellet implantation, marked by a sharp increase in latency to tail flick. Mice receiving saline gavage treatments (10 day SAL) developed tolerance to this effect, evidenced by a return of tail flick latency to baseline values by day 3 and a lack of response to acute morphine challenge (10 mg/kg, subcutaneously) on the final testing day. Mice receiving 5 days of vancomycin treatment (5 day VAN, i.e., VAN and morphine pellet [MP] initiated simultaneously) demonstrated a significant increase in latency to tail flick on day 2 (p < 0.001 versus 10 day SAL), but ultimately developed tolerance as well. In contrast, tolerance development was prevented in mice receiving 10 days of vancomycin treatment (10 day VAN), manifested by a significant increase in latency to tail flick on all test days (p < 0.001 versus 10day SAL) and an additional increase in latency with acute morphine challenge on the final test day. This tolerance prevention was further enhanced in mice receiving 15 days of vancomycin treatment (15 day VAN), demonstrated by a significant increase in tail-flick latencies on all test days (p < 0.001 versus 10 day VAN).
Figure 1.

Antinociceptive Morphine Tolerance in Mice Is Prevented by Oral Vancomycin in a Manner Dependent on the Duration of Treatment
(A) Schematic of the treatment regimen utilized for each cohort in this study.
(B) Tail-immersion assay for mice receiving 5, 10, or 15 days of oral vancomycin (VAN, 10 mg/kg). Control subjects received 10 days of oral saline (SAL). Following morphine pellet (MP) implantation, the 10 day SAL and 5 day VAN cohorts demonstrated a progressive reduction in tail-flick latency and lacked a response to acute morphine challenge on day 5 (10 mg/kg, subcutaneously), indicating tolerance development. The 10 day VAN treatment resulted in a significant increase in tail-flick latency throughout the testing period (versus 10 day SAL) and an additional increase with acute morphine challenge on day 5, indicating tolerance prevention. This tolerance prevention was significantly enhanced with 15 day VAN treatment (versus 10 day VAN). 10 day SAL (N = 8), 5 day VAN (N = 10), 10 day VAN (N = 14), 15 day VAN (N = 10); significance indicated versus 10 day SAL (filled symbols, p < 0.05) and versus 10 day VAN (†p < 0.001, ‡p < 0.0001) by two-way repeated-measures ANOVA with Bonferroni post hoc analysis; data expressed as mean ± SEM (see Table S1 for additional values).
Oral Vancomycin Prevents Tolerance to Morphine-Mediated Reductions of Excitability in Small Diameter DRG Neurons
Our previous investigations have demonstrated the ability of an antibiotic cocktail to prevent cellular level tolerance development in small diameter primary afferent neurons isolated from L5-S1 DRG (Kang et al., 2017). In current clamp, neurons from non-tolerant mice demonstrated a reduction of excitability with acute morphine challenge (i.e., depolarization of the action potential threshold), whereas neurons from mice with chronic morphine exposure displayed no significant response. This chronic morphine-induced tolerance was prevented in mice receiving concurrent treatment with broad-spectrum antibiotics. We thereby hypothesized that our behavioral observations with vancomycin alone would similarly be reflected by these cellular level tolerance studies.
Responses to acute morphine challenge (3 μM) in vitro were assessed in small diameter (<30 pF) DRG neurons isolated from mice receiving chronic morphine exposure and 10 days of oral vancomycin treatment (Figures 2A and 2B and Table S2). Whole-cell current-clamp experiments demonstrated a reduction of excitability following acute morphine challenge, evidenced by a significant decrease in the number of action potentials at double rheobase (1.7 ± 0.2 at baseline versus 1.1 ± 0.1 with morphine, p < 0.01) and depolarization of the action potential threshold (−14.5 ± 1.2 mV at baseline versus −11.3 ± 1.4 mV with morphine, p < 0.001).
Figure 2.

Characterization of Responses to Acute Morphine Challenge in Small Diameter DRG Neurons
(A and B) Oral vancomycin treatment (VAN, 10 mg/kg) for 10 days blocks cellular level morphine tolerance in DRG neurons. Representative raw traces (left) and individual observations (right) indicate the number of action potentials at double rheobase (A) and action potential threshold (B) at baseline (black) and following acute morphine challenge (3 μM, red). Morphine perfusion significantly reduced the number of action potentials at double rheobase and depolarized the action potential threshold. (N = 7, n = 18), **p < 0.01, ***p < 0.001 by two-tailed paired Student's t test; data expressed as mean ± SEM.
(C) Acute morphine challenge is unable to reduce the excitability of DRG neurons in the presence of naloxone (1 μM). The action potential threshold (left) and number of action potentials at double rheobase (right) are presented at baseline (black) and following acute morphine challenge (3 μM, red). No significant shift in either parameter was noted. N = 4, n = 9; ns, not significant by two-tailed paired Student's t test; data expressed as mean ± SEM.
(D) Morphine-induced depolarizations of action potential threshold persist in the complete absence of internal and external Ca2+. The thresholds for each cell are presented at baseline (black) and following acute morphine challenge (3 μM, red) and indicate a significant depolarization with morphine perfusion. N = 5, n = 7; *p < 0.05 by two-tailed paired Student's t test (see Table S2 for additional values).
Acute Morphine-Induced Reductions of Excitability in Small Diameter DRG Neurons Are Mitigated by the MOR Antagonist Naloxone
To examine whether our observations with acute morphine challenge in vitro are mediated by action at μ-opioid receptors (MORs), morphine responses were assessed in small diameter DRG neurons from naive mice in the presence of the MOR antagonist naloxone (1 μM; Figure 2C and Table S2). Whole-cell current-clamp experiments revealed no significant alteration of excitability following acute morphine challenge (3 μM), manifested by a lack of change in the number of action potentials at double rheobase (1.8 ± 0.3 at baseline versus 1.8 ± 0.3 with morphine) or the action potential threshold (−12.9 ± 1.5 mV at baseline versus −12.2 ± 1.6 mV with morphine).
Acute Morphine-Induced Depolarization of Action Potential Threshold in Small Diameter DRG Neurons Occurs in the Absence of Ca2+
There is a significant body of evidence documenting the modulation of Ca2+ currents by opioids in DRG neurons (Hamill et al., 1981, Schroeder and McCleskey, 1993, Nomura et al., 1994). To examine whether acute morphine-induced shifts in action potential threshold are wholly mediated by modulation of Ca2+ currents, morphine responses were recorded in small diameter DRG neurons from naive mice in the complete absence of internal and external Ca2+ (Figure 2D and Table S2). Whole-cell current-clamp experiments demonstrated a significant depolarization of the action potential threshold following acute morphine challenge (3 μM; −12.3 ± 2.9 mV at baseline versus −8.8 ± 3.2 mV with morphine, p < 0.05). This suggests the involvement of a Ca2+-independent signaling mechanism in mediating morphine-induced shifts of the action potential threshold.
Acute Morphine Challenge in Small Diameter DRG Neurons Reduces the Magnitude of TTX-R Na+ Currents by Enhancing Channel Inactivation
Previous investigations in mice suggest a potential role for TTX-R Na+ currents in mediating cellular level responses to acute morphine challenge in vitro. To assess this, morphine responses were recorded in small diameter DRG neurons from mice receiving chronic exposure to an MP or a placebo pellet (PP) and 10 days of vancomycin (VAN) or saline (SAL) by oral gavage. TTX-R Na+ currents were isolated, and whole-cell voltage-clamp experiments were performed utilizing a double-pulse protocol (see Transparent Methods). Representative raw traces from the conditioning pulse (Figure 3) and current-voltage (I-V) relationships (Figure 4) indicate a significant reduction of peak TTX-R Na+ current densities in neurons from PP + SAL and PP + VAN mice in the −10 to +20 mV range (p < 0.001 and p < 0.01, respectively). This effect was mitigated in neurons from MP + SAL mice (indicating tolerance development), but preserved in neurons from MP + VAN mice (indicating tolerance prevention; p < 0.0001) (see Figure S1 for non-normalized I-V curves). In concurrence with previous investigations (Chen et al., 2012), non-normalized current densities (Figure S2) demonstrated a trend toward enhanced magnitude in mice with chronic morphine exposure (MP) compared with placebo (PP). However, this effect was not statistically significant in this study.
Figure 3.

Representative Voltage-Clamp Traces of TTX-R Na+ Currents in the Absence and Presence of Acute Morphine Challenge
Raw traces for voltage steps from −10 to +20 mV are presented at baseline (black) and following acute morphine challenge (3 μM, red). Neurons from PP + SAL and PP + VAN mice demonstrate a significant reduction in the magnitude of inward currents following morphine perfusion. This effect is mitigated in neurons from MP + SAL mice (indicating tolerance development), but preserved in neurons from MP + VAN mice (indicating tolerance prevention). A notable trend toward enhancement of inward current magnitude was noted in cohorts administered morphine pellet (MP) when compared with cohorts given placebo (PP). However, this effect was not statistically significant in this study.
Figure 4.

Acute Morphine Challenge Results in Reduction of TTX-R Na+ Current Magnitude That Is Prevented by Chronic Morphine Exposure In Vivo, but Preserved with Concurrent Oral Vancomycin Treatment
Current-voltage (I-V) relationships of TTX-R Na+ channels in voltage-clamped DRG neurons are shown at baseline (black) and following acute morphine challenge (3 μM, red). Neurons from PP + SAL and PP + VAN mice demonstrate a significant reduction of current density for steps from −10 to +20 mV. This effect was mitigated in neurons from MP + SAL mice (indicating tolerance development), but preserved in neurons from MP + VAN mice (indicating tolerance prevention). PP + SAL (N = 7, n = 7), PP + VAN (N = 4, n = 6), MP + SAL (N = 7, n = 9), MP + VAN (N = 5, n = 7), *p < 0.05, #p < 0.01, †p < 0.001, ‡p < 0.0001 by two-way repeated-measures ANOVA with Bonferroni post hoc analysis; data expressed as mean ± SEM.
To test whether acute morphine challenge (3 μM) modulates the voltage dependence of TTX-R Na+ channel activation, the peak current densities utilized for construction of I-V curves were transformed to relative conductances (G/Gmax) and fit with a Boltzmann function (see Transparent Methods). No significant shifts in the voltage of half-maximum (V1/2) activation or slope factor (k) were noted in any of the treatment cohorts with acute morphine challenge (Figure 5 and Table S3). Boltzmann fit models within each treatment group were compared by ordinary least squares non-linear regression. No significant difference in fit parameters was detected in any of the treatment cohorts following acute morphine challenge (Table S3). This is reflected by the near-zero output values for the F-ratio test statistic (F) and difference in Akaike information criterion (ΔAIC).
Figure 5.

Acute Morphine Challenge Does Not Alter the Voltage Dependence of Activation of TTX-R Na+ Channels
Relative conductances (G/Gmax) of TTX-R Na+ channels at baseline (black) and following acute morphine challenge (3 μM, red). Values transformed from I-V curves are plotted as a function of the step potential and fit with a Boltzmann function (see Transparent Methods). No significant shifts in the voltage of half-maximum (V1/2) activation, slope factor (k), or Boltzmann fit parameters are noted in any of the treatment cohorts following acute morphine challenge. PP + SAL (N = 7, n = 7), PP + VAN (N = 4, n = 6), MP + SAL (N = 7, n = 9), MP + VAN (N = 5, n = 7), analyzed by two-way repeated-measures ANOVA with Bonferroni post hoc analysis and ordinary least squares non-linear regression; data expressed as mean ± SEM (see Table S3 for additional values).
To examine the effects of acute morphine challenge (3 μM) on steady-state inactivation, the relative peak current density (I/Imax) elicited by the test pulse was plotted as a function of the conditioning pulse potential and fit with a Boltzmann function (Figure 6 and Table S4). Acute morphine challenge reduced I/Imax at all potentials tested in neurons from PP + SAL and PP + VAN mice, indicating enhanced inactivation. This effect was mitigated in neurons from MP + SAL mice (indicating tolerance development), but preserved in neurons from MP + VAN mice (indicating tolerance prevention). Notably, the voltage of half-maximum (V1/2) inactivation and slope factor (k) did not vary with acute morphine in any of the treatment cohorts. Boltzmann fit models within each treatment group were compared by ordinary least squares non-linear regression. Neurons from PP + SAL and PP + VAN mice demonstrated a significant difference in fit parameters following acute morphine challenge (p < 0.0001; Table S4). This effect was again mitigated in neurons from MP + SAL mice, but preserved in neurons from MP + VAN mice (p < 0.0001). These findings are reflected by the diminutive values of F and ΔAIC in MP + SAL mice compared with all other groups. To assess whether the time-dependent rate of TTX-R Na+ channel inactivation is affected by acute morphine challenge (3 μM), inactivation time constants (τinactiv) were estimated by fitting a monoexponential function to the falling phase of the conditioning pulse current traces at various potentials (Figure S3). Although a general trend toward reduction of τinactiv with acute morphine challenge was noted, no significant modulation was detected in any cohort in this study.
Figure 6.

Acute Morphine Challenge Enhances Steady-State Inactivation of TTX-R Na+ Channels
Relative peak current density (I/Imax) of TTX-R Na+ channels at baseline (black) and following acute morphine challenge (3 μM, red). Values obtained during the test pulse are plotted as a function of the conditioning pulse potential and fit with a Boltzmann function (see Transparent Methods). Boltzmann fit comparisons indicate that acute morphine challenge reduced I/Imax (i.e., enhanced inactivation) at all potentials tested in neurons from PP + SAL and PP + VAN mice. This effect was mitigated in neurons from MP + SAL mice (indicating tolerance development), but preserved in neurons from MP + VAN mice (indicating tolerance prevention). No significant shifts in the voltage of half-maximum (V1/2) inactivation and slope factor (k) were noted in any of the treatment cohorts with acute morphine challenge. PP + SAL (N = 7, n = 7), PP + VAN (N = 4, n = 6), MP + SAL (N = 7, n = 9), MP + VAN (N = 5, n = 7), analyzed by two-way repeated-measures ANOVA with Bonferroni post hoc analysis and ordinary least squares non-linear regression; data expressed as mean ± SEM (see Table S4 for additional values).
Mediators in the Colon of Mice with Chronic Morphine Exposure Induce Tolerance in Naïve Small Diameter DRG Neurons That Is Prevented by Concurrent Oral Vancomycin Treatment
To test whether mediators in the colon microenvironment can independently induce tolerance in DRG neurons, colonic supernatants were collected from each treatment cohort and applied to naive DRG neuron cultures (see Transparent Methods; Figures 7, 8, 9, and S4 and Table S5). Whole-cell current-clamp experiments were performed to monitor the effect of acute morphine challenge (3 μM) on cell excitability. The number of action potentials at double rheobase is shown for each cohort in Figures 7 and 9. Neurons in supernatants from MP + SAL mice demonstrated significant hyperexcitability at baseline, manifested as an increase in the number of observed action potentials (2.1 ± 0.3 versus PP + SAL 1.2 ± 0.1, p < 0.05). Excitability in these cells was unaltered by acute morphine challenge (2.1 ± 0.3 at baseline versus 1.9 ± 0.3 with morphine), indicating tolerance development. Neurons in supernatants from MP + VAN mice also demonstrated hyperexcitability at baseline (2.0 ± 0.3 versus PP + VAN 1.1 ± 0.1, p < 0.05), but responded to acute morphine challenge with a reduction in the number of observed action potentials (2.0 ± 0.3 at baseline versus 1.1 ± 0.1 with morphine, p < 0.001), indicating tolerance prevention. Neurons in supernatants from PP + SAL and PP + VAN mice largely demonstrated a single action potential at baseline, creating a floor effect that prevented detection of excitability reductions with acute morphine challenge (PP + SAL 1.2 ± 0.1 at baseline versus 1.1 ± 0.1 with morphine; PP + VAN 1.1 ± 0.1 at baseline versus 1.1 ± 0.1 with morphine).
Figure 7.

Representative Current-Clamp Traces for the Number of Action Potentials at Double Rheobase in Colonic Supernatant Experiments
Current-clamp traces at double rheobase are presented at baseline (black) and following acute morphine challenge (3 μM, red) for naive DRG neurons exposed to colonic supernatants from each treatment cohort. Neurons in supernatants from PP + SAL and PP + VAN mice demonstrated low excitability at baseline, most often characterized by a single action potential at double rheobase. Owing to a floor effect, no reduction could be detected with morphine perfusion. Neurons in supernatants from MP + SAL mice demonstrated enhanced excitability at baseline, characterized by a greater number of action potentials at double rheobase. No reduction was detected with acute morphine challenge, indicating tolerance development. Neurons in supernatants from MP + VAN mice were also hyperexcitable at baseline, but demonstrated a reduction in the number of action potentials at double rheobase with acute morphine challenge, indicating tolerance prevention (see Figure 9 for individual observations).
Figure 8.

Representative Current-Clamp Traces for Action Potential Threshold in Colonic Supernatant Experiments
Action potential threshold (Vt) is presented at baseline (black) and following acute morphine challenge (3 μM, red) for naive DRG neurons exposed to colonic supernatants from each treatment cohort. Neurons in supernatants from PP + SAL and PP + VAN mice demonstrated low excitability at baseline, characterized by depolarization of Vt. Morphine perfusion significantly depolarized Vt further, indicating a lack of tolerance development. Neurons in supernatants from MP + SAL mice demonstrated enhanced excitability at baseline, characterized by hyperpolarization of Vt. No depolarization was noted with acute morphine challenge, indicating tolerance development. Neurons in supernatants from MP + VAN mice were also hyperexcitable at baseline, but demonstrated depolarization of Vt with acute morphine challenge, indicating tolerance prevention (see Figure 9 for individual observations).
Figure 9.

Mediators in the Gut Wall of Mice with Chronic Morphine Exposure Induce Tolerance in Naive DRG Neurons That Is Prevented by Oral Vancomycin Treatment
Individual observations of naive DRG neurons exposed to colonic supernatants from each treatment cohort. Action potentials at double rheobase and action potential threshold (Vt) are presented at baseline (black) and following acute morphine challenge (3 μM, red). Neurons in supernatants from PP + SAL and PP + VAN mice demonstrated low excitability at baseline, characterized by fewer action potentials at double rheobase and depolarization of the action potential threshold (Vt). A significant depolarization of Vt was noted with morphine perfusion; however, no alteration in the number of action potentials at double rheobase could be detected owing to a floor effect. Neurons in supernatants from MP + SAL mice demonstrated enhanced excitability at baseline, characterized by more action potentials at double rheobase and hyperpolarization of Vt. No shift in either parameter was noted with acute morphine challenge, indicating tolerance development. Neurons in supernatants from MP + VAN mice also demonstrated enhanced excitability at baseline, but responded to acute morphine challenge with reductions in the number of action potentials at double rheobase and depolarizations of Vt. PP + SAL (N = 8, n = 9), PP + VAN (N = 6, n = 8), MP + SAL (N = 5, n = 9), MP + VAN (N = 7, n = 8); ns, not significant; *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001 by two-way repeated-measures ANOVA with Bonferroni post hoc analysis; data expressed as mean ± SEM (see Table S5 for additional values).
The action potential threshold (Vt) is shown for each cohort in Figures 8 and 9. For neurons in supernatants from MP + SAL mice, hyperexcitability at baseline manifested as hyperpolarization of Vt (−20.1 ± 1.3 mV versus PP + SAL -10.8 ± 2.4 mV, p < 0.05). Again, excitability in these cells was unaltered by acute morphine challenge (−20.1 ± 1.3 mV at baseline versus −19.7 ± 1.3 mV with morphine), indicating tolerance development. Neurons in supernatants from MP + VAN mice were also hyperexcitable at baseline (−21.2 ± 2.1 mV versus PP + VAN -9.2 ± 1.6 mV, p < 0.01) but responded to acute morphine challenge with depolarizations of Vt (−21.2 ± 2.1 mV at baseline versus −17.4 ± 2.3 mV with morphine), indicating tolerance prevention. Reductions of excitability with acute morphine challenge were revealed in neurons with supernatants from PP + SAL and PP + VAN mice, manifested by depolarizations of Vt (PP + SAL −10.8 ± 2.4 mV at baseline versus −6.9 ± 2.9 mV with morphine, p < 0.001; PP + VAN -9.2 ± 1.6 mV at baseline versus −4.5 ± 2.6 mV with morphine, p < 0.0001).
Discussion
The purpose of this study was to investigate the role of TTX-R Na+ channels and gut-derived mediators in morphine action and tolerance in small diameter DRG neurons. The salient findings are that (1) oral vancomycin prevents antinociceptive morphine tolerance in a manner dependent on the duration of treatment, (2) oral vancomycin prevents cellular level morphine tolerance in small diameter DRG neurons, (3) acute morphine challenge in small diameter DRG neurons results in inactivation of TTX-R Na+ channels, (4) acute morphine-induced inhibition of TTX-R Na+ channels is mitigated in cells from tolerant mice, (5) mediators in the gut microenvironment of mice with chronic morphine exposure can induce tolerance and hyperexcitability in naive small diameter DRG neurons, and (6) induction of tolerance (but not hyperexcitability) by gut mediators is mitigated by concurrent treatment with oral vancomycin.
Owing to the clinical and social ramifications of physical dependence and addiction, the mechanisms underlying opioid tolerance development have been studied for decades. Recent evidence suggests that morphine exposure in mice results in dysbiosis of resident gastrointestinal microflora, marked by alterations of microbial composition and compromise of gut epithelial tight junction integrity (Meng et al., 2013). The latter, occurring through mechanisms that involve toll-like receptors (TLRs) 2 and 4, results in gut epithelial “leakiness,” which allows translocation of luminal bacteria to the gut wall and circulation. The subsequent release of bacterial products and pro-inflammatory cytokines may modulate the development of morphine tolerance in nociceptive primary afferent neurons. There is some evidence that morphine can non-stereoselectively bind and activate TLR4 receptors in microglia (Watkins et al., 2009, Hutchinson et al., 2010), whereas the role of this binding in tolerance and OIH remains unresolved (Corder et al., 2017), and both have been noted to develop in TLR4 knockout mice (Fukagawa et al., 2013, Mattioli et al., 2014). In addition, we previously demonstrated that antinociceptive tolerance following chronic morphine exposure in mice could be prevented by concurrent treatment with oral vancomycin, inherently suggesting mechanisms beyond direct TLR4 binding (Kang et al., 2017). In the present study, this tolerance prevention was noted to occur in a manner dependent on the duration of treatment and was recapitulated at the cellular level in isolated DRG neurons. The virtual 0% bioavailability of oral vancomycin suggests a role for gastrointestinal microbiota in this process.
The DRG contain the neuronal soma of both somatic and visceral nociceptive primary afferents, serving as a “relay station” to the central nervous system (CNS). It has been suggested that opioid receptors on these neurons account for 50%–100% of the antinociceptive effect of systemic opioids (Stein and Machelska, 2011). Seminal investigations of opioid action in CNS neurons noted hyperpolarization of resting membrane potential, decrease of action potential duration, and decrease in neurotransmitter release following opioid exposure in myriad cell populations (Schulman, 1981, North and Williams, 1983, Yoshimura and North, 1983). Although significant heterogeneity was noted among cell populations (Duggan and North, 1983), these observations were primarily attributed to increases in K+ efflux and decreases in Ca2+ influx. DRG neurons have not demonstrated the hyperpolarization of resting membrane potential and modulation of K+ conductance that many central neurons display (Akins and McCleskey, 1993). Therefore early investigations in these cells primarily focused on the modulation of Ca2+ currents (Hamill et al., 1981, Schroeder and McCleskey, 1993, Nomura et al., 1994). The vast majority of such studies isolated Ca2+ currents, in part, by replacement of external Na+ with non-permeable tetraethylammonium (TEA). This approach may have undermined an important role held by Na+ channels in mediating opioid responses in DRG neurons (Gold et al., 1996a, Gold et al., 1996b, Chen et al., 2012, Ross et al., 2012). Indeed, some early CNS investigations observed inhibition of Na+ currents (North and Williams, 1985, Aghajanian and Wang, 1987) and the present study indicated that morphine-induced depolarization of action potential threshold in DRG neurons occurs even in the complete absence of Ca2+. The action potential threshold is defined as the value of the membrane potential at which the magnitudes of inward and outward currents contributing to depolarization are exactly equal. Once a triggering event depolarizes the membrane in excess of this value, the positive feedback loop of voltage-gated inward current activation closes and the action potential is observed. The major drivers of this phenomenon have been classically identified as voltage-gated Na+ and K+ channels. Given that previous investigations in DRG neurons have not noted modulation of K+ conductance with opioid exposure (Akins and McCleskey, 1993), there is great precedent for studying Na+ channels.
Our voltage-clamp investigations of isolated TTX-R Na+ currents indicated a significant reduction of current magnitude following acute morphine challenge. Reflecting previous behavioral observations, chronic morphine exposure resulted in tolerance to this effect, which was prevented by concurrent treatment with oral vancomycin. No modulation of the voltage dependence of activation was noted in these records, suggesting that the TTX-R Na+ channel open probability (Popen) curve is unaffected by acute morphine. In contrast, steady-state inactivation studies indicated an enhancement of TTX-R Na+ channel inactivation with acute morphine. Chronic morphine exposure again resulted in tolerance to this effect, which was prevented by concurrent treatment with oral vancomycin. Notably, the voltage of half-maximum (V1/2) inactivation, slope factor (k), and inactivation time constant (τinactiv) did not vary with acute morphine in any of the treatment cohorts. This implies that morphine induces complete inactivation in a subset of TTX-R Na+ channels (effectively eliminating them from the population available for activation), while leaving the inactivation kinetics of the remaining channels unaltered. It is as of yet unclear whether this inactivation effect is saturable by elevating the challenge dose of morphine or if a subpopulation of TTX-R Na+ channels will demonstrate resistance to this effect at all doses.
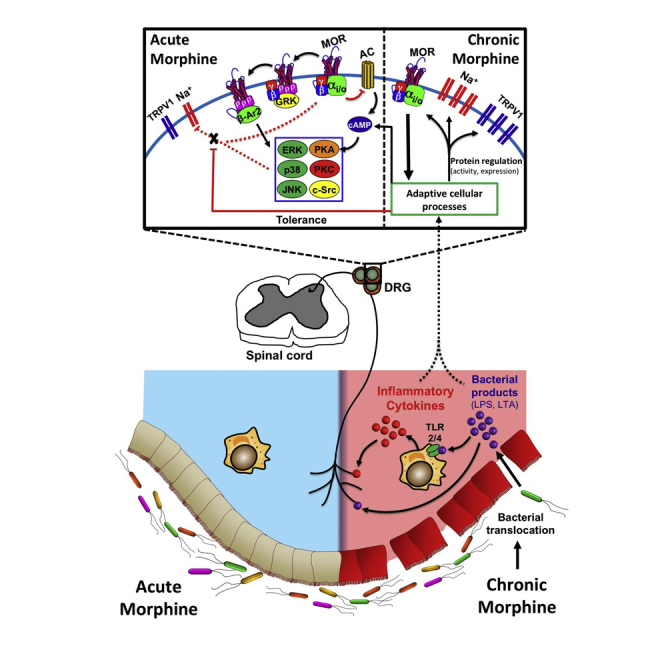
The mechanism by which acute morphine challenge enhances TTX-R Na+ channel inactivation remains elusive. (A model of our working hypothesis is presented in Figure 10.) MORs are classical Gi/o-coupled receptors, activation of which stimulates the exchange of GDP for GTP on the Gα subunit. The subsequent dissociation of Gα and Gβγ subunits promotes modulation of ion channel currents and activation of various kinase cascades. The Gα subunit interacts with adenylyl cyclase to inhibit the cyclic AMP (cAMP)/protein kinase A (PKA) pathway. The Gβγ subunit (1) activates GIRK channels (Torrecilla et al., 2002, Torrecilla et al., 2008, Sadja et al., 2003), (2) inhibits voltage-gated Ca2+ channels (Schroeder et al., 1991, Moises et al., 1994), (3) induces recruitment of GRK2/3, (4) activates the mitogen-activated protein kinase (MAPK) cascade (Ras, Raf, MEK, ERK1/2, JNK, p38) in a PI3K-/c-Src-dependent manner (Polakiewicz et al., 1998a, Polakiewicz et al., 1998b, Williams et al., 2001), (5) activates the phospholipase C/protein kinase C pathway, and (6) stimulates Ca2+/calmodulin-dependent protein kinase II (Raehal et al., 2011). Inactivation of Na+ channels involves protein conformational changes (e.g., of the fast inactivation gate at the S3-S4 intracellular loop; Catterall, 2002) that result in occlusion of the conduction pore. Others have noted a direct block of sodium channels with the peripherally restricted opioid loperamide (Wu et al., 2017), whereas our studies with naloxone alternatively suggest an MOR-dependent mechanism for morphine in DRG neurons. The observation of tolerance development with chronic morphine exposure supports this, as this process would not likely impede direct inhibition. Inactivation is therefore plausibly propagated by direct Gβγ inhibition or modification of intracellular kinase activity that results in a phosphorylation/dephosphorylation event. Gβγ is of particular interest, given its known role in modulating the conductance of various ion channels, including Nav1.1 and 1.2 (Mantegazza et al., 2005).
Figure 10.

General Schematic of the Working Hypothesis
Morphine acting acutely on small diameter DRG neurons results in inhibition of TTX-R Na+ channels. This action is MOR dependent, likely occurring via direct Gβγ inhibition or a downstream phosphorylation/dephosphorylation event. Chronic morphine exposure compromises gut epithelial tight junction integrity, allowing translocation of luminal bacteria and the subsequent release of bacterial products and pro-inflammatory cytokines. These act on small diameter DRG neurons, producing tolerance to the inhibitory action of acute morphine on TTX-R Na+ channels. Elimination of Gram-positive gut microbiota by oral vancomycin prevents this tolerance, but does not prevent the upregulation of TTX-R Na+ and TRPV1 channels induced by chronic morphine.
The trend toward enhancement of TTX-R Na+ currents with chronic morphine exposure in this study has been observed to be significant by others (Chen et al., 2012). Prior investigations have additionally demonstrated a trend toward enhancement of TTX-R Na+ channel mRNA expression in whole DRG isolates from these mice (Ross et al., 2012). Although this effect was not significant, the authors rightly noted that the expression might be selectively enhanced in cellular subpopulations (e.g., nociceptors) and that single-cell PCR experiments should be conducted. Furthermore, it is possible that TTX-R Na+ channel expression is upregulated without notable changes in mRNA concentration. Indeed, Chen and colleagues demonstrated that TRPV1 surface expression is increased in these neurons without an associated increase in TRPV1 mRNA expression (Chen et al., 2008), suggesting modulation of other cellular mechanisms (e.g., translation, splicing, or trafficking). A comprehensive investigation of this topic with regard to TTX-R Na+ currents is undoubtedly necessary.
Gβγ-induced GRK2/3 recruitment to the MOR results in phosphorylation of the agonist-bound receptor, producing a conformational change in the MOR that increases its affinity for β-arrestin2 (β-Ar2). Binding of β-Ar2 (1) creates steric hindrance that prevents coupling of the MOR to G proteins, (2) promotes receptor internalization by targeting the MOR to clathrin-coated pits, and (3) serves as a scaffolding protein for the activation of various intracellular kinase cascades (e.g., MAPK). Importantly, a prerequisite for β-Ar2 activation of the MAPK cascade is receptor internalization (Khokhlatchev et al., 1998). Morphine is a poor stimulator of internalization compared with the other opioids, making MAPK activation by this pathway rather ineffective (Williams et al., 2001). For this reason, it is unlikely that inhibition of TTX-R Na+ channels by acute morphine challenge occurs via this pathway. However, it should be noted that chronic morphine exposure has been demonstrated to result in adaptive mechanisms that promote the activation of MAPK by β-Ar2 (Atkins et al., 1998). MAPK can phosphorylate multiple cytoplasmic and nuclear targets (e.g., CREB) to modulate transcriptional events. As discussed, chronic morphine exposure results in hyperexcitability of DRG neurons associated with enhancement of TTX-R Na+ and TRPV1 currents (Chen et al., 2008, Ross et al., 2012, Smith et al., 2012). It is therefore quite possible that this β-Ar2/MAPK pathway is involved in the cellular adaptive processes that take place with chronic morphine exposure to modulate excitability and tolerance. Supporting this, Chen and colleagues demonstrated that chronic morphine exposure has been linked to an upregulation of TRPV1 in DRG neurons, which is associated with increased phosphorylation of MAPKs (p38, ERK, and JNK; Chen et al., 2008). In this study, inhibition of the MAPK pathway prevented tolerance and hypernociception induced by chronic morphine exposure. It is therefore possible that MAPK is also involved in the modulation of TTX-R Na+ channel surface expression and tolerance. Indeed, p38 has been observed to directly phosphorylate Nav1.8 channels to increase current density (Hudmon et al., 2008). Adding to the complexity of this issue, MOR β-Ar2 recruitment per se can modulate channel activity by diverting β-Ar2 away from other molecular targets. This effect has been noted to enhance the sensitivity of TRPV1 channels (Rowan et al., 2014). Whether this mechanism could also affect TTX-R Na+ currents (directly or indirectly) remains to be determined.
Our investigations with colon tissue supernatants suggest that tolerance and hyperexcitability associated with chronic morphine exposure develop independently and as indirect actions of morphine, presumably downstream of peripheral MOR activation (Corder et al., 2017). The efficacy of oral vancomycin in preventing tolerance development indicates the involvement of Gram-positive microbiota, including translocation and the secondary release of bacterial products and pro-inflammatory cytokines. In this regard, inflammation has been shown to result in significant plasticity of opioid signaling, including receptor expression, G-protein signaling, and receptor trafficking (Zhang et al., 2014). Vancomycin did not alter the development of hyperexcitability, indicating a mechanism independent of Gram-positive microbiota. This does not, however, rule out the possibility for involvement of Gram-negative gut microbes or inflammatory mechanisms independent of bacterial translocation. Certainly, direct opioid action on immune cells is well documented, as is the role of TRPV1 in both opioid- and inflammation-induced hypernociception (Caterina et al., 2000, Caterina and Julius, 2001, Ji et al., 2002, Chen et al., 2008, Ross et al., 2012, Smith et al., 2012).
Further characterization of the mechanisms underlying opioid action and tolerance in DRG neurons is undoubtedly necessary. Although visceral sensory afferents account for less than 10% of all DRG neurons (Beyak et al., 2006), our investigations did not discriminate for those with gut innervation, implying relevance to somatic sensory afferents as well. Communication with somatic afferents may result from systemically circulating mediators or local activation of satellite glia by visceral afferents. Furthermore, the extent to which morphine-induced inhibition of Nav1.8 and 1.9 specifically mediates depolarization of action potential threshold in DRG neurons remains unclear. It is quite possible that inhibition is subtype selective, or even that TTX-sensitive Na+ channels are involved. Elucidating such details could greatly affect clinical pain management and improve patient outcomes.
Methods
All methods can be found in the accompanying Transparent Methods supplemental file.
Acknowledgments
This study was supported by the National Institutes of Health grants F30 DA042542 to R.A.M., R01 DA036975 to H.I.A. and W.L.D., P30 DA033934, and T32 DA007027.
Author Contributions
R.A.M. and H.I.A. intellectually conceived and designed the experiments. R.A.M. conducted the experiments. H.I.A. and W.L.D. supervised the project. R.A.M. produced the manuscript, which H.I.A. edited and all authors approved.
Declaration of Interests
All authors declare no conflict of interest or competing financial interests.
Published: March 22, 2018
Footnotes
Supplemental Information includes Transparent Methods, four figures, and five tables and can be found with this article online at https://doi.org/10.1016/j.isci.2018.03.003.
Supplemental Information
References
- Aghajanian G., Wang Y. Common alpha-2 and opiate effector mechanisms in the locus ceruleus: intracellular studies in brain slices. Neuropharmacology. 1987;26:789–800. doi: 10.1016/0028-3908(87)90054-2. [DOI] [PubMed] [Google Scholar]
- Akbar A., Yiangou Y., Facer P., Walters J.R., Anand P., Ghosh S. Increased capsaicin receptor TRPV1-expressing sensory fibres in irritable bowel syndrome and their correlation with abdominal pain. Gut. 2008;57:923–929. doi: 10.1136/gut.2007.138982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akins P., McCleskey E. Characterization of potassium currents in adult rat sensory neurons and modulation by opioids and cyclic AMP. Neuroscience. 1993;56:759–769. doi: 10.1016/0306-4522(93)90372-m. [DOI] [PubMed] [Google Scholar]
- Amaya F., Decosterd I., Samad T.A., Plumpton C., Tate S., Mannion R.J., Costigan M., Woolf C.J. Diversity of expression of the sensory neuron-specific TTX-resistant voltage-gated sodium ion channels SNS and SNS2. Mol. Cell. Neurosci. 2000;15:331–342. doi: 10.1006/mcne.1999.0828. [DOI] [PubMed] [Google Scholar]
- Angst M.S., Clark J.D. Opioid-induced hyperalgesia: a qualitative systematic review. Anesthesiology. 2006;104:570–587. doi: 10.1097/00000542-200603000-00025. [DOI] [PubMed] [Google Scholar]
- Atkins C.M., Selcher J.C., Petraitis J.J., Trzaskos J.M., Sweatt J.D. The MAPK cascade is required for mammalian associative learning. Nat. Neurosci. 1998;1:602–609. doi: 10.1038/2836. [DOI] [PubMed] [Google Scholar]
- Barbara G., Stanghellini V., De Giorgio R., Cremon C., Cottrell G.S., Santini D., Pasquinelli G., Morselli-Labate A.M., Grady E.F., Bunnett N.W. Activated mast cells in proximity to colonic nerves correlate with abdominal pain in irritable bowel syndrome. Gastroenterology. 2004;126:693–702. doi: 10.1053/j.gastro.2003.11.055. [DOI] [PubMed] [Google Scholar]
- Beyak M., Bulmer D., Jiang W., Keating C., Rong W., Grundy D. Extrinsic Sensory Afferent Nerves Innervating the Gastrointestinal Tract. In: Johnson L.R., Barrett K.E., Ghishan F.K., Merchant J.L., Said H.M., Wood J.D., editors. 4th edn. vol. 1. McGraw-Hill; 2006. pp. 685–726. (Physiology of the Gastrointestinal Tract). [Google Scholar]
- Caterina M.J., Leffler A., Malmberg A.B., Martin W.J., Trafton J., Petersen-Zeitz K.R., Koltzenburg M., Basbaum A.I., Julius D. Impaired nociception and pain sensation in mice lacking the capsaicin receptor. Science. 2000;288:306–314. doi: 10.1126/science.288.5464.306. [DOI] [PubMed] [Google Scholar]
- Caterina M., Julius D. The vanilloid receptor: a molecular gateway to the pain pathway. Annu. Rev. Neurosci. 2001;24:487–517. doi: 10.1146/annurev.neuro.24.1.487. [DOI] [PubMed] [Google Scholar]
- Catterall W.A. Molecular mechanisms of gating and drug block of sodium channels. Novartis Found. Symp. 2002;241:206–232. [PubMed] [Google Scholar]
- Cenac N., Andrews C.N., Holzhausen M., Chapman K., Cottrell G., Andrade-Gordon P., Steinhoff M., Barbara G., Beck P., Bunnett N.W. Role for protease activity in visceral pain in irritable bowel syndrome. J. Clin. Invest. 2007;117:636–647. doi: 10.1172/JCI29255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J., Gong Z.H., Yan H., Qiao Z., Qin B.Y. Neuroplastic alteration of TTX-resistant sodium channel with visceral pain and morphine-induced hyperalgesia. J. Pain Res. 2012;5:491–502. doi: 10.2147/JPR.S27751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y., Geis C., Sommer C. Activation of TRPV1 contributes to morphine Tolerance: involvement of the mitogen-activated protein kinase signaling pathway. J. Neurosci. 2008;28:5836–5845. doi: 10.1523/JNEUROSCI.4170-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collett B.J. Opioid tolerance: the clinical perspective. Br. J. Anaesth. 1998;81:58–68. doi: 10.1093/bja/81.1.58. [DOI] [PubMed] [Google Scholar]
- Corder G., Tawfik V.L., Wang D., Sypek E.I., Low S.A., Dickinson J.R., Sotoudeh C., Clark J.D., Barres B.A., Bohlen C.J., Scherrer G. Loss of μ opioid receptor signaling in nociceptors, but not microglia, abrogates morphine tolerance without disrupting analgesia. Nat. Med. 2017;23:164–173. doi: 10.1038/nm.4262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dowell D., Haegerich T.M., Chou R. CDC guideline for prescribing opioids for chronic pain — United States, 2016. MMWR Recomm. Rep. 2016;65:1–49. doi: 10.15585/mmwr.rr6501e1. [DOI] [PubMed] [Google Scholar]
- Duggan A., North R. Electrophysiology of opioids. Pharmacol. Rev. 1983;35:219–281. [PubMed] [Google Scholar]
- Elliott A., Elliott J. Characterization of TTX-sensitive and TTX-resistant sodium currents in small cells from adult rat dorsal root ganglia. J. Physiol. 1993;463:39–56. doi: 10.1113/jphysiol.1993.sp019583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukagawa H., Koyama T., Kakuyama M., Fukuda K. Microglial activation involved in morphine tolerance is not mediated by toll-like receptor 4. J. Anesth. 2013;27:93–97. doi: 10.1007/s00540-012-1469-4. [DOI] [PubMed] [Google Scholar]
- Fukuda J., Kameyama M. Tetrodotoxin-sensitive and tetrodotoxin-resistant sodium channels in tissue-cultured spinal ganglion neurons from adult mammals. Brain Res. 1980;182:191–197. doi: 10.1016/0006-8993(80)90844-6. [DOI] [PubMed] [Google Scholar]
- Gold M., Reichling D.B., Shuster M.J., Levine J.D. Hyperalgesic agents increase a tetrodotoxin-resistant Na+ current in nociceptors. Proc. Natl. Acad. Sci. USA. 1996;93:1108–1112. doi: 10.1073/pnas.93.3.1108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gold M.S., Shuster M.J., Levine J.D. Characterization of six voltage-gated K+ currents in adult rat sensory neurons. J. Neurophysiol. 1996;75:2629–2646. doi: 10.1152/jn.1996.75.6.2629. [DOI] [PubMed] [Google Scholar]
- Hamill O., Marty A., Neher E., Sakmann B., Sigworth F.J. Improved patch-clamp techniques for high-resolution current recordings from cells and cell-free membrane patches. Pflugers Arch. 1981;391:85–100. doi: 10.1007/BF00656997. [DOI] [PubMed] [Google Scholar]
- Hudmon A., Choi J.S., Tyrrell L., Black J.A., Rush A.M., Waxman S.G., Dib-Hajj S.D. Phosphorylation of sodium channel Na(v)1.8 by p38 mitogen-activated protein kinase increases current density in dorsal root ganglion neurons. J. Neurosci. 2008;28:3190–3201. doi: 10.1523/JNEUROSCI.4403-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hutchinson M.R., Zhang Y., Shridhar M., Evans J.H., Buchanan M.M., Zhao T.X., Slivka P.F., Coats B.D., Rezvani N., Wieseler J. Evidence that opioids may have toll like receptor 4 and MD-2 effects. Brain Behav. Immun. 2010;24:83–95. doi: 10.1016/j.bbi.2009.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji R.R., Samad T.A., Jin S.X., Schmoll R., Woolf C.J. p38 MAPK activation by NGF in primary sensory neurons after inflammation increases TRPV1 levels and maintains heat hyperalgesia. Neuron. 2002;36:57–68. doi: 10.1016/s0896-6273(02)00908-x. [DOI] [PubMed] [Google Scholar]
- Kang M., Mischel R.A., Bhave S., Komla E., Cho A., Huang C., Dewey W.L., Akbarali H.I. The effect of gut microbiome on tolerance to morphine mediated antinociception in mice. Sci. Rep. 2017;7:42658. doi: 10.1038/srep42658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khokhlatchev A.V., Canagarajah B., Wilsbacher J., Robinson M., Atkinson M., Goldsmith E., Cobb M.H. Phosphorylation of the MAP kinase ERK2 promotes its homodimerization and nuclear translocation. Cell. 1998;93:605–615. doi: 10.1016/s0092-8674(00)81189-7. [DOI] [PubMed] [Google Scholar]
- Mantegazza M., Yu F.H., Powell A.J., Clare J.J., Catterall W.A., Scheuer T. Molecular determinants for modulation of persistent sodium current by G-protein betagamma subunits. J. Neurosci. 2005;25:3341–3349. doi: 10.1523/JNEUROSCI.0104-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuda Y., Yoshida S., Yonezawa T. Tetrodotoxin sensitivity and Ca2+ component of action potentials of mouse dorsal root ganglion cells cultured in vitro. Brain Res. 1978;154:69–82. doi: 10.1016/0006-8993(78)91052-1. [DOI] [PubMed] [Google Scholar]
- Mattioli T.A., Leduc-Pessah H., Skelhorne-Gross G., Nicol C.J., Milne B., Trang T., Cahill C.M. Toll-like receptor 4 mutant and null mice retain morphine-induced tolerance, hyperalgesia, and physical dependence. PLoS One. 2014;9:e97361. doi: 10.1371/journal.pone.0097361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meng J., Yu H., Ma J., Wang J., Banerjee S., Charboneau R., Barke R.A., Roy S. Morphine induces bacterial translocation in mice by compromising intestinal barrier function in a TLR-dependent manner. PLoS One. 2013;8:e54040. doi: 10.1371/journal.pone.0054040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moises H., Rusin K., Macdonald R. mu-opioid receptor-mediated reduction of neuronal calcium current occurs via a Go-type GTP-binding protein. J. Neurosci. 1994;14:3842–3851. doi: 10.1523/JNEUROSCI.14-06-03842.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nomura K., Reuveny E., Narahashi T. Opioid inhibition and desensitization of calcium channel currents in rat dorsal root ganglion neurons. J. Pharmacol. Exp. Ther. 1994;270:466–474. [PubMed] [Google Scholar]
- North R., Williams J. Opiate activation of potassium conductance inhibits calcium action potentials in rat locus ceoruleus neurones. Br. J. Pharmacol. 1983;80:225–228. doi: 10.1111/j.1476-5381.1983.tb10023.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- North R., Williams J. On the potassium conductance increased by opioids in rat locus coeruleus neurones. J. Physiol. 1985;364:265–280. doi: 10.1113/jphysiol.1985.sp015743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogata N., Tatebayashi H. Ontogenic development of the TTX-sensitive and TTX-insensitive Na+ channels in neurons of the rat dorsal root ganglia. Brain Res. Dev. Brain Res. 1992;65:93–100. doi: 10.1016/0165-3806(92)90012-l. [DOI] [PubMed] [Google Scholar]
- Ogata N., Tatebayashi H. Kinetic analysis of two types of Na+ channels in rat dorsal root ganglia. J. Physiol. 1993;466:9–37. [PMC free article] [PubMed] [Google Scholar]
- Polakiewicz R.D., Schieferl S.M., Dorner L.F., Kansra V., Comb M.J. A mitogen-activated protein kinase pathway is required for u-opioid receptor desensitization. J. Biol. Chem. 1998;273:12402–12406. doi: 10.1074/jbc.273.20.12402. [DOI] [PubMed] [Google Scholar]
- Polakiewicz R.D., Schieferl S.M., Gingras A.C., Sonenberg N., Comb M.J. mu-opioid receptor activates signaling pathways implicated in cell survival and translational control. J. Biol. Chem. 1998;273:23534–23541. doi: 10.1074/jbc.273.36.23534. [DOI] [PubMed] [Google Scholar]
- Raehal K.M., Schmid C.L., Groer C.E., Bohn L.M. Functional selectivity at the u-opioid receptor: implications for understanding opioid analgesia and tolerance. Pharmacol. Rev. 2011;63:1001–1019. doi: 10.1124/pr.111.004598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rizzo M., Kocsis J., Waxman S. Slow sodium conductances of dorsal root ganglion neurons: intraneuronal homogeneity and interneuronal heterogeneity. J. Neurophysiol. 1994;72:2796–2815. doi: 10.1152/jn.1994.72.6.2796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ross G.R., Gade A.R., Dewey W.L., Akbarali H.I. Opioid-induced hypernociception is associated with hyperexcitability and altered tetrodotoxin-resistant Na+ channel function of dorsal root ganglia. Am. J. Physiol. Cell Physiol. 2012;302:C1152–C1161. doi: 10.1152/ajpcell.00171.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rowan M.P., Bierbower S.M., Eskander M.A., Szteyn K., Por E.D., Gomez R., Veldhuis N., Bunnett N.W., Jeske N.A. Activation of mu opioid receptors sensitizes transient receptor potential vanilloid type 1 (TRPV1) via β-arrestin-2-mediated cross-talk. PLoS One. 2014;9:e93688. doi: 10.1371/journal.pone.0093688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roy M., Narahashi T. Differential properties of tetrodotoxin-sensitive and tetrodotoxin-resistant sodium channels in rat dorsal root ganglion neurons. J. Neurosci. 1992;12:2104–2111. doi: 10.1523/JNEUROSCI.12-06-02104.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sadja R., Alagem N., Reuveny E. Gating of GIRK channels: details of an intricate, membrane-delimited signaling complex. Neuron. 2003;39:9–12. doi: 10.1016/s0896-6273(03)00402-1. [DOI] [PubMed] [Google Scholar]
- Schroeder J.E., Fischbach P.S., Zheng D., McCleskey E.W. Activation of u opioid receptors inhibits transient high- and low-threshold Ca2+ currents, but spares a sustained current. Neuron. 1991;6:13–20. doi: 10.1016/0896-6273(91)90117-i. [DOI] [PubMed] [Google Scholar]
- Schroeder J., McCleskey E. Inhibition of Ca2+ currents by a u-opioid in a defined subset of rat sensory neurons. J. Neurosci. 1993;13:867–873. doi: 10.1523/JNEUROSCI.13-02-00867.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulman J.A. Anatomical distribution and physiological effects of enkephalin in rat inferior olive. Regul. Pept. 1981;2:125–137. doi: 10.1016/0167-0115(81)90006-9. [DOI] [PubMed] [Google Scholar]
- Smith T.H., Grider J.R., Dewey W.L., Akbarali H.I. Morphine decreases enteric neuron excitability via inhibition of sodium channels. PLoS One. 2012;7:e45251. doi: 10.1371/journal.pone.0045251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stein C., Machelska H. Modulation of peripheral sensory neurons by the immune system: implications for pain therapy. Pharmacol. Rev. 2011;63:860–881. doi: 10.1124/pr.110.003145. [DOI] [PubMed] [Google Scholar]
- Torrecilla M., Marker C.L., Cintora S.C., Stoffel M., Williams J.T., Wickman K. G-protein-gated potassium channels containing Kir3.2 and Kir3.3 subunits mediate the acute inhibitory effects of opioids on locus ceruleus neurons. J. Neurosci. 2002;22:4328–4334. doi: 10.1523/JNEUROSCI.22-11-04328.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torrecilla M., Quillinan N., Williams J.T., Wickman K. Pre- and postsynaptic regulation of locus coeruleus neurons after chronic morphine treatment: a study of GIRK-knockout mice. Eur. J. Neurosci. 2008;28:618–624. doi: 10.1111/j.1460-9568.2008.06348.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watkins L.R., Hutchinson M.R., Rice K.C., Maier S.F. The “toll” of opioid-induced glial activation: Improving the clinical efficacy of opioids by targeting glia. Trends Pharmacol. Sci. 2009;30:581–591. doi: 10.1016/j.tips.2009.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams J.T., Christie M., Manzoni O. Cellular and synaptic adaptations mediating opioid dependence. Physiol. Rev. 2001;81:299–343. doi: 10.1152/physrev.2001.81.1.299. [DOI] [PubMed] [Google Scholar]
- Wu Y., Zou B., Liang L., Li M., Tao Y.X., Yu H., Wang X., Li M. Loperamide inhibits sodium channels to alleviate inflammatory hyperalgesia. Neuropharmacology. 2017;117:282–291. doi: 10.1016/j.neuropharm.2017.02.010. [DOI] [PubMed] [Google Scholar]
- Xu Y., Xie Z., Wang H., Shen Z., Guo Y., Gao Y., Chen X., Wu Q., Li X., Wang K. Bacterial diversity of intestinal microbiota in patients with substance use disorders revealed by 16S rRNA gene deep sequencing. Sci. Rep. 2017;7:3628. doi: 10.1038/s41598-017-03706-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshimura M., North R. Substantia gelatinosa neurones in vitro hyperpolarised by enkephalin. Nature. 1983;305:266–277. doi: 10.1038/305529a0. [DOI] [PubMed] [Google Scholar]
- Zhang X., Bao L., Li S. Opioid receptor trafficking and interaction in nociceptors. Br. J. Pharmacol. 2014;172:364–374. doi: 10.1111/bph.12653. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.