

Abstract
Transcription activator proteins typically contain two functional domains: a DNA binding domain (DBD) that binds to DNA with sequence specificity and an activation domain (AD) whose established function is to recruit RNA polymerase. In this report, we show that purified recombinant nuclear factor κB (NF-κB) RelA dimers bind specific κB DNA sites with an affinity significantly lower than that of the same dimers from nuclear extracts of activated cells, suggesting that additional nuclear cofactors might facilitate DNA binding by the RelA dimers. Additionally, recombinant RelA binds DNA with relatively low affinity at a physiological salt concentration in vitro. The addition of p53 or RPS3 (ribosomal protein S3) increases RelA:DNA binding affinity 2- to >50-fold depending on the protein and ionic conditions. These cofactor proteins do not form stable ternary complexes, suggesting that they stabilize the RelA:DNA complex through dynamic interactions. Surprisingly, the RelA-DBD alone fails to bind DNA under the same solution conditions even in the presence of cofactors, suggesting an important role of the RelA-AD in DNA binding. Reduced RelA:DNA binding at a physiological ionic strength suggests that multiple cofactors might be acting simultaneously to mitigate the electrolyte effect and stabilize the RelA:DNA complex in vivo. Overall, our observations suggest that the RelA-AD and multiple cofactor proteins function cooperatively to prime the RelA-DBD and stabilize the RelA:DNA complex in cells. Our study provides a mechanism for nuclear cofactor proteins in NF-κB-dependent gene regulation.
Graphical Abstract
The nuclear factor specific for the κ light chain gene enhancer in B cells (NF-κB) is a family of dimeric transcription factors that regulate the expression levels of hundreds of diverse target genes influencing a broad range of cellular programs from immune and inflammatory responses to survival and death.1 Five distinct polypeptides, RelA/p65, RelB, c-Rel, p50, and p52, comprise the mammalian NF-κB family. These five subunits share a highly conserved stretch of roughly 300 amino acids, known as the Rel homology region (RHR) (Figure 1a). The RHR is responsible for the DNA binding and thus is essentially the DNA binding domain (DBD). RelA is the most well-studied family member because of its ubiquitous expression in nearly all cell types as well as its involvement in almost all cellular functions shown to result from NF-κB activity.2 RelA functions primarily as a heterodimer with p50. The prototypical NF-κB p50:RelA heterodimer is kept inactive through its noncovalent association with IκBα, one of the IκB family inhibitor proteins. A wide variety of environmental and stress response stimuli, including proinflammatory cytokines, rapidly activates the p50:RelA heterodimer through targeted proteolytic degradation of IκBα. In the absence of sufficient p50, RelA assembles into homodimers, which regulate genes whose inducible expression usually is controlled by the p50:RelA heterodimer.3,4 The RelA homodimer is present at low levels in normal cells where it is preferentially inhibited by IκBβ.5 RelA is also capable of forming conditional heterodimers with c-Rel or p52, which display cell-type and stimulus-specific activities.
Figure 1.
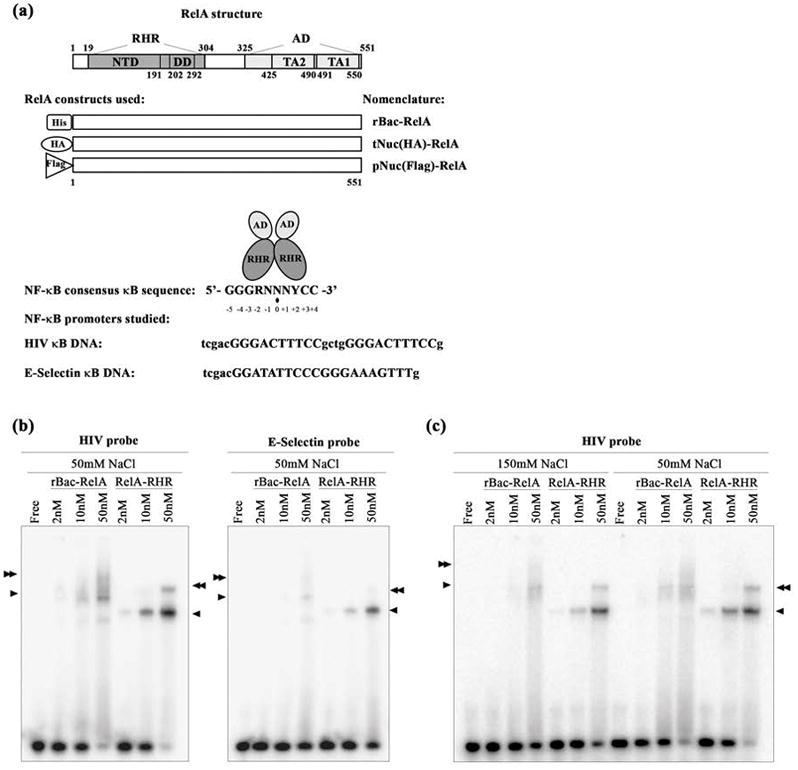
DNA binding by recombinant NF-κB dimers. (a) The top panel shows a schematic representation of RelA. Residues of the full length protein as well as different domains are indicated. Abbreviations: RHR, Rel homology region; AD, activation domain; NTD, N-terminal domain; DD, dimerization domain; TA2, transactivation domain 2; TA1, transactivation domain 1. The middle panel shows a schematic representation of the three RelA proteins studied in this work. Abbreviations: rBac-RelA, recombinant full length His-RelA expressed in insect Sf9 cells using a baculovirus system; tNuc(HA)-RelA, full length HA-RelA overexpressed in HEK 293T cells and present in their total nuclear extracts; pNuc(Flag)-RelA, full length Flag-RelA overexpressed in HEK 293T cells and purified from nuclear extracts by two steps of protein purification. The bottom panel shows the consensus κB DNA sequence with the residue separating two half-sites denoted by a black dot. R, N, and Y denote purine, any base, and pyrimidine, respectively. HIV and E-Selectin DNA sequences studied in this work are shown. κB sites are indicated in uppercase letters. (b) EMSA showing the RelA-RHR homodimer binds two different κB DNAs more strongly than rBac-RelA does. Single and double arrowheads indicate the specific RelA:DNA complexes formed. (c) EMSA showing the difference in binding when the RelA:DNA complex is formed at two different salt concentrations.
NF-κB dimers are sequence-specific DNA binding factors that regulate target gene expression levels by binding to DNA response elements known collectively as “κB sites” or “κB DNAs” (Figure 1a). The fundamental principles that direct NF-κB:DNA complex formation have been determined through elucidation of three-dimensional structures of several NF-κB:DNA complexes,2,6,7 identification of cis-acting regulatory sequences coupled to reporter activity assays, in vitro DNA binding activity using an electrophoretic mobility shift assay (EMSA), and gene expression microarray experiments. The κB site consensus is 5'-GGGRNNNYCC-3' (R, N, and Y denote purine, any base, and pyrimidine, respectively), where each NF-κB dimer subunit binds either 4 or 5 bp half-sites separated by a single base pair (Figure 1a). Although this consensus accommodates many DNA sequences, NF-κB dimers have repeatedly been found to bind DNA that falls outside the consensus, as well. For example, NF-κB dimers have been shown to bind DNA that retains only one half-site consensus.8–10 The modular architecture of the NF-κB subunit DNA binding domain allows NF-κB dimers to bind DNA sequences with minimal sequence conservation. These differences in binding mode also affect target gene expression levels. Even a single nucleotide variation within a κB site can switch a gene regulatory program from activation to repression.11,12 This suggests that κB sites are not merely passive placeholders that function only to tether NF-κB to a promoter. Instead, κB sites play an active role in influencing gene regulation. Most NF-κB responsive genes contain more than one κB site, and different dimers can target these sites with variable specificity.12,13 Furthermore, the distribution of multiple κB sites differs significantly among different target genes. They can span a vast genomic region, covering both promoter and enhancer regions. They can be clustered within the promoter region, near the transcription start site. Or they may reside within the enhancer, several thousand base pairs from the start site.13,14
Genomewide chromatin immunoprecipitation followed by sequencing (ChIP-seq) studies revealed other unexpected modes of DNA binding by NF-κB. The first genomewide study reported by Martone et al. investigated RelA targets within chromosome 22.15 This investigation uncovered two surprises: more than one-third of the RelA target genes do not contain a κB site, and often RelA binds stably to intergenic regions far from an active gene. Since that report, several other genomewide ChIP-seq studies have been performed with RelA as well as with several other transcription factors.16,17 Like RelA, all the transcription factors tested were detected at nonconsensus DNA binding sites. The degree of nonconsensus site binding varies from 15 to 50% depending on the transcription factor, stimulus, and cell type.
Consistent with these previous reports, a recent study of the NF-κB DNA binding landscape in lymphoid B cell lines (LCL) revealed that one-third of all bound NF-κB was at non-κB sites.18 The nonconsensus NF-κB binding sites were, however, enriched for recognized gene regulatory elements such as E-box, ZNF143, PU.1/IRF4, and CTCF motifs. Such DNAs are known to recruit their own respective direct binding transcription factor proteins, and it is possible that NF-κB acts at these sites as a non-DNA binding cofactor to regulate target gene expression. The LCL study also determined that recruitment of NF-κB to a significant fraction of κB sites requires the accessory transcription factor FOXM1. Reduction of FOXM1 levels correlates with a reduced level of expression of the NF-κB target genes. Thus, it appears that FOXM1 acts as a cofactor of NF-κB.
E2F1 was reported as the first global NF-κB cofactor in a study by Lim et al.19 That study indicated that E2F1 plays an important role in NF-κB-mediated activation of inflammatory genes in response to bacterial lipopolysaccharide (LPS) treatment in HeLa cells. Some of the target genes contain both a κB site and the binding site for E2F1 within their promoters, but many others carried only the κB site. In E2F1 knockdown HeLa cells or E2F1 knockout cells, the level of expression of several of these NF-κB target genes was significantly reduced. Similarly, ribosomal protein S3 (RPS3) and p53 have been found to enhance activation of specific NF-κB target genes in response to stimuli.20–22 Overall, these studies suggest that many transcription factors, or nucleic acid binding proteins in general, can function in a manner independent of their own DNA binding propensities as cofactors that influence direct DNA binding by other transcription factors. Although the in vivo functional significance for the “cofactor” activity of NF-κB, FOXM1, and E2F1 is beginning to come to light, a mechanism for how one transcription factor might act as a cofactor to modulate the DNA binding potential of another has not been thoroughly investigated.
In this study, we provide evidence that the NF-κB RelA subunit relies upon diverse cofactor proteins for its DNA binding. We show that in the absence of cofactors such as RPS3 and p53, RelA interacts with κB site DNA only weakly in solution under nearly physiological binding conditions. It is not expected that such low-affinity RelA dimers would be able to bind their DNA sites in vivo. We propose a new model for cofactor-dependent NF-κB:DNA binding.
MATERIALS AND METHODS
Plasmids, Antibodies, and Reagents
pRC-HA-hRelA (1–551), His-p50 (1–435), GST-RelA-AD (325–551), and GST-RelA-AD (429–551) have been previously described.23 Wild-type RelA was subcloned into a plasmid to express RelA as a Flag fusion protein. The HA-RelA construct was used as the template to generate a RelA-RHR fragment and insert it into the pET15b plasmid that expresses protein as N-terminal His fusion protein. All Flag-p53 constructs were a kind gift from S. Lauberth and were used as the template to amplify the inserts and subclone them into the pET15b His-tagged plasmid. The p53-AD (1–95) and p53-DBD (96–292) plasmids were generated using the His-p53 wild-type (wt) construct as the template followed by their subcloning into the pET24d His-tagged plasmid. His-RPS3, His-OGG1, and His-HMGA1 [formerly HMGI(Y)] plasmids were a kind gift obtained from M. Lenardo and T. Hazra. E-Selectin and HIV luciferase reporters containing the specific κB DNA promoter have been previously described.23 The antibodies recognizing RelA (sc-372), p50 (sc-7178), and actin (sc-1616) were purchased from Santa Cruz. Anti-HA.11 (901501) was obtained from BioLegend. Anti-p84 (C1C3) was purchased from GeneTex. Anti-His, anti-p53, and anti-tubulin antibodies and TNF-α were gifts from BioBharati Life Science (Kolkata, India).
Protein Expression and Purification
His-RelA full length protein (rBac-RelA) expression was performed in Sf9 insect cells using a baculovirus system as previously described.23
Recombinant His-RelA-RHR (19–304) wt was expressed and purified with modifications of a previously published protocol.24 Briefly, cells were lysed with lysis buffer [25 mM Tris-HCl (pH 7.5), 150 mM NaCl, 0.5 mM phenyl-methanesulfonyl fluoride (PMSF), 2 mM β-mercaptoethanol (BME), 10% (v/v) glycerol, and 10 mM imidazole] and sonicated. The lysate was cleared by centrifugation at 13000 rpm for 30 min at 4 °C. The supernatant was decanted into an ice-cold beaker, and 1/9 of 10% (v/v) streptomycin was added while the mixture was being gently stirred. The sample was left to stir for an additional 20 min at 4 °C and cleared by centrifugation at 13000 rpm for 30 min at 4 °C. The supernatant was loaded onto a nickel-NTA resin column (a gift from BioBharati Life Science) pre-equilibrated with lysis buffer. Beads were washed with 10 column volumes of wash buffer [25 mM Tris-HCl (pH 7.5), 150 mM NaCl, 10% (v/v) glycerol, and 10 mM imidazole]. Finally, protein was eluted by adding 10 mL of elution buffer [25 mM Tris-HCl (pH 7.5), 150 mM NaCl, 10% (v/v) glycerol, and 250 mM imidazole]. Peak fractions were spun down and loaded through a Superdex 200 size exclusion column (Amersham Biosciences) pre-equilibrated with size exclusion column (SEC) buffer [25 mM Tris-HCl (pH 7.5), 200 mM NaCl, 1 mM dithiothreitol (DTT), and 5% (v/v) glycerol] at 4 °C. Peak fractions were pooled and concentrated using a Centriprep 10 kDa cutoff membrane concentrator unit (Millipore).
Recombinant GST-RelA-AD (325–551) and GST-RelA-AD (429–551) were expressed in Escherichia coli Rosetta cells by growing cells to an A600 of 0.2 followed by induction with 0.2 mM isopropyl β-D-1-thiogalactopyranoside (IPTG) for 4 h at 16°C. Cells were lysed with lysis buffer [20 mM HEPES-KOH (pH 7.5), 250 mM KCl, 0.1% (v/v) NP-40, 10% (v/v) glycerol, 0.5 mM ethylenediaminetetraacetic acid (EDTA), 0.2 mM PMSF, and 1 mM (DTT)] and sonicated. The lysate was clarified by centrifugation at 13000 rpm for 30 min at 4 °C. The supernatant was loaded onto a Glutathione Sepharose resin column (gift from BioBharati Life Science) pre-equilibrated with lysis buffer at 4 °C. Then, resin was washed with 10 column volumes of wash buffer [20 mM HEPES-KOH (pH 7.5), 400 mM KCl, 0.1% (v/v) NP-40, 10% (v/v) glycerol, 0.5 mM EDTA, and 1 mM DTT], and protein was eluted with elution buffer [20 mM HEPES-KOH (pH 7.5), 250 mM KCl, 0.1% (v/v) NP-40, 10% (v/v) glycerol, 0.5 mM EDTA, 1 mM DTT, and 25 mM glutathione]. Peak fractions were pooled and filtered through a 0.22 μm filter. Then, the sample was loaded onto a Superdex 75 size exclusion column (Amersham Biosciences) pre-equilibrated with SEC buffer [20 mM HEPES-KOH (pH 7.5), 250 mM KCl, 10% (v/v) glycerol, 1 mM DTT, and 0.5 mM EDTA]. Peak fractions were pooled and concentrated using a Centriprep 30 kDa cutoff membrane concentrator unit (Millipore).
Recombinant His-p50 (1–435) was expressed in E. coli Rosetta cells by growing cells to an A600 of 0.6 followed by induction with 1 mM IPTG overnight at room temperature. Cells were lysed with lysis buffer [25 mM Tris-HCl (pH 8.0), 200 mM NaCl, 5% (v/v) glycerol, 2 mM BME, and 10 mM imidazole] and sonicated. The lysate was clarified by centrifugation at 13000 rpm for 30 min at 4 °C. The supernatant was loaded onto a nickel-NTA resin column pre-equilibrated with lysis buffer. Then, the resin was washed with 10 column volumes of wash buffer (same as lysis buffer but with 30 mM imidazole), and protein was eluted with elution buffer (same as lysis buffer but with 250 mM imidazole). Peak fractions were pooled, spun, and concentrated using a Centriprep 30 kDa cutoff membrane concentrator unit (Millipore). The sample was loaded through a Superdex 200 size exclusion column (Amersham Biosciences) pre-equilibrated with SEC buffer [10 mM Tris (pH 7.5), 200 mM NaCl, 10 mM DTT, and 5% (v/v) glycerol] at 4 °C. Peak fractions were pooled and concentrated using a Centriprep 30 kDa cutoff membrane concentrator unit (Millipore).
The p50:RelA NF-κB heterodimer was formed by mixing in a 1:1 ratio both purified proteins diluted to 0.1 μM in a buffer consisting of 20 mM Tris-HCl (pH 7.5), 100 mM NaCl, 10 mM BME, and 5% (v/v) glycerol for 1 h at 4 °C. Then, the NF-κB heterodimer was concentrated using a Centriprep 30 kDa cutoff membrane concentrator unit (Millipore).
Recombinant His-RPS3 was expressed in E. coli Rosetta cells by growing cells to an A600 of 0.2 followed by induction with 1 mM IPTG overnight at 16 °C. Cells were lysed with lysis buffer [40 mM Tris-HCl (pH 8.0), 150 mM NaCl, 10% (v/v) glycerol, 0.2% (v/v) Triton X-100, 2.5 mM BME, 1 mM PMSF, and 10 mM imidazole] and sonicated. The lysate was clarified by centrifugation at 13000 rpm for 30 min at 4 °C. The supernatant was mixed with nickel-NTA resin pre-equilibrated with lysis buffer in batch for 2 h at 4 °C in a rotary shaker. Then, the resin was washed four times with wash buffer (same as lysis buffer but with 30 mM imidazole), and protein was eluted with elution buffer [40 mM Tris-HCl (pH 8.0), 150 mM NaCl, 10% (v/v) glycerol, 2.5 mM BME, and 400 mM imidazole]. Peak fractions were pooled and stored at 4 °C.
Recombinant His-p53 and oncogenic mutants were expressed in E. coli Rosetta cells by growing cells to an A600 of 0.2 followed by induction with 0.1 mM IPTG overnight at room temperature. Cells were lysed with lysis buffer [25 mM sodium phosphate (pH 7.5), 300 mM NaCl, 5% (v/v) glycerol, 0.2% (v/v) Triton X-100, 10 mM imidazole, and 0.5 mM PMSF] and sonicated. The lysate was clarified by centrifugation at 13000 rpm for 30 min at 4 °C. The supernatant was loaded onto a nickel- NTA resin column pre-equilibrated with lysis buffer. Then, the resin was washed with 10 column volumes of wash buffer (same as lysis buffer but with 500 mM NaCl and 45 mM imidazole), and protein was eluted with elution buffer (same as lysis buffer but without PMSF and with 250 mM imidazole). Peak fractions were pooled and filtered through a 0.22 μm filter. Then, the sample was loaded onto a Superdex 200 size exclusion column (Amersham Biosciences) pre-equilibrated with SEC buffer [25 mM sodium phosphate (pH 7.5), 300 mM NaCl, 5% (v/v) glycerol, 5 mM DTT, and 0.5 mM EDTA]. Peak fractions were pooled and concentrated using a Centriprep 30 kDa cutoff membrane concentrator unit (Millipore).
Recombinant His-p53-AD (1–95) and His-p53-DBD (96–292) were expressed in E. coli Rosetta cells by growing cells to an A600 of 0.3–0.4 followed by induction with 0.5 mM IPTG overnight at room temperature. Cells were lysed with lysis buffer [25 mM Tris-HCl (pH 8.0), 500 mM NaCl, 10% (v/v) glycerol, 0.2% (v/v) Triton X-100, 2.5 mM PMSF, 5 mM BME, 10 mM imidazole, and protease inhibitor cocktail], incubated on ice for 30 min, and sonicated. The lysate was clarified by centrifugation at 12000 rpm for 45 min at 4 °C. The supernatant was loaded onto a nickel-NTA resin column pre-equilibrated with lysis buffer. Then, the resin was washed with 10 column volumes of the same lysis buffer, and protein was eluted with elution buffer [25 mM Tris-HCl (pH 8.0), 150 mM NaCl, 10% (v/v) glycerol, 0.2% (v/v) Triton X-100, 5 mM BME, and 250 mM imidazole].
Recombinant Flag-p53 was expressed in E. coli Rosetta cells by growing cells to an A600 of 0.1–0.3 followed by induction with 0.1 mM IPTG overnight at room temperature. Cells were lysed with lysis buffer [25 mM sodium phosphate (pH 7.5), 300 mM NaCl, 5% (v/v) glycerol, 0.2% (v/v) Triton X-100, and 0.5 mM PMSF] and sonicated. The lysate was clarified by centrifugation at 13000 rpm for 15 min at 4 °C. The supernatant was mixed with 20 μL of anti-Flag M2 affinity beads (Sigma) for 2–3 h at 4 °C. Then, beads were washed four times with lysis buffer, and after the last wash, beads were resuspended in lysis buffer supplemented with 0.5 mM EDTA and protease inhibitor cocktail. Flag-p53-bound beads were stored at 4 °C.
Recombinant His-OGG1 was expressed in E. coli Rosetta cells by growing cells to an A600 of 0.6 followed by induction with 0.2 mM IPTG overnight at room temperature. Cells were lysed with lysis buffer [40 mM Tris-HCl (pH 8.0), 150 mM NaCl, 10% (v/v) glycerol, 0.2% (v/v) Triton X-100, 2.5 mM BME, 1 mM PMSF, 10 mM imidazole, and protease inhibitor cocktail] and sonicated. The lysate was clarified by centrifugation at 13000 rpm for 30 min at 4 °C. The supernatant was mixed with nickel-NTA resin pre-equilibrated with lysis buffer in batch for 3 h at 4 °C in a rotary shaker. Then, the resin was washed four times with wash buffer [40 mM Tris-HCl (pH 8.0), 150 mM NaCl, 10% (v/v) glycerol, 0.2% (v/v) Triton X-100, and 20 mM imidazole], and protein was eluted with elution buffer (same as wash buffer but with 250 mM imidazole). Peak fractions were pooled.
Recombinant His-HMGA1 was expressed in E. coli BL21 cells by growing cells to an A600 of 0.6–0.7 followed by induction with 1 mM IPTG for 3 h at 37 °C. Cells were lysed with lysis buffer [25 mM Tris-HCl (pH 8.0), 150 mM NaCl, 7 M urea, 10 mM imidazole, 10% (v/v) glycerol, 2.5 mM PMSF, 0.2% (v/v) Triton X-100, 5 mM BME, and protease inhibitor cocktail] and sonicated. The lysate was clarified by centrifugation at 12000 rpm for 45 min at 4 °C. The supernatant was loaded onto a nickel-NTA resin column pre-equilibrated with lysis buffer. Then, the resin was washed with 30 column volumes of wash1 buffer (same as lysis buffer but with 20 mM imidazole) and 20 column volumes of wash2 buffer (same as wash1 buffer but without 7 M urea). Finally, protein was eluted with elution buffer (same as wash2 buffer but with 250 mM imidazole).
All protein concentrations were determined using Bradford (Bio-Rad) reagent and were snap-frozen in liquid N2 for long-term storage at –80 °C (except His-RPS3 that was stored at 4 °C).
Electrophoretic Mobility Assays
Electrophoretic mobility shift assays (EMSAs) were performed as previously described.25 Briefly, HIV and E-Selectin probes were radio-labeled and incubated with the proteins under study for 20 min at room temperature in binding buffer [10 mM Tris-HCl (pH 7.5), 50 mM NaCl, 10% (v/v) glycerol, 1% (v/v) NP-40, 1 mM EDTA, and 0.1 mg/mL PolydIdC]. When needed, proteins were diluted in dilution buffer [20 mM Tris-HCl (pH 7.5), 50 mM NaCl, 10% (v/v) glycerol, 1 mM DTT, and 0.2 mg/mL bovine serum albumin (BSA)]. Samples were run in TGE buffer (24.8 mM Tris base, 190 mM glycine, and 1 mM EDTA) at 200 V for 1 h, and the gel was dried. In the supershift reactions, all proteins and antibodies were incubated simultaneously for 20 min in binding buffer in the presence of the probe under study. The amount of protein was quantified by the Bradford assay. Protein complexes were analyzed by native electrophoresis on a 4% (w/v) nondenatured polyacrylamide gel. Bands were quantified using ImageJ software. Probes used are indicated in Figure 1a.
Trypsin Digestion
Total 5 μM HIV-κB DNA double-stranded probe was annealed in annealing buffer [25 mM Tris-HCl (pH 7.5), 0.5 mM EDTA, and 50 mM NaCl] by denaturing the DNA strands in boiling water followed by slow cooling to room temperature. Then, 2.5 μM rBac-RelA with or without 12.5 μM His-RPS3 protein was added and the mixture was incubated for 20 min at room temperature to establish the DNA:protein complexes. Next, 0.00005% (v/v) trypsin (Corning) was added, and digestion was performed at 37 °C for the indicated times. Sample digestions were resolved by Western blot.
Whole Cell Extracts and Nuclear/Cytoplasmic Fractionations
HEK 293T cells were cultured in DMEM supplemented with 10% (v/v) fetal bovine serum (FBS) and 1% (v/v) penicillin/streptomycin/glutamine. Then, cells were transiently transfected with empty, HA-RelA, or Flag-RelA plasmids using polyethylenimine PEI (PolySciences) following the manufacturer’s protocol. After being transfected for 48 h, cells were stimulated with TNF-α (10 ng/mL) for 1 h when indicated and then harvested.
To prepare whole cell extracts, cells were lysed in RIPA buffer [20 mM Tris-HCl (pH 8.0), 200 mM NaCl, 1% (v/v) Triton X-100, 2 mM DTT, 5 mM 4-nitrophenyl phosphate di(tris) salt, 2 mM sodium orthovanadate, 1 mM PMSF, and protease inhibitor cocktail] for 30 min at 4 °C. Then, cells were centrifuged at 13000 rpm for 15 min at 4 °C, and supernatants containing the whole cell protein extracts were measured by the Bradford assay to determine the total amount of protein.
To prepare nuclear and cytoplasmic protein extracts, cells were lysed in 10 mM HEPES (pH 7.9), 1.5 mM MgCl2, 10 mM KCl, 0.5 mM DTT, 0.05% (v/v) NP-40, and protease inhibitor cocktail for 10 min on ice and spun at 3000 rpm and 4 °C. The supernatant containing the cytoplasmic fraction was measured by the Bradford assay to determine the total amount of protein. Pellets were resuspended in the same buffer once and centrifuged again at 3000 rpm for 10 min at 4 °C. Then, pellets containing the nuclei were resuspended in RIPA buffer and subjected to three lysis cycles (freeze at –80 °C and thaw at 37 °C). Finally, samples were centrifuged at 13000 rpm and 4 °C for 10 min, and supernatants containing the soluble nuclear fraction were measured by the Bradford assay to determine the total amount of protein. Nuclear and cytoplasmic extracts were aliquoted and kept at –80 °C.
Immunoprecipitations
Upon transfection of Flag-RelA, the soluble nuclear extracts obtained as indicated above were subjected to a first purification step using anti-Flag M2 affinity resin (Sigma) pre-equilibrated with RIPA buffer. Samples were immunoprecipitated for 2 h at 4 °C in a rotary shaker. Then, beads were washed four times with RIPA buffer, and Flag-tagged RelA proteins were eluted three times using 0.1 mg/mL 3× Flag peptide. These three eluted fractions were pooled and subjected to a second purification step using Q Sepharose chromatography. Briefly, samples were diluted to decrease the NaCl concentration to 50 mM, and then they were incubated with the Q Sepharose resin for 1 h at 4 °C in a rotary shaker. Subsequently, proteins were eluted after performing a salt gradient (from 50 to 500 mM NaCl). Typically, Flag-RelA was eluted with 250–500 mM NaCl. Purified nuclear Flag-tagged RelA [pNuc(Flag)-RelA] was snap-frozen in liquid N2 for long-term storage at –80 °C.
M2 beads bound to Flag-p53 (M2_Flag-p53) proteins were used to co-immunoprecipitate different RelA protein fragments. Briefly, M2_Flag-p53 protein amounts were estimated visually by running the samples in a sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS–PAGE) gel stained using Coomassie Brilliant Blue. Then, M2_Flag-p53:RelA proteins were mixed in a 1:3 ratio for 30 min at 4 °C in binding buffer [20 mM HEPES-KOH (pH 7.9), 150 mM NaCl, and 0.1% (v/v) NP-40]. Beads were washed five times in wash buffer [20 mM Tris-HCl (pH 7.9), 150 mM KCl, 20% (v/v) glycerol, 0.1 mM EDTA, and 0.1% (v/v) NP-40], boiled in Laemmli loading buffer, and analyzed by Western blot.
Docking Studies
The RelA homodimer system with DNA was prepared with the maestro protein preparation wizard to assign bond orders, add hydrogens, create zero-order bonds to metals, and create disulfide bonds. After that, the hydrogens added were minimized through the H bond assignment tool at a pH of 7.4 as determined using PROPKA. Following hydrogen bond assignment, a restrained minimization was performed on the full dimeric structure to a root-mean-square deviation of 0.3 Å. The final structure underwent APBS calculations and docking using the ClusPro2.0 algorithm with the RelA activation domain (AD) structures. We used ZDOCK to dock the RelA-AD into the dimeric RelA structure with DNA bound without letting ZDOCK sample structures in which the AD interacts with the DNA.
Fluorescence Anisotropy Assays
RelA dimers or other proteins were titrated against 1 nM fluorescein-labeled HIV-κB DNA [F-HIV-κB DNA (Figure 1a)]. Fluorescence anisotropy was measured on a nonbinding 96-well black bottom plate (Greiner) using a Tecan Safire 2 plate reader in polarization mode with an excitation wavelength of 470 nm and an emission wavelength of 520 nm with a 20 nm bandwidth. Most measurements were taken in anisotropy buffer containing 25 mM Tris-HCl (pH 7.5), 150 mM NaCl, 5% (v/v) glycerol, 0.25 mg/mL BSA, 200 nM nonspecific DNA, and 0.5% (v/v) NP-40, with an incubation of 15 min at room temperature. Measurements with KCl, KOAc, and KGlu were performed in anisotropy buffer, except with NaCl substituted with the corresponding salt. For heat-inactivated RPS3, His-RPS3 was diluted in anisotropy buffer without nonspecific DNA and incubated for 10 min at 90 °C, followed by cooling on ice for 5 min. All p53-containing samples and its control samples were measured in buffer containing 25 mM Tris-HCl (pH 8.0), 125 mM NaCl, 5% (v/v) glycerol, 0.25 mg/mL BSA, 200 nM nonspecific DNA, and 0.1% (v/v) NP-40, with a 90 min incubation at 4 °C. The sequence ATCGTGCATATTGCTA-CTAGCGTTTTTGGA was used as nonspecific DNA for the binding assays, and the fluorescein-labeled single-stranded DNA ATCGTGGGAAAGTTTCTGGATATTCCCTTGGA was used as a negative control. To quantify the binding affinity between RelA and DNA, the anisotropy data from each binding assay were normalized to the initial value without protein, plotted, and fit to the quadratic equation below26 to calculate the equilibrium dissociation constant (KD) as described previously. GraphPad PRISM 4.0 (GraphPad Software, San Diego, CA) was used to perform the curve fits. All experiments were performed a minimum of two or three times to determine the standard deviations.
RESULTS
Full Length RelA Binds κB DNA Weaker Than RelA-RHR Does
It has long been known that the in vitro DNA binding affinity of the full length NF-κB RelA homodimer is significantly lower than that exhibited by the RelA-RHR alone.23,27,28 However, those previous experiments had never been performed with highly purified recombinant full length RelA. We prepared full length RelA from baculovirus-infected Sf9 insect cells (rBac-RelA; see Figure 1a for further details on the nomenclature used throughout this paper), tested its binding to two different target κB DNAs, HIV and E-Selectin, and compared it with RelA-RHR using an electrophoretic mobility shift assay (EMSA) (Figure 1a,b).
Consistent with previous observations, rBac-RelA binds DNA with an efficiency that is lower than that of RelA-RHR as judged by the absence of significant amounts of compact rBac-RelA:DNA complex band(s) (Figure 1b). This difference in protein:DNA complex stability was attributed to an inhibitory effect of the carboxy (C)-terminal RelA-AD toward its DNA binding RHR. In the EMSA experiments, both the binding reaction and electrophoresis were performed at low ionic strengths (50 mM NaCl for binding and <50 mM glycine for electrophoresis). We repeated the EMSA under the more physiologically relevant ionic strength binding conditions of 150 mM NaCl and found that both full length RelA and RelA-RHR:DNA complexes were weakened at higher salt concentrations (Figure 1c). However, because electrophoresis needed to be performed at low ionic strengths, the full impact of a high salt concentration on RelA:DNA binding could not be fully captured by this assay. Despite this technical limitation, our EMSA results still conform to the existence of a clear difference in the DNA binding of rBac-RelA and RelA-RHR.
Nuclear RelA Dimers Bind DNA with an Affinity That Is Higher Than That of Recombinant RelA Dimers
Because all the RelA that enters the nucleus in response to stimulation of mammalian cells is full length and nuclear RelA in stimulated cells is expected to bind DNA optimally to regulate gene expression, we hypothesized that some mechanism exists that allows full length nuclear RelA to bind to DNA with an affinity similar to that observed for the RelA-RHR alone. Of note, we recently reported that in vitro DNA binding of RelA does not correlate well with its transcriptional activity.23 To test this conjecture, we compared the DNA binding activities of recombinant rBac-RelA to those of nuclear RelA expressed in HEK 293T cells. Two different versions of nuclear RelA from HEK 293T cells were used: full length RelA expressed as HA fusion protein to use as unpurified protein in the total nuclear extract [tNuc(HA)-RelA] and a Flag fusion protein to prepare pure RelA from the nuclear extract [pNuc(Flag)-RelA] (see Figure 1a for details on the nomenclature).
Because our goal is to understand the activity of both the RelA homodimer and the p50:RelA heterodimer, we tested the DNA binding ability of these dimers in TNF-α-treated cells. As shown previously, endogenous RelA is mostly present as the prototypical NF-κB p50:RelA heterodimer that binds to κB site DNA upon activation (Figure 2a, lane 2, and Figure S1a, lanes 1 and 11).
Figure 2.
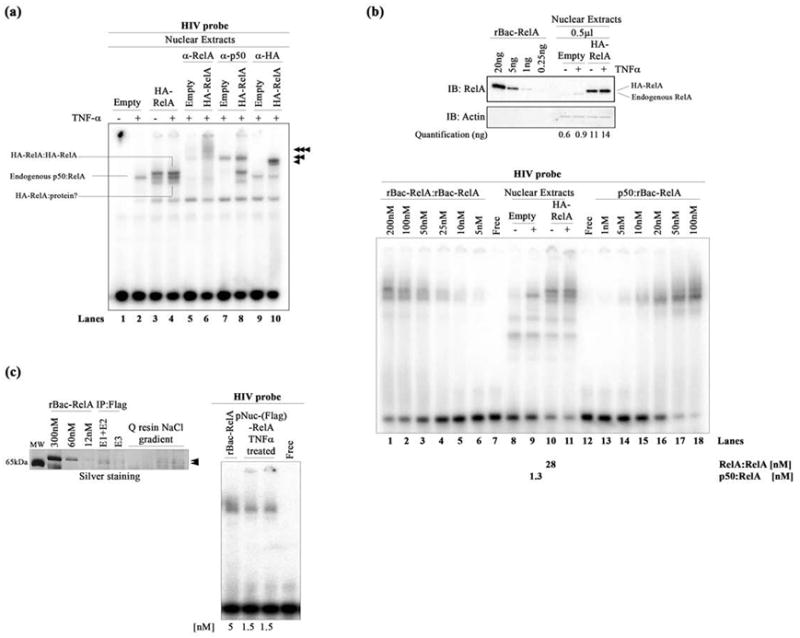
DNA binding by nuclear NF-κB dimers. (a) EMSA showing HIV-κB DNA binding by the exogenous HA-RelA homodimer (top band), endogenous p50:RelA heterodimer (middle band), and additional HA-RelA:protein complex (bottom band) present in transfected HA-RelA nuclear extracts unstimulated or stimulated with TNF-α. Single, double, and triple arrowheads indicate the supershifted complexes when using anti-RelA, anti-p50, and anti-HA antibodies, respectively. (b) The top panel shows a Western blot showing the quantitation of endogenous and exogenous HA-RelA in transfected nuclear extracts. rBac-RelA was used as a standard to perform this estimation. Bands were subjected to densitometry using ImageJ software. Actin was used as the loading control for this experiment. The bottom panel shows an EMSA showing the comparative DNA binding activities of the recombinant rBac-RelA homodimer and p50:rBac-RelA heterodimer and also the homo- and heterodimers present in transfected HA-RelA nuclear extracts. A titration of the recombinant proteins was used to estimate the concentration of NF-κB dimers present in the nucleus. Quantitations obtained are shown at the bottom of the figure. (c) The left panel shows an SDS–PAGE gel stained with silver to estimate the amount of protein present in the pNuc(Flag)-RelA sample. rBac-RelA was used as a standard. Samples that were analyzed show all the steps of the purification of the Flag-RelA protein present in the nuclear fraction. E1–E2 and E3 are the samples obtained after eluting the proteins from the anti-Flag M2 affinity resin. Then, the sample was bound to Q-Sepharose resin and eluted after performing a NaCl gradient. Arrows indicate the pNuc(Flag)-RelA after purification. The right panel shows an EMSA showing the equivalent binding between rBac-RelA and pNuc(Flag)-RelA when using similar amounts of protein (5 and 1.5 nM, respectively).
Thus, to understand DNA binding of the RelA homodimer, RelA was overexpressed by transfecting HEK 293T cells with the HA-RelA expression vector. Nuclear extracts of unstimulated cells containing exogenous HA-RelA exhibited two distinguishable bands above and below the endogenous p50:RelA:DNA complex located in the middle (Figure 2a, lane 3, and Figure S1a, lane 12). TNF-α stimulation resulted in the clear DNA binding activity of both the exogenous HA-RelA-containing dimers and the endogenous p50:RelA heterodimer showing three discrete bands (Figure 2a, lane 4, and Figure S1a, lanes 2 and 13). Antibody supershifts confirmed that the upper band corresponds to the exogenous HA-RelA homodimer in complex with DNA whereas the middle band contains solely the endogenous p50:RelA:DNA complex. The identity of the bottom band is unclear. Possibly it contains the exogenous HA-RelA in complex with an unknown additional protein or a truncated version of the RelA homodimer where one of the two RelA subunits has lost the AD (Figure 2a and Figure S1a). The bottom band cannot be the native like RelA homodimer because it is known that the endogenous RelA homodimer migrates slower than the endogenous p50:RelA homodimer. Remarkably, TNF-α stimulation did not alter the DNA binding affinity of the exogenous HA-RelA homodimer.
Next, we measured the concentration of RelA in nuclear extracts by quantitative Western blot using rBac-RelA as a standard (Figure 2b). On the basis of the analysis of three independent experiments, the nuclear exogenous HA-RelA concentration in our transfected HEK 293T cells varied between ~20 and 30 ng/μL. The concentration of endogenous nuclear RelA after stimulation registered as ~2 ng/μL (Figure 2b, top panel). It should be noted that most endogenous RelA remained in the cytoplasm, even after stimulation by TNF-α, with only ~20–30% mobilizing to the nucleus (Figure S1b). In contrast, a significant fraction of exogenous HA-RelA localized to the nucleus with or without stimulation (Figure S1b).
We next investigated the DNA binding of the recombinant rBac-RelA homodimer and p50:rBac-RelA heterodimer in comparison with the exogenous HA-RelA homodimer and endogenous p50:RelA heterodimer present in nuclear extracts (Figure 2b, bottom panel). On the basis of several independent experiments, we observed that the nuclear HA-RelA homo-dimer binds DNA with an affinity roughly 5–10-fold greater than that of the recombinant rBac-RelA homodimer (Figure 2b, bottom panel, compare lanes 10 and 1). Similarly, the endogenous nuclear p50:RelA heterodimer is also observed to bind DNA ~10–20-fold more tightly than the recombinant p50:rBac-RelA heterodimer does (Figure 2b, bottom panel, compare lanes 9 and 16). Overall, these data suggest that the NF-κB p50:RelA heterodimer and RelA homodimer present in nuclear extracts are capable of binding κB DNA with an affinity significantly higher than that of the recombinant dimers alone.
To test whether the enhanced DNA binding activity observed in RelA from nuclear extracts [tNuc(HA)-RelA] is due to post-translational modifications of the protein, overexpressed Flag-RelA was purified by two chromatographic steps from the nuclear extracts of TNF-α-stimulated cells transfected with the Flag-RelA expression vector to produce pNuc(Flag)-RelA (Figure 2c, left panel). An EMSA revealed that pure pNuc(Flag)-RelA binds with an affinity similar to that of the recombinant rBac-RelA purified from baculovirus-infected cells (Figure 2c, right panel). The absence of a shifted band similar to the p50:RelA:DNA complex suggests that indeed pNuc-(Flag)-RelA was mostly pure devoid of any detectable p50 subunit. This suggests that RelA post-translational modification in HEK 293T cells is not primarily responsible for the enhancement of DNA binding activity observed with the NF-κB dimers present in the nucleus (Figure 2b, bottom panel). We conclude that some additional factor(s) present in the nuclear extracts is responsible for enhanced DNA binding by the RelA homodimer.
RPS3 and p53 Augment DNA Binding of the Recombinant RelA Homodimer
Several factors have been shown to interact with RelA in cells, and some of these have been proposed to facilitate RelA:DNA binding.20–22,29,30 One of the factors identified was RPS3. We tested if RPS3 augments binding of RelA to DNA by titrating increasing amounts of RPS3 protein to fixed levels of the recombinant RelA:DNA complex (Figure 3a).
Figure 3.
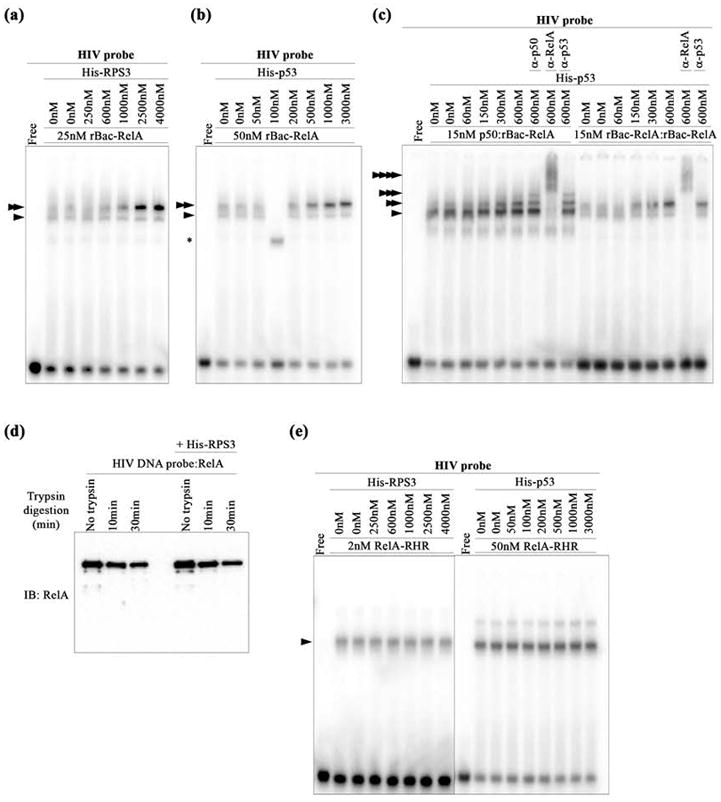
p53 and RPS3 facilitate DNA binding of rBac-RelA. (a) EMSA showing HIV-κB DNA binding by rBac-RelA in the presence of increasing concentrations of His-RPS3. Single and double arrowheads indicate the protein:DNA complexes detected. (b) EMSA showing HIV-κB DNA binding by rBac-RelA in the presence of increasing concentrations of His-p53. Single and double arrowheads indicate the protein:DNA complexes detected. The asterisk shows the formation of an unspecific complex. (c) EMSA showing HIV-κB DNA binding by the recombinant rBac-RelA homodimer and p50:rBac-RelA heterodimer in the presence of increasing concentrations of His-p53. Single and multiple arrowheads indicate the supershifted complexes detected when using anti-p50, anti-RelA, and anti-p53 antibodies. (d) Western blot analysis of the protease (trypsin) sensitivity of the RelA:DNA complex in the absence and presence of RPS3. The anti-RelA antibody (sc-372) was used to monitor RelA. (e) EMSA showing HIV-κB DNA binding by RelA-RHR in the presence of increasing concentrations of His-RPS3 or His-p53. The single arrowhead indicates the main protein:DNA complex detected.
In agreement with a previous report,20 we observe that RPS3 augments DNA binding activity of recombinant rBac-RelA in vitro. We next tested the ability of p53 to influence DNA binding of RelA and noted that, like RPS3, p53 also enhances DNA binding by the rBac-RelA homodimer (Figure 3b). Neither RPS3 nor p53 binds κB site DNA on its own (Figure S2a). Thus, the enhancement of rBac-RelA:DNA binding activity observed in the presence of RPS3 or p53 must be a result of protein–protein interaction between RelA and those cofactors. We also found that p53 enhanced DNA binding of the recombinant p50:rBac-RelA heterodimer, although the effect is somewhat less prominent as compared to that of the recombinant rBac-RelA homodimer (Figure 3c).
It is apparent from the EMSA experiment that, in the presence of RPS3 or p53, the NF-κB:DNA complex runs as a band that is more compact than the band of the more diffuse binary complex alone. This suggests that these cofactors might form a ternary complex with RelA on DNA. However, antibody supershift experiments did not provide any clear indication that p53 was present as a stable ternary complex (Figure 3c). To further test the nature of the interaction between RelA and the cofactor, we performed limited protease digestion of the RelA:DNA complex in the absence or presence of an excess of RPS3 and monitored the reaction by Western blot. No difference was observed in the limited trypsin proteolysis pattern of RelA under the two binding conditions (Figure 3d). These results suggest that the interactions between the cofactors and RelA are transient in nature. Furthermore, neither p53 nor RPS3 was observed to enhance DNA binding of the RelA-RHR (Figure 3e). These observations suggest that the interaction with p53 increases the kinetic stability of rBac-RelA for its target DNA in a manner that does not rely upon formation of a stable p53:RelA:DNA ternary complex.
We next tested whether cofactors affect DNA binding of tNuc(HA)-RelA by adding exogenous p53 to the nuclear extracts of TNFα-stimulated HEK 293T cells (Figure S2b). The EMSA results suggest that because the nuclear extracts already contained endogenous nuclear cofactors, exogenous p53 showed no further effect on the DNA binding by RelA (Figure S2b). Importantly, the p53 (and RPS3) enhancement effect toward rBac-RelA:DNA binding is specific because it was not observed when using an irrelevant protein such as BSA (Figure S2c). Overall, these observations suggest that the high-affinity DNA binding of NF-κB homo- and heterodimers observed in the cell requires support from other endogenous factors.
We tested two other DNA binding factors, OGG1 (8-oxo-guanine glycosylase 1) and HMGA1 (high-mobility group protein 1), for their ability to stimulate DNA binding of recombinant rBac-RelA because both of these factors were reported to be part of in vivo NF-κB:DNA complexes. Neither of these proteins was able to stimulate binding of RelA to HIV-κB DNA in vitro (Figure S 2d). An earlier report showed that OGG1 stimulates NF-κB:DNA complex formation by binding to a damaged site located near the κB site.29 Because no damaged base is present in our probe, OGG1 failed to show its effect. HMGA1 was shown to cooperate with RelA by binding to the minor groove of the A-T rich segment of a κB site.30 It appears that the mode of cooperation between HMGA1 and RelA is more complex and, therefore, not well recapitulated in our in vitro system or it binds highly specific DNA containing A/T rich sequences in the central segment of the κB DNA.
Oncogenic p53 Mutants Facilitate DNA Binding of RelA
We were particularly drawn to the enhancement of formation of the RelA:DNA complex by p53 because recent reports suggest that some amount of p53 oncogenic activity results from its ability to constitutively activate inflammatory genes in association with RelA.31,32 We first tested whether the entire p53 protein is necessary or if one particular structural domain of p53 is sufficient to enhance recombinant rBac-RelA:DNA binding. EMSA analysis revealed that the p53 DNA binding domain (DBD) is effective in stimulating the DNA binding activity of RelA, although the full length protein appeared to be most effective (Figure 4a,b).
Figure 4.
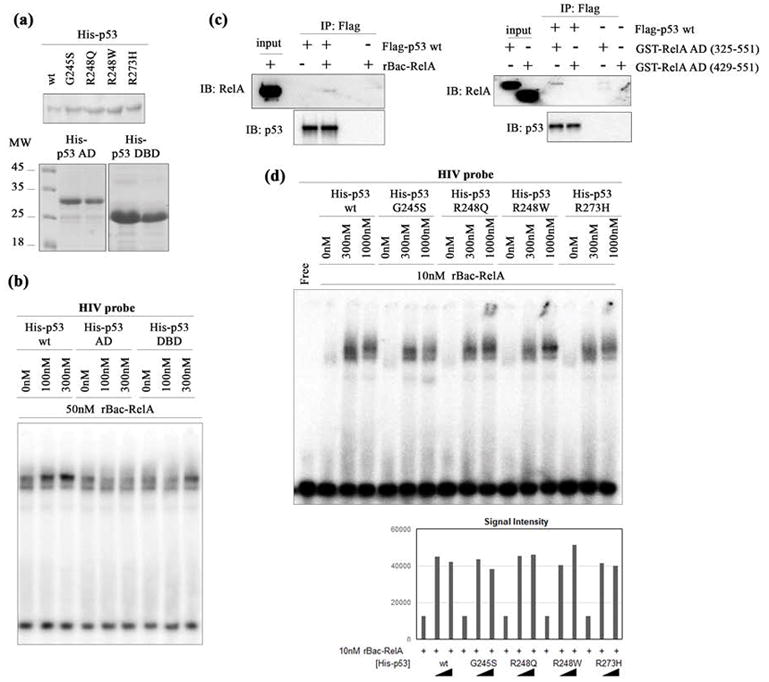
Oncogenic p53 mutants facilitate κB DNA binding by rBac-RelA. (a) Western blot analysis of all His-p53 proteins used in this study. The top panel shows the His-p53 wt protein along with four oncogenic p53 variants. The bottom panel shows the two truncated p53 proteins analyzed (His-p53-DBD, including only the p53 DNA binding domain, and His-p53-AD, including only the p53 activation domain). Two different amounts of protein were loaded. Molecular weight markers indicated in kilodaltons are shown on the left. (b) EMSA showing that HIV-κB DNA binding by rBac-RelA is enhanced by His-p53 wt and His-p53-DBD but not His-p53-AD. (c) The left panel shows a Western blot showing that rBac-RelA physically interacts with Flag-p53 wt by co-immunoprecipitation. The right panel shows a Western blot showing that RelA-AD (325–551) but not RelA-AD (429–551) mediates interaction of RelA with Flag-p53 wt detected by co-immunoprecipitation. (d) The top panel shows an EMSA showing HIV-κB DNA binding by rBac-RelA in the presence of His-p53 wt and oncogenic mutants. The bottom panel shows a graphical representation of the quantification of the bands using ImageJ software.
Co-immunoprecipitation experiments showed that both full length RelA and the AD of RelA could interact with p53 (Figure 4c). For detection of interaction, we used an antibody specific to the C-terminus of RelA. However, it is possible that the RHR of RelA also interacts with p53.
We next tested whether oncogenic mutations that map to the p53-DBD affect the DNA binding activity of recombinant rBac-RelA (Figure 4d). As evidenced by an EMSA, all four oncogenic p53 mutant proteins, G245S, R248Q, R248W, and R273H, are effective in augmenting the DNA binding activity of RelA. Previous reports had already shown that these oncogenic p53 proteins bind RelA like p53 wt.32 These data suggest that oncogenic versions of p53 that are defective in their DNA binding might retain their ability to interact with RelA and, consequently, influence the expression of RelA target genes by enhancing the DNA binding of NF-κB, even when nuclear RelA levels are low.
Profound Influence of RelA-AD and RPS3 on DNA Binding by RelA-RHR in Solution
We aimed to further characterize RelA:DNA complex formation in solution under conditions that more closely mimic cellular salt concentrations. Previous studies using fluorescence anisotropy showed that the RHR of the p50 homodimer and p50:RelA heterodimer bound κB DNA very weakly at a near physiological salt concentration of 150 mM NaCl and that of the RelA homodimer could not be measured.33 We performed binding affinity measurements using fluorescence anisotropy in which the HIV-κB DNA was labeled with a fluorescein molecule at the 5'-end (F-HIV-κB DNA) (Figure 5).
Figure 5.
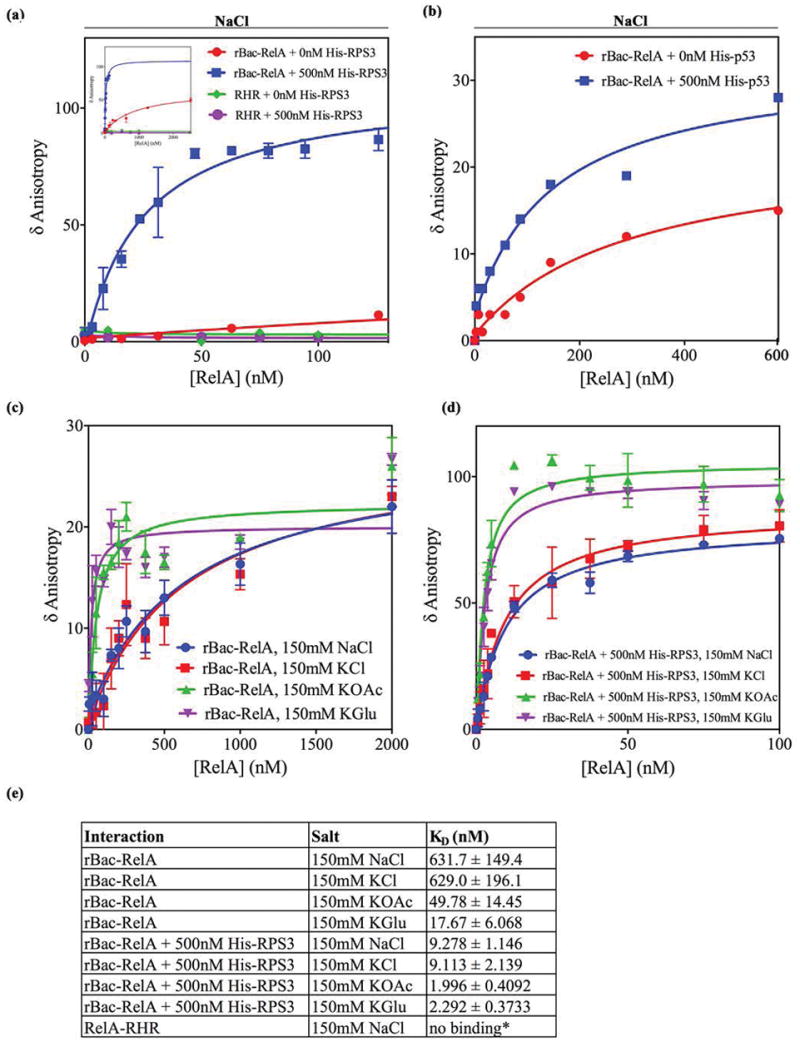
DNA binding by rBac-RelA and RelA-RHR in the absence or presence of cofactors in solution. (a) Graphical representation of the binding of 1 nM F-HIV-κB DNA to increasing amounts of rBac-RelA or RelA-RHR with or without 500 nM His-RPS3: red for rBac-RelA, blue for rBac-RelA and His-RPS3, green for RelA-RHR, and purple for RelA-RHR and His-RPS3. These bindings were performed in the presence of 150 mM NaCl. Data, given as normalized anisotropy values, were analyzed with GraphPad PRISM 4.0 and fitted using a nonlinear regression model to a hyperbolic dose–response curve. (b) Graphical representation of the binding of 1 nM F-HIV-κB DNA to increasing amounts of rBac-RelA with or without 500 nM His-p53: red for rBac-RelA and blue for rBac-RelA and His-p53. These bindings were performed in the presence of 150 mM NaCl. As indicated before, data were fitted using a nonlinear regression model to a hyperbolic dose–response curve. (c) Graphical representation of the binding of rBac-RelA to F-HIV-κB DNA in the presence of different salts at a concentration of 150 mM. (d) Same as panel c but in the presence of 500 nM His-RPS3. (e) Summary of apparent KD values obtained in this study.
We observed that the F-HIV-κB DNA does not bind BSA (Figure S3a). Therefore, all binding experiments were performed in the presence of 4 μM BSA and a >200-fold molar excess of a nonspecific DNA. Consistent with previous reports, we found that the RelA-RHR homodimer does not bind HIV-κB DNA at room temperature in a solution containing 150 mM NaCl, at pH 7.5. The presence of 500 nM RPS3 in the solution also had no impact on DNA binding by RelA-RHR (Figure 5a)
We then examined DNA binding by rBac-RelA, which has never been tested in solution. At 150 mM NaCl/KCl, rBac-RelA bound DNA weakly (KD ~ 630 nM) but significantly better than RelA-RHR did (Figure 5a, inset, and Figure 5c). This was a surprise because, according to our EMSA results, the RelA-AD was expected to inhibit DNA binding by the RHR in rBac-RelA (Figure 1b,c). We then tested the effect of RPS3 on DNA binding by rBac-RelA. In the presence of constant 500 nM RPS3, rBac-RelA bound DNA with a KD of ~10 nM, whereas in its absence, the KD was only ~630 nM. This represents a nearly 65-fold enhancement in the affinity (Figure 5a,e). Anisotropy did not change when a single-stranded fluoresceinated DNA containing the κB site was used for titration against increasing concentrations of rBac-RelA, suggesting that the fluorophore itself does not interact with the protein (Figure S3b). Heat-inactivated RPS3 had no effect on RelA:DNA complex formation, suggesting that the interaction between RPS3 and RelA is specific (Figure S3c).
We next tested the effect of p53. Surprisingly, we found that p53 could enhance the affinity of rBac-RelA by only 2–4-fold (Figure 5b,e). Moreover, we needed to use slightly different binding conditions to obtain an optimal effect of p53 (see details in the corresponding section of Materials and Methods). These results suggest that p53 has much less impact on the stability of the RelA:DNA complex in solution. It is possible that the effect of the cofactors is DNA-specific, where RPS3 might affect binding of RelA to a broad spectrum of κB DNA sequences and p53 affects a narrower subclass of sequences.
Although NaCl and KCl are popularly used to test the effect of the electrolyte on protein:DNA complex formation, a high chloride (Cl−) concentration might pose a problem for the complex to form. The cellular Cl− ion concentration is much lower than 150 mM in most cells, and Cl− is known to impose a negative effect on the binding of protein to DNA. K+ ion, which is present at levels higher than that of Na+, partly associates with glutamate and acetate ions in vivo. While differential effects between glutamate and chloride ions are established for prokaryotic processes, eukaryotic biochemical processes such as splicing are also reported to be more efficient in acetate and glutamate ions than in chloride.34–36 Therefore, we tested the effects of potassium chloride (KCl), potassium glutamate (KGlu), and potassium acetate (KOAc) on RelA:DNA complex formation in the presence and absence of RPS3. DNA binding by rBac-RelA in KCl was nearly identical to that in NaCl. However, binding was significantly enhanced in 150 mM KOAc or KGlu, even in the absence of RPS3 (Figure 5c). The binding was even further improved in the presence of RPS3 (Figure 5d). In all cases, the affinity was enhanced by ~8- to >50-fold when RPS3 was present in the binding buffer (Figure 5e). Therefore, both KGlu and KOAc enabled DNA binding more efficiently than NaCl and KCl did. This suggests that the glutamate and acetate ions somehow stabilize the interactions between RPS3 and RelA such that RelA could bind DNA better as reported.
The precise ion composition and the concentration of each ion in mammalian cells are not known. Cellular glutamate and acetate concentrations are only around 20 mM, which is much lower than the level of 150 mM used in our assay. Therefore, the high affinity of the RelA:DNA complex observed here in the presence of 150 mM KOAc or KGlu does not represent the true in vivo affinity. Similarly, in the complex ionic environment in vivo, RPS3 alone might not be sufficient for optimal RelA:DNA complex formation. It is likely that other cofactors work along with RPS3 to fill this role.
In summary, our binding studies show that RelA binds DNA differently under different salt conditions. Surprisingly, full length RelA binds better than RelA-RHR does in solution, suggesting that the RelA-AD does not inhibit full length RelA:DNA binding as we and others initially reported on the basis of EMSA experiments.23,27 Moreover, cofactors such as RPS3 and, to a lesser extent, p53 impact the stability of the RelA:DNA complex in solution, and the RelA-AD participates as a key partner in the process of complex stabilization. In other words, cofactor proteins and RelA-AD cooperate to act on the RelA-DBD for stabilization of the RelA:DNA complex in solution.
An Interaction Interface between the AD and RHR of RelA
To further unravel the mechanism of how the RelA-AD might facilitate DNA binding by its own DBD, we performed in silico docking experiments with ZDOCK37 using as our models the X-ray crystal structure of the RelA-RHR(DBD):DNA complex38 and the recently determined nuclear magnetic resonance (NMR) solution structure of the TA1 segment of RelA-AD (Figure 1a).39,40 Analysis of the docking results reveals a preferred specific binding mode in which the RelA-TA1 can contact its RHR (Figure 6a and Figure S4a,b).
Figure 6.
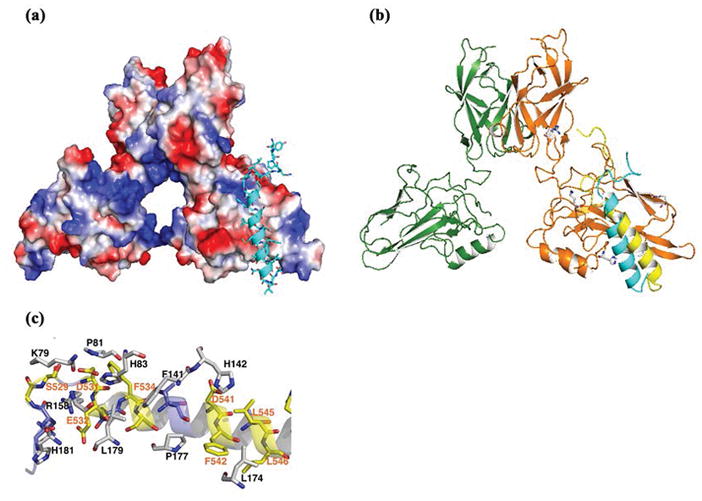
Structural modeling showing interdomain interactions between the RelA-RHR and RelA-AD. (a) Interaction between the RelA-RHR and RelA-AD as determined by a docking program (ZDOCK). The RelA-RHR is represented as an electrostatic surface model, and the helical RelA-AD-TA1 is presented as a ribbon (cyan). (b) Comparison of the RHR-TA1 interaction interface generated by two ways presented as ribbons. Manual modeling was prepared on the basis of the charge and shape complementarity (cyan) and using a docking program as shown in panel A (yellow). Both models reveal that the RelA-AD-TA1 prefers the same docking site on the RelA-RHR. (c) Ribbon presentation of detailed interaction between the RelA-RHR and RelA-AD-TA1.
We also manually created a RHR-TA1 interaction model based purely on surface charge and shape complementarity. In both cases, the resulting model consists of a negatively charged patch of acidic amino acid residues within the RelA-TA1 that engages basic residues along a surface of the RelA-RHR (Figure 6b). The negatively charged RelA-TA1 segment is interspersed with nonpolar residues. In our model, these nonpolar residues mediate closely packed van der Waals contacts with the DNA binding RelA-RHR. Thus, the proposed protein domain–domain interaction interface is supported by both electrostatic and van der Waals interactions (Figure 6c). Although the structure of RelA-TA2 bound to CBP is known,41 we were unsuccessful in modeling the RelA-TA2 region onto its RHR. Therefore, at present, it is unclear whether RelA-TA2 supports interdomain interactions by direct cooperative binding with the RelA-RHR or indirectly influencing the RHR binding potential of RelA-TA1.
When expressed and purified as separate recombinant proteins, a direct binding interaction between the RelA-RHR and RelA-AD was not detected in pull-down experiments monitored by Western blot (data not shown), although the interaction was reported by a pull-down assay of the separate protein fragments monitored by a more sensitive autoradiography-based approach.28 These observations suggest that the interaction between the RelA-RHR and RelA-AD is likely weak and transient. However, the fact that the two domains are covalently linked within the same polypeptide significantly increases the probability of domain–domain interaction in full length RelA. RPS3 and other cofactors might interact with AD-bound RHR to exert their effect. Future experiments are required to better understand how the AD and cofactors function together to facilitate DNA binding by NF-κB RelA.
DISCUSSION
We initially set out to determine whether the NF-κB dimers generated recombinantly for structural and biochemical studies differ with regard to their abilities to bind κB site DNA from those same proteins as they appear endogenously in the mammalian cell nucleus. We found that recombinant, full length RelA derived from either baculovirus-infected insect cells (rBac-RelA) or two-step-purified nuclear RelA [pNuc(Flag)-RelA] is a poorer binder of κB DNA than RelA present in the total nuclear extracts of HA-RelA-transfected mammalian cells [tNuc(HA)-RelA] (Figures 1 and 2). Differential binding suggests that efficient DNA binding by native RelA dimers requires assistance from other nuclear factors.
In this study, we show that both RPS3 and p53 are capable of significantly enhancing NF-κB:DNA complex stability. Although both the observed deficiency in κB site DNA binding by rBac-RelA and the involvement of RPS3 and p53 in NF-κB-dependent gene expression have been reported, neither the basic biochemical mechanism nor the biological implications of differential DNA binding by the rBac-RelA or RelA-RHR alone had been investigated. EMSA-based binding studies performed at very low ionic strengths revealed nearly identical effects of either p53 or RPS3 in enhancing the stability of the RelA:DNA complexes (Figure 3). Solution-based binding studies performed at near physiologic ionic strength, however, showed differential impacts of these two cofactors. RPS3 was found to be significantly more efficient (Figure 5). Contrary to the EMSA-based conclusions, the AD of RelA does not negatively impact the DNA binding of the RHR at 150 mM NaCl in solution; actually, it showed a positive effect because the rBac-RelA:DNA complex is stronger than the RHR:DNA complex at 150 mM NaCl (Figure 5). Thus, the ionic strength of the solution has a marked role in influencing RelA:DNA complex formation and stability. Although EMSA allows direct visualization of the complex in a gel, the low ionic strength and caging effects might artificially stabilize the macromolecular complex. Even under EMSA conditions, when the RelA:DNA complex was formed in the presence of 150 mM NaCl, the complex was clearly destabilized (Figure 1c). This does not suggest that the direct contacts between RelA-RHR and κB DNA observed in the X-ray crystal structures are incorrect. Rather, our results indicate that to efficiently form the proper contacts, protein and DNA must be in the right environment. It should be noted here that NF-κB:DNA complexes could be detected by an EMSA only under low-salt conditions. Although this fact has been known since the discovery of NF-κB, the effect of salt on DNA binding by native length NF-κB has rarely been addressed by previous experiments.
Because protein:DNA interactions are mostly ionic and polar in nature, an increased ionic strength serves to progressively weaken direct ionic/polar contacts between the protein and DNA through charge shielding effects. At this stage, we do not know the precise mechanism by which the cofactors enhance the stability of the RelA:DNA complex and mitigate the ionic shielding effect. We propose that the cofactors weakly interact with the RelA-AD and promote transient contacts on the surface of the RelA-RHR (Figure 7). The presence of the charged RelA-AD and cofactors at the surface of RelA and DNA might then serve to “activate” the counterions present at the binding surfaces, clearing the way for these polar surfaces of protein and DNA to form a competent binding complex. Moreover, transient contacts between cofactors and RelA could induce conformational changes in the DNA-contacting residues of RelA in a way that would favor contacts between the RelA-RHR and DNA observed in the X-ray structures. Further proof is required to establish these or some alternative mechanisms.
Figure 7.
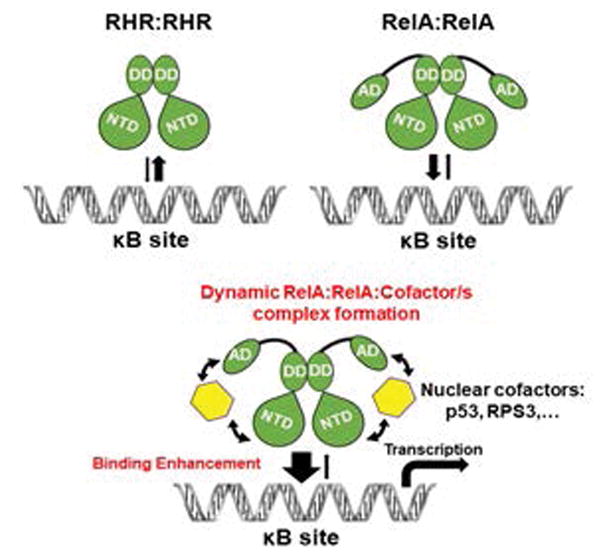
Schematic representation of RelA:DNA complex formation in solution under near physiological conditions (150 mM NaCl). The top panel shows full length RelA protein binds stronger than RelA-RHR does. The bottom panel shows enhanced binding of full length RelA in the presence of a cofactor such as RPS3 or p53. Cofactors used the RelA-AD to transiently interact with the RelA-RHR, resulting in facilitated DNA binding by the RHR.
Genomewide and limited promoter studies had previously suggested that several DNA binding transcription factors such as E2F1, FOXM1, p53, and IRF3 facilitate DNA binding of RelA at sites where these other transcription factors do not directly bind DNA.18,19,31,32,43 Conversely, RelA, as well as other NF-κB subunits, can modulate the DNA binding properties of other transcription factors.42,43 Thus, in addition to their well-studied direct DNA binding properties, each of these transcription factors appears to function in the nucleus as a signaling cofactor that influences the DNA binding and transcriptional potential of their binding partners even at locations within the genome where no consensus DNA binding site for the cofactor is present.
In addition to DNA binding transcription factors, other nucleic acid binding proteins such as RPS3 and OGG1 were also shown to serve as cofactors of NF-κB.20,29 Although RPS3 acts like p53, OGG1 was reported to use a different mechanism. OGG1 binds to a damaged site at a distance from the κB site and exerts its effect from a distance.29 Another transcription factor, Kruppel-like factor 6 (KLF-6), also augments RelA-driven transcription.44 However, KLF-6 presumably functions by a different mechanism whereby it makes contact with both DNA and RelA to facilitate DNA binding. A histone chaperone, nucleophosmin/NPM1, has been shown recently to facilitate RelA:DNA binding through weak but direct interaction with the RelA-RHR.45 Despite the fact that they are weak and transient, the interactions of nuclear cofactors with transcription factors are specific. For example, BSA does not influence RelA:DNA binding. Moreover, the nuclear HMGA1 protein, which has been shown to function synergistically with RelA on the interferon-β promoter,30 shows no ability to affect binding of RelA to HIV-κB site DNA (panels c and d of Figure S2, respectively).
Various cofactor proteins, including RPS3, were reported to play positive roles in supporting RelA-mediated transcription from overlapping but selective promoters.19,20,29 Currently, we do not recognize the source of this promoter specificity. It is possible that, within the context of chromatin, target site DNA plays a more active role in directing the interaction of transcription factors for specific cofactors. However, the interactions between nuclear factors cannot be too specific, because it is already apparent that several factors can interact with and influence RelA:DNA binding and transcriptional activity. The observation that RelA itself can also act as a cofactor for other transcription factors suggests the intriguing possibility that all transcription factors and other nucleic acid binding proteins, in general, might possess some level of ability to function as cofactors. This could explain the basis for the generation of myriads of functioning transcription factors from the DNA binding domain of yeast transcription factor Gal4 and the activation domain of viral transcription factor VP16 fused to similar domains of other DNA binding transcription factors and activation domains.43,46,47 Additional experiments are required to unravel the detailed mechanisms of assisted DNA binding by transcription factors acting in concert as cofactors.
Genetic studies revealed that depletion of any one of multiple cofactors reduces the transcriptional activity of RelA.19,20,29 This raises the question of whether multiple cofactors work in coordination to fine-tune RelA transcriptional activity at a particular binding site or if specific cofactors function at specific DNA sites. At this stage, we do not yet know enough to give a precise answer. Several aspects should be considered before reaching a conclusion about the exact mechanism for nuclear cofactor activity. First, the physical interaction between effectors and NF-κB subunits is apparently very weak. Second, the effective concentration of any of the known cofactor proteins is low (~105 molecules).48 Third, not all cofactors are present within the nucleus under any given cellular condition. Finally, there are effectively thousands of binding sites for each transcription factor (~1–2 × 104). On the basis of these considerations, one can speculate that for a DNA binding transcription factor such as RelA, a broad set of cofactors that can act to influence DNA binding affinity and, consequently, gene transcription exist. A corollary to this hypothesis is that NF-κB itself likely behaves similarly as a non-DNA binding cofactor and contributes to modulation of the nuclear activities of other transcription factors. The combined effects of multiple cofactors acting independently or in concert must reach a threshold level to induce the full functional activity of NF-κB. Targeted deactivation or removal of any of several cofactors then would decrease the effective concentration of the cofactors halting further activation. An alternative possibility is that only a limited number of cofactors act on RelA to bind to a specific promoter containing a κB site. That is, the target DNA in conjunction with a neighboring nucleosome may play essential roles in guiding cofactor-induced high-affinity interactions between NF-κB and DNA.
Because the interactions involving the AD, DNA binding RHR, and cofactor are dynamic, the AD can easily alter or simultaneously contact its other downstream effectors required for initiation of transcription, including histone acetylase proteins such as CBP/p300, the Mediator complex, and RNA polymerase II.40,41,49,50 An additional potential role of the interaction of nuclear cofactors with RelA can be envisioned on the basis of the work of van Essen et al.49 That study revealed that the RelA-AD is not essential for transcription of all RelA-dependent genes. Transcriptional activation from these promoters requires other factors in addition to RelA, such as the members of the AP1 transcription factor family. We are tempted to speculate not only that interaction between NF-κB and AP1 on these sites promotes RelA:DNA binding but also that the AP1-AD cooperates to recruit RNA polymerase and initiate target gene transcription. Our work thus leads to several novel ideas surrounding the potential transcriptional cofactor activities of nuclear protein factors. The hypotheses will direct the design of new experiments aimed at addressing these poorly understood but fundamental processes in the initiation of eukaryotic transcription.
Supplementary Material
Acknowledgments
Funding
This work was supported by National Institutes of Health Grants GM085490 to G.G. and DP2 OD007237 and P41 GM103426 to R.E.A. V.Y.-F.W. was supported by a Start-up Research Grant (SRG2016-00084-FHS) from University of Macau. Biochemistry research at San Diego State University (SDSU) is supported in part by the California Metabolic Research Foundation.
The authors thank Dr. Shannon Lauberth and Homa Rahnamoun for the Flag-p53 expression plasmids. The authors also thank Dr. Tapas Hazra, Dr. Michael Lenardo, and Dr. Steve Buldogh for plasmids, reagents, and discussion. The authors thank Dr. Simpson Joseph for help in fluorescence anisotropy and Youssie Atar in Dr. Joseph’s group for use of the instrument.
Footnotes
Notes
The authors declare the following competing financial interest(s): R.E.A. is a cofounder of, has equity interest in, and is on the scientific advisory board of Actavalon, Inc. The rest of authors declare no competing financial interest.
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.bio-chem.8b00158.
Four supplementary figures (Figures S1–S4) (PDF)
References
- 1.Hayden MS, Ghosh S. NF-kappaB, the first quarter-century: remarkable progress and outstanding questions. Genes Dev. 2012;26:203–234. doi: 10.1101/gad.183434.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ghosh G, Wang VY, Huang DB, Fusco A. NF-kappaB regulation: lessons from structures. Immunol Rev. 2012;246:36–58. doi: 10.1111/j.1600-065X.2012.01097.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hoffmann A, Leung TH, Baltimore D. Genetic analysis of NF-kappaB/Rel transcription factors defines functional specificities. EMBO J. 2003;22:5530–5539. doi: 10.1093/emboj/cdg534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sha WC, Liou HC, Tuomanen EI, Baltimore D. Targeted disruption of the p50 subunit of NF-kappa B leads to multifocal defects in immune responses. Cell. 1995;80:321–330. doi: 10.1016/0092-8674(95)90415-8. [DOI] [PubMed] [Google Scholar]
- 5.Tsui R, Kearns JD, Lynch C, Vu D, Ngo KA, Basak S, Ghosh G, Hoffmann A. IkappaBbeta enhances the generation of the low-affinity NFkappaB/RelA homodimer. Nat Commun. 2015;6:7068. doi: 10.1038/ncomms8068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chen FE, Ghosh G. Regulation of DNA binding by Rel/NF-kappaB transcription factors: structural views. Oncogene. 1999;18:6845–6852. doi: 10.1038/sj.onc.1203224. [DOI] [PubMed] [Google Scholar]
- 7.Chen FE, Huang DB, Chen YQ, Ghosh G. Crystal structure of p50/p65 heterodimer of transcription factor NF-kappaB bound to DNA. Nature. 1998;391:410–413. doi: 10.1038/34956. [DOI] [PubMed] [Google Scholar]
- 8.Chen YQ, Ghosh S, Ghosh G. A novel DNA recognition mode by the NF-kappa B p65 homodimer. Nat Struct Biol. 1998;5:67–73. doi: 10.1038/nsb0198-67. [DOI] [PubMed] [Google Scholar]
- 9.Cheng CS, Feldman KE, Lee J, Verma S, Huang DB, Huynh K, Chang M, Ponomarenko JV, Sun SC, Benedict CA, Ghosh G, Hoffmann A. The specificity of innate immune responses is enforced by repression of interferon response elements by NF-kappaB p50. Sci Signaling. 2011;4:ra11. doi: 10.1126/scisignal.2001501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Siggers T, Chang AB, Teixeira A, Wong D, Williams KJ, Ahmed B, Ragoussis J, Udalova IA, Smale ST, Bulyk ML. Principles of dimer-specific gene regulation revealed by a comprehensive characterization of NF-kappaB family DNA binding. Nat Immunol. 2011;13:95–102. doi: 10.1038/ni.2151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Leung TH, Hoffmann A, Baltimore D. One nucleotide in a kappaB site can determine cofactor specificity for NF-kappaB dimers. Cell. 2004;118:453–464. doi: 10.1016/j.cell.2004.08.007. [DOI] [PubMed] [Google Scholar]
- 12.Wang VY, Huang W, Asagiri M, Spann N, Hoffmann A, Glass C, Ghosh G. The transcriptional specificity of NF-kappaB dimers is coded within the kappaB DNA response elements. Cell Rep. 2012;2:824–839. doi: 10.1016/j.celrep.2012.08.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Xing Y, Yang Y, Zhou F, Wang J. Characterization of genome-wide binding of NF-kappaB in TNFalpha-stimulated HeLa cells. Gene. 2013;526:142–149. doi: 10.1016/j.gene.2013.05.001. [DOI] [PubMed] [Google Scholar]
- 14.Tong AJ, Liu X, Thomas BJ, Lissner MM, Baker MR, Senagolage MD, Allred AL, Barish GD, Smale ST. A Stringent Systems Approach Uncovers Gene-Specific Mechanisms Regulating Inflammation. Cell. 2016;165:165–179. doi: 10.1016/j.cell.2016.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Martone R, Euskirchen G, Bertone P, Hartman S, Royce TE, Luscombe NM, Rinn JL, Nelson FK, Miller P, Gerstein M, Weissman S, Snyder M. Distribution of NF-kappaB-binding sites across human chromosome 22. Proc Natl Acad Sci U S A. 2003;100:12247–12252. doi: 10.1073/pnas.2135255100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Heinz S, Romanoski CE, Benner C, Allison KA, Kaikkonen MU, Orozco LD, Glass CK. Effect of natural genetic variation on enhancer selection and function. Nature. 2013;503:487–492. doi: 10.1038/nature12615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jin F, Li Y, Dixon JR, Selvaraj S, Ye Z, Lee AY, Yen CA, Schmitt AD, Espinoza CA, Ren B. A high-resolution map of the three-dimensional chromatin interactome in human cells. Nature. 2013;503:290–294. doi: 10.1038/nature12644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhao B, Barrera LA, Ersing I, Willox B, Schmidt SC, Greenfeld H, Zhou H, Mollo SB, Shi TT, Takasaki K, Jiang S, Cahir-McFarland E, Kellis M, Bulyk ML, Kieff E, Gewurz BE. The NF-kappaB genomic landscape in lymphoblastoid B cells. Cell Rep. 2014;8:1595–1606. doi: 10.1016/j.celrep.2014.07.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lim CA, Yao F, Wong JJ, George J, Xu H, Chiu KP, Sung WK, Lipovich L, Vega VB, Chen J, Shahab A, Zhao XD, Hibberd M, Wei CL, Lim B, Ng HH, Ruan Y, Chin KC. Genome-wide mapping of RELA(p65) binding identifies E2F1 as a transcriptional activator recruited by NF-kappaB upon TLR4 activation. Mol Cell. 2007;27:622–635. doi: 10.1016/j.molcel.2007.06.038. [DOI] [PubMed] [Google Scholar]
- 20.Wan F, Anderson DE, Barnitz RA, Snow A, Bidere N, Zheng L, Hegde V, Lam LT, Staudt LM, Levens D, Deutsch WA, Lenardo MJ. Ribosomal protein S3: a KH domain subunit in NF-kappaB complexes that mediates selective gene regulation. Cell. 2007;131:927–939. doi: 10.1016/j.cell.2007.10.009. [DOI] [PubMed] [Google Scholar]
- 21.Choy MK, Movassagh M, Siggens L, Vujic A, Goddard M, Sanchez A, Perkins N, Figg N, Bennett M, Carroll J, Foo R. High-throughput sequencing identifies STAT3 as the DNA-associated factor for p53-NF-kappaB-complex-dependent gene expression in human heart failure. Genome Med. 2010;2:37. doi: 10.1186/gm158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kawauchi K, Araki K, Tobiume K, Tanaka N. Activated p53 induces NF-kappaB DNA binding but suppresses its transcriptional activation. Biochem Biophys Res Commun. 2008;372:137– 141. doi: 10.1016/j.bbrc.2008.05.021. [DOI] [PubMed] [Google Scholar]
- 23.Mulero MC, Huang DB, Nguyen HT, Wang VY, Li Y, Biswas T, Ghosh G. DNA-binding affinity and transcriptional activity of the RelA homodimer of nuclear factor kappaB are not correlated. J Biol Chem. 2017;292:18821–18830. doi: 10.1074/jbc.M117.813980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Huxford T, Malek S, Ghosh G. Preparation and crystallization of dynamic NF-kappa B.Ikappa B complexes. J Biol Chem. 2000;275:32800–32806. doi: 10.1074/jbc.M006037200. [DOI] [PubMed] [Google Scholar]
- 25.Moorthy AK, Huang DB, Wang VY, Vu D, Ghosh G. X-ray structure of a NF-kappaB p50/RelB/DNA complex reveals assembly of multiple dimers on tandem kappaB sites. J Mol Biol. 2007;373:723–734. doi: 10.1016/j.jmb.2007.08.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pollard TD. A guide to simple and informative binding assays. Mol Biol Cell. 2010;21:4061–4067. doi: 10.1091/mbc.E10-08-0683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Nolan GP, Ghosh S, Liou HC, Tempst P, Baltimore D. DNA binding and I kappa B inhibition of the cloned p65 subunit of NF-kappa B, a rel-related polypeptide. Cell. 1991;64:961–969. doi: 10.1016/0092-8674(91)90320-x. [DOI] [PubMed] [Google Scholar]
- 28.Zhong H, Voll RE, Ghosh S. Phosphorylation of NF-kappa B p65 by PKA stimulates transcriptional activity by promoting a novel bivalent interaction with the coactivator CBP/ p300. Mol Cell. 1998;1:661–671. doi: 10.1016/s1097-2765(00)80066-0. [DOI] [PubMed] [Google Scholar]
- 29.Pan L, Zhu B, Hao W, Zeng X, Vlahopoulos SA, Hazra TK, Hegde ML, Radak Z, Bacsi A, Brasier AR, Ba X, Boldogh I. Oxidized Guanine Base Lesions Function in 8-Oxoguanine DNA Glycosylase-1-mediated Epigenetic Regulation of Nuclear Factor kappaB-driven Gene Expression. J Biol Chem. 2016;291:25553–25566. doi: 10.1074/jbc.M116.751453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yie J, Liang S, Merika M, Thanos D. Intra- and intermolecular cooperative binding of high-mobility-group protein I(Y) to the beta-interferon promoter. Mol Cell Biol. 1997;17:3649–3662. doi: 10.1128/mcb.17.7.3649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cooks T, Pateras IS, Tarcic O, Solomon H, Schetter AJ, Wilder S, Lozano G, Pikarsky E, Forshew T, Rozenfeld N, Harpaz N, Itzkowitz S, Harris CC, Rotter V, Gorgoulis VG, Oren M. Mutant p53 prolongs NF-kappaB activation and promotes chronic inflammation and inflammation-associated color-ectal cancer. Cancer Cell. 2013;23:634–646. doi: 10.1016/j.ccr.2013.03.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rahnamoun H, Lu H, Duttke SH, Benner C, Glass CK, Lauberth SM. Mutant p53 shapes the enhancer landscape of cancer cells in response to chronic immune signaling. Nat Commun. 2017;8:754. doi: 10.1038/s41467-017-01117-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Phelps CB, Sengchanthalangsy LL, Malek S, Ghosh G. Mechanism of kappa B DNA binding by Rel/NF-kappa B dimers. J Biol Chem. 2000;275:24392–24399. doi: 10.1074/jbc.M003784200. [DOI] [PubMed] [Google Scholar]
- 34.Reichert V, Moore MJ. Better conditions for mammalian in vitro splicing provided by acetate and glutamate as potassium counterions. Nucleic Acids Res. 2000;28:416–423. doi: 10.1093/nar/28.2.416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sengupta R, Pantel A, Cheng X, Shkel I, Peran I, Stenzoski N, Raleigh DP, Record MT., Jr Positioning the Intracellular Salt Potassium Glutamate in the Hofmeister Series by Chemical Unfolding Studies of NTL9. Biochemistry. 2016;55:2251–2259. doi: 10.1021/acs.biochem.6b00173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cheng X, Guinn EJ, Buechel E, Wong R, Sengupta R, Shkel IA, Record MT., Jr Basis of Protein Stabilization by K Glutamate: Unfavorable Interactions with Carbon, Oxygen Groups. Biophys J. 2016;111:1854–1865. doi: 10.1016/j.bpj.2016.08.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pierce BG, Wiehe K, Hwang H, Kim BH, Vreven T, Weng Z. ZDOCK server: interactive docking prediction of protein-protein complexes and symmetric multimers. Bioinformatics. 2014;30:1771–1773. doi: 10.1093/bioinformatics/btu097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chen YQ, Sengchanthalangsy LL, Hackett A, Ghosh G. NF-kappaB p65 (RelA) homodimer uses distinct mechanisms to recognize DNA targets. Structure. 2000;8:419–428. doi: 10.1016/s0969-2126(00)00123-4. [DOI] [PubMed] [Google Scholar]
- 39.Schmitz ML, dos Santos Silva MA, Altmann H, Czisch M, Holak TA, Baeuerle PA. Structural and functional analysis of the NF-kappa B p65 C terminus. An acidic and modular transactivation domain with the potential to adopt an alpha-helical conformation. J Biol Chem. 1994;269:25613–25620. [PubMed] [Google Scholar]
- 40.Lecoq L, Raiola L, Chabot PR, Cyr N, Arseneault G, Legault P, Omichinski JG. Structural characterization of interactions between transactivation domain 1 of the p65 subunit of NF-kappaB and transcription regulatory factors. Nucleic Acids Res. 2017;45:5564–5576. doi: 10.1093/nar/gkx146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mukherjee SP, Behar M, Birnbaum HA, Hoffmann A, Wright PE, Ghosh G. Analysis of the RelA: CBP/p300 interaction reveals its involvement in NF-kappaB-driven transcription. PLoS Biol. 2013;11:e1001647. doi: 10.1371/journal.pbio.1001647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Freaney JE, Kim R, Mandhana R, Horvath CM. Extensive cooperation of immune master regulators IRF3 and NFkappaB in RNA Pol II recruitment and pause release in human innate antiviral transcription. Cell Rep. 2013;4:959–973. doi: 10.1016/j.celrep.2013.07.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ogawa S, Lozach J, Benner C, Pascual G, Tangirala RK, Westin S, Hoffmann A, Subramaniam S, David M, Rosenfeld MG, Glass CK. Molecular determinants of crosstalk between nuclear receptors and toll-like receptors. Cell. 2005;122:707–721. doi: 10.1016/j.cell.2005.06.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zhang Y, Lei CQ, Hu YH, Xia T, Li M, Zhong B, Shu HB. Kruppel-like factor 6 is a co-activator of NF-kappaB that mediates p65-dependent transcription of selected downstream genes. J Biol Chem. 2014;289:12876–12885. doi: 10.1074/jbc.M113.535831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lin J, Kato M, Nagata K, Okuwaki M. Efficient DNA binding of NF-kappaB requires the chaperone-like function of NPM1. Nucleic Acids Res. 2017;45:3707–3723. doi: 10.1093/nar/gkw1285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sadowski I, Ma J, Triezenberg S, Ptashne M. GAL4-VP16 is an unusually potent transcriptional activator. Nature. 1988;335:563–564. doi: 10.1038/335563a0. [DOI] [PubMed] [Google Scholar]
- 47.Lin YS, Carey M, Ptashne M, Green MR. How different eukaryotic transcriptional activators can cooperate promiscuously. Nature. 1990;345:359–361. doi: 10.1038/345359a0. [DOI] [PubMed] [Google Scholar]
- 48.Hottiger MO, Felzien LK, Nabel GJ. Modulation of cytokine-induced HIV gene expression by competitive binding of transcription factors to the coactivator p300. EMBO J. 1998;17:3124–3134. doi: 10.1093/emboj/17.11.3124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.van Essen D, Engist B, Natoli G, Saccani S. Two modes of transcriptional activation at native promoters by NF-kappaB p65. PLoS Biol. 2009;7:e1000073. doi: 10.1371/journal.pbio.1000073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Malik S, Roeder RG. Dynamic regulation of pol II transcription by the mammalian Mediator complex. Trends Biochem Sci. 2005;30:256–263. doi: 10.1016/j.tibs.2005.03.009. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.