

Abstract
The most common forms of acquired epilepsies arise following acute brain insults such as traumatic brain injury, stroke, or CNS infections. Treatment is effective for only 60-70% of patients and remains symptomatic despite decades of effort to develop epilepsy prevention therapies. Recent preclinical efforts are focused on likely primary drivers of epileptogenesis, namely inflammation, neuron loss, plasticity and circuit reorganization. This review suggests a path to identify neuronal and molecular targets for clinical testing of specific hypotheses about epileptogenesis and its prevention or modification. Acquired human epilepsies with different etiologies share some features with animal models. We identify these commonalities and discuss their relevance to development of successful epilepsy prevention or disease modification strategies. Risk factors for developing epilepsy that appear common to multiple acute injury etiologies include intracranial bleeding, disruption of the blood-brain barrier, more severe injury and early seizures within a week of injury. In diverse human epilepsies and animal models, seizures appear to propagate within a limbic or thalamocortical/cortico-cortical network. Common histopathology features of epilepsy of diverse and mostly focal origin are microglial activation and astrogliosis, heterotopic neurons in the white matter, loss of neurons, and the presence of inflammatory cellular infiltrates. Astrocytes exhibit smaller K+ conductances and lose gap junction coupling in many animal models as well as in sclerotic hippocampi from temporal lobe epilepsy patients. There is increasing evidence that epilepsy can be prevented or aborted in preclinical animal models of acquired epilepsy by interfering with processes that appear common to multiple acute injury etiologies, e.g., in post-status epilepticus models of focal epilepsy by transient treatment with a trkB/PLCγ1 inhibitor, isoflurane or HMGB1 antibodies and by topical administration of adenosine, in the cortical fluid percussion injury model by focal cooling, and in the albumin post-traumatic epilepsy model by losartan. Preclinical studies further highlight the roles of mTOR1 pathways, JAK-STAT3, IL-1R/TLR4 signaling and other inflammatory pathways in the genesis or modulation of epilepsy after brain injury. The wealth of commonalities, diversity of molecular targets identified preclinically, and likely multidimensional nature of epileptogenesis argue for a combinatorial strategy in prevention therapy. Going forward, the identification of impending epilepsy biomarkers to allow better patient selection, together with better alignment with multi-site preclinical trials in animal models, should guide the clinical testing of new hypotheses for epileptogenesis and its prevention.
Keywords: Epileptogenesis, acquired epilepsy, antiepileptogenesis, traumatic brain injury, stroke, status epilepticus, CNS infections
Introduction and clinical context
Approximately 20-60% of all epilepsy is caused by acute CNS insults1. In the U.S., traumatic brain injury (TBI) causes approximately 6% of all epilepsies, cerebrovascular accident (CVA) 11%, infections 4%, and new onset cryptogenic status epilepticus (SE) <1%2.
The ability to prevent epilepsy after brain injury or reduce its severity is one of the great unmet needs in neurology3. A latent period of days to years often exists between the acute insult and the onset of clinically obvious epilepsy4 (Fig. 1). This post-injury interval offers an opportunity to intervene with treatment to prevent or delay epilepsy. No preventive treatment exists, yet few preventive studies have been conducted during the last 25 years despite the availability of a variety of drugs marketed for non-epilepsy indications that may have antiepileptogenic or disease-modifying effects in animal models.3,5,6,7 One reason for this is that -in the absence of prospective invasive EEG data- the incidence of epilepsy after acute epileptogenic insults appears relatively low (e.g., 20-50% after moderate to severe TBI), and the clinical diagnosis of spontaneous (“late”) seizures can take years, making clinical trials logistically challenging. Furthermore, there is uncertainty about whether the results of animal studies can be extrapolated to humans. The most commonly used animal models of epileptogenesis are post-SE models of temporal lobe epilepsy (TLE) and kindling models. The validity of applying results from these models to human epilepsy developing after TBI, CVA or infections has not been established.
Fig. 1. Epileptogenic processes and risk factors involved in development of epilepsy after acute brain insults: a conceptual view.
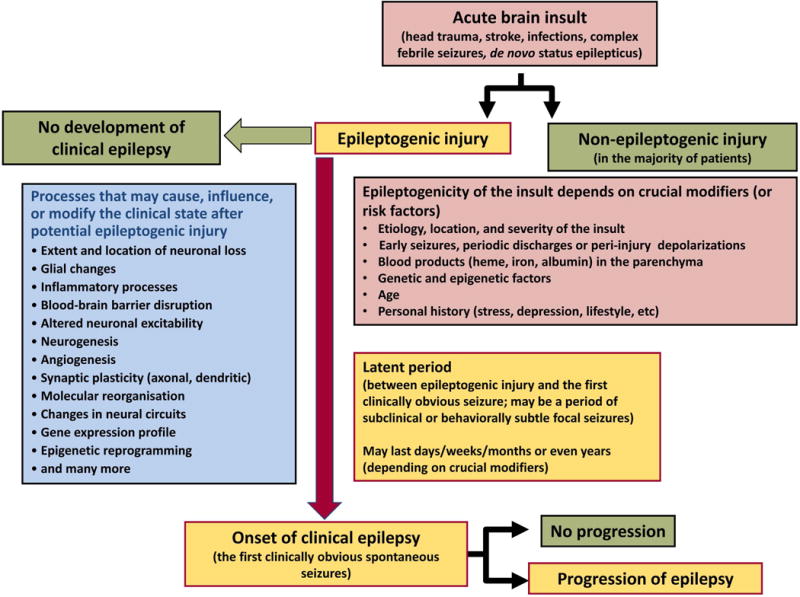
Possibly depending on crucial modifiers or risk factors, the same brain injury can be epileptogenic or not. In the majority of patients, brain insults do not cause epilepsy as discussed in the text. Furthermore, as illustrated in the figure, not all epileptogenic processes, once initiated, result in epilepsy, i.e. complete their course to clinically obvious disease. The term epileptogenesis includes processes that render the brain susceptible to spontaneous recurrent seizures and processes that intensify seizures and make them more refractory to therapy (progression). During epileptogenesis, multiple brain alterations occur, including altered excitability of neurons and/or neuronal circuits, activation of microglia, astrocyte dysfunction, alterations in expression and function of receptors and ion channels (in part recapitulating ontogenesis), loss of neurons, neurogenesis, axonal and dendritic sprouting, gliosis, inflammatory processes, and more. It is important to note that some of these alterations may be related to post-injury repair or recovery and not suited as targets to halt the epileptogenic process. The figure has been modified from previous versions.3,15,276
In some implementations of fluid percussion injury (FPI; a model of TBI) with invasive-EEG monitoring, only 20-50% of the animals appear to develop epilepsy and their epileptic seizures are infrequent.8,9 However, optimization of the FPI model to maximize incidence and speed of epileptogenesis, and EEG montage to detect focal perilesional neocortical seizures, has resulted in a post-traumatic epilepsy (PTE) model that produces rapid epileptogenesis (>90% incidence of spontaneous seizures within 1 month of brain injury) and frequent seizures, including focal neocortical seizures that do not spread to the hippocampus, and this model has been successfully deployed in anti-epileptogenesis studies.10,11
Clinical evidence points to common elements among the acute brain injuries that lead to human epilepsy (Fig. 1), which may provide insights into common mechanisms of epileptogenesis. (Box 1 and Supplementary Text 1). Although only a small proportion of patients with TBI, CVA and infections develop epilepsy, certain subgroups of patients have a higher risk. Risk factors (Table 1) may provide insights into common mechanisms of epileptogenesis. The highest risk factors for epilepsy after acute injury include extravasation of blood due to breach of blood vessels (TBI with intracranial hemorrhage, CVA due to cerebral hemorrhage or ischemic CVA with hemorrhagic conversion), severe injury, disruption of blood-brain barrier (BBB) with cerebral edema (encephalitis, febrile SE), early seizures occurring within one week after injury, and involvement of frontal or temporal lobes (Table 1). All but locus of injury are associated with an inflammatory response; other converging mechanisms by which these risk factors may promote epilepsy are discussed in Supplementary Text 1. The focal epilepsies resulting from such brain insults, e.g., TLE with hippocampal alterations or neocortical epilepsy, which can also involve hippocampal abnormalities, are often similar and reinforce the notion of commonalities in epileptogenic processes.
Box 1. Is the etiology of acquired epilepsy a major prognostic factor for response to treatment?
Observations on the basis of individual case series support the hypothesis that
children with acquired vs. congenital etiology of seizures have higher seizure frequency and a less favorable outlook for terminal seizure remission,
diffuse brain damage is more likely to result in pharmacoresistance than other substrates for focal epilepsy,
posttraumatic and post-infectious epilepsy are commonly severe and refractory to treatment,
epilepsy develops sooner and with higher incidence in children following SE or non-febrile cause than for febrile SE, and
drug response is surprisingly similar for epilepsy after stroke, vascular malformation, and tumor.
For further details see Supplementary Table 5 and Supplementary Text 1.
Table 1. Clinical risk factors for developing epilepsy after TBI, CVA and infection.
Data are from Englander et al.321, Annegers et al.322, Graham et al.323, Annegers et al.324, and Haapaniemi et al.325
| Risk Factor | % Epilepsy Risk | ||
|---|---|---|---|
| PTE | Early post-traumatic seizures | 26 | |
| Glasgow coma scale (GCS) | 3-8 | 17 | |
| Glasgow coma scale | 9-12 | 24 | |
| Glasgow coma scale | 13-15 | 8 | |
| Mid-line shift 1-5 mm | 15-18 | ||
| Mid-Line shift > 5 mm | 26 | ||
| Contusion, Cortical | multiple | 25 | |
| Contusion, Cortical | single | 8 | |
| Contusion, Subcortical | single | 16 | |
| Contusion, Subcortical | multiple | 33 | |
| Contusions, Frontal | unilateral | 20 | |
| bilateral | 26 | ||
| Contusion, Temporal | unilateral | 16 | |
| bilateral | 31 | ||
| Contusion, Parietal | unilateral | 19 | |
| bilateral | 66 | ||
| Subdural hematoma (SDH) | 15 | ||
| SDH requiring surgery | 28-44 | ||
| SDH + contusions | 35 | ||
| Surgery, single | 14 | ||
| Multiple | 37 | ||
| Fragment penetration, | bone | 0 | |
| metal | 25 | ||
| bone & metal | 63 | ||
| Depressed skull fracture | 27 | ||
| CVA | Total Anterior Circulation | 29 | |
| Early seizures (after ICH) | 27 | ||
| Intracranial haemorrhage (ICH) | 18 | ||
| Cortical location | 19-25 | ||
| Partial anterior circulation | 13 | ||
| Age < 65 years | 16 | ||
| GCS < 8 | 28 | ||
| 9-12 | 25 | ||
| 13-15 | 11 | ||
| Barthel Index severe | 13 | ||
| mild | 8 | ||
| Subarachnoid hemorrhage | 22 | ||
| Infection | Bacterial meningitis, | with early seizure | 13 |
| without early seizure | 2 | ||
| Viral encephalitis, | with early seizure | 22 | |
| without early seizure | 10 | ||
The purpose of this review is to examine the evidence for possible commonalities in epileptogenic processes among different causes of acute injury in humans, and among different animal models of acquired epileptogenesis. We attempt to answer the question whether evidence from diverse animal models can be applied across the different common causes of human epilepsy after acute injury, and be utilized in translational studies of epilepsy prevention.
Clinical evidence for commonalities in focal epilepsies resulting from different acute injuries in adults and children is described in more detail in Supplementary Text 1.
Acute brain injury EEG findings and how they relate to later epilepsy
The similarities in acute EEG findings in a variety of acute brain injuries are much more striking than the differences. Nonconvulsive seizures are seen in 13-25% overall, and are increasingly seen with more cortical involvement, more intraparenchymal blood, acute clinical seizures, and the presence of coma.12,13 Similarly, spreading depolarizations (a form of peri-injury spreading depression seen on intracranial EEG) are common in a variety of acute brain injuries, including subarachnoid hemorrhage, TBI, spontaneous intraparenchymal hemorrhage, and malignant ischemic stroke.14 Acute nonconvulsive seizures, periodic discharges and spreading depolarizations have been independently correlated with adverse physiological effects and worse neurological outcomes in numerous studies.15
Only a few investigations have looked at the later development of epilepsy based on acute EEG findings, but they seem to have predictive value as well. For example, a study of children undergoing continuous EEG monitoring in the pediatric ICU showed that 47% of those with acute symptomatic electrographic SE went on to develop epilepsy, versus 11% in those without this EEG finding.16 Electrographic SE had an odds ratio of 13.3 for development of epilepsy; it was also associated with worse functional outcome and quality of life. In a study of >100 survivors who had lateralized periodic discharges (LPDs) or nonconvulsive seizures (NCSzs), new development of epilepsy was seen in 17% of those with LPDs only, 38% with NCSzs only, and 48% with both LPDs and NCSzs.17 Thus, patients with these acute EEG findings may be an appropriate cohort for epilepsy-prevention trials.
In critically ill patients, invasive EEG reveals seizures and PDs in a higher percentage of patients than can be seen on scalp EEG, some of which appear to be causing metabolic distress.18,19 Whether these findings will predict later epilepsy, or whether detection of acute high-frequency oscillations (HFOs) with this type of recording will be as predictive as they are in animal models20, remains to be determined.
Thus, there appear to be commonalities in the acute EEG findings and in their prognostic significance, regardless of the specific type of acute brain injury. However, there has been minimal work on whether serial scalp EEGs can detect those patients that are in the process of developing epilepsy, or whether the specific type of acute brain injury matters once a given pattern is seen.
Genetic aspects in patients
There is little doubt that genetic factors have a role in the likelihood of developing epilepsy after acquired acute brain insults. The study of the genetic basis of familial epilepsies with Mendelian inheritance and the epileptic encephalopathies has clearly established the role of genes expressing ion channels, components of synaptic machinery, and other cellular determinants of network excitability.21,22 Some of these same genetic variants (e.g., DEPDC5, LGI1, PCDH19, SCN1A, GRIN2A, KCNQ2, GABRG2, and others) are weakly associated with common forms of non-acquired epilepsy where a genetic basis is suspected given there is no identifiable insult or lesion.23 Thus, as suggested decades ago24, it is highly likely that variants in these and other genes in normal humans influence the likelihood of developing epilepsy after acute brain injury. This concept is supported by the observation that the genetic background of rodents can be a major determinant in the development of chronic, recurrent seizures following a variety of brain insults25. The observation that coexpression of two different genes with epilepsy-causing mutations can mask the severity of epilepsy caused by each acting individually26 likely contributes to the variable penetrance of epilepsy risk genes. However, it is not yet known whether any of the mechanisms of network dysfunction associated with the myriad of genetic epilepsies overlap with the underlying molecular basis of the acquired epilepsies.
Few clinical or epidemiological studies of epilepsy following brain injury have investigated the role of genetics. Jennett reported that a family history of epilepsy was more common in young patients with posttraumatic epilepsy27, but other studies showed no association in patients of any age.28,29,30 Ottman et al. found no increased family history of epilepsy in patients who developed chronic seizures after stroke or meningitis/encephalitis. In the most definitive study of TBI to date, Christensen and colleagues31 followed more than 1.6 million Danish children and adults for up to 30 years after TBI, and found that patients with a family history of epilepsy had a notably higher risk of epilepsy after mild (Relative Risk 5.75) and severe (10.1) TBI. In epilepsy developing after SE, twin studies have suggested a clear genetic influence in both febrile and afebrile SE32,33, but no large-scale studies in the general population have been completed to date.
With regard to the identification of specific genetic variants associated with the development of epilepsy following brain injury in humans, most findings come from studies of TBI. A recent review by Bennett et al.34 summarizes the results of 31 separate studies describing polymorphisms in 24 genes, including genes involved in the inflammatory response (e.g. pro- and anti-inflammatory cytokines such as tumor necrosis factor(TNF)-α and interleukin(IL)-6), neuronal plasticity (e.g. brain-derived neurotrophic factor, BDNF), and cognitive function (e.g. catechol-O-methyl transferase). Some of these genes are of particular interest given the roles of inflammatory factors and growth factors in epileptogenesis.35,36 Other reports have implicated glutamic acid decarboxylase37, glutamate transport genes38, and the adenosine A1 receptor39. Only two studies have reported genetic associations with PSE; one identified a polymorphism in a TNF receptor, CD440, and the other in mitochondrial aldehyde dehydrogenase 241. Much more work is required to verify these findings as well as their biological relevance.
Common neuropathology findings in focal human epilepsies
The neuropathology in 9,523 patients with focal epilepsy and epilepsy surgery in 36 European centers was recently reviewed (European Epilepsy Brain Bank; for details see Blümcke et al.42). 36 histopathology diagnoses were described, classified into seven categories. Hippocampal sclerosis, brain tumors, and malformations of cortical development (MCD) were most common, accounting for 78.8% of the series. 72% of lesions were located in the temporal lobe, compared to 13% in frontal and 9% in multiple lobes.
The following structural commonalities were identified:
Reactive astrogliosis is always visible in epilepsy surgery specimens.43 In the neocortex of patients with epilepsy, astrocytes are typically activated at the BBB or blood-CSF-barrier (Fig. 2A). In subcortical white matter, fibrillary (or spider) gliosis can be seen. It is difficult to microscopically identify single astrocytes in this condition as they are covered and hidden by a dense meshwork of spider-like ramifying processes (Fig. 2B). This pattern is the likely cause for neuroimaging changes in white matter of patients with TLE.44 Astrocytes may play a significant role in epileptogenesis (see separate section below).
Excess of heterotopic neurons in white matter can be seen in many epilepsy-surgery specimens, such as hippocampal sclerosis or MCD (Fig. 3). This is referred to as “mild malformation of cortical development” when detected only as microscopic abnormality45,46 Heterotopic neurons are rare in other brain diseases, but consistently reported in epilepsy.45,48,49 No pathomechanistic hypotheses have been developed yet from this observation.
White matter angiopathy is also common50, seemingly unrelated to a specific disease category, but associated with early seizure onset and long-seizure duration.
Inflammatory cellular infiltrates are frequently found. Lymphocytic infiltration can be provoked during peri-surgical manipulation of brain tissue. Intracranial neurophysiology techniques applied during pre-surgical evaluation can also induce a mixed lymphocytic-microglial signature.
Fig. 2. Common patterns of astrogliosis in epileptic human brain tissue.
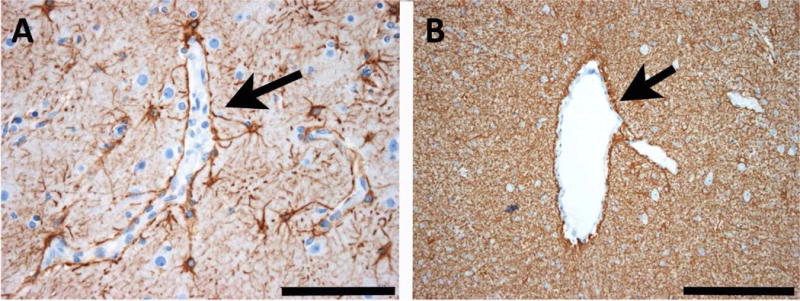
A: Reactive astrogliosis in the neocortex of human epilepsy surgery brain specimens is commonly seen along cortical capillaries (arrow). B: In white matter, there is another common pattern of astrogliosis built by a dense glial fibrillary meshwork. The arrow points towards an enlarged venous vessel. Scale bar in A = 100 μm, in B = 200 μm. GFAP immunohistochemistry (brownish color) with bluish hematoxylin counterstaining.
Fig. 3. Common patterns of heterotopic neurons in the white matter of epileptic human brain tissue.
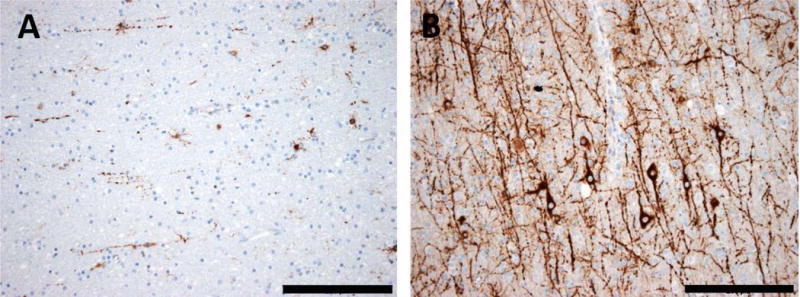
A: The white matter of human temporal lobe usually contains only single heterotopic neurons. B: In many epilepsy specimens of human temporal lobe, there is a vast excess of heterotopic neurons with ramifying neuronal processes in white matter. Scale bar in A and B = 200 μm. MAP2 immunohistochemistry (brownish color) with bluish hematoxylin counterstaining.
A common feature of these histopathology findings is anatomical localization in the white matter which, in epilepsy, is an under-studied anatomical compartment. The epileptogenic lesion is connected via axonal projections to its target regions, together referred to as the seizure network. In focal epilepsy, synchronized activation within the seizure network produces the clinical semiology. As yet, we have neither basic nor clinical evidence for seizures arising directly from white matter.
In addition to these histopathology findings, epigenetic alterations may be involved in epileptogenesis.51 Electrical membrane activity transmits directly into the cell nucleus, thereby driving and adjusting a neuron’s response, including molecular memories that underlie long-lasting response to the environment, i.e. the molecular memory of a neuron.52 Epigenetic histone code modification and long-lasting DNA methylation are a hallmark of molecular memory.53 Increased DNA methylation patterns occur in various animal models of epilepsy and in drug-resistant human epileptic brain tissue.51, 54-57 It can be hypothesized that sustained epileptic activity alters the molecular memory of a neuron, promoting the development of a chronic epileptic state, which can be termed “epileptogenic memory”.
Commonalities in routes of seizure propagation from different epileptic foci
Commonalities in the pathways of seizure propagation are likely a function of the underlying network architecture - with circuits permissive to re-entrant activity being particularly vulnerable to ictogenesis.58 In some models of epilepsy (e.g., amygdala kindling, intra-hippocampal kainate injection) primary injury is localized, whereas in others (e.g., SE) injury is diffuse, yet generally, the seizure phenotype is the same.59 Injuries after TBI, hypoxia/ischemia, or infection may likewise be focal or diffuse, and similarly engage two primary networks when propagating beyond the initial focus: (1) the limbic seizure network seen in TLE, and (2) a thalamocortical network similar to that seen in absence epilepsies, characterized by bilateral spike-and-wave discharges (SWDs) and behavioral arrest. Simply put, because the routes of seizure propagation are constrained by anatomy60, so are the electrographic and behavioral responses to epileptogenic insults (Fig. 4).
Fig. 4. Schematic illustrating components of the limbic (A) and thalamocortical (B) seizure circuits.
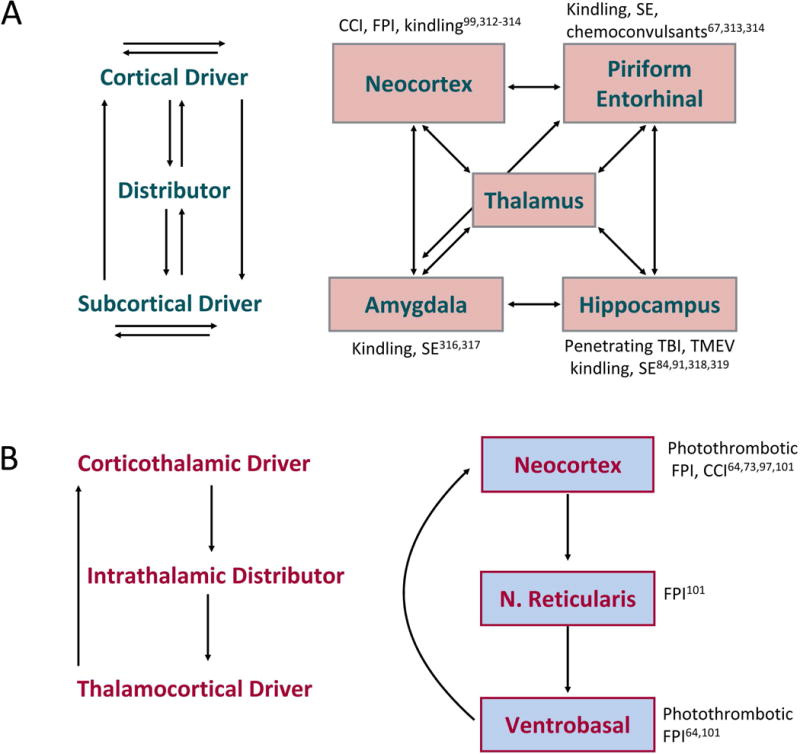
Areas impacted by particular epileptogenic insults (both classic and newer models of acquired epilepsy) are noted near particular network nodes. Arrows indicate interconnections between the nodes. Due to the distributed nature of these circuits, many sites may trigger, lead, or participate in seizure activity.308,309
In most SE models, subcortical and temporal cortical damage is thought to drive seizure generating network(s).61 In many of the TBI and stroke induced epilepsy models, however, the seizure onset zone is likely located in the cortico-thalamo-cortical network, although comprehensive studies remain to be done.62-66 The hippocampus, amygdala, piriform and entorhinal cortices are low-threshold entry points into the limbic seizure circuit.67,68 Conceptually, nodes of this network can be thought of as drivers (e.g., cortical regions, hippocampus, amygdala) and distributors (e.g., midline thalamus) that relay ictal activity from the driver focus to a broader network (Fig. 4A). The thalamocortical network has been best examined with absence seizures: corticothalamic drivers recruit intrathalamic distributors which in turn recruit thalamocortical drivers (Fig. 4B).
While the nonconvulsive focal seizures reported in acquired models typically have a higher frequency than that of genetic absence bilateral seizures, they might engage this same circuits.64, 69, 70 In both of these circuits, primary components may be modulated by inputs that, while not central to ictogenesis, can influence patterns of excitation and/or gate seizure activity.
Perilesional focal seizures or pathological high frequency oscillations (HFOs) at the site of injury often predominate in the early post-injury phase and precede the development of limbic or thalamocortical spreading seizures.20,71,72 While HFOs are behaviorally silent (i.e., purely electrographic), non-spreading perilesional focal seizures induce behavioral arrest when occurring in the frontal neocortex63, but appear behaviorally silent when occurring in the parietal or occipital neocortex72,73. The emergence of seizure activity beyond the injury site may be thought of as secondary epileptogenesis.
Seizure activity evoked by direct activation of cortical drivers gains rapid access to the limbic seizure network by virtue of cortico-cortical projections, cortico-thalamic projections and projections from cortex to subcortical drivers.74-77 Similarly, subcortical foci recruit cortical seizure involvement through direct projections and through distributors.78,79 Cortical foci may likewise recruit the thalamocortical circuit.80 In many models for example, focal suppression of activity in the thalamus is potently anticonvulsant64, 79, 81, 82 and the same is true in patients with drug resistant types of focal epilepsy83.
Direct injury to cortical drivers after TBI - frontal, parietal, or occipital-directed FPI- all trigger pathology in hippocampus, and are associated with both limbic seizures and thalamocortical-like seizures that present -just like in humans- different clinical correlates depending on the ictal sites.70, 71,73 Suppression of ventrobasal neuron activity after cortical infarct inhibits seizures both in the cortex and thalamus64, suggesting that just as in some evoked models (e.g., cortical penicillin), activity in the thalamus is necessary to maintain cortical seizures.80
Direct injury to sub-cortical drivers (e.g. hippocampus after TBI) is unsurprisingly associated with primary seizure onset zones in the hippocampus. However, seizure onset has also been reported in other nodes of the network, including cortical drivers such as piriform cortex.84 This pattern is similar in rats and humans, where even within a single subject, seizures may initiate at different foci.85-87 Direct injury to cortical drivers after TBI is particularly informative - frontal, parietal, or occipital-directed FPI all trigger pathology in hippocampus, and are all associated with both limbic seizures and thalamocortical-like seizures.71,73 Primary injury to the hippocampus after focal herpes encephalitis88-90 or Theiler’s murine viral encephalitis91-93 is likewise associated with hippocampal seizures94. Secondary injury to subcortical drivers is also seen after TBI, controlled cortical impact (CCI), and photothrombotic CVA lesions. These changes in hippocampus along with primary cortical injury may underlie the decreased resting state BOLD95 and the decreased cortical and hippocampal glucose utilization reported after FPI96.
In addition to the temporal lobe seizure phenotype seen in some models, several models of acquired epilepsy display non-convulsive focal seizures. Brief focal seizures, with accompanying behavioral arrest have been reported after FPI, ballistic injury, and ischemic injury in rats.64, 97-100 These are associated with injury to cortical drivers62, 63,70 and alterations in gene expression and physiology in intrathalamic distributors and drivers.101 The relevance of these discharges to acquired epilepsies in humans is debated.72,102,103
Cases in which seizures originate from sites distal to the injury are perhaps the most interesting and suggest that insults can exert epileptogenic effects by engaging a constrained set of networks. While a great deal is known regarding these networks in evoked seizures and classic models, little network mapping has been conducted following TBI, hypoxia/ischemia, or infection. Even fewer studies have directly manipulated the primary seizure circuitry in these models64, and no studies have manipulated modulators (e.g., basal ganglia, cerebellum) after these insults, although they gate a wide variety of seizures in other models.104-107 These areas of future exploration may prove essential to determining the degree of network overlap among these models and other models of acute or acquired epilepsy, and may likewise guide network level interventions for acquired epilepsies.
Commonalities in circuit reorganization after acquired brain injuries
Acquired (but also inherited) brain injuries result in complex network reorganizations. To paraphrase Eccles, Ito and Szentágothai’s classic book, one can see “Epilepsy as a neuronal plasticity machine”. In fact, it is difficult to pinpoint a network parameter that is not changed in epilepsy. Moreover, other than those with epileptic encephalopathies, seizures are usually rare events even in people with severe epilepsy (a person with daily seizures will still spend 99.9% of the time not having clinical seizures), pointing to the existence of compensatory mechanisms, which, most of the time, prevent clinical seizures from occurring. Electrophysiological changes in epileptogenesis and ictogenesis are therefore difficult to interpret, as they could constitute compensatory mechanisms, reflecting a different organization of neuronal circuits to carry the same tasks.108
The most obvious reorganizations commonly found in patients and animal SE models of epilepsy are cell loss, particularly of GABAergic interneurons109-111 reactive synaptogenesis and axonal sprouting, in particular from glutamatergic neurons112-115, and the molecular reorganization of glutamate and GABA receptor subunits.116-118 Many of these features are also found in TBI models, such as the sprouting of excitatory axons, the increase in excitatory drive and decrease in inhibitory drive.119,120 However, some changes, for example, GABA subunit changes during epileptogenesis, may be insult-specific.121 Whether these modifications are similar in animal models and patients is difficult to address. In humans, the lack of proper control tissue (peritumoral non-epileptogenic tissue is usually used as control) prevents an appropriate comparison. Despite this caveat, commonalities are clearly apparent. Do these changes cause similar network consequences?
In rodent models and humans, a loss of interneurons results in decreased GABAergic synaptic (phasic) inputs.122,123 However, phasic inhibition is decreased in the dendrites but increased in the soma in pyramidal cells123, despite a reduction in GABA quantal release.124 GABA also exerts tonic inhibition of neurons via activation of extrasynaptic receptors. In animal post-SE epilepsy, TBI and stroke models and in human epilepsy, there is a preservation (or indeed an increase) in tonic inhibition, despite the decrease in phasic inhibition.125-128 The computational consequences of the shift of GABAergic inhibition from dendritic to somatic results in inhibitory drive being more capable of synchronising principal cells, and the shift of inhibition from phasic to tonic results in an increase in the gain of neurons, whilst preserving their baseline.123,129 The predicted net result is an increase in synchronisation and excitability during periods of increased network activity. Large scale realistic computer models now exist to test this prediction directly.130
The inhibitory action of GABA depends upon the reversal potential of Cl− currents (ECl). A minority of neurons in human slices show altered ECl making GABA depolarizing131,132, a result reproduced in rodent models but only before animals developed spontaneous seizures133. Interictal activity appears to depend upon depolarizing GABA in vitro.131,132 In experimental models in vivo, interictal activity originates from interneuron firing.134 Interneurons may play opposing roles during the progression and spread of seizure activity, and these roles may be determined both by the synchronising effects of somatic inhibition and the propensity for inhibition to become depolarising during excessive activity.135,136 Glutamatergic drive is also increased in epileptogenic tissue from humans137 and rodents115, with an increased contribution of NMDA receptors, which contribute to the bursting response.138-140
Finally, acquired channelopathies, which could change the excitability and integrative properties of cells, have been described in experimental models.141,142 Interestingly, genetic overexpression of Kv1.1 K+ channels can be disease modifying in experimental models.143 These issues remain to be investigated in human tissue.
Commonalities in electrophysiological alterations extend to seizures themselves. Seizure dynamics appear to follow general rules across species (from flies to humans) and brain regions144, but whether electrophysiological commonalities can “explain” epilepsy is not known, as, for most, no causal link has been established. There are many different biophysical paths to seizures in any given neuronal circuit144, and it is likely that, in most cases, the electrophysiological changes that occur just reflect lowered threshold for seizure genesis and/or facilitated seizure propagation.145
Common hippocampal and parahippocampal circuits and structural lesions
Many animal models of epileptogenesis involve structural etiologies, e.g., TBI, stroke, and cortical dysplasia.146 A variety of seemingly unrelated insults, including head trauma, prolonged febrile seizures, vascular abnormalities and encephalitis, can cause similar hippocampal pathology43,147-149 and the same seizure disorder, i.e. acquired mesial TLE with hippocampal sclerosis.150,151 The hippocampal and temporal cortical damage after experimental SE resembles that found in resected tissue of patients with refractory TLE of different etiologies, including loss of dentate hilar and hippocampal pyramidal cells, layer III damage in the entorhinal cortex, gliosis, and mossy fiber sprouting.152-154 Notably, dentate hilar neuron loss (endfolium sclerosis) was reported to be the single common pathology in epilepsy patients with any detectable hippocampal pathology.147
TBI models induced with CCI or lateral FPI result in loss of hilar cells, mossy fiber sprouting, gliosis and neurogenesis, whereas granule and pyramidal cell damage is less severe than in SE models.155 Hippocampal inhibitory neurons appear particularly vulnerable to lateral FPI.156 In correspondence with animal models, a subpopulation of patients with TBI shows progressive hippocampal volume loss and white matter atrophy in MRI.157,158 Taken together, hippocampal atrophy is often seen in acquired epilepsy patients and experimental models. However no study has compared hippocampal pathology between experimental models or humans with or without post-SE, post-TBI or post-stroke epilepsy, which is critical to pinpoint epileptogenesis-specific pathologies.8
In some rodent models, prolonged dentate granule cell discharge selectively destroys vulnerable hippocampal neurons that control granule cell inhibition109,159, thus weakening the normal gating function of the dentate gyrus.161-165 Mild to moderate fluid percussion injury can also cause selective hilar neuron loss and granule cell hyperexcitability.166 Why do dissimilar insults sometimes produce similar pathology and the same neurological condition?
A central role has been proposed for excitatory dentate hilar mossy cells in establishing granule cell inhibition via activation of inhibitory neurons159,160, and for mossy cell loss in granule cell disinhibition109,152,165. Optogenetic activation or silencing of dentate granule cells promoted or suppressed seizures in a unilateral hippocampal kainate mouse model, respectively106, in support of the dentate gate hypothesis. Similar studies capable of selectively exciting or silencing residual mossy cells are needed to refute or confirm the “dormant basket cell” hypothesis.152,167 Regardless of the network mechanism underlying injury-related granule cell hyperexcitability, the selective loss of entorhinal cortex Layer III neurons168 and dentate hilar neurons159 may be a common pathology produced by different insults.
An alternate perspective holds that epileptogenesis requires prolonged molecular and/or structural changes triggered by acute brain injury, because epilepsy develops only after a seizure-free period that can last for decades.169 In this scenario, during the period from insult to clinical epilepsy the epileptogenic mechanism gradually elaborates.66,170,172 Hippocampal transcriptomics and miRNA changes in SE and TBI models and in human TLE have been studied in an effort to identify critical pathways (Fig. 5 and Supplementary Tables 1 and 2). Very few similarities have been identified in hippocampal transcriptomics and miRNA networks among the various structural models, even when data from the same etiology (e.g., SE models) are compared, or between the experimental data and the sparse data available from human epileptic hippocampus. However, comparison of these findings has been difficult because of variability in cell types, tissue sampling, timing of tissue sampling during the course of epileptogenic process, analysis platforms, brain regions, methods for inducing injury, age and species of animals, and unknown epilepsy phenotype of animals sampled at early time points. Dingledine et al. overcame some of the methodological difficulties.172 They performed a well-powered transcriptomics analysis of laser-dissected dentate granule cells in three different SE models, each model generated in two laboratories, used the same analysis platform, and found only 4.5% concordance of differentially expressed genes among the models.
Fig. 5. Word clouds summarizing altered biological processes in the hippocampus in experimental models of structural epilepsies and in human drug-refractory temporal lobe epilepsy (TLE).

The analysis included (A) genome-wide (GW) changes in gene expression and (B) predicted targets of regulated miRNAs. Studies included in the analysis are listed in Supplementary Tables 2 and 3. Biological functions/processes were collected from the gene ontology (GO) or pathway analyses performed in each study. The data was organized to align with different phases of epileptogenic process (acute post-injury time point, latency phase, and epilepsy phase). Word clouds were generated using Wordcloud for R Package. For the sake of clarity, the words: ‘regulation’, ‘cell’, ‘cells’, ‘process’ and stop words (‘English’) were removed from the figures. All other words presented in original articles were included (minimum frequency=1). Larger the word font size in the cloud, more often the word appeared e.g. in the DAVID or IPA® analyses. Note that there were differences in the original analyses, for example, in the terminology used to name different classes of biological functions and use of cut-off p-values (e.g., analyses performed by DAVID or IPA®). Also, the word cloud generated from the human data in panel B is not unbiased (asterisk) as it was extracted from the text (no pathway or GO analysis was presented by authors). Major differences between models were observed, especially when predicted miRNA-target regulated processes were compared (B). SE models appeared more similar with each other than with TBI.
The same hippocampal network that can serve as a primary seizure source when the hippocampal formation is injured may play a secondary role when seizure activity from extratemporal foci spreads to, and recruits, the hippocampal formation.173 Several brain regions innervate and influence the hippocampal formation, including non-temporal neocortex, thalamus, amygdala, and septum174, and it is likely that both the damaged and undamaged hippocampus can generate the same clinical signs regardless of where seizures start. Seizures that originate from temporal neocortical- and even extra-temporal neocortical foci may propagate to the hippocampal formation in both humans175,176 and rodents71.
In sum, qualitative similarities in hippocampal pathology are evident among the SE, TBI, and stroke models as well as between experimental and human TLE triggered by these etiologies. These deserve further analysis in the context of search for biomarkers (Box 2, Supplementary Table 3 and Text 2) and treatment targets of epileptogenesis.
Box 2. Biomarkers of epileptogenesis after different acquired acute brain insults.
A biomarker is a defined characteristic that is measured as an indicator of normal biological processes, pathogenic processes, or responses to an exposure or intervention, including therapeutic interventions. Categories of biomarkers include:
susceptibility/risk biomarkers (e.g., epilepsy susceptibility genes or polymorphisms),
diagnostic biomarkers (e.g., molecular, electrophysiological, or MRI biomarkers),
prognostic biomarkers (e.g., molecular and occurrence of pHFOs and rHFOSs).
For further details see Supplementary Table 3 and Supplementary Text 2.
Dysfunctional astrocytes and microglia: commonalities in epileptogenic processes
A common pathological hallmark in the sequence of events converting a normal brain into an epileptic brain after an acquired insult is microglial activation and subsequent astrogliosis, which results from inflammatory processes.177-180 Astrocytes regulate and amplify immune-mediated mechanisms implicated in epileptogenesis.177,181-186 Evidence from experimental epileptogenesis studies and from studies of established human epilepsy shows that astrocytes undergo functional changes including dysregulation of astroglial Kir and gap junction channels, which together alter glio-neuronal communication and, by impairing uptake and redistribution of extracellular K+ accumulated during neuronal activity, can contribute to or cause seizures.178,185-189 Downregulation of astrocytic Kir channels appears to be a common response to brain injury and has been observed after TBI190, CVA191, BBB damage192, entorhinal cortex lesions193, and freeze-lesion induced cortical dysplasia194,195, and in association with experimentally induced posttraumatic epilepsy.70 Similarly, patch clamp analysis in neurosurgical specimens from TLE patients reveals reduction in astroglial K+ currents in sclerotic vs. non-sclerotic hippocampi.196 Reduced glial K+ conductances and loss of astrocyte gap junction coupling compromise the clearance of extracellular K+ and impair K+ homeostasis. Loss of coupling is mediated by pro-inflammatory cytokines and precedes alterations in neurons and K+ clearance, suggesting a causal role of dysfunctional K+ buffering by astrocytes in the etiology of different epilepsies.185,186 Reduced astrocyte coupling was also reported after acute CVA in situ.197 Whether glial coupling is affected after TBI is unknown. One study observed enhanced phosphorylation of Cx43 protein in astrocytes after experimental TBI198 but functional coupling was not assessed.
Albumin extravasation into the brain following focal injury causes inflammation and activation of a TGF-β pathway in astrocytes, which impairs K+ homeostasis and leads to epilepsy. Importantly, treatment with losartan, a clinically used angiotensin II type I receptor inhibitor, prevented epilepsy in most mice.199 In addition to its known action as an angiotensin II type I receptor inhibitor, losartan exhibits effects on inflammatory pathways and potassium channels200 that might contribute to its antiepileptogenesis effect.
Astrocytes control the availability of the brain’s endogenous anticonvulsant, adenosine, through expression of ADK.201 As a molecular link between energy homeostasis and metabolic demand, ADK expression undergoes acute downregulation followed by chronic overexpression, triggered by ischemia or TBI, and chemically or electrically induced SE.57, 202-207 Overexpression of ADK is linked to astrogliosis and has been reported in human TLE surgical specimens with hippocampal sclerosis and in tumor-associated epilepsy.205,208-210 Increased ADK lowers tissue levels of adenosine, thereby increasing neuronal excitability and precipitating seizures.203,206,211 In addition, increased ADK promotes DNA methylation as an epigenetic contributor to epileptogenesis.55,57 Overexpression of ADK has been associated with progression and worsening of epilepsy.205 Conversely, therapeutic adenosine augmentation212,213 suppresses not only seizures but also prevents development and progression of epilepsy in systemic kainate and pilocarpine models57,214.
In conclusion, dysfunction of glial Kir channels and gap junctions, and subsequent disruption of K+ and adenosine homeostasis are common responses of the brain to TBI, CVA, SE, BBB damage, cortical lesion and dysplasia, and may contribute to or initiate epileptogenesis. Aquaporin 4, expressed exclusively in astrocytes, is translocated from perivascular astrocytic end-feet to the parenchyma in cortical tissue resected during epilepsy surgery.215 A role for aquaporin 4 in the disruption of water balance during epileptogenesis is worth following up.216-218
Inflammatory processes after acquired brain injuries
Brain inflammation, or neuroinflammation219, is common after TBI, CNS infections, CVA and SE in humans and animals. Common to all these different acute injuries is the generation of an immune/inflammatory response in CNS-specific cells and cellular components of the BBB. A key common brain response is the rapid activation of microglia220, which promptly release an array of frontline inflammatory molecules including damage-associated molecular patterns (e.g. HMGB1, ATP, S100β), cytokines (IL-1β, TNF-α, IL-6), various chemokines221 and related effector pathways including COX-2/PGE2 and complement factors.36 This shared tissue injury response might be triggered to activate homeostatic programs. However, if the ensuing inflammatory cascade persists, it leads to CNS dysfunction and neuropathology.220 Non-immune cells like astrocytes and neurons, together with the BBB, play an important role in the immune responses to injuries.36,179,222 As various inflammatory molecules contribute to the mechanisms of acute and chronic seizures by altering synaptic transmission and neuronal excitability223 (Fig. 6), it is conceivable that neuroinflammation contributes to the development of various forms of acquired epilepsies.36,175,224-226 Inflammatory mediators commonly present in human brain and related animal models after acute epileptogenic injuries are shown in Supplementary Table 4. Type and severity of injury determine the relative contribution of innate immunity and circulating leukocytes to increased inflammatory molecules, acute or secondary brain damage227-229, and neuronal network excitability. While HMGB1 and IL-1β contribute to hyperexcitability and epilepsy progression after SE in rodents230-232, IL-6 has a pivotal role in epileptogenesis induced by murine viral encephalitis233. While antiinflammatory drugs dampen epileptogenesis in animal models of SE or viral encephalitis231-235, there is no information whether inflammation is involved in epileptogenesis after stroke or TBI236,237, except for focal cooling preventing both cortical IL-1 induction and epilepsy after TBI in rats62. There is, however, strong evidence that inflammation plays either a pathologic or beneficial role following TBI or stroke depending on the post-injury phase238,239. While TNF-α or CX3CR1 knock-out mice showed reduced functional deficits compared to wild-type mice acutely post-TBI, their functional recovery over time was impaired and cortical damage was increased240-242. Similarly, TNF-α was involved in both secondary brain damage and regenerative recovery after experimental stroke.243-245 The pathologic involvement of inflammation is supported by evidence that excessive brain levels of IL-1β, IL-6, TNF-α (exceeding several times their serum changes) or HMGB1232,246,247 contribute to experimental acute and chronic seizures and SE-induced epileptogenesis36,179,226. Importantly, HMGB1 antibodies have been shown to prevent epileptogenesis in the mouse intrahippocampal kainate model.248 Studies of neuroinflammatory changes in postmortem human brain after CVA/stroke, TBI, infections and SE are limited, although they are essential to validate the potential pathophysiological role of injury-induced inflammation in epileptogenesis. Encephalitis-induced epilepsy caused by neurotropic viruses226 is associated with activation of Toll-like receptors, which drives neuronal damage but may also promote tissue repair.249 The precise relationship between viruses, inflammation and seizure development requires further studies in animals and humans.
Fig. 6. Pathophysiological immune/inflammatory sequelae triggered by various acute brain injuries.
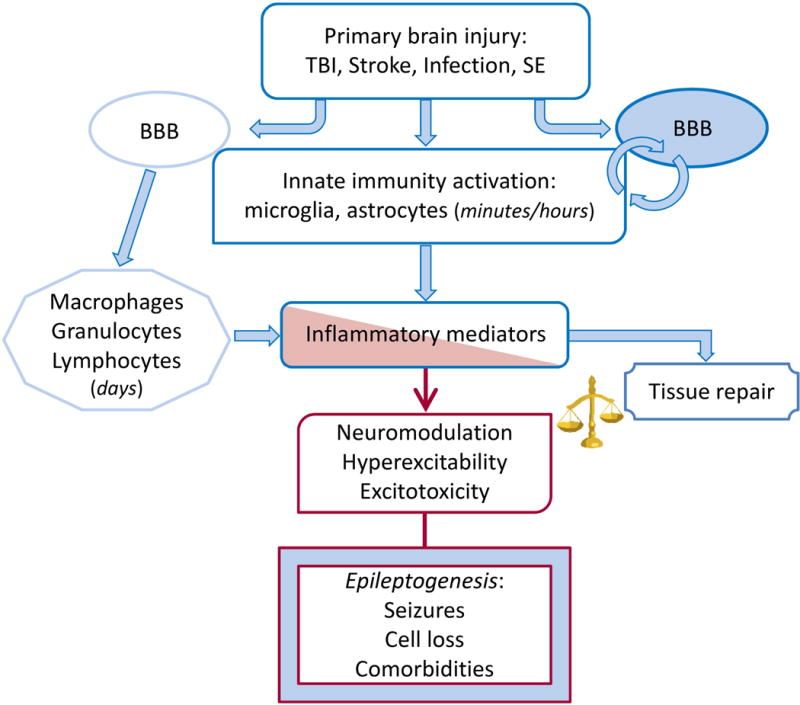
Rapid activation of brain resident innate immunity cells at the site of injury and BBB functional and structural alterations result in the generation of the inflammatory cascade in seizure-prone brain areas. Leukocytes may contribute to perpetuate the inflammatory cascade following interactions with the brain vasculature. The time-locked sequence of these events likely depends on the type of injury. Inflammatory mediators can promote brain damage and dysfunction or contribute to tissue repair depending on their levels and persistence. If the inflammatory milieu exceeds the homeostatic threshold it may lead to hyperexcitability, decrease seizure threshold and promote neuropathology thereby contributing to disease progression. These pathologic events depend on the ability of inflammatory molecules such as cytokines, DAMPs and prostaglandins to alter expression and function of gap junctions, voltage-gated or receptor-coupled ion channels thus contributing to acquired channelopathies, to modify GABA and glutamate release and re-uptake176,177,185 and to alter BBB permeability. BBB dysfunction results in albumin extravasation which compromises homeostatic astrocytic functions and induces excitatory synaptogenesis by activating the TGF-β/ALK5 signaling.310,311,320
We need to better understand the difference between pathologic vs. repair and recovery responses, and evaluate more thoroughly potential commonalities across brain injuries, in order to design drugs with broad therapeutic potential. The term “inflammation” covers many pathways some of which are beneficial after injury238,239, such that targeted intervention at the right time could succeed when global suppression has failed.
Signaling pathway commonalities
Four pathways activated after several types of cerebral insults that are associated with epileptogenesis or epilepsy progression are Janus Kinase/Signal Transducer and Activator of Transcription (JAK/STAT), mammalian Target of Rapamycin Complex (mTORC), BDNF/tyrosine receptor kinase B/phospholipase Cγ1 (BDNF/trkB/PLCγ1), and IL-1R1/TLR4 pathways (Fig. 7). These signaling pathways mediate cellular physiological mechanisms that are critical for memory formation, long-term depression (LTD) and long-term potentiation (LTP).
Fig. 7. Signaling pathway commonalities.
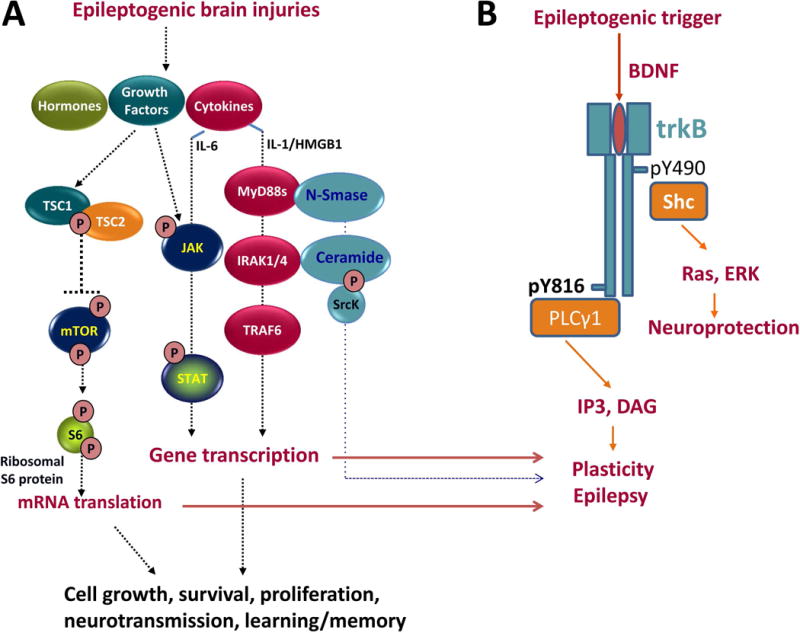
Epileptogenic brain injuries, including TBI, stroke, and SE, increase levels of growth factors, cytokines and hormones that activate the (A) JAK/STAT, IL-1R1/TLR4 and mTORC1 signaling and (B) BDNF/trkB/PLCγ signaling pathways resulting in altered expression of genes and proteins involved in cell growth, cell survival, cell proliferation, neurotransmission, learning and memory, potentially contributing to epileptogenesis and cognitive co-morbidities.
The JAK/STAT pathway (Fig. 7A) is activated after many types of epileptogenic injuries in experimental models, including TBI, SE and stroke.250-255 It regulates expression of genes critical for multiple essential functions including cell proliferation, differentiation, neurogenesis, learning and memory. JAK/STAT mediates decreases in α1 subunit-containing GABAA receptors in hippocampus that occur after experimental SE and TBI250,252 and may contribute to the increased hyperexcitability observed in the hippocampus of injured animals and to subsequent development of epilepsy. Inhibition of STAT3 phosphorylation after SE-induced brain injury reduces the severity of subsequent epilepsy in the rat pilocarpine model256, suggesting that JAK-STAT activation after SE may contribute to epileptogenesis. In the CCI model of PTE in mice, the JAK/STAT pathway is differentially activated depending on the severity of brain injury, and correlates with the presence and severity of subsequent cognitive co-morbidities.257 Interestingly, transient inhibition of the JAK/STAT pathway immediately after CCI improved long-term vestibular motor recovery after injury but did not significantly affect memory performance.257
In rodent models, epileptogenic brain injuries including SE and TBI trigger an immediate and long-lasting increase in the phosphorylation of the S6 ribosomal protein in the hippocampus, which is a downstream marker of an excessively activated mTORC1 pathway (Fig. 7A).258-260 Similarly, structural or dysplastic lesions associated with infantile spasms in rodents or humans also demonstrate overactivation of the mTORC1 pathway in the epileptogenic area.261 Treatment with mTORC1 inhibitors is anti-seizure262-264 and in some models of acquired epilepsy, particularly tuberous sclerosis, antiepileptogenic.259,265-267 Excessive activation of the mTORC1 pathway in acquired models of epilepsy is also associated with cognitive and behavioral deficits, which may be reversed following treatment with the mTORC inhibitor rapamycin.258,266 Taken together, these findings suggest that JAK-STAT and mTORC1 signaling pathway activation following a variety of brain injuries may contribute to development of acquired epilepsy and co-morbid hippocampal-dependent spatial learning and memory deficits.
The interaction of BDNF with receptor trkB is involved in fundamental cellular processes including neuronal proliferation, differentiation and survival as well as neurotransmitter release and synaptic plasticity. Abnormal BDNF-mediated activation of TrkB has been reported in various neurological and psychiatric disorders.268 Altered TrkB signaling (Fig. 7B) underlies epileptogenesis in both the kindling model35 and after SE, as demonstrated by the ability of TrkB inhibitors targeting the PLCγ1 signaling pathway that were administered during the latent period to prevent the development of acquired epilepsy in some animals.100,269 The IL-1R1/TLR4 pathway (Fig. 7A), activated by IL-1β and HMGB1, contributes not only to acute and chronic seizure recurrence270 and kindling271 but also to disease progression post-SE in rodents.231, 232,247
The pharmacology of animal models of acquired epilepsy
The rat amygdala kindling model (AKM) of TLE has been extensively studied6,272,273, although kindled rats do not exhibit SRS, and therefore lack a defining feature of human TLE274. Nonetheless, drugs effective in human TLE are effective against seizures in fully kindled rats.275 The efficacy of various anti-seizure drugs (ASDs) in suppressing seizures in fully kindled rats is remarkably similar to their efficacy in reducing the frequency of SRS in post-SE TLE models(Table 2).276 About 20% of kindled rats are resistant to high doses of ASDs such as phenytoin. Similarly, ASD-resistant rats can be selected from post-SE models of TLE.275 The pharmacology of SRS developing in models of TBI, stroke or viral encephalitis is largely unknown, with some exceptions.277-279 In models of TBI and encephalitis, valproate reduced seizure burden while carbamazepine was ineffective, indicating pharmacologic commonalities.277,279
Table 2. Efficacy of clinically approved antiseizure drugs to block different types of seizures in acute seizure models (MES, PTZ), the amygdala kindling model, post-SE models of TLE, and patients with focal epilepsy.
Data from post-SE models are from rats in which SE was induced by either systemic pilocarpine or kainate, or electrical stimulation of the basolateral amygdala, or from mice in which SE was induced by intrahippocampal administration of kainate. Data are from Löscher.275, 276 Note that the kindling model correctly predicts efficacy of drugs against focal seizures, whereas the MES test is less predictive in this regard; with some exception, doses of antiseizure drugs to suppress kindled seizures are higher than those effective in the MES test. Furthermore, note the correspondence between drug effects on induced amygdala kindled seizures and spontaneously recurrent seizures in post-SE models. NE, not effective.
| Drug | Antiseizure effect in rodents (rats or mice) on | Antiseizure effect on spontaneous focal seizures in patients with epilepy | |||
|---|---|---|---|---|---|
| MES (generalized tonic seizures) | PTZ (generalized clonic seizures) | Amygdala kindled focal and/or secondarily generalized tonic-clonic seizures | Spontaneous focal and/or secondarily generalized seizures in post-SE models | ||
| Benzodiazepines | + | + | + (but tolerance) | + | + (but tolerance) |
| Brivaracetam | + | + | + | + | |
| Carbamazepine | + | NE | +3 | + | + |
| Eslicarbazepine acetate | + | NE | + | + | |
| Ethosuximide | NE | + | NE | NE | NE |
| Felbamate | + | + | +3 | + | |
| Gabapentin | + | + | +3 | + | |
| Lacosamide | + | NE | +1 | + | |
| Lamotrigine | + | NE | +3 | +2 | + |
| Levetiracetam | NE | NE | + | +2 | + |
| Oxcarbazepine | + | NE | + | + | |
| Perampanel | + | + | + | + | |
| Phenobarbital | + | + | +3 | +2 | + |
| Phenytoin | + | NE | +3 | +2 | + |
| Pregabalin | + | + | +1 | + | + |
| Primidone | + | + | + | + | |
| Retigabine (ezogabine) | + | + | + | + | |
| Tiagabine | NE | + | + | + | + |
| Topiramate | + | NE | +3 | + | + |
| Valproate | + | + | +3 | + | + |
| Vigabatrin | NE | ± | +3 | + | + |
| Zonisamide | + | NE | + | + | |
Drug treatment during amygdala kindling acquisition has been used to detect disease-modifying efficacy by administering drug before each amygdala stimulation. Retardation of kindling development may indicate a true antiepileptogenic action or simply suppression of the effects of the stimulation. For most ASDs the latter is true, whereas for some drugs, including valproate, levetiracetam, and brivaracetam, effects on kindling acquisition (e.g., reduced seizure duration) persist long after drug wash-out, suggesting enduring disease modification.6,280-282 The disease-modifying effects of such drugs have been confirmed in post-SE models of TLE6, suggesting that there are shared mechanisms of epileptogenesis in the kindling and SE models.
A variety of experimental and clinically approved drugs has been evaluated for antiepileptogenic effects in post-SE models of TLE.6, 275, 282, 283 Among clinically used ASDs, disease-modifying effects (e.g., neuroprotection, prevention of behavioral comorbidities, reduced frequency, duration or severity of SRS) were only detected after treatment with valproate, levetiracetam, brivaracetam or topiramate, although none of these drugs prevented epilepsy.6, 282 While SE models involve significant neuronal loss, neuroprotection alone may be disease-modifying but not antiepileptogenic.6 Some studies found that antiinflammatory drugs such as celecoxib or immunosupressive drugs such as rapamycin exert antiepileptogenic activity in post-SE models, but these effects appear to be very sensitive to the antiinflammatory drugs used and the timing of drug administration.6, 261, 282, 284 Interestingly, bumetanide, which inhibits the Na-K-2Cl importer NKCC1285 and thus increases the hyperpolarizing action of GABA, was not antiepileptogenic in the lithium-pilocarpine SE model in adult rats286 although it was reported to have antiepileptogenic actions in a model of complex febrile seizures in rats287 and in a mouse model of genetic epilepsy288. By contrast, in SE models a trkB signaling inhibitor100,269, the multitargeted drug isoflurane, which exhibits antiinflammatory, antioxidative, antiapoptotic, and neuroprotective effects289, and HMGB1 antibodies248 can be antiepileptogenic, providing crucial proof-of-principle that antiepileptogenesis is possible. In addition, inhibition of the IL-1R1/TLR4 pathway, and the associated ROS production, has been shown to mediate disease modifying effects in different post-SE models.231,232,247
Only a few studies used TBI models to evaluate possible PTE prevention by drug treatment during the latent period.9 When PTE was induced by rostral parasagittal FPI in rats, 5 weeks of mild focal cooling of the perilesional neocortex almost completely prevented the development of epilepsy62, whereas he investigational drug carisbamate exerted no antiepileptogenic efficacy278, indicating that the model is capable of detecting antiepileptogenic activity. As noted, losartan prevented epileptogenesis in a PTE model, albeit in high doses.199
Other studies report antiepileptogenic efficacy of statins in different models290, and of minocycline in an encephalomyelitis virus infection model291. The preclinical effects of statins were translated to a clinical study. Guo et al.259 reported that acute statin use reduces the risk of poststroke early-onset seizures, and may also prevent the progression of initial poststroke seizures to chronic epilepsy, findings that, if reproducible, would be the first proof-of-concept of antiepileptogenesis in humans.292 Antiepileptogenic effects of statins are also suggested by two other clinical studies.293, 294 In addition to effects on serum cholesterol levels, statins exhibit immunomodulatory, antiinflammatory, and anti-excitotoxic properties290, which could explain the antiepileptogenic effects that have been observed both experimentally and clinically.
Consequences for translation and development of antiepileptogenic therapies
There are three key questions. First, which of the commonalities described in neuropathology, EEG parameters, and molecular and inflammatory pathways are causes rather than consequences of acquired epilepsy? Second, are these commonalities able to clearly distinguish the patients who develop epilepsy from those who have a similar brain insult, but do not develop epilepsy? Third, to what extent do potential causal or susceptibility genes impact the propensity for developing acquired epilepsy? Biomarkers of impending epilepsy are needed to design feasible clinical trials to address these questions. Nonetheless, the richness of commonalities described in this review supports the potential benefit of combinatorial drug treatment, as already demonstrated in preclinical antiepileptogenesis studies.295 Furthermore, these commonalities also appear to emphasize the promise of systems and network approaches towards identification of master regulators controlling epileptogenesis that may provide drug targets for future antiepileptogenic therapies.296-298
The first step in clinical development of antiepileptogenic therapies would be assessment of their safety and tolerability in healthy volunteers, a standard approach for Phase I studies. It will be advantageous to demonstrate target engagement in the brain before moving into later stages, in order to reduce the risk of development failure.299,300 A newly developed synaptic density marker appears promising to measure neurodegeneration and neuronal network changes during epileptogenesis.301 Other indirect target engagement measures could include quantitative EEG or MEG techniques302 or assessment of specific molecular biomarkers in plasma that are clearly linked with drug activity in the brain. Development of such clinical biomarkers (Box 2) should be performed in parallel with the preclinical drug development of antiepileptogenic drug candidates.
Antiepileptogenesis studies should be performed in homogenous patient cohorts sharing similar clinical features such as, for example, severity of injury, accumulation of intracranial blood and incidence of early seizures, because these appear to be common and most likely predictors of focal epilepsy development (Table 3). Other important stratification factors should include the commonality in early EEG parameters (e.g. HFOs) and similarities in the involvement of the epileptogenic limbic structures (e.g. MRI). Furthermore, whenever possible, stratification based on molecular biomarkers should be applied (Box 2). Subjects sharing involvement of similar mechanisms such as dysregulation of the JAK-STAT, mTOR, or BDNF/trkB pathways and neuroinflammation are preferred for antiepileptogenesis trials. The importance and availability of relevant disease-specific and drug response biomarkers is critical for success in clinical development. A recent analysis of outcome of more than 1000 clinical development programs, performed between 2006 and 2015, showed that clinical development success leading to regulatory approval is three times higher when biomarkers permit patient stratification.303 Finally, specific causal or susceptibility genes, if properly validated in enabling studies, should be the ultimate stratification factors since their value was already demonstrated in trials targeting disease modification in other neurodegenerative diseases.304
Table 3.
Summary of commonalities in epileptogenic processes from different acquired acute brain insults.
| Commonalities resulting from acquired acute brain insults | Comments | |
|---|---|---|
| Partial epilepsies resulting from acquired acute brain insults in adults |
|
|
| Partial epilepsies resulting from acquired acute brain insults in children |
|
Febrile status and MTS relationship |
| Post-insult acute EEG alterations (clinical) |
|
Invasive EEG findings in critically ill patients show that an even higher percentage of patients have nonconvulsive seizures and periodic discharges than can be seen on scalp EEG (regardless of the specific type of acute brain injury). |
| Genetic aspects (clinical) |
|
There have essentially been no comparative studies to date. |
| Pathologies (clinical) |
|
Difference to non-epileptic lesions with gliosis uncertain; No appropriate animal models for heterotopic neurons |
| Routes of seizure propagation |
|
|
| Circuit reorganization |
|
|
| Role of hippocampus and parahippocampal circuits and structural lesions |
|
|
| Dysfunctional astrocytes and microglia |
|
Downregulation of Kir4.1 conductance and block of Cx43- mediated gap junction coupling compromise K+ homeostasis |
| Inflammatory processes |
|
Type of acute injury determines cellular sources of inflammatory mediators and pathophysiological role of immune mediators; Scarse information on:
|
| Signaling pathways |
|
Pathway inhibition is antiepileptic in some models and antiepileptogenic in others |
| Biomarkers |
|
|
| Treatment response in patients |
|
Limitations of the current evidence on drug response in the early days of acquired epilepsy are manifold. |
| Animal models of acquired epilepsy |
|
|
| Pharmacology of animal models of acquired epilepsy |
|
Only few data on the pharmacology of models of TBI, stroke, or viral encephalitis (see text). |
Depending on the clinical development strategy, proof-of-concept studies should accumulate evidence that the onset of epilepsy is delayed after brain injury (ideally prevention of epilepsy) or that core clinical features of an ongoing epilepsy are substantially altered (disease modification). The standard ASD add-on trial design and seizure outcome may be used, the difference being that the follow-up period may need to be much longer in prevention trials to demonstrate a therapeutic effect after withdrawal of the antiepileptogenic agent.3,283, 305
Clinical development of drug combinations is associated with a number of significant challenges, therefore the Food and Drug Administration (FDA) issued practical guidelines to facilitate this process.306,307 It is certainly more straightforward to develop a combination treatment that consists of already approved medications, than for two or more investigational drugs used in combination. However, regardless of the approval status of the drug constituents, the New Drug Application (NDA) submitted to the FDA should contain appropriate data to establish the overall safety and effectiveness for the new dosing regimen or indication as proposed in the combination product. Even in the case of already approved drugs such data may not be easily obtainable or compatible with the proposed drug combination, new indication or treatment regimen.
The biggest challenge in development of combinatorial drug treatments is the need to demonstrate that each constituent contributes to the efficacy. This typically involves the necessity to include single drug arms in the trial, which considerably increases the complexity of the design, overall study duration and costs. It is even more challenging if the optimal doses of each drug are not yet defined, which may require additional dose-finding studies for each drug alone or in combination. Finally, even before embarking on clinical trials additional preclinical data may be needed, e.g., safety/toxicology, pharmacokinetics (drug-drug interactions), target/biomarker interactions, etc.
Lastly, there may be legal, intellectual property and freedom-to-operate limitations, particularly for approved drugs used in combination. This may further complicate the development and commercialization of the new combinatorial product.
Conclusions and future directions
The data reviewed indicate that there are a number of commonalities among the three main acute CNS injuries that lead to epilepsy in humans – TBI, CVA and infection. At the clinical level, the presence of intracranial blood, breakdown of the BBB, severe injury, early clinical seizures and EEG seizures, and periodic discharges on early EEG increase the risk for epilepsy in all three. Common anatomical modifications include astrocytosis, white matter abnormalities impacting seizure network and propagation, and neuronal heterotopia. Common elements in animal models include involvement of anatomically constrained networks that relay and distribute seizures remotely from the initial locus of injury, which can explain the common phenomenon of secondary epileptogenic involvement of the temporal lobe, activation of astrocytes with impairment of extracellular K+ buffering and of adenosine homeostasis, selective death of inhibitory interneurons and disinhibition/overinhibition of primary excitatory cells, and activation of the JAK/STAT and mTORC1 pathways. On the other hand transcriptomics have as yet shown few shared gene expression changes across (or within) models, and the role of neuroinflammation and BDNF/Trk signaling in epileptogensis has been demonstrated in SE and kindling models but has not yet been evaluated in animal PTE and PSE. Whether the causes of epilepsy share common mechanisms and features with the causes of the most common comorbidities (anxiety, depression) is an important question to pursue.
Review of the data suggests several conclusions:
The three common causes of epilepsy after acute injury, TBI, CVA and infection, share enough risk factors, electrophysiology, and pathology features to possibly consider them, in epileptogenesis, as one broad category (with individual variations). This has major implication for preventive therapeutic development. To validate or disprove this hypothesis, we should target comparison of epileptogenic processes as well as potential biomarker discovery and therapeutic intervention in both clinical and animal studies comparatively across the three types of injury - particularly for TBI and CVA where clinical intervention is logistically most feasible. If the hypothesis proves correct, this would allow a broader approach to development of biomarkers of epileptogenesis and of preventive treatments.
It is still unclear whether data obtained from the logistically easier models of SE and kindling are applicable to the human conditions of TBI, CVA and infection because relevant data do not exist. This calls for a concerted effort to systematically evaluate the SE and kindling model findings in the PTE, PSE and infection models. Answering this question will determine whether biomarker and therapeutic intervention studies, which can more easily be done in SE and kindling models, have relevance for human preventive treatment discovery. If they do, this would facilitate translation from animal to clinical studies.
Finally, this review identifies a plethora of potential biomarkers, as well as preclinical evidence for several preventive therapeutic strategies that can be followed in human studies. Clinical preventive studies may be significantly strengthened if the preclinical studies were systematically evaluated across the different animal models. Perhaps epileptogenic features, biomarkers and positive treatments common to all the animal models may have the greatest chance of clinical success, and lead, at long last, to a breakthrough in the preventive and/or disease modifying treatment for epilepsy.
Supplementary Material
Key points.
Epileptogenesis after common acute brain insults shares enough features to consider it as one broad category
We identify clinical and preclinical commonalities in epileptogenic processes and their relevance to successful epilepsy prevention strategies
Status epilepticus (SE) model findings should be corroborated in trauma and stroke models to see whether in future SE data can be used to support human studies
Preclinical evidence for several recently-identified preventive therapeutic strategies can be followed in human studies
Acknowledgments
We thank Dr. Rai D’Ambrosio for critical reading of a prefinal version of the manuscript and Nick Pirolli for help with the references. A.V., A.P., W.L., E.A., and M.C.W. acknowledge support from the European Union’s Seventh Framework Programme (FP7) under grant agreement 602102 (EPITARGET). W.L. acknowledges support from the German Research Foundation (DFG, Bonn, Germany; grant # LO 274/1 - LO 274/16), the National Institutes of Health (NIH; Bethesda, MD, USA: grant R21 NS049592), and the Niedersachsen-Research Network on Neuroinfectiology (N-RENNT) of the Ministry of Science and Culture of Lower Saxony in Germany. R.D. receives research support from the NIH (grants U01 NS058158, R01NS097776 and R21 NS03364). A.B.K. acknowledges NIH grants R01 NS051710 and R21 NS083057 and DOD grant W81XWH-11-1-0501. J.E.Jr. acknowledges NIH grants NS002808, NS033310, NS042372, NS065877, NS071048, NS080181, NS100064, and a grant from the Resnick Family Foundation. P.L.P. receives research support from the NIH (R01NS082286) and the SSADH Foundation. D.B. acknowledges NIH grants R01 NS065957, R01 NS084920, R21 NS088024, and R01 NS061844. M.A.R. acknowledges NIH Grant U54 NS079202. A.V. acknowledges grants from Citizen United for Epilepsy Research (CURE) and Fondazione Italiana Ricerca Epilessia-Associazione Italiana Contro l’ Epilessia (FIRE-AICE). I.B. receives research support from European Union’s Seventh Framework Programme (FP7) under grant agreement 602531 (DESIRE). C.S. acknowledges support from the DFG (grant # STE 552/3), Network of European Funding for Neuroscience Research (ERA-NET NEURO project BrIE), and European Commission Horizon 2020 Programme (H2020-MSCA-ITN EU-GliaPhD). A.P. received research support from The Academy of Finland (Grant #273909 and #272249). M.C.W. acknowledges grants from the MRC (G0400136, G0802158, MR/L01095X/1) and the Wellcome Trust (#083163). P.A.F. acknowledges funding by the NIH (KL2TR001432 from NCATS, R01NS097762 from NINDS). The funding agencies had no role in the design, preparation or writing of this manuscript.
Footnotes
DR. INGMAR BLUMCKE (Orcid ID: 0000-0001-8676-0788)
PROF. MARTIN J BRODIE (Orcid ID: 0000-0003-1781-2892)
DR. MICHAEL A. ROGAWSKI (Orcid ID: 0000-0002-3296-8193)
Disclosure
P.K. has served on advisory boards of Lundbeck and UCB Pharma, has acted as a consultant to Eisai, Lundbeck and UCB Pharma, is on Speaker’s Bureau of Eisai, Sunovion and UCB Pharma, and has received research grants from Eisai and Lundbeck. P.L.P. receives royalties from Demos Medical Publishers for the books, Inherited Metabolic Epilepsies and Neuro-Logic: A Primer on Localization. D.B. has acted as consultant to UCB Pharma and Roche. M.J.B. serves on the scientific advisory boards of Eisai, UCB Pharma, GlaxoSmithKline, Lundbeck, Bial, GW Pharmaceuticals and Takeda, is on the speakers’ bureau for Eisai, UCB Pharma, GlaxoSmithKline, Lundbeck, Newbridge, Sanofi Aventis and Abbott and has accepted travel grants for scientific meetings from Eisai, UCB Pharma and Lundbeck. L.J.H. has received research support to Yale University for investigator-initiated studies from Eisai and Upsher-Smith; consultation fees for advising from Ceribell, Monteris, Neuropace, Sun Neuroscience, and Engage Therapeutics; royalties for authoring chapters for UpToDate-Neurology, chapters for Medlink—Neurology, and from Wiley for co-authoring the book “Atlas of EEG in Critical Care”, by Hirsch and Brenner; and honoraria for speaking from Neuropace. A.P. has received research funding from Neurotrauma Sciences LLC. A.V. has acted as consultant to UCB Pharma and Takeda. H.K. and R.M.K. are employees of UCB Pharma. M.C.W. has served on advisory boards of Eisai, and UCB Pharma, has acted as a consultant to GSK, Pfizer, is on Speaker’s Bureau of Eisai, UCB Pharma and SAGE, and has received research grants from Vitaflo. W.L. has served on advisory boards of Grünenthal and UCB Pharma, has acted as a consultant to Pfizer, Lundbeck, GSK, Schering Plough, Boehringer-Ingelheim, UCB Pharma, Bayer, and AWD/Elbion, and has received research grants from UCB Pharma, AWD/Elbion, Merz, Boehringer-Ingelheim, Johnson & Johnson, Pfizer, Novartis, Merz, Desitin, and Piqur. ABK has served on the Executive Committee and Board of American Epilepsy Society and Scientific Advisory Board of CURE, and has acted as a consultant for Pfizer. M.S., J.E.Jr., R.D., I.B., E.A., C.S., R.S.S., C.B., P.A.F., K.K., D.H.L., N.P., M.A.R. and D.S. report no conflict of interest. The mentioned companies or agencies had no role in the design, preparation or writing of this manuscript. We confirm that we have read the Journal’s position on issues involved in ethical publication and affirm that this review is consistent with those guidelines.
Supporting Information
Additional Supporting Information may be found in the online version of this article
References
- 1.Banerjee PN, Filippi D, Allen Hauser W. The descriptive epidemiology of epilepsy-a review. Epilepsy Res. 2009;85:31–45. doi: 10.1016/j.eplepsyres.2009.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hauser WA, Annegers JF, Kurland LT. Incidence of epilepsy and unprovoked seizures in Rochester, Minnesota: 1993. Epilepsia. 34:453–68. doi: 10.1111/j.1528-1157.1993.tb02586.x. [DOI] [PubMed] [Google Scholar]
- 3.Löscher W, Klitgaard H, Twyman RE, et al. New avenues for anti-epileptic drug discovery and development. Nat Rev Drug Discov. 2013;12:757–76. doi: 10.1038/nrd4126. [DOI] [PubMed] [Google Scholar]
- 4.Pitkänen A, Lukasiuk K, Dudek FE, et al. Epileptogenesis. Cold Spring Harb Perspect Med. 2015;5:a022822. doi: 10.1101/cshperspect.a022822. pii. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Temkin NR. Preventing and treating posttraumatic seizures: the human experience. Epilepsia. 2009;50(Suppl 2):10–3. doi: 10.1111/j.1528-1167.2008.02005.x. [DOI] [PubMed] [Google Scholar]
- 6.Löscher W, Brandt C. Prevention or modification of epileptogenesis after brain insults: experimental approaches and translational research. Pharmacol Rev. 2010;62:668–700. doi: 10.1124/pr.110.003046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Klein P, Tyrlikova I. Prevention of Epilepsy: Should We Be Avoiding Clinical Trials? Epilepsy Behav. 2017;72:188–94. doi: 10.1016/j.yebeh.2017.05.024. [DOI] [PubMed] [Google Scholar]
- 8.Pitkänen A, Kharatishvili I, Karhunen H, et al. Epileptogenesis in experimental models. Epilepsia. 2007;48(Suppl 2):13–20. doi: 10.1111/j.1528-1167.2007.01063.x. [DOI] [PubMed] [Google Scholar]
- 9.Pitkänen A, Bolkvadze T, Immonen R. Anti-epileptogenesis in rodent post-traumatic epilepsy models. Neurosci Lett. 2011;497:163–171. doi: 10.1016/j.neulet.2011.02.033. [DOI] [PubMed] [Google Scholar]
- 10.Curia G, Eastman CL, Miller JW, et al. Translational Research in Traumatic Brain Injury. Boca Raton (FL): CRC Press/Taylor and Francis Group; 2016. Modeling Post-Traumatic Epilepsy for Therapy Development. 2016. Chapter 10. [PubMed] [Google Scholar]
- 11.Eastman CL, Fender JS, Temkin NR, et al. Optimized methods for epilepsy therapy development using an etiologically realistic model of focal epilepsy in the rat. Exp Neurol. 2015;264:150–62. doi: 10.1016/j.expneurol.2014.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sinha S, Hirsch L, Ebersole J, et al. EEG monitoring in the ICU. Current Practice of Clinical EEG Wolters Kluwer Health. 2014:543–598. [Google Scholar]; Siniscalchi A, Mintzer S. Statins for poststroke seizures: the first antiepileptogenic agent? Neurology. 2015;85:661–2. doi: 10.1212/WNL.0000000000001878. [DOI] [PubMed] [Google Scholar]
- 13.Herman ST, Abend NS, Bleck TP, et al. Consensus statement on continuous EEG in critically ill adults and children, part I: indications. J Clin Neurophysiol. 2015;32:87–95. doi: 10.1097/WNP.0000000000000166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dreier JP, Fabricius M, Ayata C, et al. Recording, analysis, and interpretation of spreading depolarizations in neurointensive care: Review and recommendations of the COSBID research group. J Cereb Blood Flow Metab. 2016 Jan 1; doi: 10.1177/0271678X16654496. 271678X16654496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Löscher W, Hirsch LJ, Schmidt D. The enigma of the latent period in the development of symptomatic acquired epilepsy - Traditional view versus new concepts. Epilepsy Behav. 2015;52:78–92. doi: 10.1016/j.yebeh.2015.08.037. [DOI] [PubMed] [Google Scholar]
- 16.Wagenman KL, Blake TP, Sanchez SM, et al. Electrographic status epilepticus and long-term outcome in critically ill children. Neurology. 2014;82:396–404. doi: 10.1212/WNL.0000000000000082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Punia V, Garcia CG, Hantus S. Incidence of recurrent seizures following hospital discharge in patients with LPDs (PLEDs) Epilepsy Behav. 2015 Aug;49:250–4. doi: 10.1016/j.yebeh.2015.06.026. [DOI] [PubMed] [Google Scholar]
- 18.Vespa P, Tubi M, Claassen J, et al. Metabolic crisis occurs with seizures and periodic discharges after brain trauma. Ann Neurol. 2016;79:579–90. doi: 10.1002/ana.24606. [DOI] [PubMed] [Google Scholar]
- 19.Waziri A, Claassen J, Stuart RM, et al. Intracortical electroencephalography in acute brain injury. Ann Neurol. 2009;66:366–77. doi: 10.1002/ana.21721. [DOI] [PubMed] [Google Scholar]
- 20.Bragin A, Li L, Almajano J, et al. Pathologic electrographic changes after experimental traumatic brain injury. Epilepsia. 2016;57:735–45. doi: 10.1111/epi.13359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.El Achkar CM, Olson HE, Poduri A, et al. The genetics of the epilepsies. Curr Neurol Neurosci Rep. 2015;15:39. doi: 10.1007/s11910-015-0559-8. [DOI] [PubMed] [Google Scholar]
- 22.McTague A, Howell KB, Cross JH, et al. The genetic landscape of the epileptic encephalopathies of infancy and childhood. Lancet Neurol. 2016;15:304–16. doi: 10.1016/S1474-4422(15)00250-1. [DOI] [PubMed] [Google Scholar]
- 23.Epi4K. Ultra-rare genetic variation in common epilepsies: a case-control sequencing study. Lancet Neurol. 2017;16:135–143. doi: 10.1016/S1474-4422(16)30359-3. [DOI] [PubMed] [Google Scholar]
- 24.Caveness WF, Meirowsky AM, Rish BL, et al. The nature of posttraumatic epilepsy. J Neurosurg. 1979;50:545–53. doi: 10.3171/jns.1979.50.5.0545. [DOI] [PubMed] [Google Scholar]
- 25.Frankel WN. Genetics of complex neurological disease: challenges and opportunities for modeling epilepsy in mice and rats. Trends Genet. 2009;25:361–7. doi: 10.1016/j.tig.2009.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Glasscock E, Qian J, Yoo Jong, et al. Masking epilepsy by combining two epilepsy genes. Nature Neuroscience. 2007;10:1554–1558. doi: 10.1038/nn1999. [DOI] [PubMed] [Google Scholar]
- 27.Jennett B. Epilepsy after non-missile head injuries. 2nd. London: Heinemann Medical; 1975. [DOI] [PubMed] [Google Scholar]
- 28.Salazar AM, Jabbari B, Vance SC, et al. Epilepsy after penetrating head injury. I. Clinical correlates: a report of the Vietnam Head Injury Study. Neurology. 1985;35:1406–14. doi: 10.1212/wnl.35.10.1406. [DOI] [PubMed] [Google Scholar]
- 29.Schaumann BA, Annegers JF, Johnson SB, et al. Family history of seizures in posttraumatic and alcohol-associated seizure disorders. Epilepsia. 35:48–52. doi: 10.1111/j.1528-1157.1994.tb02911.x. [DOI] [PubMed] [Google Scholar]
- 30.Ottman R, Lee JH, Risch N, et al. Clinical indicators of genetic susceptibility to epilepsy. Epilepsia. 1996;37:353–61. doi: 10.1111/j.1528-1157.1996.tb00571.x. [DOI] [PubMed] [Google Scholar]
- 31.Christensen J, Pedersen MG, Pedersen CB, et al. Lancet. London, England: Long-term risk of epilepsy after traumatic brain injury in children and young adults: a population-based cohort study. [DOI] [PubMed] [Google Scholar]
- 32.Corey LA, Pellock JM, DeLorenzo RJ. Status epilepticus in a population-based Virginia twin sample. Epilepsia. 2004;45:159–65. doi: 10.1111/j.0013-9580.2004.34303.x. [DOI] [PubMed] [Google Scholar]
- 33.Eckhaus J, Lawrence KM, Helbig I, et al. Genetics of febrile seizure subtypes and syndromes: a twin study. Epilepsy Res. 2013;105:103–9. doi: 10.1016/j.eplepsyres.2013.02.011. [DOI] [PubMed] [Google Scholar]
- 34.Bennett ER, Reuter-Rice K, Laskowitz DT. Genetic Influences in Traumatic Brain Injury. In: Laskowitz D, Grant G, editors. Translational Research in Traumatic Brain Injury. Boca Raton (FL): [Google Scholar]
- 35.McNamara JO, Scharfman HE. Temporal Lobe Epilepsy and the BDNF Receptor, TrkB. In: Noebels JL, Avoli M, Rogawski MA, Olsen RW, Delgado-Escueta AV, editors. Jasper’s Basic Mechanisms of the Epilepsies [Internet] 4th. Bethesda (MD): [PubMed] [Google Scholar]
- 36.Vezzani A, Friedman A, Dingledine RJ. The role of inflammation in epileptogenesis. Neuropharmacology. 2013b;69:16–24. doi: 10.1016/j.neuropharm.2012.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Darrah SD, Miller MA, Ren D, et al. Genetic variability in glutamic acid decarboxylase genes: associations with post-traumatic seizures after severe TBI. Epilepsy Res. 2013;103:180–94. doi: 10.1016/j.eplepsyres.2012.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ritter AC, Kammerer CM, Brooks MM, et al. Genetic variation in neuronal glutamate transport genes and associations with posttraumatic seizure. Epilepsia. 2016;57:984–93. doi: 10.1111/epi.13397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wagner AK, Miller MA, Scanlon J, et al. Adenosine A1 receptor gene variants associated with post-traumatic seizures after severe TBI. Epilepsy Res. 2010;90:259–272. doi: 10.1016/j.eplepsyres.2010.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhang B, Chen M, Yang H, et al. Evidence for involvement of the CD40/CD40L system in post-stroke epilepsy. Neurosci Lett. 2014;567:6–10. doi: 10.1016/j.neulet.2014.03.003. [DOI] [PubMed] [Google Scholar]
- 41.Yang H, Song Z, Yang G-P, et al. The ALDH2 rs671 Polymorphism Affects Post-Stroke Epilepsy Susceptibility and Plasma 4-HNE Levels. PLoS One. 2014;9:e109634. doi: 10.1371/journal.pone.0109634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Blümcke I, Spreafico R, Haaker G, et al. Histopathological findings in brain tissue obtained from epilepsy surgery. New Engl J Med. 2017;377:1648–1656. doi: 10.1056/NEJMoa1703784. [DOI] [PubMed] [Google Scholar]
- 43.Blümcke I, Sarnat HB, Coras R. Surgical Neuropathology of Focal Epilepsies: Textbook and Atlas. Montrouge, France: John Libbey Eurotext; 2015. [Google Scholar]
- 44.Garbelli R, Milesi G, Medici V, et al. Blurring in patients with temporal lobe epilepsy: clinical, high-field imaging and ultrastructural study. Brain. 2012;135:2337–49. doi: 10.1093/brain/aws149. [DOI] [PubMed] [Google Scholar]
- 45.Blümcke I, Thom M, Aronica E, et al. The clinicopathologic spectrum of focal cortical dysplasias: a consensus classification proposed by an ad hoc Task Force of the ILAE Diagnostic Methods Commission. Epilepsia. 2011;52:158–74. doi: 10.1111/j.1528-1167.2010.02777.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Palmini A, Najm I, Avanzini G, et al. Terminology and classification of the cortical dysplasias. Neurology. 2004;62:S2–8. doi: 10.1212/01.wnl.0000114507.30388.7e. [DOI] [PubMed] [Google Scholar]
- 47.Emery JA, Roper SN, Rojiani AM. White matter neuronal heterotopia in temporal lobe epilepsy: a morphometric and immunohistochemical study. J Neuropathol Exp Neurol. 1997;56:1276–82. doi: 10.1097/00005072-199712000-00002. [DOI] [PubMed] [Google Scholar]
- 48.Hildebrandt M, Pieper T, Winkler P, et al. Neuropathological spectrum of cortical dysplasia in children with severe focal epilepsies. Acta Neuropathol. 2005;110:1–11. doi: 10.1007/s00401-005-1016-6. [DOI] [PubMed] [Google Scholar]
- 49.Rojiani AM, Emery JA, Anderson KJ, et al. Distribution of heterotopic neurons in normal hemispheric white matter: a morphometric analysis. J Neuropathol Exp Neurol. 1996;55:178–83. doi: 10.1097/00005072-199602000-00006. [DOI] [PubMed] [Google Scholar]
- 50.Hildebrandt M, Amann K, Schröder R, et al. White matter angiopathy is common in pediatric patients with intractable focal epilepsies. Epilepsia. 2008;49:804–15. doi: 10.1111/j.1528-1167.2007.01514.x. [DOI] [PubMed] [Google Scholar]
- 51.Kobow K, Blümcke I. The methylation hypothesis: do epigenetic chromatin modifications play a role in epileptogenesis? Epilepsia. 2011;52(Suppl 4):15–9. doi: 10.1111/j.1528-1167.2011.03145.x. [DOI] [PubMed] [Google Scholar]
- 52.Fischer A, Sananbenesi F, Wang X, et al. Recovery of learning and memory is associated with chromatin remodelling. Nature. 2007;447:178–82. doi: 10.1038/nature05772. [DOI] [PubMed] [Google Scholar]
- 53.Peleg S, Sananbenesi F, Zovoilis A, et al. Altered histone acetylation is associated with age-dependent memory impairment in mice. Science. 2010;328:753–6. doi: 10.1126/science.1186088. [DOI] [PubMed] [Google Scholar]
- 54.Dębski KJ, Pitkänen A, Puhakka N, et al. Etiology matters - Genomic DNA Methylation Patterns in Three Rat Models of Acquired Epilepsy. Sci Rep. 2016;6:25668. doi: 10.1038/srep25668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Williams-Karnesky RL, Sandau US, Lusardi TA, et al. Epigenetic changes induced by adenosine augmentation therapy prevent epileptogenesis. J Clin Invest. 2013;123:3552–63. doi: 10.1172/JCI65636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kobow K, Jeske I, Hildebrandt M, et al. Increased reelin promoter methylation is associated with granule cell dispersion in human temporal lobe epilepsy. J Neuropathol Exp Neurol. 2009;68:356–64. doi: 10.1097/NEN.0b013e31819ba737. [DOI] [PubMed] [Google Scholar]
- 57.Kobow K, Kaspi A, Harikrishnan KN, et al. Deep sequencing reveals increased DNA methylation in chronic rat epilepsy. Acta Neuropathol. 2013;126:741–56. doi: 10.1007/s00401-013-1168-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Da Silva FHL, Gorter JA, Wadman WJ. Epilepsy as a dynamic disease of neuronal networks. Handb Clin Neurol. 2012;107:35–62. doi: 10.1016/B978-0-444-52898-8.00003-3. [DOI] [PubMed] [Google Scholar]
- 59.Kandratavicius L, Balista PA, Lopes-Aguiar C, et al. Animal models of epilepsy: use and limitations. Neuropsychiatr Dis Treat. 2014;10:1693–705. doi: 10.2147/NDT.S50371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Proix T, Bartolomei F, Guye M, et al. Individual brain structure and modelling predict seizure propagation. Brain. 2017;140:641–654. doi: 10.1093/brain/awx004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Goldberg EM, Coulter DA. Mechanisms of epileptogenesis: a convergence on neural circuit dysfunction. Nat Rev Neurosci. 2013;14:337–49. doi: 10.1038/nrn3482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.D’Ambrosio R, Eastman CL, Darvas F, et al. Mild passive focal cooling prevents epileptic seizures after head injury in rats. Ann Neurol. 2013;73:199–209. doi: 10.1002/ana.23764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.D’Ambrosio R, Hakimian S, Stewart T, et al. Functional definition of seizure provides new insight into post-traumatic epileptogenesis. Brain. 2009;132:2805–21. doi: 10.1093/brain/awp217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Paz JT, Davidson TJ, Frechette ES, et al. Closed-loop optogenetic control of thalamus as a tool for interrupting seizures after cortical injury. Nat Neurosci. 2013;16:64–70. doi: 10.1038/nn.3269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Paz JT, Huguenard JR. Microcircuits and their interactions in epilepsy: is the focus out of focus? Nat Neurosci. 2015;18:351–9. doi: 10.1038/nn.3950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Bragin A, Wilson CL, Engel J. Chronic epileptogenesis requires development of a network of pathologically interconnected neuron clusters: a hypothesis. Epilepsia. 2000;41(Suppl 6):S144–52. doi: 10.1111/j.1528-1157.2000.tb01573.x. [DOI] [PubMed] [Google Scholar]
- 67.McIntyre DC, Kelly ME, Dufresne C. FAST and SLOW amygdala kindling rat strains: comparison of amygdala, hippocampal, piriform and perirhinal cortex kindling. Epilepsy Res. 1999;35:197–209. doi: 10.1016/s0920-1211(99)00012-1. [DOI] [PubMed] [Google Scholar]
- 68.Vismer MS, Forcelli PA, Skopin MD, et al. The piriform, perirhinal, and entorhinal cortex in seizure generation. Front Neural Circuits. 2015;9:27. doi: 10.3389/fncir.2015.00027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.D’Ambrosio R, Fairbanks JP, Fender JS, et al. Post-traumatic epilepsy following fluid percussion injury in the rat. Brain. 2004;127:304–14. doi: 10.1093/brain/awh038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Stewart TH, Eastman CL, Groblewski PA, et al. Chronic dysfunction of astrocytic inwardly rectifying K+ channels specific to the neocortical epileptic focus after fluid percussion injury in the rat. J Neurophysiol. 2010;104:3345–60. doi: 10.1152/jn.00398.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.D’Ambrosio R, Fender JS, Fairbanks JP, et al. Progression from frontal-parietal to mesial-temporal epilepsy after fluid percussion injury in the rat. Brain. 2005;128:174–88. doi: 10.1093/brain/awh337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Reid AY, Bragin A, Giza CC, et al. The progression of electrophysiologic abnormalities during epileptogenesis after experimental traumatic brain injury. Epilepsia. 2016;57:1558–1567. doi: 10.1111/epi.13486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Curia G, Levitt M, Fender JS, et al. Impact of Injury Location and Severity on Posttraumatic Epilepsy in the Rat: Role of Frontal Neocortex. Cereb Cortex. 2011;21:1574–1592. doi: 10.1093/cercor/bhq218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Poggio GF. The Propagation of Cortical-After-Discharge Through Subcortical Structures. Arch Neurol Psychiatry. 1956;75:350. doi: 10.1001/archneurpsyc.1956.02330220014002. [DOI] [PubMed] [Google Scholar]
- 75.Udvarhelyi GB. Dissemination of Acute Focal Seizures In the Monkey. Arch Neurol. 1965;12:333. doi: 10.1001/archneur.1965.00460280003001. [DOI] [PubMed] [Google Scholar]
- 76.Louis ED, Williamson PD, Darcey TM. Chronic focal epilepsy induced by microinjection of tetanus toxin into the cat motor cortex. Electroencephalogr Clin Neurophysiol. 1990;75:548–57. doi: 10.1016/0013-4694(90)90141-6. [DOI] [PubMed] [Google Scholar]
- 77.Halonen T, Tortorella A, Zrebeet H, et al. Posterior piriform and perirhinal cortex relay seizures evoked from the area tempestas: role of excitatory and inhibitory amino acid receptors. Brain Res. 1994;652:145–148. doi: 10.1016/0006-8993(94)90328-x. [DOI] [PubMed] [Google Scholar]
- 78.Walker AE. Dissemination of Acute Focal Seizures In the Monkey Arch. Neurol. 1965;12:357. doi: 10.1001/archneur.1965.00460280027002. [DOI] [PubMed] [Google Scholar]
- 79.Bertram EH, Zhang D, Williamson JM. Multiple roles of midline dorsal thalamic nuclei in induction and spread of limbic seizures. Epilepsia. 2008;49:256–68. doi: 10.1111/j.1528-1167.2007.01408.x. [DOI] [PubMed] [Google Scholar]
- 80.Avoli M, Gloor P. Interaction of cortex and thalamus in spike and wave discharges of feline generalized penicillin epilepsy. Exp Neurol. 1982;76:196–217. doi: 10.1016/0014-4886(82)90112-1. [DOI] [PubMed] [Google Scholar]
- 81.Cassidy RM, Gale K. Mediodorsal thalamus plays a critical role in the development of limbic motor seizures. J Neurosci. 1998;18:9002–9. doi: 10.1523/JNEUROSCI.18-21-09002.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Wicker E, Forcelli PA. Chemogenetic silencing of the midline and intralaminar thalamus blocks amygdala-kindled seizures. Exp Neurol. 2016;283:404–12. doi: 10.1016/j.expneurol.2016.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Fisher RS, Velasco AL. Electrical brain stimulation for epilepsy. Nat Rev Neurol. 2014;10:261–70. doi: 10.1038/nrneurol.2014.59. [DOI] [PubMed] [Google Scholar]
- 84.Kendirli MT, Rose DT, Bertram EH. A model of posttraumatic epilepsy after penetrating brain injuries: effect of lesion size and metal fragments. Epilepsia. 2014;55:1969–77. doi: 10.1111/epi.12854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Bertram EH. Functional anatomy of spontaneous seizures in a rat model of limbic epilepsy. Epilepsia. 1997;38:95–105. doi: 10.1111/j.1528-1157.1997.tb01083.x. [DOI] [PubMed] [Google Scholar]
- 86.Wennberg R, Arruda F, Quesney LF, et al. Preeminence of Extrahippocampal Structures in the Generation of Mesial Temporal Seizures: Evidence from Human Depth Electrode Recordings. Epilepsia. 2002;43:716–726. doi: 10.1046/j.1528-1157.2002.31101.x. [DOI] [PubMed] [Google Scholar]
- 87.Toyoda I, Bower MR, Leyva F, et al. Early activation of ventral hippocampus and subiculum during spontaneous seizures in a rat model of temporal lobe epilepsy. J Neurosci. 2013;33:11100–15. doi: 10.1523/JNEUROSCI.0472-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Schlitt M, Bucher AP, Stroop WG, et al. Neurovirulence in an experimental focal herpes encephalitis: relationship to observed seizures. Brain Res. 1988;440:293–8. doi: 10.1016/0006-8993(88)90998-5. [DOI] [PubMed] [Google Scholar]
- 89.Stroop WG, Schaefer DC. Neurovirulence of two clonally related herpes simplex virus type 1 strains in a rabbit seizure model. J Neuropathol Exp Neurol. 1989;48:171–83. doi: 10.1097/00005072-198903000-00004. [DOI] [PubMed] [Google Scholar]
- 90.Chen S-F, Huang C-C, Wu H-M, et al. Seizure, neuron loss, and mossy fiber sprouting in herpes simplex virus type 1-infected organotypic hippocampal cultures. Epilepsia. 2004;45:322–32. doi: 10.1111/j.0013-9580.2004.37403.x. [DOI] [PubMed] [Google Scholar]
- 91.Libbey JE, Kirkman NJ, Smith MCP, et al. Seizures following picornavirus infection. Epilepsia. 2008;49:1066–1074. doi: 10.1111/j.1528-1167.2008.01535.x. [DOI] [PubMed] [Google Scholar]
- 92.Stewart K-AA, Wilcox KS, Fujinami RS, et al. Development of postinfection epilepsy after Theiler’s virus infection of C57BL/6 mice. J Neuropathol Exp Neurol. 2010;69:1210–9. doi: 10.1097/NEN.0b013e3181ffc420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Bröer S, Käufer C, Haist V, et al. Brain inflammation, neurodegeneration and seizure development following picornavirus infection markedly differ among virus and mouse strains and substrains. Exp Neurol. 2016;279:57–74. doi: 10.1016/j.expneurol.2016.02.011. [DOI] [PubMed] [Google Scholar]
- 94.Smeal RM, Stewart K-A, Iacob E, et al. The activity within the CA3 excitatory network during Theiler’s virus encephalitis is distinct from that observed during chronic epilepsy. J Neurovirol. 2012;18:30–44. doi: 10.1007/s13365-012-0082-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Mishra AM, Bai X, Sanganahalli BG, et al. Decreased resting functional connectivity after traumatic brain injury in the rat. PLoS One. 2014;9:e95280. doi: 10.1371/journal.pone.0095280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Shultz SR, Cardamone L, Liu YR, et al. Can structural or functional changes following traumatic brain injury in the rat predict epileptic outcome? Epilepsia. 2013;54:1240–50. doi: 10.1111/epi.12223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Kelly KM, Kharlamov A, Hentosz TM, et al. Photothrombotic brain infarction results in seizure activity in aging Fischer 344 and Sprague Dawley rats. Epilepsy Res. 2001;47:189–203. doi: 10.1016/s0920-1211(01)00294-7. [DOI] [PubMed] [Google Scholar]
- 98.Hartings JA, Williams AJ, Tortella FC. Occurrence of nonconvulsive seizures, periodic epileptiform discharges, and intermittent rhythmic delta activity in rat focal ischemia. Exp Neurol. 2003;179:139–49. doi: 10.1016/s0014-4886(02)00013-4. [DOI] [PubMed] [Google Scholar]
- 99.D’Ambrosio R, Fairbanks JP, Fender JS, et al. Post-traumatic epilepsy following fluid percussion injury in the rat. Brain. 2004;127:304–14. doi: 10.1093/brain/awh038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Liu G, Gu B, He X-P, et al. Transient inhibition of TrkB kinase after status epilepticus prevents development of temporal lobe epilepsy. Neuron. 2013;79:31–8. doi: 10.1016/j.neuron.2013.04.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Huusko N, Pitkänen A. Parvalbumin immunoreactivity and expression of GABAA receptor subunits in the thalamus after experimental TBI. Neuroscience. 2014;267:30–45. doi: 10.1016/j.neuroscience.2014.02.026. [DOI] [PubMed] [Google Scholar]
- 102.Rodgers KM, Dudek FE, Barth DS. Progressive, Seizure-Like, Spike-Wave Discharges Are Common in Both Injured and Uninjured Sprague-Dawley Rats: Implications for the Fluid Percussion Injury Model of Post-Traumatic Epilepsy. J Neurosci. 2015;35:9194–204. doi: 10.1523/JNEUROSCI.0919-15.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.D’Ambrosio R, Eastman CL, Miller JW. Inadequate experimental methods and erroneous epilepsy diagnostic criteria result in confounding acquired focal epilepsy with genetic absence epilepsy. :2015. [Google Scholar]
- 104.Iadarola MJ, Gale K. Substantia nigra: site of anticonvulsant activity mediated by gamma-aminobutyric acid. Science. 1982;218:1237–40. doi: 10.1126/science.7146907. [DOI] [PubMed] [Google Scholar]
- 105.Bröer S, Backofen-Wehrhahn B, Bankstahl M, et al. Vigabatrin for focal drug delivery in epilepsy: bilateral microinfusion into the subthalamic nucleus is more effective than intranigral or systemic administration in a rat seizure model. Neurobiol Dis. 2012;46:362–76. doi: 10.1016/j.nbd.2012.01.017. [DOI] [PubMed] [Google Scholar]
- 106.Krook-Magnuson E, Szabo GG, Armstrong C, et al. Cerebellar Directed Optogenetic Intervention Inhibits Spontaneous Hippocampal Seizures in a Mouse Model of Temporal Lobe Epilepsy. eNeuro. 2014 Dec;1(1):e.2014. doi: 10.1523/ENEURO.0005-14.2014. pii. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Soper C, Wicker E, Kulick CV, et al. Optogenetic activation of superior colliculus neurons suppresses seizures originating in diverse brain networks. Neurobiol Dis. 2016;87:102–15. doi: 10.1016/j.nbd.2015.12.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Marder E, Taylor AL. Multiple models to capture the variability in biological neurons and networks. Nat Neurosci. 2011;14:133–8. doi: 10.1038/nn.2735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Sloviter RS. Decreased hippocampal inhibition and a selective loss of interneurons in experimental epilepsy. Science. 1987;235:73–6. doi: 10.1126/science.2879352. [DOI] [PubMed] [Google Scholar]
- 110.de Lanerolle NC, Kim JH, Robbins RJ, et al. Hippocampal interneuron loss and plasticity in human temporal lobe epilepsy. Brain Res. 1989;495:387–95. doi: 10.1016/0006-8993(89)90234-5. [DOI] [PubMed] [Google Scholar]
- 111.Houser CR, Esclapez M. Vulnerability and plasticity of the GABA system in the pilocarpine model of spontaneous recurrent seizures. Epilepsy Res. 1996;26:207–18. doi: 10.1016/s0920-1211(96)00054-x. [DOI] [PubMed] [Google Scholar]
- 112.Tauck DL, Nadler JV. Evidence of functional mossy fiber sprouting in hippocampal formation of kainic acid-treated rats. J Neurosci. 1985;5:1016–22. doi: 10.1523/JNEUROSCI.05-04-01016.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Sutula T, Cascino G, Cavazos J, et al. Mossy fiber synaptic reorganization in the epileptic human temporal lobe. Ann Neurol. 1989;26:321–30. doi: 10.1002/ana.410260303. [DOI] [PubMed] [Google Scholar]
- 114.Ben-Ari Y, Represa A. Brief seizure episodes induce long-term potentiation and mossy fibre sprouting in the hippocampus. Trends Neurosci. 1990;13:312–8. doi: 10.1016/0166-2236(90)90135-w. [DOI] [PubMed] [Google Scholar]
- 115.Esclapez M, Hirsch JC, Ben-Ari Y, et al. Newly formed excitatory pathways provide a substrate for hyperexcitability in experimental temporal lobe epilepsy. J Comp Neurol. 1999;408:449–60. doi: 10.1002/(sici)1096-9861(19990614)408:4<449::aid-cne1>3.0.co;2-r. [DOI] [PubMed] [Google Scholar]
- 116.Tsunashima K, Schwarzer C, Kirchmair E, et al. GABA(A) receptor subunits in the rat hippocampus III: altered messenger RNA expression in kainic acid-induced epilepsy. Neuroscience. 1997;80:1019–32. doi: 10.1016/s0306-4522(97)00144-9. [DOI] [PubMed] [Google Scholar]
- 117.Brooks-Kayal AR, Shumate MD, Jin H, Rikhter TY, et al. Selective changes in single cell GABA(A) receptor subunit expression and function in temporal lobe epilepsy. Nat Med. 1998;4:1166–72. doi: 10.1038/2661. [DOI] [PubMed] [Google Scholar]
- 118.Loup F, Wieser HG, Yonekawa Y, et al. Selective alterations in GABAA receptor subtypes in human temporal lobe epilepsy. J Neurosci. 2000;20:5401–19. doi: 10.1523/JNEUROSCI.20-14-05401.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Hunt RF, Scheff SW, Smith BN. Regionally localized recurrent excitation in the dentate gyrus of a cortical contusion model of posttraumatic epilepsy. J Neurophysiol. 2010;103:1490–500. doi: 10.1152/jn.00957.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Cantu D, Walker K, Andresen L, et al. Traumatic Brain Injury Increases Cortical Glutamate Network Activity by Compromising GABAergic Control. Cereb Cortex. 2015;25:2306–20. doi: 10.1093/cercor/bhu041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Drexel M, Puhakka N, Kirchmair E, et al. Expression of GABA receptor subunits in the hippocampus and thalamus after experimental traumatic brain injury. Neuropharmacology. 2015;88:122–133. doi: 10.1016/j.neuropharm.2014.08.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Williamson A, Patrylo PR, Spencer DD. Decrease in inhibition in dentate granule cells from patients with medial temporal lobe epilepsy. Ann Neurol. 1999;45:92–9. doi: 10.1002/1531-8249(199901)45:1<92::aid-art15>3.0.co;2-n. [DOI] [PubMed] [Google Scholar]
- 123.Cossart R, Dinocourt C, Hirsch JC, et al. Dendritic but not somatic GABAergic inhibition is decreased in experimental epilepsy. Nat Neurosci. 2001;4:52–62. doi: 10.1038/82900. [DOI] [PubMed] [Google Scholar]
- 124.Hirsch JC, Agassandian C, Merchán-Pérez A, Ben-Ari Y, DeFelipe J, Esclapez M, et al. Deficit of quantal release of GABA in experimental models of temporal lobe epilepsy. Nat Neurosci. 1999;2:499–500. doi: 10.1038/9142. [DOI] [PubMed] [Google Scholar]
- 125.Scimemi A, Semyanov A, Sperk G, et al. Multiple and plastic receptors mediate tonic GABAA receptor currents in the hippocampus. J Neurosci. 2005;25:10016–24. doi: 10.1523/JNEUROSCI.2520-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Scimemi A, Andersson A, Heeroma JH, et al. Tonic GABA(A) receptor-mediated currents in human brain. Eur J Neurosci. 2006;24:1157–60. doi: 10.1111/j.1460-9568.2006.04989.x. [DOI] [PubMed] [Google Scholar]
- 127.Clarkson AN, Huang BS, Macisaac SE, et al. Reducing excessive GABA-mediated tonic inhibition promotes functional recovery after stroke. Nature. 2010;468:305–9. doi: 10.1038/nature09511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Pavlov I, Huusko N, Drexel M, et al. Progressive loss of phasic, but not tonic, GABAA receptor-mediated inhibition in dentate granule cells in a model of post-traumatic epilepsy in rats. Neuroscience. 2011;194:208–19. doi: 10.1016/j.neuroscience.2011.07.074. [DOI] [PubMed] [Google Scholar]
- 129.Pavlov I, Savtchenko LP, Kullmann DM, et al. Outwardly rectifying tonically active GABAA receptors in pyramidal cells modulate neuronal offset, not gain. J Neurosci. 2009;29:15341–50. doi: 10.1523/JNEUROSCI.2747-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Alexander A, Maroso M, Soltesz I. Organization and control of epileptic circuits in temporal lobe epilepsy. Prog Brain Res. 2016;226:127–54. doi: 10.1016/bs.pbr.2016.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Cohen I, Navarro V, Clemenceau S, et al. On the origin of interictal activity in human temporal lobe epilepsy in vitro. Science. 2002;298:1418–21. doi: 10.1126/science.1076510. [DOI] [PubMed] [Google Scholar]
- 132.Huberfeld G, Wittner L, Clemenceau S, et al. Perturbed chloride homeostasis and GABAergic signaling in human temporal lobe epilepsy. J Neurosci. 2007;27:9866–73. doi: 10.1523/JNEUROSCI.2761-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Pathak HR, Weissinger F, Terunuma M, et al. Disrupted dentate granule cell chloride regulation enhances synaptic excitability during development of temporal lobe epilepsy. J Neurosci. 2007 Dec 19;27(51):14012–22. doi: 10.1523/JNEUROSCI.4390-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Muldoon SF, Villette V, Tressard T, et al. GABAergic inhibition shapes interictal dynamics in awake epileptic mice. Brain. 2015;138:2875–90. doi: 10.1093/brain/awv227. [DOI] [PubMed] [Google Scholar]
- 135.Ellender TJ, Raimondo JV, Irkle A, et al. Excitatory effects of parvalbumin-expressing interneurons maintain hippocampal epileptiform activity via synchronous afterdischarges. J Neurosci. 2014;34:15208–22. doi: 10.1523/JNEUROSCI.1747-14.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Sessolo M, Marcon I, Bovetti S, et al. Parvalbumin-Positive Inhibitory Interneurons Oppose Propagation But Favor Generation of Focal Epileptiform Activity. J Neurosci. 2015;35:9544–57. doi: 10.1523/JNEUROSCI.5117-14.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Isokawa M. Preservation of dendrites with the presence of reorganized mossy fiber collaterals in hippocampal dentate granule cells in patients with temporal lobe epilepsy. Brain Res. 1997;744:339–43. doi: 10.1016/S0006-8993(96)01067-0. [DOI] [PubMed] [Google Scholar]
- 138.Avoli M, Olivier A. Bursting in human epileptogenic neocortex is depressed by an N-methyl-D-aspartate antagonist. Neurosci Lett. 1987;76:249–54. doi: 10.1016/0304-3940(87)90724-5. [DOI] [PubMed] [Google Scholar]
- 139.Isokawa M, Levesque MF. Increased NMDA responses and dendritic degeneration in human epileptic hippocampal neurons in slices. Neurosci Lett. 1991;132:212–6. doi: 10.1016/0304-3940(91)90304-c. [DOI] [PubMed] [Google Scholar]
- 140.Turner DA, Wheal HV. Excitatory synaptic potentials in kainic acid-denervated rat CA1 pyramidal neurons. J Neurosci. 1991;11:2786–94. doi: 10.1523/JNEUROSCI.11-09-02786.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Bernard C, Anderson A, Becker A, et al. Acquired dendritic channelopathy in temporal lobe epilepsy. Science. 2004;305:532–5. doi: 10.1126/science.1097065. [DOI] [PubMed] [Google Scholar]
- 142.Beck H, Yaari Y. Plasticity of intrinsic neuronal properties in CNS disorders. Nat Rev Neurosci. 2008;9:357–69. doi: 10.1038/nrn2371. [DOI] [PubMed] [Google Scholar]
- 143.Wykes RC, Heeroma JH, Mantoan L, et al. Optogenetic and potassium channel gene therapy in a rodent model of focal neocortical epilepsy. Sci Transl Med. 2012;4:161ra152. doi: 10.1126/scitranslmed.3004190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Jirsa VK, Stacey WC, Quilichini PP, et al. On the nature of seizure dynamics. Brain. 2014;137:2210–30. doi: 10.1093/brain/awu133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Proix T, Bartolomei F, Chauvel P, et al. Permittivity coupling across brain regions determines seizure recruitment in partial epilepsy. J Neurosci. 2014;34:15009–21. doi: 10.1523/JNEUROSCI.1570-14.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Galanopoulou AS, Pitkänen A, Buckmaster PS, et al. Animal Models of Seizures and Eplilepsy. Amsterdam: Elsevier; 2017. What should models model? What needs to be modeled? pp. 1107–1119. [Google Scholar]
- 147.Margerison JH, Corsellis JA. Epilepsy and the temporal lobes. A clinical, electroencephalographic and neuropathological study of the brain in epilepsy, with particular reference to the temporal lobes. Brain. 1966;89:499–530. doi: 10.1093/brain/89.3.499. [DOI] [PubMed] [Google Scholar]
- 148.Bruton CJ, Stevens JR, Frith CD. Epilepsy, psychosis, and schizophrenia: clinical and neuropathologic correlations. Neurology. 1994;44:34–42. doi: 10.1212/wnl.44.1.34. [DOI] [PubMed] [Google Scholar]
- 149.Karhunen H, Jolkkonen J, Sivenius J, et al. Epileptogenesis after experimental focal cerebral ischemia. Neurochem Res. 2005;30:1529–42. doi: 10.1007/s11064-005-8831-y. [DOI] [PubMed] [Google Scholar]
- 150.Engel J. Introduction to temporal lobe epilepsy. Epilepsy Res. 1996;26:141–50. doi: 10.1016/s0920-1211(96)00043-5. [DOI] [PubMed] [Google Scholar]
- 151.Borelli P, Shorvon SD, Stevens JM, et al. Extratemporal ictal clinical features in hippocampal sclerosis: their relationship to the degree of hippocampal volume loss and to the outcome of temporal lobectomy. Epilepsia. 2008;49:1333–9. doi: 10.1111/j.1528-1167.2008.01694.x. [DOI] [PubMed] [Google Scholar]
- 152.Sloviter RS. Permanently altered hippocampal structure, excitability, and inhibition after experimental status epilepticus in the rat: the ‘dormant basket cell’ hypothesis and its possible relevance to temporal lobe epilepsy. Hippocampus. 1991;1:41–66. doi: 10.1002/hipo.450010106. [DOI] [PubMed] [Google Scholar]
- 153.Scharfman HE. Epileptogenesis in the parahippocampal region. Parallels with the dentate gyrus. Ann N Y Acad Sci. 2000;911:305–27. doi: 10.1111/j.1749-6632.2000.tb06734.x. [DOI] [PubMed] [Google Scholar]
- 154.Swartz BE, Houser CR, Tomiyasu U, et al. Hippocampal cell loss in posttraumatic human epilepsy. Epilepsia. 2006;47:1373–82. doi: 10.1111/j.1528-1167.2006.00602.x. [DOI] [PubMed] [Google Scholar]
- 155.Pitkänen A, McIntosh TK. Animal models of post-traumatic epilepsy. J Neurotrauma. 2006;23:241–61. doi: 10.1089/neu.2006.23.241. [DOI] [PubMed] [Google Scholar]
- 156.Huusko N, Römer C, Ndode-Ekane XE, et al. Loss of hippocampal interneurons and epileptogenesis: a comparison of two animal models of acquired epilepsy. Brain Struct Funct. 2013;220:153–91. doi: 10.1007/s00429-013-0644-1. [DOI] [PubMed] [Google Scholar]
- 157.Diaz-Arrastia R, Agostini MA, Madden CJ, et al. Posttraumatic epilepsy: the endophenotypes of a human model of epileptogenesis. Epilepsia. 2009;50(Suppl 2):14–20. doi: 10.1111/j.1528-1167.2008.02006.x. [DOI] [PubMed] [Google Scholar]
- 158.Brezova V, Moen KG, Skandsen T, et al. Prospective longitudinal MRI study of brain volumes and diffusion changes during the first year after moderate to severe traumatic brain injury. NeuroImage Clin. 2014;5:128–40. doi: 10.1016/j.nicl.2014.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Zappone CA, Sloviter RS. Translamellar disinhibition in the rat hippocampal dentate gyrus after seizure-induced degeneration of vulnerable hilar neurons. J Neurosci. 2004;24:853–64. doi: 10.1523/JNEUROSCI.1619-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Jinde S, Zsiros V, Jiang Z, et al. Hilar mossy cell degeneration causes transient dentate granule cell hyperexcitability and impaired pattern separation. Neuron. 2012;76:1189–200. doi: 10.1016/j.neuron.2012.10.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Winson J, Abzug C. Gating of neuronal transmission in the hippocampus: efficacy of transmission varies with behavioral state. Science. 1977;196:1223–5. doi: 10.1126/science.193192. [DOI] [PubMed] [Google Scholar]
- 162.Winson J, Abzug C. Neuronal transmission through hippocampal pathways dependent on behavior. J Neurophysiol. 1978;41:716–32. doi: 10.1152/jn.1978.41.3.716. [DOI] [PubMed] [Google Scholar]
- 163.Heinemann U, Beck H, Dreier JP, et al. The dentate gyrus as a regulated gate for the propagation of epileptiform activity. Epilepsy Res Suppl. 1992;7:273. [PubMed] [Google Scholar]
- 164.Lothman EW, Stringer JL, Bertram EH. The dentate gyrus as a control point for seizures in the hippocampus and beyond. Epilepsy Res Suppl. 1992;7:301–13. [PubMed] [Google Scholar]
- 165.Sloviter RS. The functional organization of the hippocampal dentate gyrus and its relevance to the pathogenesis of temporal lobe epilepsy. Ann Neurol. 1994;35:640–54. doi: 10.1002/ana.410350604. [DOI] [PubMed] [Google Scholar]
- 166.Lowenstein DH, Thomas MJ, Smith DH, et al. Selective vulnerability of dentate hilar neurons following traumatic brain injury: a potential mechanistic link between head trauma and disorders of the hippocampus. J Neurosci. 1992;12:4846–53. doi: 10.1523/JNEUROSCI.12-12-04846.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Sloviter RS, Zappone CA, Harvey BD, et al. ‘Dormant basket cell’ hypothesis revisited: relative vulnerabilities of dentate gyrus mossy cells and inhibitory interneurons after hippocampal status epilepticus in the rat. J Comp Neurol. 2003;459:44–76. doi: 10.1002/cne.10630. [DOI] [PubMed] [Google Scholar]
- 168.Du F, Whetsell WO, Abou-Khalil B, et al. Preferential neuronal loss in layer III of the entorhinal cortex in patients with temporal lobe epilepsy. Epilepsy Res. 1993;16:223–33. doi: 10.1016/0920-1211(93)90083-j. [DOI] [PubMed] [Google Scholar]
- 169.Pitkänen A, Engel J. Past and Present Definitions of Epileptogenesis and Its Biomarkers Neurotherapeutics. 2014;11:231–241. doi: 10.1007/s13311-014-0257-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Wasterlain CG, Mazarati AM, Shirasaka Y, et al. Seizure-induced hippocampal damage and chronic epilepsy: a hebbian theory of epileptogenesis. Adv Neurol. 1999;79:829–43. [PubMed] [Google Scholar]
- 171.Dingledine R, Varvel NH, Dudek FE. When and how do seizures kill neurons, and is cell death relevant to epileptogenesis? Adv Exp Med Biol. 2014;813:109–22. doi: 10.1007/978-94-017-8914-1_9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Dingledine R, Coulter DA, Fritsch B, et al. Transcriptional profile of hippocampal dentate granule cells in four rat epilepsy models. Scientific Data. 2017;4:170061. doi: 10.1038/sdata.2017.61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Kahane P, Barba C, Rheims S, et al. The concept of temporal ‘plus’ epilepsy. Rev Neurol (Paris) 2015;171:267–72. doi: 10.1016/j.neurol.2015.01.562. [DOI] [PubMed] [Google Scholar]
- 174.Amaral D, Lavenex P. The Hippocampus Book. New York: Oxford University Press; 2007. Hippocampal neuroanatomy; p. 872. [Google Scholar]
- 175.Babb TL, Halgren E, Wilson C, et al. Neuronal firing patterns during the spread of an occipital lobe seizure to the temporal lobes in man. Electroencephalogr Clin Neurophysiol. 1981;51:104–7. doi: 10.1016/0013-4694(81)91513-3. [DOI] [PubMed] [Google Scholar]
- 176.Shihabuddin B, Abou-Khalil B, Delbeke D, et al. Orbito-frontal epilepsy masquerading as temporal lobe epilepsy-a case report. Seizure. 2001;10:134–8. doi: 10.1053/seiz.2000.0477. [DOI] [PubMed] [Google Scholar]
- 177.Devinsky O, Vezzani A, Najjar S, et al. Glia and epilepsy: excitability and inflammation. Trends Neurosci. 2013;36:174–84. doi: 10.1016/j.tins.2012.11.008. [DOI] [PubMed] [Google Scholar]
- 178.Seifert G, Steinhäuser C. Neuron-astrocyte signaling and epilepsy. Exp Neurol. 2013;244:4–10. doi: 10.1016/j.expneurol.2011.08.024. [DOI] [PubMed] [Google Scholar]
- 179.Vezzani A, Aronica E, Mazarati A, et al. Epilepsy and brain inflammation. Exp Neurol. 2013a;244:11–21. doi: 10.1016/j.expneurol.2011.09.033. [DOI] [PubMed] [Google Scholar]
- 180.Boison D, Aronica E. Comorbidities in Neurology: Is adenosine the common link? Neuropharmacology. 2015;97:18–34. doi: 10.1016/j.neuropharm.2015.04.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Farina C, Aloisi F, Meinl E. Astrocytes are active players in cerebral innate immunity. Trends Immunol. 2007;28:138–45. doi: 10.1016/j.it.2007.01.005. [DOI] [PubMed] [Google Scholar]
- 182.Vezzani A, Ravizza T, Balosso S, et al. Glia as a source of cytokines: implications for neuronal excitability and survival. Epilepsia. 2008;49(Suppl 2):24–32. doi: 10.1111/j.1528-1167.2008.01490.x. [DOI] [PubMed] [Google Scholar]
- 183.Wetherington J, Serrano G, Dingledine R. Astrocytes in the epileptic brain. Neuron. 2008;58:168–78. doi: 10.1016/j.neuron.2008.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Aronica E, Ravizza T, Zurolo E, et al. Astrocyte immune responses in epilepsy. Glia. 2012;60:1258–68. doi: 10.1002/glia.22312. [DOI] [PubMed] [Google Scholar]
- 185.Bedner P, Dupper A, Hüttmann K, et al. Astrocyte uncoupling as a cause of human temporal lobe epilepsy. Brain. 2015;138:1208–22. doi: 10.1093/brain/awv067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Khan D, Dupper A, Deshpande T, et al. Experimental febrile seizures impair interastrocytic gap junction coupling in juvenile mice. J Neurosci Res. 2016;94:804–13. doi: 10.1002/jnr.23726. [DOI] [PubMed] [Google Scholar]
- 187.Seifert G, Schilling K, Steinhäuser C. Astrocyte dysfunction in neurological disorders: a molecular perspective. Nat Rev Neurosci. 2006;7:194–206. doi: 10.1038/nrn1870. [DOI] [PubMed] [Google Scholar]
- 188.Seifert G, Carmignoto G, Steinhäuser C. Astrocyte dysfunction in epilepsy. Brain Res Rev. 2010;63:212–21. doi: 10.1016/j.brainresrev.2009.10.004. [DOI] [PubMed] [Google Scholar]
- 189.Steinhäuser C, Seifert G, Bedner P. Astrocyte dysfunction in temporal lobe epilepsy: K+ channels and gap junction coupling. Glia. 2012;60:1192–202. doi: 10.1002/glia.22313. [DOI] [PubMed] [Google Scholar]
- 190.D’Ambrosio R, Maris DO, Grady MS, et al. Impaired K(+) homeostasis and altered electrophysiological properties of post-traumatic hippocampal glia. J Neurosci. 1999;19:8152–62. doi: 10.1523/JNEUROSCI.19-18-08152.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Köller H, Schroeter M, Jander S, et al. Time course of inwardly rectifying K(+) current reduction in glial cells surrounding ischemic brain lesions. Brain Res. 2000;872:194–8. doi: 10.1016/s0006-8993(00)02434-3. [DOI] [PubMed] [Google Scholar]
- 192.David Y, Cacheaux LP, Ivens S, et al. Astrocytic dysfunction in epileptogenesis: consequence of altered potassium and glutamate homeostasis? J Neurosci. 2009;29:10588–99. doi: 10.1523/JNEUROSCI.2323-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Schröder W, Hager G, Kouprijanova E, et al. Lesion-induced changes of electrophysiological properties in astrocytes of the rat dentate gyrus. Glia. 1999;28:166–74. [PubMed] [Google Scholar]
- 194.Bordey A, Hablitz JJ, Sontheimer H. Reactive astrocytes show enhanced inwardly rectifying K+ currents in situ. Neuroreport. 2000;11:3151–5. doi: 10.1097/00001756-200009280-00022. [DOI] [PubMed] [Google Scholar]
- 195.Bordey A, Lyons SA, Hablitz JJ, et al. Electrophysiological characteristics of reactive astrocytes in experimental cortical dysplasia. J Neurophysiol. 2001;85:1719–31. doi: 10.1152/jn.2001.85.4.1719. [DOI] [PubMed] [Google Scholar]
- 196.Hinterkeuser S, Schröder W, Hager G, et al. Astrocytes in the hippocampus of patients with temporal lobe epilepsy display changes in potassium conductances. Eur J Neurosci. 2000;12:2087–96. doi: 10.1046/j.1460-9568.2000.00104.x. [DOI] [PubMed] [Google Scholar]
- 197.Cotrina ML, Kang J, Lin JH, et al. Astrocytic gap junctions remain open during ischemic conditions. J Neurosci. 1998;18:2520–37. doi: 10.1523/JNEUROSCI.18-07-02520.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Ohsumi A, Nawashiro H, Otani N, et al. Temporal and spatial profile of phosphorylated connexin43 after traumatic brain injury in rats. J Neurotrauma. 2010;27:1255–63. doi: 10.1089/neu.2009.1234. [DOI] [PubMed] [Google Scholar]
- 199.Bar-Klein G, Cacheaux LP, Kamintsky L, et al. Losartan prevents acquired epilepsy via TGF-β signaling suppression. Ann Neurol. 2014;75:864–75. doi: 10.1002/ana.24147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Michel MC, Foster C, Brunner HR, et al. A systematic comparison of the properties of clinically used angiotensin II type 1 receptor antagonists. Pharmacol Rev. 2013;65:809–48. doi: 10.1124/pr.112.007278. [DOI] [PubMed] [Google Scholar]
- 201.Boison D. Adenosine kinase: exploitation for therapeutic gain. Pharmacol Rev. 2013;65:906–43. doi: 10.1124/pr.112.006361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Gouder N, Scheurer L, Fritschy J-M, et al. Overexpression of adenosine kinase in epileptic hippocampus contributes to epileptogenesis. J Neurosci. 2004;24:692–701. doi: 10.1523/JNEUROSCI.4781-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Li T, Ren G, Lusardi T, et al. Adenosine kinase is a target for the prediction and prevention of epileptogenesis in mice. J Clin Invest. 2008;118:571–82. doi: 10.1172/JCI33737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Pignataro G, Maysami S, Studer FE, et al. Downregulation of hippocampal adenosine kinase after focal ischemia as potential endogenous neuroprotective mechanism. J Cereb Blood Flow Metab. 2008;28:17–23. doi: 10.1038/sj.jcbfm.9600499. [DOI] [PubMed] [Google Scholar]
- 205.Aronica E, Zurolo E, Iyer A, et al. Upregulation of adenosine kinase in astrocytes in experimental and human temporal lobe epilepsy. Epilepsia. 2011;52:1645–55. doi: 10.1111/j.1528-1167.2011.03115.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Li T, Lytle N, Lan J-Q, et al. Local disruption of glial adenosine homeostasis in mice associates with focal electrographic seizures: a first step in epileptogenesis? Glia. 2012;60:83–95. doi: 10.1002/glia.21250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Lusardi TA, Lytle NK, Szybala C, et al. Caffeine prevents acute mortality after TBI in rats without increased morbidity. Exp Neurol. 2012;234:161–8. doi: 10.1016/j.expneurol.2011.12.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Masino SA, Li T, Theofilas P. A ketogenic diet suppresses seizures in mice through adenosine A₁ receptors. J Clin Invest. 2011;121:2679–83. doi: 10.1172/JCI57813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.de Groot M, Iyer A, Zurolo E, et al. Overexpression of ADK in human astrocytic tumors and peritumoral tissue is related to tumor-associated epilepsy. Epilepsia. 2012;53:58–66. doi: 10.1111/j.1528-1167.2011.03306.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Aronica E, Sandau US, Iyer A, et al. Glial adenosine kinase–a neuropathological marker of the epileptic brain. Neurochem Int. 2013;63:688–95. doi: 10.1016/j.neuint.2013.01.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Li T, Quan Lan J, Fredholm BB, et al. Adenosine dysfunction in astrogliosis: cause for seizure generation? Neuron Glia Biol. 2007;3:353–66. doi: 10.1017/S1740925X0800015X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Boison D, Huber A, Padrun V, et al. Seizure suppression by adenosine-releasing cells is independent of seizure frequency. Epilepsia. 2002;43:788–96. doi: 10.1046/j.1528-1157.2002.33001.x. [DOI] [PubMed] [Google Scholar]
- 213.Fedele DE, Koch P, Scheurer L, et al. Engineering embryonic stem cell derived glia for adenosine delivery. Neurosci Lett. 2004;370:160–5. doi: 10.1016/j.neulet.2004.08.031. [DOI] [PubMed] [Google Scholar]
- 214.Lusardi TA, Akula KK, Coffman SQ, et al. Ketogenic diet prevents epileptogenesis and disease progression in adult mice and rats. Neuropharmacology. 2015;99:500–9. doi: 10.1016/j.neuropharm.2015.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Eid T, Lee TS, Thomas MJ, et al. Loss of perivascular aquaporin 4 may underlie deficient water and K+ homeostasis in the humanepileptogenic hippocampus. Proc Natl Acad Sci U S A. 2005;102:1193–1198. doi: 10.1073/pnas.0409308102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Strohschein S, Hüttmann K, Gabriel S, et al. Impact of aquaporin-4 channels on K+ buffering and gap junction coupling in the hippocampus. Glia. 2011;59:973–980. doi: 10.1002/glia.21169. [DOI] [PubMed] [Google Scholar]
- 217.Binder DK, Nagelhus EA, Ottersen OP. Aquaporin-4 and epilepsy. Glia. 2012;60:1203–14. doi: 10.1002/glia.22317. [DOI] [PubMed] [Google Scholar]
- 218.Hubbard JA, Szu JI, Yonan JM, et al. Regulation of astrocyte glutamate transporter-1 (GLT1) and aquaporin-4 (AQP4) expression in a model of epilepsy. Exp Neurol. 2016;283(Pt A):85–96. doi: 10.1016/j.expneurol.2016.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Filiou MD, Arefin AS, Moscato P, et al. ‘Neuroinflammation’ differs categorically from inflammation: transcriptomes of Alzheimer’s disease, Parkinson’s disease, schizophrenia and inflammatory diseases compared. Neurogenetics. 2014;15:201–212. doi: 10.1007/s10048-014-0409-x. [DOI] [PubMed] [Google Scholar]
- 220.Kim JY, Kim N, Yenari MA. Mechanisms and potential therapeutic applications of microglial activation after brain injury. CNS Neurosci Ther. 2015;21:309–19. doi: 10.1111/cns.12360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Fabene PF, Bramanti P, Constantin G. The emerging role for chemokines in epilepsy. J Neuroimmunol. 2010;224:22–7. doi: 10.1016/j.jneuroim.2010.05.016. [DOI] [PubMed] [Google Scholar]
- 222.Friedman A, Bar-Klein G, Serlin Y, et al. Should losartan be administered following brain injury? Expert Rev Neurother. 2014;14:1365–75. doi: 10.1586/14737175.2014.972945. [DOI] [PubMed] [Google Scholar]
- 223.Vezzani A, Viviani B. Neuromodulatory properties of inflammatory cytokines and their impact on neuronal excitability. Neuropharmacology. 2015;96:70–82. doi: 10.1016/j.neuropharm.2014.10.027. [DOI] [PubMed] [Google Scholar]
- 224.Aronica E, Crino P. Inflammation in Epilepsy: Clinical Observations. Epilepsia. 2011 May;52(Suppl 3):26–32. doi: 10.1111/j.1528-1167.2011.03033.x. [DOI] [PubMed] [Google Scholar]
- 225.Dedeurwaerdere S1, Friedman A, Fabene PF, et al. Finding a better drug for epilepsy: antiinflammatory targets. Epilepsia. 2012 Jul;53(7):1113–8. doi: 10.1111/j.1528-1167.2012.03520.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.Vezzani A, Fujinami RS, White HS, et al. Infections, inflammation and epilepsy. Acta Neuropathol. 2016;131:211–34. doi: 10.1007/s00401-015-1481-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227.Wraith DC, Nicholson LB. The adaptive immune system in diseases of the central nervous system. J Clin Inv. 2012;122:1172–1179. doi: 10.1172/JCI58648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228.Chiu CC, Liao YE, Yang LY, et al. Neuroinflammation in animal models of traumatic brain injury. J Neurosci Methods. 2016;272:38–49. doi: 10.1016/j.jneumeth.2016.06.018. 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229.Kim JY, Park JP, Chang JY, et al. Inflammation after Ischemic Stroke: The Role of Leukocytes and Glial Cells. Exp Neurobiol. 2016;25:241–251. doi: 10.5607/en.2016.25.5.241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230.Iori V, Maroso M, Rizzi M, et al. Receptor for Advanced Glycation Endproducts is upregulated in temporal lobe epilepsy and contributes to experimental seizures. Neurobiol Dis. 2013;58:102–14. doi: 10.1016/j.nbd.2013.03.006. [DOI] [PubMed] [Google Scholar]
- 231.Iori V, Iyer A, Ravizza T, et al. Blockade of the IL-1R1/TLR4 pathway mediates disease-modification therapeutic effects in a model of acquired epilepsy. Neurobiol Dis. 2017;99:12–23. doi: 10.1016/j.nbd.2016.12.007. [DOI] [PubMed] [Google Scholar]
- 232.Walker LE, Frigerio F, Ravizza T, et al. Molecular isoforms of HMGB1 are novel mechanistic biomarkers for epilepsy. J Clin Invest. 2017;127:2118–2132. doi: 10.1172/JCI92001. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 233.Libbey JE, Kennett NJ, Wilcox KS, et al. Interleukin-6, Produced by Resident Cells of the Central Nervous System and Infiltrating Cells, Contributes to the Development of Seizures following Viral Infection. J Virol. 2011;85:6913–6922. doi: 10.1128/JVI.00458-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234.Kwon YS, Pineda E, Auvin S, et al. Neuroprotective and antiepileptogenic effects of combination of anti-inflammatory drugs in the immature brain. J Neuroinflammation. 2013;10:30. doi: 10.1186/1742-2094-10-30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235.Dey A, Kang X, Qiu J, et al. Anti-Inflammatory Small Molecules To Treat Seizures and Epilepsy: From Bench to Bedside. Trends Pharmacol Sci. 2016;37:463–84. doi: 10.1016/j.tips.2016.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236.Hunt RF, Boychuk JA, Smith BN. Neural circuit mechanisms of post-traumatic epilepsy. Front Cell Neurosci. 2013;7:89. doi: 10.3389/fncel.2013.00089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237.Zelano J. Poststroke epilepsy: update and future directions. Ther Adv Neurol Disord. 2016;9:424–35. doi: 10.1177/1756285616654423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238.Russo MV, McGavern DB. Inflammatory neuroprotection following traumatic brain injury. Science. 2016;353:783–5. doi: 10.1126/science.aaf6260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239.Sordillo PP, Sordillo LA, Helson L. Bifunctional role of pro-inflammatory cytokines after traumatic brain injury. Brain Inj. 2016;30:1043–53. doi: 10.3109/02699052.2016.1163618. [DOI] [PubMed] [Google Scholar]
- 240.Febinger HY, Thomasy HE, Pavlova MN, et al. Time-dependent effects of CX3CR1 in a mouse model of mild traumatic brain injury. J Neuroinflamm. 2015;12:154. doi: 10.1186/s12974-015-0386-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241.Scherbel U, Raghupathi R, Nakamura M, et al. Differential acute and chronic responses of tumor necrosis factor-deficient mice to experimental brain injury. Proc Natl Acad Sci U S A. 1999;96:8721–6. doi: 10.1073/pnas.96.15.8721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 242.Zanier ER, Marchesi F, Ortolano F, et al. Fractalkine Receptor Deficiency Is Associated with Early Protection but Late Worsening of Outcome following Brain Trauma in Mice. J Neurotrauma. 2016;33:1060–72. doi: 10.1089/neu.2015.4041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 243.Chamorro Á, Meisel A, Planas AM, et al. The immunology of acute stroke. Nat Rev Neurol. 2012;8:401–10. doi: 10.1038/nrneurol.2012.98. [DOI] [PubMed] [Google Scholar]
- 244.Clausen BH, Degn M, Sivasaravanaparan M, et al. Conditional ablation of myeloid TNF increases lesion volume after experimental stroke in mice, possibly via altered ERK1/2 signaling. Sci Rep. 2016;6:29291. doi: 10.1038/srep29291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 245.Mracsko E, Veltkamp R. Neuroinflammation after intracerebral hemorrhage. Front Cell Neurosci. 2014;8:388. doi: 10.3389/fncel.2014.00388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 246.Parker TM, Nguyen AH, Rabang JR, et al. The danger zone: Systematic review of the role of HMGB1 danger signalling in traumatic brain injury. Brain Inj. 2017;31:2–8. doi: 10.1080/02699052.2016.1217045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 247.Pauletti A, Terrone G, Ahmad-hekh T, et al. Targeting oxidative stress improves disease outcomes in a rat model of acquired epilepsy. Brain. 2017;140:1885–1899. doi: 10.1093/brain/awx117. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 248.Zhao J, Wang Y, Xu C, et al. Therapeutic potential of an anti-high mobility group box-1 monoclonal antibody in epilepsy. Brain Behav Immun. 2017 Feb 3; doi: 10.1016/j.bbi.2017.02.002. pii: S0889-1591(17)30024-7. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- 249.Carty M, Reinert L, Paludan SR, et al. Innate antiviral signalling in the central nervous system. Trends Immunol. 2014;35:79–87. doi: 10.1016/j.it.2013.10.012. [DOI] [PubMed] [Google Scholar]
- 250.Lund IV, Hu Y, Raol YH, et al. BDNF selectively regulates GABAA receptor transcription by activation of the JAK/STAT pathway. Sci Signal. 2008;1(41):ra9. doi: 10.1126/scisignal.1162396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 251.Oliva AA, Kang Y, Sanchez-Molano J, et al. STAT3 signaling after traumatic brain injury. J Neurochem. 2012;120:710–20. doi: 10.1111/j.1471-4159.2011.07610.x. [DOI] [PubMed] [Google Scholar]
- 252.Raible DJ, Frey LC, Cruz Del Angel Y, et al. GABA(A) receptor regulation after experimental traumatic brain injury. J Neurotrauma. 2012;29:2548–54. doi: 10.1089/neu.2012.2483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 253.Satriotomo I, Bowen KK, Vemuganti R. JAK2 and STAT3 activation contributes to neuronal damage following transient focal cerebral ischemia. J Neurochem. 2006;98:1353–68. doi: 10.1111/j.1471-4159.2006.04051.x. [DOI] [PubMed] [Google Scholar]
- 254.Suzuki S, Tanaka K, Nogawa S, et al. Phosphorylation of signal transducer and activator of transcription-3 (Stat3) after focal cerebral ischemia in rats. Exp Neurol. 2001;170:63–71. doi: 10.1006/exnr.2001.7701. [DOI] [PubMed] [Google Scholar]
- 255.Zhao J, Zhang Y, Li G, et al. Activation of JAK2/STAT pathway in cerebral cortex after experimental traumatic brain injury of rats. Neurosci Lett. 2011;498:147–52. doi: 10.1016/j.neulet.2011.05.001. [DOI] [PubMed] [Google Scholar]
- 256.Grabenstatter HL, Cruz Del Angel Y, Carlsen J, et al. The effect of STAT3 inhibition on status epilepticus and subsequent spontaneous seizures in the pilocarpine model of acquired epilepsy. Neurobiol Dis. 2014;62:73–85. doi: 10.1016/j.nbd.2013.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 257.Raible DJ, Frey LC, Cruz Del Angel Y, et al. JAK/STAT pathway regulation of GABAA receptor expression after differing severities of experimental TBI. Exp Neurol. 2015;271:445–56. doi: 10.1016/j.expneurol.2015.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 258.Brewster AL, Lugo JN, Patil VV, et al. Rapamycin reverses status epilepticus-induced memory deficits and dendritic damage. PLoS One. 2013;8:e57808. doi: 10.1371/journal.pone.0057808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 259.Guo D, Zeng L, Brody DL, Wong M. Rapamycin attenuates the development of posttraumatic epilepsy in a mouse model of traumatic brain injury. PLoS One. 2013;8:e64078. doi: 10.1371/journal.pone.0064078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 260.Zeng L-H, Rensing NR, Wong M. The mammalian target of rapamycin signaling pathway mediates epileptogenesis in a model of temporal lobe epilepsy. J Neurosci. 2009;29:6964–72. doi: 10.1523/JNEUROSCI.0066-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 261.Galanopoulou AS, Gorter JA, Cepeda C. Finding a better drug for epilepsy: the mTOR pathway as an antiepileptogenic target. Epilepsia. 2012;53:1119–30. doi: 10.1111/j.1528-1167.2012.03506.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 262.Huang X, Zhang H, Yang J, et al. Pharmacological inhibition of the mammalian target of rapamycin pathway suppresses acquired epilepsy. Neurobiol Dis. 2010;40(1):193–9. doi: 10.1016/j.nbd.2010.05.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 263.Butler CR, Boychuk JA, Smith BN. Effects of Rapamycin Treatment on Neurogenesis and Synaptic Reorganization in the Dentate Gyrus after Controlled Cortical Impact Injury in Mice. Front Syst Neurosci. 2015;9 doi: 10.3389/fnsys.2015.00163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 264.Drion CM, Borm LE, Kooijman L, et al. Effects of rapamycin and curcumin treatment on the development of epilepsy after electrically induced status epilepticus in rats. Epilepsia. 2016;57:688–697. doi: 10.1111/epi.13345. [DOI] [PubMed] [Google Scholar]
- 265.McDaniel SS, Wong M. Therapeutic role of mammalian target of rapamycin (mTOR) inhibition in preventing epileptogenesis. Neurosci Lett. 2011;497:231–239. doi: 10.1016/j.neulet.2011.02.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 266.Raffo E, Coppola A, Ono T, et al. A pulse rapamycin therapy for infantile spasms and associated cognitive decline. Neurobiol Dis. 2011;43:322–9. doi: 10.1016/j.nbd.2011.03.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 267.Rensing N, Han L, Wong M. Intermittent dosing of rapamycin maintains antiepileptogenic effects in a mouse model of tuberous sclerosis complex. Epilepsia. 2015;56:1088–1097. doi: 10.1111/epi.13031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 268.Boulle F, Kenis G, Cazorla M, et al. TrkB inhibition as a therapeutic target for CNS-related disorders. Prog Neurobiol. 2012;98:197–206. doi: 10.1016/j.pneurobio.2012.06.002. [DOI] [PubMed] [Google Scholar]
- 269.Gu B, Huang YZ, He X-P, et al. A Peptide Uncoupling BDNF Receptor TrkB from Phospholipase Cγ1 Prevents Epilepsy Induced by Status Epilepticus. Neuron. 2015;88:484–91. doi: 10.1016/j.neuron.2015.09.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 270.Vezzani A, Maroso M, Balosso S, et al. IL-1 receptor/Toll-like receptor signaling in infection, inflammation, stress and neurodegeneration couples hyperexcitability and seizures. Brain Behav Immun. 2011;25:1281–9. doi: 10.1016/j.bbi.2011.03.018. [DOI] [PubMed] [Google Scholar]
- 271.Ravizza T, Noe F, Zardoni D, et al. Interleukin Converting Enzyme inhibition impairs kindling epileptogenesis in rats by blocking astrocytic IL-1beta production. Neurobiol Dis. 2008;31:327–33. doi: 10.1016/j.nbd.2008.05.007. [DOI] [PubMed] [Google Scholar]
- 272.Bertram E. The relevance of kindling for human epilepsy. Epilepsia. 2007;48(Suppl 2):65–74. doi: 10.1111/j.1528-1167.2007.01068.x. [DOI] [PubMed] [Google Scholar]
- 273.Gorter JA, van Vliet EA, Lopes da Silva FH. Which insights have we gained from the kindling and post-status epilepticus models? J Neurosci Methods. 2016;260:96–108. doi: 10.1016/j.jneumeth.2015.03.025. [DOI] [PubMed] [Google Scholar]
- 274.Pitkänen A, Halonen T. Prevention of epilepsy. Trends Pharmacol Sci. 1998;19:253–5. doi: 10.1016/s0165-6147(98)01224-3. [DOI] [PubMed] [Google Scholar]
- 275.Löscher W. Fit for purpose application of currently existing animal models in the discovery of novel epilepsy therapies. Epilepsy Res. 2016;126:157–84. doi: 10.1016/j.eplepsyres.2016.05.016. [DOI] [PubMed] [Google Scholar]
- 276.Löscher W. Animal models of epilepsy for the development of antiepileptogenic and disease-modifying drugs. A comparison of the pharmacology of kindling and post-status epilepticus models of temporal lobe epilepsy. Epilepsy Res. 2002;50:105–23. doi: 10.1016/s0920-1211(02)00073-6. [DOI] [PubMed] [Google Scholar]
- 277.Eastman CL, Verley DR, Fender JS, et al. ECoG studies of valproate, carbamazepine and halothane in frontal-lobe epilepsy induced by head injury in the rat. Exp Neurol. 2010;224:369–88. doi: 10.1016/j.expneurol.2010.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 278.Eastman CL, Verley DR, Fender JS, et al. Antiepileptic and antiepileptogenic performance of carisbamate after head injury in the rat: blind and randomized studies. J Pharmacol Exp Ther. 2011;336:779–90. doi: 10.1124/jpet.110.175133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 279.Barker-Haliski ML, Dahle EJ, Heck TD, et al. Evaluating an etiologically relevant platform for therapy development for temporal lobe epilepsy: effects of carbamazepine and valproic acid on acute seizures and chronic behavioral comorbidities in the Theiler’s murine encephalomyelitis virus mouse model. J Pharmacol Exp Ther. 2015;353:318–29. doi: 10.1124/jpet.114.222513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 280.Silver JM, Shin C, McNamara JO. Antiepileptogenic effects of conventional anticonvulsants in the kindling model of epilespy. Ann Neurol. 1991;29:356–63. doi: 10.1002/ana.410290404. [DOI] [PubMed] [Google Scholar]
- 281.Löscher W, Hönack D, Rundfeldt C. Antiepileptogenic effects of the novel anticonvulsant levetiracetam (ucb L059) in the kindling model of temporal lobe epilepsy. J Pharmacol Exp Ther. 1998;284:474–9. [PubMed] [Google Scholar]
- 282.Kaminski RM, Rogawski MA, Klitgaard H. The potential of antiseizure drugs and agents that act on novel molecular targets as antiepileptogenic treatments. Neurotherapeutics. 2014;11:385–400. doi: 10.1007/s13311-014-0266-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 283.Pitkänen A, Nehlig A, Brooks-Kayal AR, et al. Issues related to development of antiepileptogenic therapies. Epilepsia. 2013;54(Suppl 4):35–43. doi: 10.1111/epi.12297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 284.Rojas A, Jiang J, Ganesh T, et al. Cyclooxygenase-2 in epilepsy. Epilepsia. 2014;55:17–25. doi: 10.1111/epi.12461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 285.Kaila K, Price TJ, Payne JA, et al. Cation-chloride cotransporters in neuronal development, plasticity and disease. Nat Rev Neurosci. 2014;15:637–54. doi: 10.1038/nrn3819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 286.Brandt C, Nozadze M, Heuchert N, et al. Disease-modifying effects of phenobarbital and the NKCC1 inhibitor bumetanide in the pilocarpine model of temporal lobe epilepsy. J Neurosci. 2010;30:8602–12. doi: 10.1523/JNEUROSCI.0633-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 287.Koyama R, Tao K, Sasaki T, et al. GABAergic excitation after febrile seizures induces ectopic granule cells and adult epilepsy. Nat Med. 2012;18:1271–8. doi: 10.1038/nm.2850. [DOI] [PubMed] [Google Scholar]
- 288.Marguet SL, Le-Schulte VTQ, Merseburg A, et al. Treatment during a vulnerable developmental period rescues a genetic epilepsy. Nat Med. 2015;21:1436–44. doi: 10.1038/nm.3987. [DOI] [PubMed] [Google Scholar]
- 289.Bar-Klein G, Klee R, Brandt C, et al. Isoflurane prevents acquired epilepsy in rat models of temporal lobe epilepsy. Ann Neurol. 2016;80:896–908. doi: 10.1002/ana.24804. [DOI] [PubMed] [Google Scholar]
- 290.Scicchitano F, Constanti A, Citraro R, et al. Statins and epilepsy: preclinical studies, clinical trials and statin-anticonvulsant drug interactions. Curr Drug Targets. 2015;16:747–56. doi: 10.2174/1389450116666150330114850. [DOI] [PubMed] [Google Scholar]
- 291.Barker-Haliski ML, Heck TD, Dahle EJ, et al. Acute treatment with minocycline, but not valproic acid, improves long-term behavioral outcomes in the Theiler’s virus model of temporal lobe epilepsy. Epilepsia. 2016;57:1958–1967. doi: 10.1111/epi.13577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 292.Siniscalchi A, Mintzer S. Statins for poststroke seizures: the first antiepileptogenic agent? Neurology. 2015;85:661–2. doi: 10.1212/WNL.0000000000001878. [DOI] [PubMed] [Google Scholar]
- 293.Pugh MJV, Knoefel JE, Mortensen EM, et al. New-onset epilepsy risk factors in older veterans. J Am Geriatr Soc. 2009;57:237–42. doi: 10.1111/j.1532-5415.2008.02124.x. [DOI] [PubMed] [Google Scholar]
- 294.Etminan M, Samii A, Brophy JM. Statin use and risk of epilepsy: a nested case-control study. Neurology. 2010;75:1496–500. doi: 10.1212/WNL.0b013e3181f96253. [DOI] [PubMed] [Google Scholar]
- 295.White HS, Löscher W. Searching for the ideal antiepileptogenic agent in experimental models: single treatment versus combinatorial treatment strategies. Neurotherapeutics. 2014 Apr;11(2):373–84. doi: 10.1007/s13311-013-0250-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 296.Johnson MR, Shkura K, Langley SR, et al. Systems genetics identifies a convergent gene network for cognition and neurodevelopmental disease. Nat Neurosci. 2016;19:223–32. doi: 10.1038/nn.4205. [DOI] [PubMed] [Google Scholar]
- 297.Delahaye-Duriez A, Srivastava P, Shkura K, et al. Rare and common epilepsies converge on a shared gene regulatory network providing opportunities for novel antiepileptic drug discovery. Genome Biol. 2016;17:245. doi: 10.1186/s13059-016-1097-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 298.Bassett DS, Sporns O. Network neuroscience. Nat Neurosci. 2017;20:353–364. doi: 10.1038/nn.4502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 299.Pangalos MN, Schechter LE, Hurko O. Drug development for CNS disorders: strategies for balancing risk and reducing attrition. Nat Rev Drug Discov. 2007;6:521–32. doi: 10.1038/nrd2094. [DOI] [PubMed] [Google Scholar]
- 300.Gershen LD, Zanotti-Fregonara P, Dustin IH, et al. Neuroinflammation in Temporal Lobe Epilepsy Measured Using Positron Emission Tomographic Imaging of Translocator Protein. JAMA Neurol. 2015;72:882–8. doi: 10.1001/jamaneurol.2015.0941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 301.Finnema SJ, Nabulsi NB, Eid T, et al. Imaging synaptic density in the living human brain. Sci Transl Med. 2016;8:348ra96. doi: 10.1126/scitranslmed.aaf6667. [DOI] [PubMed] [Google Scholar]
- 302.Falco-Walter J, Owen C, Sharma M, et al. Magnetoencephalography and New Imaging Modalities in Epilepsy. Neurotherapeutics. 2017;14:4–10. doi: 10.1007/s13311-016-0506-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 303.Thomas DW, Burns J, Audette J, et al. Clinical Development Success Rates 2006–2015. Bioindustry Analysis Report. 2016 Jun; [Google Scholar]
- 304.McGhee DJM, Ritchie CW, Zajicek JP, et al. A review of clinical trial designs used to detect a disease-modifying effect of drug therapy in Alzheimer’s disease and Parkinson’s disease. BMC Neurol. 2016;16:92. doi: 10.1186/s12883-016-0606-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 305.Schmidt D, Friedman D, Dichter MA. Anti-epileptogenic clinical trial designs in epilepsy: issues and options. Neurotherapeutics. 2014;11:401–11. doi: 10.1007/s13311-013-0252-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 306.U.S. Department of Health and Human Services, Food and Drug Administration, Center for Drug Evaluation and Research. Guidance for Industry Codevelopment of Two or More New Investigational Drugs for Use in Combination. 2013 Jun; https://www.fda.gov/ucm/groups/fdagov-public/@fdagov-drugs-gen/documents/document/ucm236669.pdf.
- 307.U.S. Department of Health and Human Services, Food and Drug Administration, Office of the Commissioner, Office of Combination Products. Guidance for Industry and FDA Staff: Early Development Considerations for Innovative Combination Products. 2006 Sep; https://www.fda.gov/downloads/RegulatoryInformation/Guidances/ucm126054.pdf.
- 308.Racine RJ, Paxinos G, Mosher JM, et al. The effects of various lesions and knife-cuts on septal and amygdala kindling in the rat. Brain Res. 1988;454:264–74. doi: 10.1016/0006-8993(88)90826-8. [DOI] [PubMed] [Google Scholar]
- 309.Meeren HKM, Pijn JPM, Van Luijtelaar ELJM, et al. Cortical focus drives widespread corticothalamic networks during spontaneous absence seizures in rats. J Neurosci. 2002;22:1480–95. doi: 10.1523/JNEUROSCI.22-04-01480.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 310.Braganza O, Bedner P, Hüttmann K, et al. Albumin is taken up by hippocampal NG2 cells and astrocytes and decreases gap junction coupling. Epilepsia. 2012;53:1898–1906. doi: 10.1111/j.1528-1167.2012.03665.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 311.Friedman A, Heinemann U. Role of Blood-Brain Barrier Dysfunction in Epileptogenesis. In: Noebels JL, Avoli M, Rogawski MA, et al., editors. Jasper’s Basic Mechanisms of the Epilepsies. 4th. Bethesda (MD): National Center for Biotechnology Information (US); 2012. pp. 1–12. [PubMed] [Google Scholar]
- 312.Majkowski J, Bilińska-Nigot B, Sobieszek A. Development of EEG epileptic activity and seizures during kindling in sensorimotor cortex in cats. Epilepsia. 1981;22:275–84. doi: 10.1111/j.1528-1157.1981.tb04110.x. [DOI] [PubMed] [Google Scholar]
- 313.Bolkvadze T, Pitkänen A. Development of post-traumatic epilepsy after controlled cortical impact and lateral fluid-percussion-induced brain injury in the mouse. J Neurotrauma. 2012;29:789–812. doi: 10.1089/neu.2011.1954. [DOI] [PubMed] [Google Scholar]
- 314.Kelly KM, Miller ER, Lepsveridze E, et al. Posttraumatic seizures and epilepsy in adult rats after controlled cortical impact. Epilepsy Res. 2015;117:104–16. doi: 10.1016/j.eplepsyres.2015.09.009. [DOI] [PubMed] [Google Scholar]
- 315.Piredda S, Gale K. A crucial epileptogenic site in the deep prepiriform cortex. Nature. 317:623–5. doi: 10.1038/317623a0. [DOI] [PubMed] [Google Scholar]
- 316.Cavalheiro EA, Naffah-Mazzacoratti MG, Mello LE, et al. Models of Seizures and Epilepsy. Elsevier; 2006. The Pilocarpine Model of Seizures; pp. 433–448. [Google Scholar]
- 317.Goddard GV. Development of Epileptic Seizures through Brain Stimulation at Low Intensity Nature. 1967;214:1020–1021. doi: 10.1038/2141020a0. [DOI] [PubMed] [Google Scholar]
- 318.Olney JW, Rhee V, Ho OL. Kainic acid: a powerful neurotoxic analogue of glutamate. Brain Res. 1974;77:507–12.A. doi: 10.1016/0006-8993(74)90640-4. [DOI] [PubMed] [Google Scholar]
- 319.Racine R, Rose PA, Burnham WM. Afterdischarge thresholds and kindling rates in dorsal and ventral hippocampus and dentate gyrus. Can J Neurol Sci. 1977;4:273–8. doi: 10.1017/s0317167100025117. [DOI] [PubMed] [Google Scholar]
- 320.Weissberg I, Wood L, Kamintsky L, et al. Albumin induces excitatory synaptogenesis through astrocytic TGF-beta/ALK5 signaling in a model of acquired epilepsy following blood-brain barrier dysfunction. Neurobiol Dis. 2015;78:115–25. doi: 10.1016/j.nbd.2015.02.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 321.Englander J, Bushnik T, Duong TT, et al. Analyzing risk factors for late posttraumatic seizures: a prospective, multicenter investigation. Arch Phys Med Rehabil. 2003;84:365–73. doi: 10.1053/apmr.2003.50022. [DOI] [PubMed] [Google Scholar]
- 322.Annegers JF, Hauser WA, Coan SP, et al. A population-based study of seizures after traumatic brain injuries. N Engl J Med. 1998;338:20–4. doi: 10.1056/NEJM199801013380104. [DOI] [PubMed] [Google Scholar]
- 323.Graham NSN, Crichton S, Koutroumanidis M, et al. Incidence and Associations of Poststroke Epilepsy: The Prospective South London Stroke Register. Stroke. 2013;44:605–611. doi: 10.1161/STROKEAHA.111.000220. [DOI] [PubMed] [Google Scholar]
- 324.Annegers JF, Hauser WA, Beghi E, et al. The risk of unprovoked seizures after encephalitis and meningitis. Neurology. 1988;38:1407–10. doi: 10.1212/wnl.38.9.1407. [DOI] [PubMed] [Google Scholar]
- 325.Haapaniemi E, Strbian D, Rossi C, et al. The CAVE Score for Predicting Late Seizures After Intracerebral Hemorrhage. Stroke. 2014;45:1971–1976. doi: 10.1161/STROKEAHA.114.004686. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.