

Incorporation of silicon atoms into nickel nanoparticles boosts the catalytic activity for (de)hydrogenation reactions.
Abstract
Hydrogenation reactions are essential processes in the chemical industry, giving access to a variety of valuable compounds including fine chemicals, agrochemicals, and pharmachemicals. On an industrial scale, hydrogenations are typically performed with precious metal catalysts or with base metal catalysts, such as Raney nickel, which requires special handling due to its pyrophoric nature. We report a stable and highly active intermetallic nickel silicide catalyst that can be used for hydrogenations of a wide range of unsaturated compounds. The catalyst is prepared via a straightforward procedure using SiO2 as the silicon atom source. The process involves thermal reduction of Si–O bonds in the presence of Ni nanoparticles at temperatures below 1000°C. The presence of silicon as a secondary component in the nickel metal lattice plays the key role in its properties and is of crucial importance for improved catalytic activity. This novel catalyst allows for efficient reduction of nitroarenes, carbonyls, nitriles, N-containing heterocycles, and unsaturated carbon–carbon bonds. Moreover, the reported catalyst can be used for oxidation reactions in the presence of molecular oxygen and is capable of promoting acceptorless dehydrogenation of unsaturated N-containing heterocycles, opening avenues for H2 storage in organic compounds. The generality of the nickel silicide catalyst is demonstrated in the hydrogenation of over a hundred of structurally diverse unsaturated compounds. The wide application scope and high catalytic activity of this novel catalyst make it a nice alternative to known general hydrogenation catalysts, such as Raney nickel and noble metal–based catalysts.
INTRODUCTION
Hydrogenations of unsaturated hydrocarbons and polar functional groups using heterogeneous catalysts represent fundamental tools for practical organic synthesis (1). Among the industrially applied catalysts, nickel-based materials are of special importance due to the abundance of the metal, its favorable cost advantage compared with noble metal–based hydrogenation catalysts (Pd, Pt, and Ru), and its high reactivity. The history of heterogeneous nickel catalysts dates back to the discovery of hydrogenations of unsaturated compounds at atmospheric pressure in 1897 by the French chemists Sabatier and Senderens (2), which was recognized with the Nobel Prize in 1912. Around the same time in Russia, Ipatieff (3) developed high-pressure hydrogenations in a liquid phase using finely divided nickel and nickel oxides catalysts. The breakthrough for today’s most important nickel catalysts came in 1927, when Murray Raney introduced a highly reactive form of nickel prepared by treatment of nickel-aluminum alloy with sodium hydroxide solution, today known as Raney nickel (4). Two years earlier, Raney reported the preparation of a highly dispersed nickel catalyst in a similar manner from a nickel-silicon alloy that can be used for hydrogenation of fats and oils but was not studied any further (5). Today, Raney nickel catalysts are recognized as state-of-the-art industrial hydrogenation catalysts, which have widespread applications in hydrogenolysis, dehydrogenation, and desulfurization reactions. More specifically, the reduction of glucose to sorbitol and adipodinitrile to hexamethylene diamine, hydrogenation of benzene to cyclohexane and 1,4-butynediol to 1,4-butanediol, and hardening of vegetable oils in the presence of Raney nickel represent established bulk-scale chemical processes (6, 7).
Despite Raney nickel’s favorable reactivity and low cost, this class of compounds has several drawbacks. Because of a large metal surface area, these catalysts are pyrophoric and have caused serious accidents in both academic and industrial laboratories (8, 9). Hence, the catalyst should be handled under a strictly inert atmosphere and is typically stored as a suspension in water, further complicating an accurate measurement of the catalyst. Furthermore, Raney nickel tends to deactivate over time, and significant loss of activity has been reported after storage. As a consequence, there is an industrial and academic demand for the development of an inexpensive, safe, stable Raney nickel substitute that can be used for a wide range of chemical transformations with a similar or superior reaction profile.
Recently, our group developed novel cobalt and iron oxide nanoparticles immobilized on a carbon support (Vulcan XC72R), which are encapsulated by a nitrogen-enriched graphene-layer matrix. The unusual structure of these core-shell catalysts results from simple pyrolysis of metal acetate complexes with nitrogen-based ligands (10, 11). So far, these Co–Co3O4/NGr@C or Fe2O3/NGr@C catalysts have been successfully used for the hydrogenation of nitroarenes, reductive amination, the synthesis of nitriles, and oxidation reactions of alcohols and N-heterocycles (12, 13). Despite the impressive selectivity of these nanoparticles, there is a great interest in developing non-noble metal–based systems that show higher activity. In this context, we envisioned the corresponding nickel catalysts especially suitable. However, known core-shell Ni–NiO/NGr@C nanoparticles (14) and mesoporous Ni–SiCN material reported by the group of Kempe (15) still require relatively harsh reaction conditions (110°C, 50 bar of H2 for both catalysts) and are limited to the reduction of nitroarenes. Herein, we report a serendipitous discovery of intermetallic nickel silicides supported on silica Ni-phen@SiO2-1000 with impressive catalytic applications making these materials of interest as a safe and benign substitute for Raney nickel and precious metal catalysts.
RESULTS AND DISCUSSION
Identification of the active catalyst
Our search for new hydrogenation catalysts started with the deposition of defined organometallic Ni complexes, obtained from nickel (II) acetate and 1,10-phenanthroline onto commercially available inorganic supports. Subsequent pyrolysis at 600°, 800°, or 1000°C led to a small library containing 18 catalysts named Ni–Ligand@Support–Tpyrolysis (Table 1). These materials were tested for benchmark applications, such as hydrogenations of benzonitrile to benzyl amine, nitrobenzene to aniline, and acetophenone to 1-phenylethanol (table S3). Unfortunately, nickel nanoparticles supported on a Vulcan carbon showed almost no activity. Nickel supported on silicon carbide was generally inefficient and was outcompeted by the rest of the catalysts. In contrast to previous catalysts, Ni-phen@CeO2-1000 demonstrated high activity for the acetophenone reduction but only marginal reactivity for benzonitrile and nitrobenzene reductions. Acetophenone was fully hydrogenated in the presence of Ni-phen@TiO2-800, and the same catalyst showed high conversions for benzonitrile reduction. Furthermore, corresponding nickel catalysts supported on alumina did not provide full conversion under identical reaction conditions. The influence of the pyrolysis temperature on the catalytic performance was most prominent for catalysts supported on fumed silica (Aerosil OX 50). The material pyrolyzed at 600°C generally exhibited low activity for all substrates, whereas the catalyst pyrolyzed at 800°C, Ni-phen@SiO2-800, was efficient for the reduction of nitro groups and nitriles. The most active catalyst in the series, Ni-phen@SiO2-1000, allowed full conversion for all the substrates of interest.
Table 1. Hydrogenation of benchmark substrates with nickel-supported catalysts.
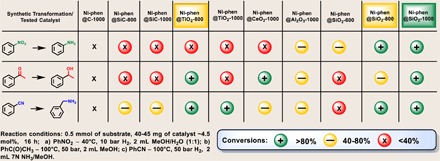
Structure of the catalysts and the mechanism of their formation
To understand what structural features of the most active Ni-phen@SiO2-1000 catalyst are responsible for this remarkable reactivity, we characterized it in detail. To our surprise, its structure is completely different compared to all the other examined materials in this series. Initial x-ray powder diffraction (XRD) analysis revealed no metallic nickel particles; instead, nickel silicides (a mixture of Ni31Si12 along with more silicon-rich Ni2Si phase) were detected (Fig. 1A). By contrast, less prominent catalysts supported on TiO2, CeO2, and the inactive Ni-phen@SiO2-600 show reflections at Bragg’s angles of 44.5°, 52°, and 76.4°, which are typically attributed to cubic metallic nickel (fig. S9). The Brunauer-Emmett-Teller (BET) surface area of Ni-phen@SiO2-1000 was found to be 70.40 m2/g and contain micro- and mesopores (table S2).
Fig. 1. Catalysts characterization.
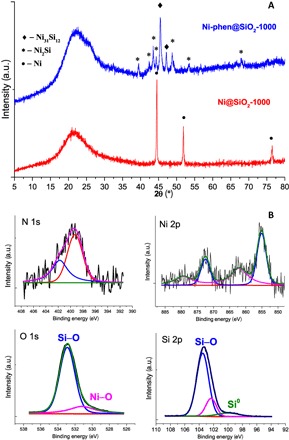
(A) XRD data for Ni-phen@SiO2-1000 (blue) and Ni@SiO2-1000 (red). (B) X-ray photoelectron spectroscopy (XPS) measurement of the intermetallic nickel silicide catalyst Ni-phen@SiO2-1000. a.u., arbitrary units.
Intrigued by this unforeseen finding, we performed transmission electron microscopy (TEM) measurement of Ni-phen@SiO2-1000. As can be seen from Fig. 2A, nickel silicide nanoparticles embedded in a graphitic matrix containing traces of silicon and oxygen were found, therefore confirming the presence of unusual Ni–Si intermetallic compounds. Most of the observed silicide particles were within a range of 20 to 70 nm; however, some particles that reach up to 150 nm exist. XPS also revealed the presence of graphitic-bonded nitrogen (Fig. 1B). Two distinct peaks are observed in the N1s spectra of Ni-phen@SiO2-1000 with an electron-binding energy of 399.3 and 401.8 eV. The lower binding-energy peak can be attributed to a pyridinic nitrogen bound to nickel, while the electron-binding energy of 401.8 eV is characteristic of a graphitic nitrogen (16, 17).
Fig. 2. Catalysts characterization.

Scanning TEM (STEM)–high-angle annular dark-field (HAADF, top), annular bright-field (ABF) images and energy-dispersive x-ray (EDX) mapping of (A) Ni-phen@SiO2-1000 (free-standing Ni-Si nanoparticle embedded in carbon) and of (B) Ni@SiO2-1000 prepared without 1,10-phenanthroline ligand (no formation of intermetallic Ni-Si nanoparticles is observed, and Ni/NiO core-shell nanoparticles are formed).
The Ni2p spectrum of Ni-phen@SiO2-1000 revealed two main features centered around 855.1 and 872.5 eV for Ni2p3/2 and Ni2p1/2, respectively, and their corresponding satellite peaks. Both binding energy and spectrum shape can be ascribed to Ni bound to oxygen atoms on the surface in the catalyst. The oxygen region shows two signals at 532.9 and 531.2 eV, which correspond to Si–O and Ni–O bonds, respectively (18). The Si2p spectrum shows two peaks at 103.3 and 99.8 eV. The first major peak originates from the silicon atoms of the support, whereas the latter reflects metallic silicon found in the graphitic layers. No signal at 97 to 98 eV, characteristic for Ni–Si bonds, was observed. Nickel-rich silicides, such as Ni3Si–Ni2Si, are known to be partially oxidized upon exposure to air (19, 20). Energy-dispersive x-ray (EDX) spectroscopy analysis of the nickel silicide particles reveals the presence of small amounts of oxygen, which is evident of nickel and silicon oxide formation on the particle surface due to storage in air. On the basis of the analytical data, the optimal catalyst Ni-phen@SiO2-1000 can be represented as Ni–Si/NixOy–SiO2 core-shell nanoparticles distributed on the surface of the silica support, covered by N-doped graphene enriched with Si atoms.
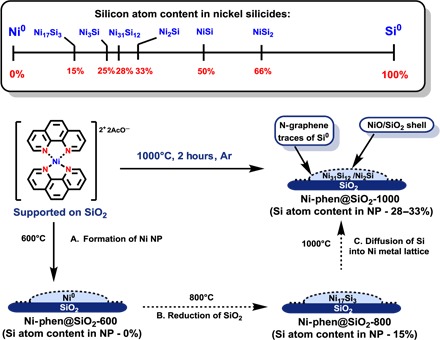
Analysis and comparison of silica-supported catalysts pyrolyzed at lower temperatures provide insight into the mechanism of the formation of the novel Ni–Si nanocatalyst. Careful examination of Ni-phen@SiO2-800 by XRD reveals the presence of Ni17Si3 phase, rather than metallic nickel (figs. S6 and S7). Therefore, a pronounced effect of the pyrolysis temperature on the degree of silicidation is observed: At 600°C, Ni(0) is formed [0 atomic % (at %) Si], and at 800°C, a silicon-poor Ni17Si3 (15 at % Si) phase is observed, finally giving Ni31Si12/Ni2Si (28 to 33 at % Si) at 1000°C. With no other silicon-containing compounds used, it is clear that Si atoms in metal silicides may only originate from the SiO2 support. It is important to point out the change in silicon oxidation states: The silica support is reduced upon pyrolysis.
Consequently, we propose the following mechanism for the formation of Ni–Si catalyst (Fig. 3). Initially, significant mass is lost due to decomposition of the organic ligand during heating up to 350°C (fig. S12). Then, up to 600°C, only a smaller loss of material is observed. During this process, metallic Ni nanoparticles are formed. Upon increasing of the temperature to 800°C, reduction of silica takes place: A silicon-poor Ni17Si3 (15 at % Si) phase is formed. We suspected that both the presence of nickel nanoparticles and nitrogen-doped carbon species are necessary for Si–O bond reduction. The rate of Si/Ni diffusion grows exponentially with temperature (21); as a result, at 1000°C, silicon atoms that formed on the surface are further distributed into the nickel lattice, giving a mixture of Ni31Si12/Ni2Si (28 to 33 at % Si). After exposure of the material to air, the surface of the Ni–Si nanoparticles is oxidized, giving a NiO/SiO2 shell. The resulting Ni-phen@SiO2-1000 is far more active compared to other catalysts in the series. The improved reactivity is the result of the incorporation of silicon atoms into the nickel crystal lattice of metals.
Fig. 3. Formation of nickel silicide nanoparticles.
In general, metal silicide catalysts have received increased interest in recent years (22). However, in contrast to other well-established heterogeneous nickel catalysts, only limited applications have been reported using nickel silicides (23–27). Apparently, one of the major obstacles that impedes progress in this important area of catalysis is the difficulty in catalyst preparation. To date, intermetallic Ni–Si catalysts are typically prepared via a multistep method introduced by Nuzzo et al. (28), which involves the preparation of Ni(0) particles with further modification using SiH4 as the Si precursor. Because of its pyrophoric nature, explosion hazard, and toxicity, utilization of SiH4 is highly undesired (29). These limitations resulted in the underdevelopment of SiH4-based methodologies and initiated the search for SiH4 substitutes (30, 31).
Effect of the ligand, metal, and silica grade
Next, the role of the ligand on the formation of nickel silicide was investigated. Thus, Ni@SiO2-1000 was prepared in an analogous manner without phenanthroline. XRD and STEM analysis of this material revealed metallic Ni nanoparticles with thin oxidic shells (Fig. 1B). No formation of elemental silicon and nickel silicides was observed, which accounts for a marked drop in reactivity (table S4). To understand the role of the ligand in more detail, by XRD, we prepared and examined a series of catalysts, using nitrogen containing ligands other than 1,10-phehanthroline. Ammonia and pyridine did not promote the formation of the nickel silicides and gave only metallic nickel nanoparticles. In the presence of tetramethylethylenediamine, a silicon-poor Ni3Si phase was formed. However, the catalytic activity of these materials toward the reduction of nitrobenzene was significantly lower, compared to the Ni-phen@SiO2-1000 (table S4).
The importance of nickel in the reduction process is shown by its comparison to supported cobalt nanoparticles used for selective semihydrogenation of alkynes (32). Although these cobalt catalysts were prepared in a similar fashion, formation of elemental silica and cobalt silicides was not observed. Instead, cobalt existed in forms of cobalt (0) and cobalt oxides. In addition to metal, ligand, and pyrolysis temperature, the grade of silica is also crucial for the formation of silicon-rich Ni–Si intermetallic compounds. Hence, pyrolysis of a Ni-phen complex at 1000°C on quartz gave only metallic nickel. On the other hand, conventional silica gel, used for column chromatography with a greater surface area, gave a mixture of Ni3Si and Ni31Si12. In conclusion, the combination of Ni and 1,10-phenanthroline together with high-surface silica is very special, allowing for a unique reduction of Si–O bonds and straightforward formation of nickel silicides.
Catalysis and applications
Hydrogenation of polar functional groups
To demonstrate the general applicability of the newly identified intermetallic Ni–Si nanocatalyst, we report the hydrogenation of more than 100 diverse unsaturated compounds. At first, the reduction of various nitroarenes, including sensitive functionalized substrates, was explored with Ni-phen@SiO2-1000 (Scheme 1A). In all cases, the corresponding anilines were obtained in good yields and selectivities with 4 mole percent (mol %) nickel catalyst under mild reaction conditions (60°C, 10 bar of H2, for 20 hours). Hydrogenation of nitro groups in the presence of reducible functional groups under mild conditions is usually achieved with noble metals (33). With Ni–Si nanocatalyst, no dehalogenation products were detected for chloro- and bromo-substituted substrates. Notably, the bromine atom and the amide function stayed intact on the nitroindoline-based substrate. In contrast to partially saturated indoline, indole-based substrate required harsher reaction conditions to give the corresponding aniline. Other heterocyclic compounds were successfully used including 3-nitropyridine and 4-nitrobenzothiadiazole. Substrates having electron-donating groups were well tolerated, whereas the presence of electron-withdrawing substituents impeded the hydrogenations process. In addition, sensitive functional groups, such as ketones, esters, and amides, remained inert under these reaction conditions.
Scheme 1. Hydrogenation of nitroarenes with intermetallic Ni-Si catalyst Ni-phen@SiO2-1000.
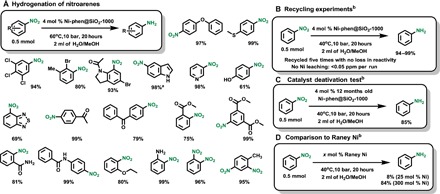
Reaction conditions: (A) Nitroarene (0.5 mmol), catalyst (40 mg; 4.0 mol % Ni), H2 (10 bar), 60°C, 20 hours, and 1:1 H2O/MeOH (2 ml). Isolated yields are reported unless otherwise indicated. (B to D) Nitrobenzene (0.5 mmol), Ni-phen@SiO2-1000 catalyst (40 mg; 4.0 mol % Ni), H2 (10 bar), 40°C, 20 hours, and 1:1 H2O/MeOH (2 ml). (D) Raney Ni catalyst (7.5 or 88 mg; 25 or 300 mol % Ni, respectively). a20 bar of H2, 80°C, 24 hours; bGC yields using n-hexadecane standard.
One advantage of heterogeneous catalysis is recyclability, which originates from the improved stability of the catalyst and is an important feature of any industrial application. In contrast to the classical Raney Ni, Ni-phen@SiO2-1000 catalyst is conveniently handled under air without any deactivation or safety hazards. Hence, we reused the catalytic material for the hydrogenation of nitrobenzene up to five times without loss of activity. As depicted in Scheme 1B, the desired aniline was obtained in excellent yields in the first five runs; however, a drop in reactivity was observed in the sixth run. The selectivity for hydrogenation remained high, no by-products were detected by gas chromatography (GC), and no metal leaching was observed (<0.05 parts per million). Unlike, Raney Ni, our nickel silicide catalyst Ni-phen@SiO2-1000 demonstrated exceptional stability for hydrogenation of nitrobenzene: No loss of activity was observed even after storage for 12 months while being exposed to air (Scheme 1C). In addition, under these mild reaction conditions, the standard Raney Ni proved only to be efficient in the stoichiometric reduction of nitrobenzene (Scheme 1D).
Next, we turned our attention to the reduction of other polar functional groups such as ketones, aldehydes, and nitriles (Scheme 2). Apart from acetophenone, diverse aromatic ketones were successfully hydrogenated in the presence of Ni-phen@SiO2-1000. To our delight, substrates having halogens, pyridine rings, and trifluoromethyl groups are reduced to corresponding secondary alcohols under mild conditions. In addition, aromatic and aliphatic aldehydes can be efficiently converted into primary alcohols with Ni-phen@SiO2-1000 at 80°C.
Scheme 2. Hydrogenation of other polar functional groups with intermetallic Ni–Si catalyst Ni-phen@SiO2-1000.
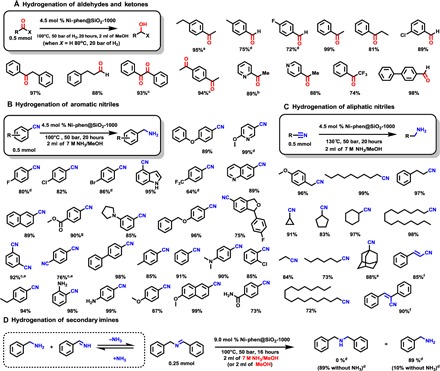
Reaction conditions: (A) Substrate (0.5 mmol), catalyst (45 mg; 4.5 mol % Ni), H2 (20 bar), 80°C (aldehydes) or 100°C (ketones), 20 hours, and 1:1 H2O/MeOH (2 ml). (B and C) Nitrile (0.5 mmol), catalyst (45 mg; 4.5 mol % Ni), H2 (50 bar), 100°C (aromatic nitriles) or 130°C (aliphatic nitriles), 20 hours, and 7 M NH3/MeOH (2 ml). Isolated yields of hydrochloride salts. (D) Imine (0.25 mmol), catalyst (45 mg; 9.0 mol % Ni), H2 (50 bar ), 100°C, 16 hours, and 7 M NH3/MeOH (2 ml) or MeOH (2 ml). Isolated yields are reported unless otherwise indicated. a110°C; b120°C; c90 mg (9.0 mol % Ni); dGC yields using n-hexadecane standard; e150°C, 48 hours; fcomplete reduction of alkene; g2 ml of MeOH as a solvent.
Next, the hydrogenation of benzonitrile was explored (fig. S15). Here, the addition of ammonia proved to be beneficial for the selective formation of the desired primary amine. Initially, the secondary imine is detected; however, with ammonia present, its concentration drops during the course of hydrogenation. Only in the absence of NH3 is the secondary imine reduced to the corresponding dibenzyl amine (Scheme 2D). Aside from standard benzonitrile, p-phenyl benzonitrile, and 2-naphthylnitrile, aromatic nitriles containing electron-donating and electron-withdrawing groups gave good to excellent yields of the desired primary amines, which in most cases were isolated as hydrochloride salts. Comparing the obtained selectivities, substrates with electron-withdrawing groups are more demanding than of those bearing electron-donating groups. Nevertheless, for substrates bearing halogen atoms (F, Br, and Cl), no dehalogenation was observed. Heterocycles, such as quinolones, indoles, and benzofurans, are well tolerated. Notably, ester functionalities remain intact, and the nickel silicide catalyst does not promote hydrogenolysis of benzyl-protected alcohols, which is of interest for multistep synthesis. Finally, hydrogenation of industrially relevant dinitriles was explored. Hence, terephthalodinitrile and 1,3-dicyanobenzene furnished the desired diamines in 76 and 92% yield, respectively. Reduction of aliphatic nitriles required a higher temperature to obtain corresponding amines. For example, hydrogenation of cyclopropyl nitrile proceeded without ring opening of the strained cycloalkane, and with sterically hindered adamantane nitrile, full conversion was achieved at 160°C. Reduction of cinnamonitrile gave a fully hydrogenated amine. This result prompted us to explore the capabilities of Ni-phen@SiO2-1000 toward hydrogenation of alkenes and alkynes.
Hydrogenation of alkenes and alkynes
Hydrogenation of carbon–carbon double and triple bonds is a typical application of Raney nickel. In general, reduction of unsaturated hydrocarbons occurs under milder reaction conditions compared to polar functional groups. Terminal and internal cis- and trans- 2-octenes were hydrogenated to fully saturated alkanes already at 40°C. Typically, hydrogenation of 1,1-disubstituted olefins is slower; thus, full conversion of 2-methylhept-1-ene was achieved at 80°C, whereas β-methylstyrene, cinnamyl alcohol, and electron-rich allyl anisole were selectively hydrogenated at 40°C. Methyl oleate, an example of bulk scale oil hydrogenation, is converted to corresponding stearate in a 99% isolated yield. A variety of alkynes were tested as well: Both terminal and internal alkynes are suitable substrates for the reaction of interest. With the reduction of alkenes and alkynes occurring at very mild temperatures, we next evaluated functional group tolerance. Aliphatic nitriles and ketones remain inert, while C–C double and triple bonds are fully hydrogenated. Even hydrogenation of conjugated 4-vinyl and 4-ethynyl benzonitriles occurred highly chemoselectively, giving p-ethyl benzonitrile in 75 to 81% yield. Notably, esters and phenols are well tolerated as well, but from the viewpoint of selectivity, the hydrogenation of the double bond of 3-vinyl benzaldehyde is most remarkable. Selective hydrogenations of α,β-unsaturated ketones and aldehydes (34) are important chemical transformations, which provide valuable intermediates for the synthesis of many fine chemicals, perfumes, and pharmaceuticals. In this respect, naturally occurring α-ionone was selectively hydrogenated with untouched trisubstituted alkene and ketone. In a similar manner, hydrogenation of cyclohexenone was highly selective toward the C==C bond. Apparently, compared to Raney nickel, these nickel silicides are not only more stable and safe but also allow for more selective transformations of functionalized substrates.
Hydrogenation and dehydrogenation of N-heterocycles
The nickel silicide catalyst Ni-phen@SiO2-1000 allowed for selective hydrogenation of imines vide supra, which consequently led us to explore hydrogenation of quinolones. In general, this process of N-containing heterocycles is initiated by reduction of C==N bonds. This transformation is important not only because of the demand for diverse heterocyclic compounds in pharmaceutical and agrochemical industries but also because, recently, it has attracted significant interest as a benign energy technology for hydrogen storage. To date, a variety of N-heterocycles have been proposed to serve as liquid organic hydrogen carriers (LOHCs) (35). To release the stored hydrogen from LOHCs, it is necessary to promote the reverse process (36). Typically, two different catalysts are used for hydrogenation and dehydrogenation reactions, many of those are based on noble metals, such as Pd, Pt, and Ir (37). Systems based on a single catalyst for both processes are extremely rare (38–41).
As depicted in Scheme 3B, hydrogenation of quinaldine and other functionalized heterocycles proceeds smoothly in the presence of supported nickel silicide. The very same material promoted the reverse reaction in the absence of an external oxidant. The identity of the released hydrogen was confirmed by GC, with only trace amounts of CO and CH4 present (Scheme 3D and fig. S19). Furthermore, this dehydrogenation can be performed at lower temperatures in the presence of air giving water as the only by-product (Scheme 3C). On the basis of the reaction conditions for different substrates shown above, the activity order can be arranged in the following sequence: alkenes-alkynes > nitro > aldehydes > ketones > aryl nitriles > quinolones > alkyl nitriles.
Scheme 3. Hydrogenation of alkenes, alkynes, and quinolines and dehydrogenation of tetrahydroquinolines with intermetallic Ni–Si catalyst Ni-phen@SiO2-1000.
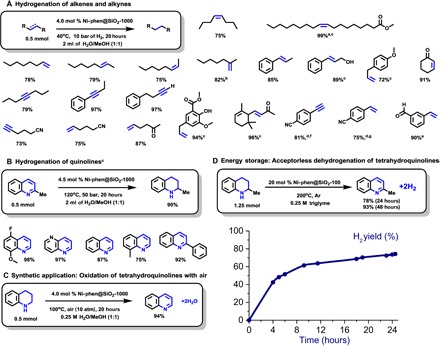
Reaction conditions: (A) Substrate (0.5 mmol), catalyst (40 mg; 4.0 mol % Ni), H2 (10 bar), 40°C, 20 hours, and 1:1 H2O/MeOH (2 ml). GC yields using n-hexadecane standard, unless otherwise indicated. (B) Quinoline (0.5 mmol), catalyst (45 mg; 4.5 mol % Ni), H2 (50 bar), 120°C, 20 hours, and 1:1 H2O/MeOH (2 ml). Isolated yields. (C) Tetrahydroquinoline (0.5 mmol), catalyst (40 mg; 4.0 mol % Ni), air (10 bar), 100°C, 20 hours, and 1:1 H2O/MeOH (2 ml). Isolated yield. a60°C; b80°C; cisolated yield of hydrogenated product; dreaction time (48 hours); ealong with 4% of m-ethylbenzyl alcohol; falong with 6% of p-ethylbenzylamine; g90% conversion, 4% of p-ethylbenzylamine is formed. (D) Tetrahydroquinaldine (1.25 mmol), catalyst (500 mg; 20 mol % Ni), 200°C, 48 hours, triglyme (5 ml), and under Ar. Yield is determined by GC analysis of the liquid phase. Hydrogen evolution was measured by a manual burette.
CONCLUSIONS
Overall, we present a safe and operationally simple preparation of supported nickel silicides. Our method offers the possibility to synthesize this and related intermetallic compounds, avoiding dangerous and toxic SiH4. Instead, the key step of our approach involves the thermal reduction of Si–O bonds in the presence of Ni nanoparticles and subsequent silicidation. The resulting material exhibits a remarkable catalytic activity for diverse (de)hydrogenation processes. The generality, long-term stability, ease of handling, and recyclability make this catalyst an attractive alternative to Raney nickel and precious metal–based catalysts.
MATERIALS AND METHODS
Catalyst preparation
A 250-ml oven-dried single-necked round-bottomed flask equipped with an Allihn reflux condenser and a Teflon-coated, egg-shaped magnetic stir bar (40 mm × 18 mm) was charged with Ni(OAc)2·4H2O (373.3 mg, 1.5 mmol, 1.0 equiv.) and 1,10-phenanthroline monohydrate (594 mg, 3.0 mmol, 2.0 equiv.) and dissolved in ethanol (60 ml). After stirring for 5 min at 25°C, the flask was immersed in an oil bath and heated at 60°C for 2 hours. To the reaction mixture, 2.10 g of silica (Aerosil OX 50) was added via a glass funnel, and the resulting heterogeneous mixture was stirred at 750 rpm for 2 hours at 60°C. The flask was taken out from the bath and cooled to ambient temperature. The solvent was removed in vacuo (180 mbar; bath temperature, 40°C; 200 rpm) and then dried under an oil pump vacuum (1.0 mmHg, 23°C) for 14 hours to give a light blue-green solid. The sample was ground to a fine powder (2.82 g), which was then transferred to a ceramic crucible (height, 20 mm; top Ø, 40 mm) and placed in an oven. The latter was evacuated to ca. 5 mbar and then flushed with argon three times. The furnace was heated to 1000°C at a rate of 25°C/min and held at 1000°C for 2 hours under argon atmosphere. After the heating was switched off, the oven was allowed to reach room temperature, giving the Ni-phen@SiO2-1000 catalyst as a black powder (2.476 g; note that during the whole process, argon was constantly passed through the oven).
General procedure for hydrogenations
An 8-ml glass vial (top Ø, 14 mm; height, 50 mm) equipped with a Teflon-coated oval magnetic stirring bar (8 mm × 5 mm) and a plastic screw cap was charged with a substrate (0.5 mmol, 1.0 equiv.), 40 to 45 mg of Ni-phen@SiO2-1000 catalyst (~4 to 4.5 mol % Ni), and 2 ml of appropriate solvent. The silicone septum was punctured with a 26-gauge syringe needle (0.45 mm × 12 mm), and the vial was placed in an aluminum plate, which was then transferred into the 300-ml autoclave. Once sealed, the autoclave was placed into an aluminum block and purged three times with hydrogen (at 5 to 10 bar). Then, it was pressurized with H2 and heated up to the required temperature under thorough stirring (700 rpm). After 16 hours, the autoclave was removed from the aluminum block and cooled to room temperature in a water bath. The remaining hydrogen was discharged, and the vials containing reaction products were removed from the autoclave. An internal standard (100 μl of n-hexadecane) was added to the crude reaction mixture, followed by the addition of 6 ml of ethyl acetate. The resulting mixture was intensively stirred for 30 s, and then, the solid catalyst was separated by centrifugation and the liquid phase was subjected to GC analysis.
Procedure for acceptorless dehydrogenation
A 25-ml Schlenk tube was charged at ambient atmosphere with the Ni-phen@SiO2-1000 catalyst (500 mg, 20 mol % Ni) and 1,2,3,4-tetrahydroquinaldine (180 μl, 1.25 mmol, 1.0 equiv.). The Schlenk tube was connected to the reaction setup and flushed with argon for approximately 20 to 30 min (fig. S16), followed by heating to 200°C in an aluminum block. The desired temperature was achieved within 20 min. The measurement of the gas evolution was started together with starting the heating. The influence of temperature on the volume was corrected by performing reactions without a catalyst. After 48 hours, a gas sample was taken to determine the identity and quantity of the gas components.
Procedure for the oxidation of 1,2,3,4-tetrahydroquinoline with air
An 8-ml glass vial (Ø, 14 mm; height, 50 mm) equipped with a Teflon-coated oval magnetic stirring bar (8 mm × 5 mm) and a plastic screw cap was charged with 1,2,3,4-tetrahydroquinoline (63 μl, 0.5 mmol, 1.0 equiv.), 40 mg of Ni-phen@SiO2-1000 (~4 mol % Ni), 1 ml of deionized water, and 1 ml of methanol. The silicone septum was punctured with a 26-gauge syringe needle (0.45 mm × 12 mm), and the vial was placed in the aluminum plate, which was then transferred into the 300-ml autoclave. Once sealed, the autoclave was placed into an aluminum block and purged three times with air (at 5 to 10 bar). Then, it was pressurized to 10 bar of air, heated up, and kept at 100°C under thorough stirring (700 rpm). After 20 hours, the autoclave was removed from the aluminum block and cooled to room temperature in a water bath. The remaining air was discharged, and the vial containing the reaction mixture was removed from the autoclave. An internal standard (100 μl of n-hexadecane) was added to the crude reaction mixture, followed by the addition of 6 ml of ethyl acetate. The resulting mixture was intensively stirred for 30 s, and then, the solid catalyst was separated by centrifugation and the liquid phase was subjected to GC analysis.
Supplementary Material
Acknowledgments
We thank A.-E. Surkus for the preparation of the catalysts; A. Lehmann for elemental analysis; C. Rautenberg and U. Bentrup for thermogravimetric–differential scanning calorimetry–mass spectrometry (TG-DSC-MS); A. Wotzka and S. Wohlrab for TGA; M. Schneider for XRD; R. Eckelt for BET analysis; and X. Cui, D. Formenti [all at Leibniz-Institut für Katalyse (LIKAT)], and A. Edwards (University of Kansas) for valuable discussions. Funding: This work is supported by the European Research Council (NoNaCat Grant). Author contributions: M.B. and P.R. planned the project. P.R. developed and prepared the catalysts and designed and performed all catalytic hydrogenation experiments. P.R., M.B., and K.J. wrote the paper. A.A. and H.J. performed catalytic dehydrogenation experiments. M.-M.P. performed TEM measurements and analysis. G.A. performed the XPS measurements and analysis. H.L. performed XRD measurements and analysis. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/4/6/eaat0761/DC1
section S1. Materials and methods
section S2. Procedure for the catalyst preparation (fig. S1).
section S3. TEM and EDX data (fig. S2 to S5)
section S4. XRD diffraction patterns and data (figs. S6 to S10)
section S5. XPS spectra and data (fig. S11)
section S6. Elemental analysis of the catalysts and BET (fig. S12)
section S7. Thermogravimetric analysis (TGA; figs. S13 and S14)
section S8. Procedures for hydrogenation reactions (figs. S15 and S16)
section S9. Product characterization
section S10. Procedures for dehydrogenation reactions (figs. S17 to S19)
section S11. Catalyst recycling
section S12. Nuclear magnetic resonance (NMR) spectral charts
fig. S1. Ni-phen@SiO2-1000 after pyrolysis.
fig. S2. TEM images of the Ni-phen@SiO2-1000.
fig. S3. ABF and HAADF-STEM images of intermetallic nickel silicide catalyst Ni-phen@SiO2-1000.
fig. S4. HAADF-STEM and EDX measurement of the intermetallic nickel silicide catalyst Ni-phen@SiO2-1000.
fig. S5. ABF-, HAADF-STEM, and EDX measurement of the catalyst Ni@SiO2-1000 prepared without ligand.
fig. S6. Powder pattern of nickel-based catalysts supported on fumed silica.
fig. S7. Powder pattern of Ni-phen@SiO2-800 measured up to 148°2θ.
fig. S8. Powder pattern of various nickel-based catalysts with different ligands (S2.3).
fig. S9. Powder pattern of various nickel-based catalysts on various supports.
fig. S10. Powder pattern of nickel catalyst on silica gel and quartz (S2.3).
fig. S11. XPS measurement of the intermetallic nickel silicide catalyst.
fig. S12. N2 adsorption-desorption isotherms and BJH desorption pore size distribution.
fig. S13. TG-DSC-MS analysis of Ni-phen@SiO2-1000.
fig. S14. TG-DSC-MS analysis of Ni-phen@SiO2-1000.
fig. S15. Autoclave and glass vials used for hydrogenations.
fig S16. Concentration/time diagram for the hydrogenation of benzonitrile.
fig. S17. Manual burette setup.
fig. S18. Gas evolution from 1,2,3,4-tetrahydroquinaldine.
fig. S19. Gas composition analysis by GC.
table S1. Elemental analysis of the studied catalysts.
table S2. BET surface area and pore volume of the samples.
table S3. Hydrogenation of the standard substrates (Table 1 in the main text).
table S4. Hydrogenation of the nitrobenzene with SiO2-supported catalysts.
table S5. Recycling experiments.
table S6. Nickel leaching in the recycling experiments.
table S7. Effect of the pH on hydrogenation of nitrobenzene.
REFERENCES AND NOTES
- 1.S. Nishimura, Handbook of Heterogeneous Catalytic Hydrogenation for Organic Synthesis (John Wiley & Sons Inc., 2001). [Google Scholar]
- 2.Sabatier P., Senderens J. B., Action du nickel sur l’éthylène. Synthèse de l’éthane. Comptes Rendus Acad. Sci. 124, 1358–1360 (1897). [Google Scholar]
- 3.V. N. Ipatieff, Catalytic Reactions at High Pressures and Temperatures (The Macmillan Co., 1936). [Google Scholar]
- 4.M. Raney, Method of producing finely-divided nickel, U.S. Patent 1,628,190 (1927).
- 5.M. Raney, Method of preparing catalytic material, U.S. Patent 1,563,587 (1925).
- 6.T. Osawa, Heterogeneous Catalysis, in Modern Organonickel Chemistry, Y. Tamaru, Ed. (Wiley-VCH, 2005), pp. 273–305. [Google Scholar]
- 7.K. Weissermel, H.-J. Arpe, Acetylene, in Industrial Organic Chemistry, (Wiley-VCH, ed. 4, 2003), pp. 91–105. [Google Scholar]
- 8.Sales J., Mushtaq F., Christou M. D., Nomen R., Study of major accidents involving chemical reactive substances: Analysis and lessons learned. Process Saf. Environ. Prot. 85, 117–124 (2007). [Google Scholar]
- 9.Roessler F., Catalytic hydrogenation in the liquid phase. Chimia 57, 791–798 (2003). [Google Scholar]
- 10.Jagadeesh R. V., Surkus A.-E., Junge H., Pohl M.-M., Radnik J., Rabeah J., Huan H., Schünemann V., Brückner A., Beller M., Nanoscale Fe2O3-based catalysts for selective hydrogenation of nitroarenes to anilines. Science 342, 1073–1076 (2013). [DOI] [PubMed] [Google Scholar]
- 11.Westerhaus F. A., Jagadeesh R. V., Wienhöfer G., Pohl M.-M., Radnik J., Surkus A.-E., Rabeah J., Junge K., Junge H., Nielsen M., Brückner A., Beller M., Heterogenized cobalt oxide catalysts for nitroarene reduction by pyrolysis of molecularly defined complexes. Nat. Chem. 5, 537–543 (2013). [DOI] [PubMed] [Google Scholar]
- 12.Jagadeesh R. V., Junge H., Beller M., Green synthesis of nitriles using non-noble metal oxides-based nanocatalysts. Nat. Commun. 5, 4123 (2014). [DOI] [PubMed] [Google Scholar]
- 13.Cui X., Li Y., Bachmann S., Scalone M., Surkus A.-E., Junge K., Topf C., Beller M., Synthesis and characterization of iron–nitrogen-doped graphene/core–shell catalysts: Efficient oxidative dehydrogenation of N-heterocycles. J. Am. Chem. Soc. 137, 10652–10658 (2015). [DOI] [PubMed] [Google Scholar]
- 14.Pisiewicz S., Formenti D., Surkus A.-E., Pohl M.-M., Radnik J., Junge K., Topf C., Bach-mann S., Scalone M., Beller M., Synthesis of nickel nanoparticles with N-doped graphene shells for catalytic reduction reactions. ChemCatChem 8, 129–134 (2016). [Google Scholar]
- 15.Hahn G., Ewert J.-K., Denner C., Tilgner D., Kempe R., A reusable mesoporous nickel nanocomposite catalyst for the selective hydrogenation of nitroarenes in the presence of sensitive functional groups. ChemCatChem 8, 2461–2465 (2016). [Google Scholar]
- 16.Casanovas J., Ricart J. M., Rubio J., Illas F., Jiménez-Mateos J. M., Origin of the large N 1s binding energy in x-ray photoelectron spectra of calcined carbonaceous materials. J. Am. Chem. Soc. 118, 8071–8076 (1996). [Google Scholar]
- 17.Pels J. R., Kapteijn F., Moulijn J. A., Zhu Q., Thomas K. M., Evolution of nitrogen functionalities in carbonaceous material during pyrolysis. Carbon 33, 1641–1653 (1995). [Google Scholar]
- 18.You Y., Mayyas M., Xu S., Mansuri I., Gaikwad V., Munroe P., Sahajwalla V., Joshi R. K., Growth of NiO nanorods, SiC nanowires and monolayer graphene via a CVD method. Green Chem. 19, 5599–5607 (2017). [Google Scholar]
- 19.Chen X., Guan J., Sha G., Gao Z., Williams C. T., Liang C., Preparation and magnetic properties of single phase Ni2Si by reverse Rochow reaction. RSC Adv. 4, 653–659 (2014). [Google Scholar]
- 20.Valeri S., Del Pennino U., Sassaroli P., Oxidation behaviour of nickel silicides investigated by AES and XPS. Surf. Sci. 134, L537–L542 (1983). [Google Scholar]
- 21.Spit F. H. M., Gupta D., Tu K. N., Diffusivity and solubility of Ni (63Ni) in monocrystalline Si. Phys. Rev. B 39, 1255–1260 (1989). [DOI] [PubMed] [Google Scholar]
- 22.Furukawa S., Komatsu T., Intermetallic compounds: Promising inorganic materials for well-structured and electronically modified reaction environments for efficient catalysis. ACS Catal. 7, 735–765 (2017). [Google Scholar]
- 23.Chen X., Zhao A., Shao Z., Li C., Williams C. T., Liang C., Synthesis and catalytic properties for phenylacetylene hydrogenation of silicide modified nickel catalysts. J. Phys. Chem. C 114, 16525–16533 (2010). [Google Scholar]
- 24.Chen X., Zhang M., Yang K., Williams C. T., Liang C., Raney Ni–Si catalysts for selective hydrogenation of highly concentrated 2-butyne-1,4-diol to 2-butene-1,4-diol. Catal. Lett. 144, 1118–1126 (2014). [Google Scholar]
- 25.Chen X., Li M., Guan J., Wang X., Williams C. T., Liang C., Nickel–silicon intermetallics with enhanced selectivity in hydrogenation reactions of cinnamaldehyde and phenylacetylene. Ing. Eng. Chem. Res. 51, 3604–3611 (2012). [Google Scholar]
- 26.Chen X., Jin J., Sha G., Li C., Zhang B., Su D., Williams C. T.,Liang C., Silicon–nickel intermetallic compounds supported on silica as a highly efficient catalyst for CO methanation. Catal. Sci. Technol. 4, 53–61 (2014). [Google Scholar]
- 27.Chen X., Liu X., Wang L., Li M., Williams C. T., Liang C. H., High sulfur tolerance of Ni–Si intermetallics as hydrodesulfurization catalysts. RSC Adv. 3, 1728–1731 (2013). [Google Scholar]
- 28.Nuzzo R. G., Dubois L. H., Bowles N. E., Trecoske M. A., Derivatized, high surface area, supported nickel catalysts. J. Catal. 85, 267–271 (1984). [Google Scholar]
- 29.Chang Y.-Y., Peng D.-J., Wu H.-C., Tsaur C.-C., Shen C.-C., Tsai H.-Y., Chen J.-R., Revisiting of a silane explosion in a photovoltaic fabrication plant. Process Saf. Prog. 26, 155–158 (2007). [Google Scholar]
- 30.Simonneau A., Oestreich M., Formal SiH4 chemistry using stable and easy-to-handle surrogates. Nat. Chem. 7, 816–822 (2015). [DOI] [PubMed] [Google Scholar]
- 31.Zaheer M.,Keenan C. D., Hermannsdörfer J., Roessler E., Motz G., Senker J., Kempe R., Robust microporous monoliths with integrated catalytically active metal sites investigated by hyperpolarized 129Xe NMR. Chem. Mater. 24, 3952–3963 (2012). [Google Scholar]
- 32.Chen F., Kreyenschulte C., Radnik J., Lund H., Surkus A.-E., Junge K., Beller M., Selective semihydrogenation of alkynes with N-graphitic-modified cobalt nanoparticles supported on silica. ACS Catal. 7, 1526–1532 (2017). [Google Scholar]
- 33.Zhao M., Deng K., He L., Liu Y., Li G., Zhao H., Tang Z., Core–shell palladium nanoparticle@metal–organic frameworks as multifunctional catalysts for cascade reactions. J. Am. Chem. Soc. 136, 1738–1741 (2014). [DOI] [PubMed] [Google Scholar]
- 34.Zhao M., Yuan K., Wang Y., Li G., Guo J., Gu L., Hu W., Zhao H., Tang Z., Metal–organic frameworks as selectivity regulators for hydrogenation reactions. Nature 539, 76–80 (2016). [DOI] [PubMed] [Google Scholar]
- 35.He T., Pei Q., Chen P., Liquid organic hydrogen carriers. J. Energy Chem. 24, 587–594 (2015). [Google Scholar]
- 36.Yamaguchi R., Ikeda C., Takahashi Y., Fujita K.-i., Homogeneous catalytic system for reversible dehydrogenation–hydrogenation reactions of nitrogen heterocycles with reversible interconversion of catalytic species. J. Am. Chem. Soc. 131, 8410–8412 (2009). [DOI] [PubMed] [Google Scholar]
- 37.Moromi S. K., Siddiki S. M. A. H., Kon K., Toyao T., Shimizu K.-i., Acceptorless dehydrogenation of N-heterocycles by supported Pt catalysts. Catal. Today 281, 507–511 (2017). [Google Scholar]
- 38.Mikami Y., Ebata K., Mitsudome T., Mizugaki T., Jitsukawa K., Kaneda K., Reversible dehydrogenation-hydrogenation of tetrahydroquinoline-quinoline using a supported cooper nanoparticle catalyst. Heterocycles 82, 1371–1377 (2011). [Google Scholar]
- 39.Chakraborty S., Brennessel W. W., Jones W. D., A molecular iron catalyst for the acceptorless dehydrogenation and hydrogenation of N-heterocycles. J. Am. Chem. Soc. 136, 8564–8567 (2014). [DOI] [PubMed] [Google Scholar]
- 40.Xu R., Chakraborty S., Yuan H., Jones W. D., Acceptorless, reversible dehydrogenation and hydrogenation of N-heterocycles with a cobalt pincer catalyst. ACS Catal. 5, 6350–6354 (2015). [Google Scholar]
- 41.Deraedt C., Ye R., Ralston W. T., Toste F. D., Somorjai G. A., Dendrimer-stabilized metal nanoparticles as efficient catalysts for reversible dehydrogenation/hydrogenation of N-heterocycles. J. Am. Chem. Soc. 139, 18084–18092 (2017). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/4/6/eaat0761/DC1
section S1. Materials and methods
section S2. Procedure for the catalyst preparation (fig. S1).
section S3. TEM and EDX data (fig. S2 to S5)
section S4. XRD diffraction patterns and data (figs. S6 to S10)
section S5. XPS spectra and data (fig. S11)
section S6. Elemental analysis of the catalysts and BET (fig. S12)
section S7. Thermogravimetric analysis (TGA; figs. S13 and S14)
section S8. Procedures for hydrogenation reactions (figs. S15 and S16)
section S9. Product characterization
section S10. Procedures for dehydrogenation reactions (figs. S17 to S19)
section S11. Catalyst recycling
section S12. Nuclear magnetic resonance (NMR) spectral charts
fig. S1. Ni-phen@SiO2-1000 after pyrolysis.
fig. S2. TEM images of the Ni-phen@SiO2-1000.
fig. S3. ABF and HAADF-STEM images of intermetallic nickel silicide catalyst Ni-phen@SiO2-1000.
fig. S4. HAADF-STEM and EDX measurement of the intermetallic nickel silicide catalyst Ni-phen@SiO2-1000.
fig. S5. ABF-, HAADF-STEM, and EDX measurement of the catalyst Ni@SiO2-1000 prepared without ligand.
fig. S6. Powder pattern of nickel-based catalysts supported on fumed silica.
fig. S7. Powder pattern of Ni-phen@SiO2-800 measured up to 148°2θ.
fig. S8. Powder pattern of various nickel-based catalysts with different ligands (S2.3).
fig. S9. Powder pattern of various nickel-based catalysts on various supports.
fig. S10. Powder pattern of nickel catalyst on silica gel and quartz (S2.3).
fig. S11. XPS measurement of the intermetallic nickel silicide catalyst.
fig. S12. N2 adsorption-desorption isotherms and BJH desorption pore size distribution.
fig. S13. TG-DSC-MS analysis of Ni-phen@SiO2-1000.
fig. S14. TG-DSC-MS analysis of Ni-phen@SiO2-1000.
fig. S15. Autoclave and glass vials used for hydrogenations.
fig S16. Concentration/time diagram for the hydrogenation of benzonitrile.
fig. S17. Manual burette setup.
fig. S18. Gas evolution from 1,2,3,4-tetrahydroquinaldine.
fig. S19. Gas composition analysis by GC.
table S1. Elemental analysis of the studied catalysts.
table S2. BET surface area and pore volume of the samples.
table S3. Hydrogenation of the standard substrates (Table 1 in the main text).
table S4. Hydrogenation of the nitrobenzene with SiO2-supported catalysts.
table S5. Recycling experiments.
table S6. Nickel leaching in the recycling experiments.
table S7. Effect of the pH on hydrogenation of nitrobenzene.