

ABSTRACT
The combination of CTLA-4 blockade ipilimumab (Ipi) with VEGF-A blocking antibody bevacizumab (Bev) has demonstrated favorable clinical outcomes in patients with advanced melanoma. Galectin-3 (Gal-3) plays a prominent role in tumor growth, metastasis, angiogenesis, and immune evasion. Here we report that Ipi plus Bev (Ipi-Bev) therapy increased Gal-3 antibody titers by 50% or more in approximately one third of treated patients. Antibody responses to Gal-3 were associated with higher complete and partial responses and better overall survival. Ipi alone also elicited antibody responses to Gal-3 at a frequency comparable to the Ipi-Bev combination. However, an association of elicited antibody responses to Gal-3 with clinical outcomes was not observed in Ipi alone treated patients. In contrast to being neutralized in Ipi-Bev treated patients, circulating VEGF-A increased by 100% or more in a subset of patients after Ipi treatment, with most having progressive disease. Among the Ipi treated patients with therapy-induced Gal-3 antibody increases, circulating VEGF-A was increased in 3 of 6 nonresponders but in none of 4 responders as a result of treatment. Gal-3 antibody responses occurred significantly less frequently (3.2%) in a cohort of patients receiving PD-1 blockade where high pre-treatment serum Gal-3 was associated with reduced OS and response rates. Our findings suggest that anti-CTLA-4 elicited humoral immune responses to Gal-3 in melanoma patients which may contribute to the antitumor effect in the presence of an anti-VEGF-A combination. Furthermore, pre-treatment circulating Gal-3 may potentially have prognostic and predictive value for immune checkpoint therapy.
KEYWORDS: antibody responses to galectin-3, anti-VEGF, immune therapy, galectin-3, melanoma
Introduction
Immune therapy targeting immune checkpoint CTLA-4 with ipilimumab (Ipi) has been demonstrated to improve overall survival of patients with metastatic melanoma.1,2 High circulating VEGF-A levels correlate with poor clinical outcomes to Ipi in melanoma patients.3 VEGF-A has known inhibitory activity on the immune system.4-8 As a result, angiogenesis is an enticing target for combination with immune checkpoint blockade.9
Ipilimumab plus bevacizumab (Ipi-Bev) triggered profound infiltration of lymphocytes into melanoma tumors in association with increased tumor endothelial activation, tumor expression of IP-10, and circulating TNFα, IL-1α, and IFN2α.9,10 Furthermore, Ipi-Bev therapy influenced immune cells and endothelial cells in the circulation, as well as many soluble factors including angiogenic factors, cytokines/chemokines and antibodies.10,11 Consistent with the suppressive role of CTLA-4 in humoral immune responses,12,13 Ipi-Bev elicited humoral immune responses to tumor and tumor stromal cell targets in patients with advanced melanoma.9-11,14 By screening human protein arrays with post-treatment plasma from Ipi-Bev treated patients, antibodies recognizing galectin-1 (Gal-1), −3 (Gal-3) and −9 (Gal-9) were detected.9,14 We have reported that Ipi-Bev induced functional humoral immunity to Gal-1 which was associated with favorable clinical outcomes.14 In the same cohort of patients, antibody responses to Gal-3 were also detected.9 Similar to Gal-1,15,16 Gal-3 belongs to the galectin family of β-galactoside-binding proteins and is expressed and secreted from tumor cells and tumor stromal cells, promoting tumor progression, angiogenesis and immune suppression in preclinical studies.17-22 Given its role in tumor biology and further support of the association of angiogenesis and immune regulation, the current study characterized humoral immune responses to Gal-3 in melanoma patients receiving Ipi, Ipi-Bev, or PD-1 blockade.
Results
Ipi and Ipi-Bev therapy elicited humoral immune responses to Gal-3
We have previously reported that Ipi-Bev therapy elicited humoral immune responses to several antigen targets in patients with metastatic melanoma.9,10 Protein arrays were screened with post-treatment plasma samples from three Ipi-Bev treated patients and one pembrolizumab (Pembro)-treated patient. Antibodies recognizing Gal-3 were detected in the Pembro-treated and two of the Ipi-Bev treated patients (Supplementary Figure S1). Gal-3 antibodies were further confirmed by immunoblot and/or ELISA analyses of pre- and post-treatment plasma samples of representative Ipi-Bev treated patients (Fig. 1A-C). We considered an increase by 50% (i.e. fold change = 1.5) in Gal-3 antibody titer as significant. This division point divides the distribution of Gal-3 antibody fold change values into the lower 3/4 and upper 1/4. Based on this cut-off, 14 (32.6%) of Ipi-Bev treated patients had antibody responses to Gal-3 as a function of the treatment (Fig. 1C). To better characterize Gal-3 antibody responses to Ipi-Bev treatment, we examined longitudinal changes in Gal-3 antibody titers in 4 patients with the longest survival.14 Similar to antibody responses to Gal-1 in these patients,14 Gal-3 antibodies increased following initial treatment of Ipi-Bev and lasted for months (Supplementary Figure S2A-D). Nonetheless, antibody responses to Gal-3 had varying dynamic patterns later in the course of treatment and Gal-3 antibody levels eventually declined. Similar dynamic changes were also observed in Gal-1 antibody responses,14 while patient-specific differences in dynamic patterns of Gal-1 and Gal-3 antibodies existed.
Figure 1.
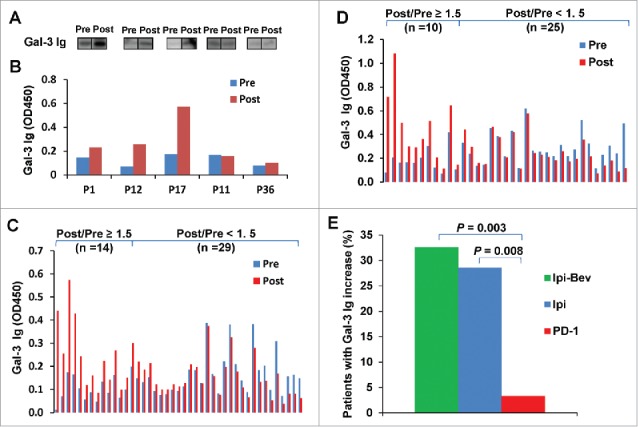
Ipi and Ipi-Bev elicited humoral immune responses to Gal-3. A and B) Immunoblot analysis (A) and ELISA (B) of Gal-3 antibodies in pre- and post-treatment plasma samples of representative melanoma patients treated with Ipi-Bev. C and D) Gal-3 antibody titers in pre- and post-treatment plasma samples of Ipi-Bev treated (C) and Ipi alone treated (D) patients. E) Proportions of Ipi-Bev, Ipi alone and PD-1 blockade treated patients with Gal-3 antibody fold changes ≥ 1.5. Patients receiving PD-1 blockade had the lowest incidence while Ipi and Ipi-Bev treated patients are comparable (Ipi-Bev vs. PD-1 blockade, P = 0.003; Ipi vs. PD-1 blockade, P = 0.008; Ipi-Bev vs. Ipi, P = 0.81).
To address the effect of anti-VEGF-A and anti-PD-1 on humoral immune responses to Gal-3, we also determined Gal-3 antibody titers in the pre- and post-treatment plasma samples from 35 Ipi treated and 31 PD-1 blockade treated patients. Increases in Gal-3 antibody titers by 50% or more as a result of treatment were seen in 10 (28.6%) Ipi treated and 1 (3.2%) PD-1 blockade treated patients (Fig. 1D and E).
We next asked if circulating Gal-3 antibodies could neutralize the biological activities of Gal-3. While Gal-3 can suppress T cell function by preventing the formation of functional secretory synapse,23 binding of Gal-3 to CD45 expressed on T cells suppresses T cell function with evidence for inducing apoptosis in T cells.24,25 We examined if detected Gal-3 antibodies from patietns post-treatment are functional in blocking binding of Gal-3 to CD45. Gal-3 was expressed in a fusion form (designated as HAS-Gal-3) with His, Avi, and SUMO tags at its N-terminus in bacterial cells in the presence of biotin to allow the Avi tag to be biotinylated. The Gal-3 sequence and biotinylation of purified HAS-Gal-3 was confirmed (Supplementary Figure S3A-C). Binding of HAS-Gal-3 to coated CD45 was confirmed to be Gal-3 and β-galactoside dependent as it was blocked by a neutralizing antibody of Gal-3 and β-lactose but not a control antibody and sucrose (Supplementary Figure S3D). To determine if endogenous Gal-3 antibodies can block the binding of Gal-3 to CD45, post-treatment plasma samples with increased Gal-3 antibody titer were used (Supplementary Figure S4 A). Incubation of the sample with coated HAS-Gal-3 protein but not BSA (as control) resulted in depletion of Gal-3 antibodies (Gal-3 Ig, Supplementary Figure S4B). We then compared the binding of HAS-Gal-3 to coated CD45 in the presence of control (BSA pre-absorbed) and Gal-3 antibody-depleted plasma samples. Higher binding of HAS-Gal-3 to CD45 was detected with Gal-3 antibody-depleted samples compared to control samples (Supplementary Figure S4C), indicating that depletion of endogenous Gal-3 antibodies increased binding of Gal-3 to CD45. Similarly, pre-absorption of Gal-3 neutralizing antibody with HAS-Gal-3 but not BSA depleted the antibody (Gal-3 Ab, Supplementary Figure S4B) and restored binding of HAS-Gal-3 to CD45 (Supplementary Figure S4D). These findings suggest that post-treatment detected Gal-3 antibodies in patients may be capable of blocking Gal-3 binding to CD45.
Antibody responses to Gal-3 correlated with clinical outcomes to Ipi-Bev therapy
The majority of Ipi-Bev patients with increased Gal-3 antibody responses (Gal-3 antibody fold change ≥ 1.5) had CR/PR or SD (Table 1; Fig. 2A). Increased antibody responses to Gal-3 occurred at a substantial higher frequency in CR/PR patients compared to SD and PD patients (Fig. 2A; Supplementary Table S1). Patients who experienced increased Gal-3 antibody responses had a significantly higher CR/PR rate than those who did not (Fig. 2B; Supplementary Table S2). The median survival of patients with no increased Gal-3 antibody responses was 73 weeks (95% CI: 55 to 83 weeks), while that of patients with increased Gal-3 antibody responses has not been reached (Fig. 2C). In Ipi alone treated patients, Gal-3 antibody responses were increased at comparable frequency among CR/PR, SD and PD patients (Fig. 2D; Supplementary Table S1). Ipi induced Gal-3 antibody responses were not associated with response rate (Fig. 2E; Supplementary Table S3) and survival (Fig. 2F).
Table 1.
Circulating VEGF-A concentrations in Ipi-Bev and Ipi treated patients with therapy-induced antibody responses to Gal-3.
| Ipi-Bev treated patients |
Ipi treated patients |
|||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Patient ID | Gal-3 Ig fold change | Pre VEGF-A (pg/ml) | Post VEGF-A (pg/ml) | VEGF-A fold change | Clinical response | Patient ID | Gal-3 Ig fold change | Pre VEGF-A (pg/ml) | Post VEGF-A (pg/ml) | VEGF-A fold change | Clinical response | |
| P12 | 3.39 | 167.9 | ULD | < 0.1 | CR | P316 | 1.53 | 494.6 | 462.9 | 0.94 | PR | |
| P1 | 1.68 | 19.4 | ULD | < 0.1 | PR | P164 | 8.56 | 238.7 | 259.6 | 1.09 | SD | |
| P5 | 2.66 | 199.5 | ULD | < 0.1 | PR | P171 | 3.37 | 314.9 | 200.4 | 0.64 | SD | |
| P6 | 1.52 | 119.3 | 10.4 | 0.1 | PR | P252 | 1.91 | 219.4 | 147.0 | 0.67 | SD | |
| P8 | 2.04 | 194.0 | ULD | < 0.1 | PR | P166 | 4.31 | 267.5 | 278.2 | 1.04 | PD | |
| P13 | 1.82 | 173.6 | ULD | < 0.1 | PR | P193 | 1.67 | 167.9 | 389.5 | 2.32 | PD | |
| P20 | 1.82 | 42.9 | ULD | < 0.1 | PR | P259 | 1.78 | 349.9 | 838.0 | 2.40 | PD | |
| P16 | 1.53 | 164.1 | ULD | < 0.1 | SD | P315 | 1.55 | 500.2 | 442.3 | 0.88 | PD | |
| P17 | 3.30 | 102.9 | 7.0 | 0.1 | SD | P318 | 1.52 | 476.3 | 431.8 | 0.91 | PD | |
| P26 | 31.25 | 143.0 | ULD | < 0.1 | SD | P192 | 1.91 | ULD | 66.4 | >2.0 | PD | |
| PBI-1 | 1.68 | 284.1 | ULD | < 0.1 | SD | |||||||
| PMGH-1 | 2.11 | 117.3 | ULD | < 0.1 | SD | |||||||
| P4 | 1.51 | 98.8 | ULD | < 0.1 | PD | |||||||
| P30 | 1.51 | 214.0 | ULD | < 0.1 | PD | |||||||
ULD, under limit of detection; CR, complete response; PR, partial response; SD, stable disease; PD, progressive disease. Patients with VEGF-A fold change ≥ 2 are indicated in red color.
Figure 2.
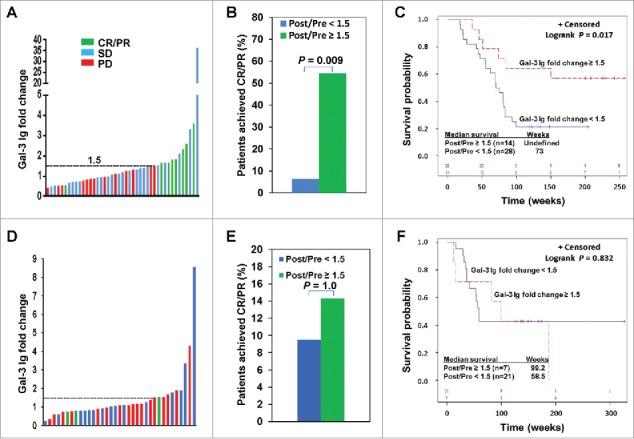
Ipi-Bev-induced humoral immune responses to Gal-3 correlated with better clinical outcomes. A) Patients were plotted based on their Gal-3 antibody fold changes. Each bar represents a patient and the color of the bar indicates clinical response of the patient. CR/PR, complete/partial responses; SD, stable disease; PD, progressive disease. B) Higher percentage of patients with Gal-3 antibody fold changes ≥ 1.5 achieved CR/PR than those with Gal-3 antibody fold changes < 1.5 (P = 0.009). C) Conditional landmark analysis (18 weeks) of patients based on Gal-3 antibody fold change ≥ 1.5 or < 1.5 (P = 0.017). The median survival of the patients with Gal-3 antibody fold change < 1.5 was 73 weeks (95% CI, 55 to 83), while that of patients with fold change ≥ 1.5 was unreached. D) Ipi treated patients were plotted based on their Gal-3 antibody fold changes. E) Ipi-induced Gal-3 antibody responses were not associated with clinical responses. F) Ipi-induced Gal-3 antibody responses were not associated with overall survival. The median survival of the patients with Gal-3 antibody fold change ≥ 1.5 and < 1.5 was 99.2 weeks (95% CI: 12.6 – 186.8) and 58.5 weeks (95% CI: 36.4 – ∞) respectively. However, the difference in survival is not statistically significant (P = 0.832).
Circulating Gal-3 was associated with clinical outcomes to PD-1 blockade
Gal-3 was expressed in all melanoma cell lines tested as well as in tumor associated endothelial cells (TEC) cultured in vitro as determined by quantitative RT-PCR (qRT-PCR) and immunoblot (Supplementary Figures S5A and B). Gal-3 was also expressed in HUVEC and HDMEC, but appears to be at lower levels compared to TEC (Supplementary Figures S5A and B). High Gal-3 expression was detected in macrophages that were differentiated from monocytes with M-CSF (Supplementary Figure S5A). Gal-3 expression did not appear to be significantly altered in melanoma cells, HDMEC and TEC by bevacizumab as revealed by both qRT-PCR and immunoblot (Supplementary Figures S5C and D). We examined Gal-3 expression in paired pre-treatment and post-treatment tumor biopsies of 8 Ipi-Bev treated patients consisting of 3 PR, 3 SD, and 2 PD patients (Supplementary Table S4). Gal-3 was expressed in pre-treatment tumor cells of all the patients, and was either increased, decreased or not changed after Ipi-Bev treatment (Fig. 3A and B; Supplementary Table S4). Gal-3 was detected in TEC of a subset of the patients prior to treatment, and was decreased or not changed by Ipi-Bev (Fig. 3A and B; Supplementary Table S4). We also measured Gal-3 in the circulation of Ipi-Bev and PD-1 blockade treated patients. In Ipi-Bev treated patients, circulating Gal-3 levels were either increased, decreased, or not altered in responses to treatment (Fig. 3C), and were comparable across patients with CR/PR, SD or PD before and after treatment (Supplementary Figure S6A and B). Furthermore, pre-treatment Gal-3 was not associated with OS in these patients (Supplementary Figure S6C). In contrast, high pre-treatment serum Gal-3 was associated with reduced OS and response in PD-1 blockade treated patients: median survival in patients with high pre-treatment Gal-3 was 12.1 months, and was not reached in patients with low pre-treatment Gal-3 (Fig. 4A). Patients with high pre-treatment Gal-3 had a hazard of death that was significantly increased almost five-fold compared to patients with lower Gal-3 ((HR: 4.8, 95% CI: 1.4 to 16.8), P = 0.01). Among patients with low pre-treatment Gal-3, 50% of the patients achieved PR compared with 24% in patients with high pre-treatment Gal-3 (Fig. 4B). Rates of disease progression were 18% compared with 57% in patients with low versus high pre-treatment Gal-3, respectively (Fig. 4B). In addition to Gal-3, high pre-treatment circulating Gal-1 was associated with reduced OS in this cohort of patients (Fig. 4C).
Figure 3.
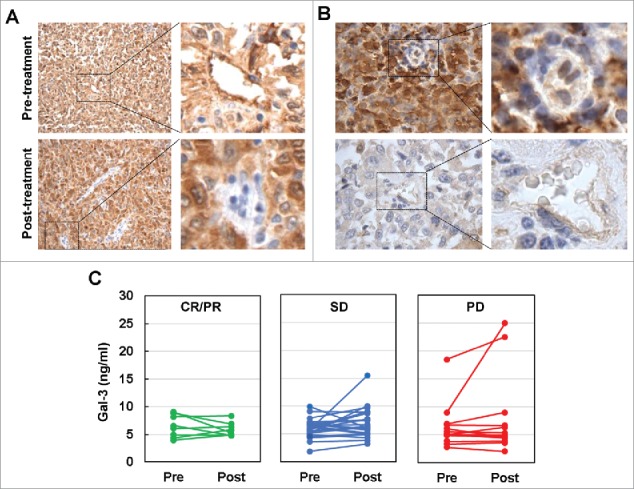
Ipi-Bev influenced Gal-3 expression in melanoma tumors and in the circulation of melanoma patients. A) Patient P4. Gal-3 expression was reduced in TEC while modestly increased in tumor cells after Ipi-Bev treatment. B) Patient P20. Gal-3 expression was downregulated in tumor cells but not in TEC as a result of treatment. C) Circulating Gal-3 levels in pre- and post-treatment plasma samples. Patients were grouped into CR/PR, SD and PD subgroups. In each subgroup, increase, decrease, or no change in Gal-3 levels were seen.
Figure 4.
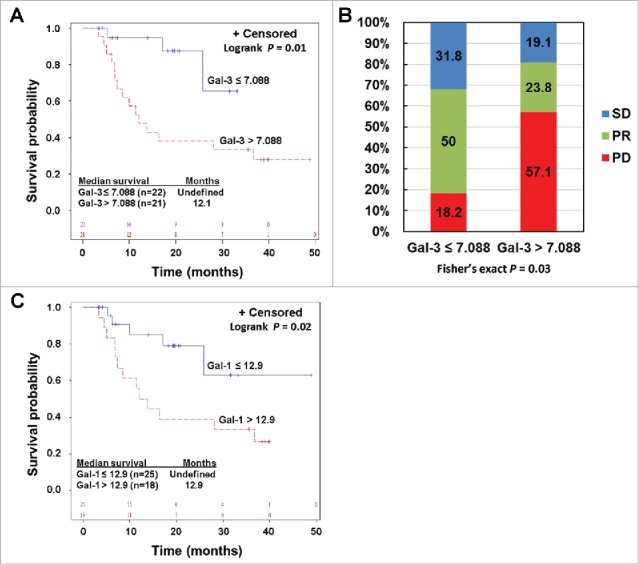
Pre-treatment circulating Gal-3 and Gal-1 is respectively associated with clinical outcomes in PD-1 blockade treated patients. A) Kaplan-Meier survival curves of patients based on pre-treatment Gal-3 levels. Median survival in patients with Gal-3 > 7.088 ng/ml was 12.1 months (95% CI: 7 to 37 months), and was not reached in patients with Gal-3 ≤ 7.088 ng/ml (P = 0.01). B) Responses to PD-1 blockade based on pre-treatment Gal-3 levels. Percentages of patients achieved PR or SD or experienced PD are also indicated. C. High pre-treatment circulating Gal-1 was associated with reduced survival in PD-1 blockade treated patients. Patients were divided into high and low groups based on a division point of circulating Gal-1 of 12.9 ng/ml. Median survival in patients with high Gal-1 was 12.9 (95% CI: 6.9 – 36.7) months, and was not reached in patients with low Gal-1 (P = 0.02).
Circulating VEGF-A levels increased in a subset of Ipi treated melanoma patients
Our previous studies revealed that circulating VEGF-A was not significantly altered in Ipi alone treated patients overall,3 but was effectively neutralized by Bev in Ipi-Bev treated patients.10 These findings suggest circulating VEGF-A levels may link to the observed discrepancy in the association of Gal-3 antibody responses and clinical outcomes between Ipi-Bev and Ipi alone treated patients since both groups of patients had similar increased Gal-3 antibody responses with treatment. We therefore measured VEGF-A concentrations in the pre- and post-treatment plasma samples of the Ipi alone treated patients whose antibody responses to Gal-3 were determined. Similar to our previous report,3 Ipi alone did not significantly alter circulating VEGF-A overall (Supplementary Table S5; Supplementary Figure S7A). The pre-treatment and post-treatment VEGF-A levels of CR, PR, and SD patients were not significantly different from those of PD patients (Supplementary Table S5; Supplementary Figure S7B). Nonetheless, 4 of 16 (25%) PD patients had VEGF-A increased by 100% or more compared to 1 in 17 (5.9%) CR/PR/SD patients (Supplementary Figure S7C). Among the Ipi treated patients with increased Gal-3 antibody reactions, PD patients and CR/PR/SD patients had comparable pre-treatment plasma VEGF-A levels (Supplementary Table S5). Although PD patients had a higher median post-treatment VEGF-A than CR/PR/SD patients, it did not reach statistical significance (Supplementary Table S5; Supplementary Figure S7D). Nonetheless, three of the 6 PD patients with increased Gal-3 antibody levels had VEGF-A levels increased by 100% in the post-treatment samples while none of the 4 CR/PR/SD patients did (Table 1). In the Ipi-Bev treated cohort, all patients with elevated Gal-3 antibody titers had pronounced reduction in serum VEGF-A with most them being undetectable post-treatment (Table 1). These findings suggest a possible link of circulating VEGF-A levels with the discrepancy in the association of Gal-3 antibody responses with clinical outcomes in Ipi and Ipi-Bev patients. However, due to small numbers of patients in each group, further validation in larger cohorts of patients are needed.
Discussion
Ipi-Bev therapy of melanoma has demonstrated durable clinical outcomes, associated with increased circulating memory effector T cells, tumor lymphocyte infiltration, and humoral immune responses to tumor and tumor stromal targets including galectins.9,10 In the current study, we found that Ipi-Bev treatment increased circulating levels of antibodies to Gal-3 in a subset of melanoma patients which was associated with improved clinical outcomes. These antibodies appear to be functional in blocking the binding of Gal-3 to CD45, although further confirmation is needed. Similarly, neutralizing Gal-3 antibodies have recently been reported to be induced in patients with pancreatic ductal adenocarcinoma (PDA) who responded to a GM-CSF-secreting allogeneic PDA vaccine.26 Our findings suggest that treatment-induced humoral immunity to Gal-3 may add an additional anti-tumor mechanism to Ipi-Bev.
We have independently identified Ipi-Bev elicited humoral antibody responses to Gal-3 and Gal-1.9,14 Both Gal-1 and Gal-3 have protumoral, proangiogenic, and immunosuppressive activities. In this context, it is interesting that antibody responses to Gal-1 and Gal-3 were elicited at comparable frequency in Ipi-Bev treated patients (Gal-1, 37.2%;14 Gal-3, 32.6%) and a higher fraction of Ipi alone treated patients had antibody responses to Gal-3 compared to Gal-1 (Gal-3, 28.6%; Gal-1, 17.1%14), while PD-1 blockade rarely induced antibody responses to both Gal-1 (3.2%)14 and Gal-3 (3.2%). This may reflect the different activities of CTLA-4 and PD-1 in regulation of humoral immune response and antibody production through differentially regulating T follicular helper and T follicular regulatory cells, as revealed in animal and in vitro studies.12,13,27,28
Like Gal-1, Gal-3 is known to promote tumor progression, angiogenesis, and immune suppression in preclinical studies.19-21 Specifically, extracellular Gal-3 negatively influences multiple components of antitumor immunity, including T cell activity,21,26,29-31 NK cell mediated immune surveillance,32-34 and DC activation and expansion.26,35,36 Serum Gal-3 levels are significantly higher in melanoma patients than normal healthy controls.37 High circulating Gal-3 levels in melanoma patients are associated with poor prognosis.38 Circulating Gal-3 levels were corelated with circulating Gal-1 pre-treatment, post-treatment, and in fold change respectively in Ipi-Bev- and PD-1 blockade treated patients (Supplementary Table S6). Consistent with their immunoinhibitory activity, the current study demonstrates that high pre-treatment circulating Gal-3 and Gal-1 was respectively associated with poor clinical outcomes to PD-1 blockade. These findings suggest that circulating Gal-3 and Gal-1 may be of prognostic or predicative value in anti-PD-1 therapy and warrant further investigation. It is intriguing that these associations were not observed in Ipi-Bev treated patients as reported here and in our previous study.14 One potential reason for this discrepancy could be that Ipi-Bev elicited antibodies to Gal-3 and Gal-114 more frequently than PD-1 blockade. Enhanced immunity to these galectins might impede their protumoral activities, weakening the association of their pre-treatment levels with clinical outcomes. Given the associated outcomes with pre-treatment Gal-3 and Gal-1 levels and lack of Gal-3 and Gal-1 humoral immune responses in PD-1 treated patients, the current data suggests an interest in future investigation of neutralizing Gal-3 and Gal-1 antibodies with anti-PD-1 therapies.
Unlike Ipi-Bev treated patients, an association of antibody responses to Gal-3 with clinical outcomes was not observed in Ipi alone treated patients. The reasons behind this discrepancy include sample bias inevitably associated with limited sample size. Of note, Ipi alone and Ipi-Bev treated patients had significant differences in serum VEGF-A levels: while circulating VEGF-A was effectively blocked in Ipi-Bev treated patients,10 serum VEGF-A was not significantly reduced in Ipi treated patients. Our findings together suggest that antibody responses to Gal-3 could be beneficial, but this benefit may be impeded by circulating VEGF-A. In addition to its prominent angiogenic activity, VEGF-A is known to have potent immunosuppressive activity via enhancing expression of immunoinhibitory molecules on activated T cells,39 promoting MDSC and Treg expansion, and inhibiting T cell trafficking, DC maturation and antigen presentation.4-8 VEGF-A and Gal-3 are also known to co-ordinate each other in promoting angiogenesis.40,41 Gal-3 has been shown to induce VEGF-A expression in ovarian cancer cells and is required for TGFβ-induced VEGF-A release from macrophages.42,43 As a result, Gal-3 neutralization may enhance the antitumor effect of anti-VEGF-A therapy. Gal-3 also has VEGF-A independent immunosuppressive activity and tumor promoting activity.21 Blocking of both Gal-3 and VEGF-A may therefore result in synergistic antitumor effects by inhibiting angiogenesis, metastasis and immunosuppression. As a result, there may not exist such clinical impact of Gal-3 blockade in the absence of anti-VEGF-A. Further investigations targeting Gal-3 as well as combinations with anti-VEGF-A, other anti-angiogenesis treatments, and immune checkpoint blockade are worthy to consider for cancer therapy.
Methods
Collection of patient plasma and serum samples
Pre-treatment and post-treatment blood samples were collected from patients with metastatic melanoma who received Ipi, Ipi-Bev or PD-1 blockade [Nivolumab (Nivo); pembrolizumab (Pembro)] on Dana-Farber/Harvard Cancer Center Institutional Review Board (IRB) approved protocols. The collection of plasma and serum samples has been previously described.10,11
Detection of Gal-3 antibody in patient plasma samples
Gal-3 antibodies were detected in screening of protein arrays with ∼9,000 distinct recombinant human proteins with post-treatment plasma from three Ipi-Bev treated patients (including 1 CR and 2 PR patients) and one pembrolizumab (Pembro) treated patient.9 Gal-3 antibodies in the plasma samples were further confirmed by immunoblot analyses and determined by ELISA using recombinant human Gal-3 (R&D Systems, 8259-GA) and the previously described protocols.10,11 ELISA of Gal-3 antibodies were run in duplicate and the mean values are taken as the titers.
Depletion of Gal-3 neutralizing antibody and Gal-3 antibodies from plasma samples
Biotinylated Gal-3 fused with His, Avi, and SUMO tags at the N-terminus (designated as HAS-Gal-3) was expressed and purified as described in the supplementary section. Recombinant HAS-Gal-3 or BSA was coated onto a 96-well plate overnight at 4°C. The plate was blocked with 2% BSA and incubated with Gal-3 neutralizing antibody (clone B2C10, Santa Cruz Biotechnologies) diluted in PBS plus 0.2% BSA or plasma diluted in PBS for 8h. This depletion step was repeated 3 times. Depletion of Gal-3 antibodies was confirmed by immunoblot analysis.
Binding of Gal-3 to CD45
Recombinant human CD45 (R&D Systems, 1430-CD) was coated onto 96-well plates (25 ng/well) at 4°C overnight followed by blocking with 2% BSA in PBS for 1 hour at RT. HAS-Gal-3 (25 ng/well) in PBS plus 0.1% BSA was added to each well coated with CD45 and incubated for 1 hour at RT, followed by washing with PBS and incubation with streptavidin-HRP diluted in PBS with 1% BSA for 1 hour at RT. The plates were thoroughly washed with PBS and developed with TBM (Sigma, 34028). The reaction was stopped with 1N HCl and OD450 and OD570 were read in a microplate reader. In some experiments, HAS-Gal-3 was pre-incubated with Gal-3 neutralizing antibody or plasma samples preabsorbed with BSA or HAS-Gal-3 prior to incubation with coated CD45. Additionally, HAS-Gal-3 was incubated with coated CD45 in the presence of 5 mM lactose or sucrose to confirm β-galactoside dependent binding of HAS-Gal-3 to CD45.
Determination of Gal-3 and VEGF-A in patient serum samples
Gal-3 and VEGF-A concentrations in serum and plasma samples were determined in duplicate using Luminex assay kits (R&D Systems). The blood samples were diluted as recommended by the manufacturer (1:50 for Gal-3 and 1:2 for VEGF-A). The instructions for Luminex assays provided by the manufacturer were followed. VEGF-A concentrations in sera of Ipi-Bev treated patients were determined using Luminex assay kits from Millipore.10
Immunohistochemistry for Gal-3
Tumor sections were deparaffinized, rehydrated and heated in a steamer for 30 minutes for antigen retrieval in citrate buffer pH 6.0. After cooling, sections were incubated with peroxidase block for five minutes, and then serum-free protein block (DAKO, X0909) for 20 minutes. Slides were then incubated at room temperature for one hour with mouse monoclonal Gal-3 antibody (Leica microsystems, NCL-GAL3, 1:250) diluted in Da Vinci green diluent (Biocare Medical, PD900). The slides were incubated with Envision anti-mouse HRP-labeled polymer (DAKO, K4000) for 30 minutes. The slides were visualized with diaminobenzidine (DAKO), washed in distilled water, hematoxylin counterstained, dehydrated and mounted. Positive and negative controls were included in each panel of staining. All the slides were evaluated and scored by a pathologist (X.L.) blinded to clinical data. All the scoring was performed twice with at least three-day interval. A subset of samples for each marker was also reviewed by a second pathologist (S.R.) unaware of other data. H-Scores were calculated by multiplying staining intensity (1, 2, 3) with staining percentage (0% –100%).
Statistical analysis
The associations of antibody responses to Gal-3 and overall survival to Ipi-Bev were assessed using two analytic techniques as we previously described.14 An 18-week conditional landmark analysis, which corresponded to the maximum number of weeks between samples, was used in the first technique. Forty-two patients who were alive at 18 weeks were classified according to whether or not they achieved a meaningful fold-change for Gal-3 antibodies and followed forward in time. The second technique was based on the extended Cox model with time-dependent covariates. All 43 patients were initially classified as not having a meaningful fold-change for Gal-3 antibodies. At the time fold change increased to ≥ 1.5, a division point corresponding to a division of the distribution of fold change values into the lower 3/4 and upper 1/4, the patient was re-classified as having achieved a meaningful change. To evaluate if an increase in Gal-3 antibody titers was associated with subsequent clinical response (RECIST) in the patients, 27 patients who were assessed for antibody changes on or before the date that response was first recorded were included. The median time between pre-treatment and post-treatment antibody assessments in these patients was 8 weeks (range: 4 to 14 weeks). Clinical response was dichotomized as complete response/partial response (CR/PR) or stable disease/progressive disease (SD/PD, unevaluable). To assess the associations between pre-treatment circulating Gal-3 or Gal-1 and clinical outcomes, an “optimum” cut point was estimated using the PD-1 dataset as a test set based on the method of Contal-O'Quigley using leave-one-out jackknife cross-validation. Each sample was divided into high and low using the relevant cut point. The distributions of overall survival based on pre-treatment Gal-3 were based on the method of Kaplan-Meier. Cox proportional hazards regression models based on the designated cut point were used to estimate hazard ratios and 95% confidence intervals. p-values are based on the Wald chi-squared test. Comparisons of response according to the optimum pre-treatment cut point or antibody change were based on Fisher's exact tests. The differences in binding of Gal-3 to CD45 were evaluated using Wilcoxon signed-rank test. P < 0.05 was considered statistically significant for all comparisons. There were no corrections for multiple comparisons.
Supplementary Material
Funding Statement
HHS | NIH | National Cancer Institute (NCI) (NIH CA143832) NIH CA143832 (F.S.H.), the Melanoma Research Alliance (F.S.H.), the Sharon Crowley Martin Memorial Fund for Melanoma Research (F.S.H.) and the Malcolm and Emily Mac Naught Fund for Melanoma Research (F.S.H.) at Dana-Farber Cancer Institute, Genentech/Roche, and Bristol-Myers Squibb.
Disclosure of potential conflicts of interest
F.S.H has served as a consultant to Genentech and has received research support to institution from Bristol-Myers Squibb. Patent application antibody responses to Gal-3 as biomarker is pending.
References
- 1.Hodi FS, O'Day SJ, McDermott DF, Weber RW, Sosman JA, Haanen JB, Gonzalez R, Robert C, Schadendorf D, Hassel JC, et al.. Improved survival with ipilimumab in patients with metastatic melanoma. N Eng J Med. 2010;363:711–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Robert C, Thomas L, Bondarenko I, O'Day S, Weber J, Garbe C, Lebbe C, Baurain JF, Testori A, Grob JJ, et al.. Ipilimumab plus dacarbazine for previously untreated metastatic melanoma. N Eng J Med. 2011;364:2517–26. [DOI] [PubMed] [Google Scholar]
- 3.Yuan J, Zhou J, Dong Z, Tandon S, Kuk D, Panageas KS, Wong P, Wu X, Naidoo J, Page DB, et al.. Pretreatment serum VEGF is associated with clinical response and overall survival in advanced melanoma patients treated with ipilimumab. Cancer Immunol Res. 2014;2:127–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ohm JE, Carbone DP. VEGF as a mediator of tumor-associated immunodeficiency. Immunol Res. 2001;23:263–72. [DOI] [PubMed] [Google Scholar]
- 5.Oyama T, Ran S, Ishida T, Nadaf S, Kerr L, Carbone DP, Gabrilovich DI. Vascular endothelial growth factor affects dendritic cell maturation through the inhibition of nuclear factor-kappa B activation in hemopoietic progenitor cells. J Immunol. 1998;160:1224–32. [PubMed] [Google Scholar]
- 6.Vanneman M, Dranoff G. Combining immunotherapy and targeted therapies in cancer treatment. Nat Rev Cancer. 2012;12:237–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Shrimali RK, Yu Z, Theoret MR, Chinnasamy D, Restifo NP, Rosenberg SA. Antiangiogenic agents can increase lymphocyte infiltration into tumor and enhance the effectiveness of adoptive immunotherapy of cancer. Cancer Res. 2010;70:6171–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Huang H, Langenkamp E, Georganaki M, Loskog A, Fuchs PF, Dieterich LC, Kreuger J, Dimberg A. VEGF suppresses T-lymphocyte infiltration in the tumor microenvironment through inhibition of NF-kappaB-induced endothelial activation. FASEB J. 2015;29:227–38. [DOI] [PubMed] [Google Scholar]
- 9.Hodi FS, Lawrence D, Lezcano C, Wu X, Zhou J, Sasada T, Zeng W, Giobbie-Hurder A, Atkins MB, Ibrahim N, et al.. Bevacizumab plus ipilimumab in patients with metastatic melanoma. Cancer Immunol Res. 2014;2:632–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wu X, Giobbie-Hurder A, Liao X, Lawrence D, McDermott D, Zhou J, Rodig S, Hodi FS. VEGF neutralization plus CTLA-4 blockade alters soluble and cellular factors associated with enhancing Lymphocyte infiltration and humoral recognition in melanoma. Cancer Immunol Res. 2016;4:858–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wu X, Giobbie-Hurder A, Liao X, Connelly C, Connolly EM, Li J, Manos MP, Lawrence D, McDermott D, Severgnini M, et al.. Angiopoietin-2 as a biomarker and target for immune checkpoint therapy. Cancer Immunol Res. 2017;5:17–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sage PT, Paterson AM, Lovitch SB, Sharpe AH. The coinhibitory receptor CTLA-4 controls B cell responses by modulating T follicular helper, T follicular regulatory, and T regulatory cells. Immunity. 2014;41:1026–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sage PT, Sharpe AH. T follicular regulatory cells in the regulation of B cell responses. Trends Immunol. 2015;36:410–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wu X, Li J, Connolly EM, Liao X, Ouyang J, Giobbie-Hurder A, Lawrence D, McDermott D, Murphy G, Zhou J, et al.. Combined Anti-VEGF and Anti-CTLA-4 therapy elicits humoral immunity to Galectin-1 which is associated with favorable clinical outcomes. Cancer Immunol. Res. 2017;5:446–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Camby I, Le Mercier M, Lefranc F, Kiss R. Galectin-1: a small protein with major functions. Glycobiology. 2006;16:137R–57R. [DOI] [PubMed] [Google Scholar]
- 16.Astorgues-Xerri L, Riveiro ME, Tijeras-Raballand A, Serova M, Neuzillet C, Albert S, Raymond E, Faivre S. Unraveling galectin-1 as a novel therapeutic target for cancer. Cancer Treatment Rev. 2014;40:307–19. [DOI] [PubMed] [Google Scholar]
- 17.Liu FT, Rabinovich GA. Galectins as modulators of tumour progression. Nat Rev Cancer. 2005;5:29–41. [DOI] [PubMed] [Google Scholar]
- 18.Thijssen VL, Poirier F, Baum LG, Griffioen AW. Galectins in the tumor endothelium: opportunities for combined cancer therapy. Blood. 2007;110:2819–27. [DOI] [PubMed] [Google Scholar]
- 19.Radosavljevic G, Volarevic V, Jovanovic I, Milovanovic M, Pejnovic N, Arsenijevic N, Hsu DK, Lukic ML. The roles of Galectin-3 in autoimmunity and tumor progression. Immunol Res. 2012;52:100–10. [DOI] [PubMed] [Google Scholar]
- 20.Cardoso AC, Andrade LN, Bustos SO, Chammas R. Galectin-3 determines tumor cell adaptive strategies in stressed tumor microenvironments. Front Oncol. 2016;6:127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ruvolo PP. Galectin 3 as a guardian of the tumor microenvironment. Biochim Et Biophys Acta. 2016;1863:427–37. [DOI] [PubMed] [Google Scholar]
- 22.Fortuna-Costa A, Gomes AM, Kozlowski EO, Stelling MP, Pavao MS. Extracellular galectin-3 in tumor progression and metastasis. Front Oncol. 2014;4:138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Petit AE, Demotte N, Scheid B, Wildmann C, Bigirimana R, Gordon-Alonso M, et al.. A major secretory defect of tumour-infiltrating T lymphocytes due to galectin impairing LFA-1-mediated synapse completion. Nat Communications. 2016;7:12242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Demetriou M, Granovsky M, Quaggin S, Dennis JW. Negative regulation of T-cell activation and autoimmunity by Mgat5 N-glycosylation. Nature. 2001;409:733–9. [DOI] [PubMed] [Google Scholar]
- 25.Xue J, Gao X, Fu C, Cong Z, Jiang H, Wang W, Chen T, Wei Q, Qin C. Regulation of galectin-3-induced apoptosis of Jurkat cells by both O-glycans and N-glycans on CD45. FEBS Lett. 2013;587:3986–94. [DOI] [PubMed] [Google Scholar]
- 26.Kouo T, Huang L, Pucsek AB, Cao M, Solt S, Armstrong T, Jaffee E. Galectin-3 shapes antitumor immune responses by suppressing CD8+ T Cells via LAG-3 and inhibiting expansion of plasmacytoid dendritic cells. Cancer Immunol Res. 2015;3:412–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sage PT, Francisco LM, Carman CV, Sharpe AH. The receptor PD-1 controls follicular regulatory T cells in the lymph nodes and blood. Nat Immunol. 2013;14:152–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sage PT, Sharpe AH. T follicular regulatory cells. Immunol Rev. 2016;271:246–59. [DOI] [PubMed] [Google Scholar]
- 29.Demotte N, Wieers G, Van Der Smissen P, Moser M, Schmidt C, Thielemans K, Squifflet JL, Weynand B, Carrasco J, Lurquin C, et al.. A galectin-3 ligand corrects the impaired function of human CD4 and CD8 tumor-infiltrating lymphocytes and favors tumor rejection in mice. Cancer Res. 2010;70:7476–88. [DOI] [PubMed] [Google Scholar]
- 30.Fukumori T, Takenaka Y, Yoshii T, Kim HR, Hogan V, Inohara H, Kagawa S, Raz A. CD29 and CD7 mediate galectin-3-induced type II T-cell apoptosis. Cancer Res. 2003;63:8302–11. [PubMed] [Google Scholar]
- 31.Chen HY, Fermin A, Vardhana S, Weng IC, Lo KF, Chang EY, Maverakis E, Yang RY, Hsu DK, Dustin ML, et al.. Galectin-3 negatively regulates TCR-mediated CD4+ T-cell activation at the immunological synapse. Proc Natl Acad Sci U S A. 2009;106:14496–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tsuboi S, Sutoh M, Hatakeyama S, Hiraoka N, Habuchi T, Horikawa Y, Hashimoto Y, Yoneyama T, Mori K, Koie T, et al.. A novel strategy for evasion of NK cell immunity by tumours expressing core2 O-glycans. EMBO J. 2011;30:3173–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wang W, Guo H, Geng J, Zheng X, Wei H, Sun R, Tian Z. Tumor-released Galectin-3, a soluble inhibitory ligand of human NKp30, plays an important role in tumor escape from NK cell attack. J Biol Chem. 2014;289:33311–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Suzuki Y, Sutoh M, Hatakeyama S, Mori K, Yamamoto H, Koie T, Saitoh H, Yamaya K, Funyu T, Habuchi T, et al.. MUC1 carrying core 2 O-glycans functions as a molecular shield against NK cell attack, promoting bladder tumor metastasis. Int J Oncol. 2012;40:1831–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Breuilh L, Vanhoutte F, Fontaine J, van Stijn CM, Tillie-Leblond I, Capron M, Faveeuw C, Jouault T, van Die I, Gosset P, et al.. Galectin-3 modulates immune and inflammatory responses during helminthic infection: impact of galectin-3 deficiency on the functions of dendritic cells. Infect Immun. 2007;75:5148–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wu SY, Yu JS, Liu FT, Miaw SC, Wu-Hsieh BA. Galectin-3 negatively regulates dendritic cell production of IL-23/IL-17-axis cytokines in infection by Histoplasma capsulatum. J Immunol. 2013;190:3427–37. [DOI] [PubMed] [Google Scholar]
- 37.Vereecken P, Zouaoui Boudjeltia K, Debray C, Awada A, Legssyer I, Sales F, Petein M, Vanhaeverbeek M, Ghanem G, Heenen M. High serum galectin-3 in advanced melanoma: preliminary results. Clin Exp Dermatol. 2006;31:105–9. [DOI] [PubMed] [Google Scholar]
- 38.Vereecken P, Awada A, Suciu S, Castro G, Morandini R, Litynska A, Lienard D, Ezzedine K, Ghanem G, Heenen M. Evaluation of the prognostic significance of serum galectin-3 in American joint committee on cancer stage III and stage IV melanoma patients. Melanoma Res. 2009;19:316–20. [DOI] [PubMed] [Google Scholar]
- 39.Voron T, Colussi O, Marcheteau E, Pernot S, Nizard M, Pointet AL, Latreche S, Bergaya S, Benhamouda N, Tanchot C, et al.. VEGF-A modulates expression of inhibitory checkpoints on CD8+ T cells in tumors. J Exp Med. 2015;212:139–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Markowska AI, Jefferies KC, Panjwani N. Galectin-3 protein modulates cell surface expression and activation of vascular endothelial growth factor receptor 2 in human endothelial cells. J Biol Chem. 2011;286:29913–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Markowska AI, Liu FT, Panjwani N. Galectin-3 is an important mediator of VEGF- and bFGF-mediated angiogenic response. J Exp Med. 2010;207:1981–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Machado CM, Andrade LN, Teixeira VR, Costa FF, Melo CM, dos Santos SN, Nonogaki S, Liu FT, Bernardes ES, Camargo AA, et al.. Galectin-3 disruption impaired tumoral angiogenesis by reducing VEGF secretion from TGFbeta1-induced macrophages. Cancer Med. 2014;3:201–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Cai G, Ma X, Chen B, Huang Y, Liu S, Yang H, Zou W. Galectin-3 induces ovarian cancer cell survival and chemoresistance via TLR4 signaling activation. Tumour Biol. 2016;37(9):11883–11891. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.