

ABSTRACT
A major advantage of immunotherapy of cancer is that effector cells induced at one site should be able to kill metastatic cancer cells in other sites or tissues. However, different tissues have unique immune components, and very little is known about whether effector T cells induced against tumors in one tissue can work against the same tumors in other tissues. Here, we used CT26 murine tumor models to investigate anti-tumor immune responses in the skin and lungs and characterized cross-protection between the two tissues. Blockade of the function of Treg cells with anti-CD25 allowed for T cell-dependent rejection of s.c. tumors. When these mice were simultaneously inoculated i.v. with CT26, they also rejected tumors in the lung. Interestingly, in the absence of s.c. tumors, anti-CD25 treatment alone had no effect on lung tumor growth. These observations suggested that T cell-mediated anti-tumor protective immunity induced against s.c. tumors can also protect against lung metastases of the same tumors. In contrast, NKT cell-deficiency in CD1d−/− mice conferred significant protection against lung tumors but had no effect on the growth of tumors in the skin, and tumor rejection induced against the CT26 in the lung did not confer protection for the same tumor cells in the skin. Thus, effector cells against the same tumor do not work in all tissues, and the induction site of the effector T cells is critical to control metastasis. Further, the regulation of tumor immunity may be different for the same tumor in different anatomical locations.
KEYWORDS: cross-protection, Immunoregulation, lung metastases, NKT cells, skin tumors, T-cell immunity, tissue-specific immunity, Treg cells
Introduction
In recent years, many therapies that harness the ability of the immune system to kill tumor cells have demonstrated dramatic results in both animal models and humans, and immunotherapy has emerged as a critical and effective component of cancer therapy. Additionally, it is now recognized that tumor immunity also plays roles in conventional treatment such as radiation and chemotherapy. It has been shown that some chemotherapeutic drugs, such as cyclophosphamide and fludarabine, enhance tumor immunosurveillance by decreasing the suppressive cells such as regulatory T cells.1,2 Localized radiation of tumors sometimes leads to responses at distant sites, referred to as an abscopal effect, presumably through the induction of anti-tumor immune responses.3 The future of cancer therapies involves integrated approaches of surgery, chemotherapy, radiation and immunotherapy to effectively clear the tumor cells.
The ability of anti-cancer therapeutics to be curative often relies on the capacity to induce protection not only at the site of the primary tumor but also against metastases in different tissues. The induction of anti-tumor immune responses capable of detecting and clearing tumor cells in any tissue would result in long-term protection against recurrence and metastasis. While cancer immunotherapy has shown dramatic results in a subset of patients, it does not work for all cancer types. Immunotherapy is hampered not only by the difficulty in mounting effective long-term memory responses but also by the suppressive immune environment that often occurs in the tumor microenvironment. Checkpoint inhibitors that target immune suppressive molecules are among the most promising anti-cancer therapeutics.4 Monoclonal antibodies to programmed death-1 (PD-1) and cytotoxic T lymphocyte associated-antigen-4 (CTLA-4) enhance immunity to cancer antigens by allowing for the generation and expansion of antitumor T cells.5,6 These therapies are most effective in patients with evidence of preexisting antitumor immunity, which suggests that these immunotherapies enable established immunity.7 However, even these immunotherapies have not had broad successes with every tumor type, suggesting that there may be tumor- or tissue-specific factors that determine success. Further understanding of which type of immunity, both anti-tumor and regulatory, predominates in which tissue will help to tailor therapies to each patient to increase chances of success.
While primary tumors can often be removed, metastases to other tissues and organs are typically more difficult to treat even though the primary tumor may not recur. This occurs even with immunotherapy, suggesting that tissue-specific immunity may be critical in determining the effectiveness of a treatment. It has been discovered in recent years that tissue specific immune responses play critical roles in many settings. However, the interplay between tissue specific and systemic immune responses against cancer is not well understood.
Multiple regulatory mechanisms of anti-tumor immunity exist, and the predominant mechanism in regulation of tumor immunity in one model does not always work in others. One of the most extensively studied regulators of tumor immunity is the regulatory T cell (Treg), defined by expression of CD4, IL-2 receptor α (CD25) and the transcription factor forkhead box p3 (Foxp3).8,9 The blockade of Tregs has been found to enhance protection against tumors in many mouse models.10-15 Our group and others have previously described a subset of natural killer T (NKT) cells that play a role in the negative regulation of antitumor immune responses.16-19 Studies in murine tumor models have illustrated that either Tregs or type II NKT cells can play a dominant role.17,18,20 Our work with a CT26 colon cancer model has shown that Tregs predominate in the skin while NKT cells are the key regulator of tumor immunosurveillance in the lung. It is unclear what determines which of these types of suppressive T cells predominates and how these cells interact. Even less is known regarding the impact of tissue environment on effector T cells and whether memory cells induced against tumors in one tissue also can target the same tumor cells at other sites.
In this study, we took advantage of this difference in immune regulation in the skin by Treg cells and lung by type II NKT cells for the same tumor to examine the ability of effector cells induced against tumors growing in the skin or lungs to induce cross protection against the same tumor at the other site in the same animal. We show here that memory T cell responses generated in the skin are capable of suppressing tumor growth at other sites; however, while removal of regulatory NKT cells can unmask T cell responses capable of clearing tumors from the lungs, this does not promote memory or protective immunity in the skin. Because one of the biggest advantages of cancer immunotherapy is the theoretical ability to induce protective immune responses at metastatic sites, it is critical to understand how cross-protective immune responses are regulated in different tissue environments.
Materials and methods
Mice
Female BALB/c mice were purchased from Animal Production Colonies, Frederick Cancer Research Facility, National Cancer Institute (NCI; Frederick, MD). BALB/c RAG1−/− mice and BALB/c CD1d−/− mice (The Jackson Laboratory, Bar Harbor, ME) were bred at the NCI under specific pathogen-free and Helicobacter-free conditions. Female mice (at least 6 weeks of age and < 6 months of age) were used for all experiments. All experimental protocols were approved by and performed under the guidelines of the NCI's Animal Care and Use Committee.
Cell lines
The CT26 colon carcinoma cell line was maintained in RPMI-1640 supplemented with 10% fetal calf serum, 100 units/ml Penicillin and 100 μg/ml Streptomycin, L-glutamine, sodium pyruvate and nonessential amino acids. Cells were cultured in an atmosphere of 37 °C and 5% CO2.
In vivo tumor assays and antibody treatment
A single cell suspension of CT26 cells in PBS was prepared and injected s.c. (5 × 104 -5 × 105 cells in 0.1 ml) or i.v. (3-5 × 105 cells in 0.2 ml) on day 0. Size of s.c. tumors was measured periodically, starting day 3, by caliper gauge. For the lung tumor model, mice were euthanized 12–22 days after tumor challenge. Lungs were stained and fixed, and metastases were enumerated as previously described.19
Antibody treatments were performed according to the schedules described for each experiment. The following doses of antibodies were administered intraperitoneally: CD4 (clone GK1.5, Harlan), 0.2 mg CD8 (clone 2.43, Harlan Laboratories), 25 µl anti-asialo GM1 (Wako Chemical Company, Richmon, VA) (sufficient to deplete ≥90% of DX5+ NK cells). Rat immunoglobulin G (IgG, Sigma-Aldrich, St. Louis, MO) or rabbit serum (Cedarlane Laboratories, Burlington, NC) were injected as a control for all antibody treatments. 0.5 mg of anti-CD25 (PC61, Harlan, Indiana, IN) was administered intravenously. Cell depletion of more than 90% was confirmed by flow cytometric staining for CD4 (clone RM4-5), CD8 (clone 53–6.7, BD Biosciences, San Jose, CA) and pan-NK cell marker (clone DX5, Thermo Fisher Scientific, Waltham, MA) at the time of tumor challenge and at the conclusion of the experiment.
T cell adoptive transfer
Splenic cells from CD1d-/- (naïve and immunized), naïve wild-type and CD25-depleted wild-type mice that rejected s.c. CT26 tumors were prepared. In some experiments, CD4+ and/or CD8+ cells were further depleted. 1 × 107 cells were transferred by i.v. injection into each RAG1−/− mouse. Recipient mice were challenged with CT26 one day after the T cell transfer.
Preparation of lung and tumor infiltrating leukocytes
Naïve, CD25-depleted or metastatic lungs as well as subcutaneous tumors were harvested for single cell analysis one day after anti-CD25 injection or ten days after tumor inoculation. Tumor infiltrating leukocytes were prepared as described previously.21 Lung leukocytes were obtained by processing with the Lung Dissociation Kit (Miltenyi Biotec, Auburn, CA), debris was removed via a nylon membrane and cells were washed before leukocytes were fractionated using Percoll (Sigma-Aldrich, St. Louis, MO).
Flow cytometry
Single cell suspensions were stained with LIVE/DEAD™ Fixable Aqua Dead Cell Stain Kit (Thermo Fisher Scientific) followed by incubation with anti-CD16/CD32 and PBS57-loaded CD1d-tetramer (NIH tetramer core facility) before surface and intranuclear staining. Fluorescently labeled monoclonal antibodies against CD45, TCR-β chain, CD8, CD4, CD25 and FoxP3 were obtained from BioLegend, San Diego, CA. True-Nuclear staining buffer (BioLegend) was utilized for intranuclear staining. Cells were analyzed for fluorescence by FACSymphony (BD Bioscience) and Flowjo (FlowJo, LLC, Ashland, Oregon).
Statistics
The data were analyzed using the nonparametric Mann–Whitney test, Kruskal-Wallis with post hoc Dunn's test or 2-way ANOVA with Tukey post hoc using GraphPad Prism (version 5 and 7; GraphPad software). Significance was determined at p < 0.05. All experiments were repeated at least twice to confirm reproducibility of results, and representative data from independent experiments are shown.
Results
CD25+ cells suppress anti-tumor immunity in the skin, not lung
We first assessed the role of CD25+ cells in regulation of anti-tumor immunity in the skin and lungs. Depletion of CD25+ cells resulted in s.c. tumor rejection in all mice (Fig. 1A). All mice developed palpable tumors that completely regressed beginning at day 10. In contrast, there was no effect on tumor growth in the lungs (Fig. 1B) although we confirmed a significant reduction of the number of CD4+CD25+Foxp3+T cells in the lungs of anti-CD25 treated mice (Fig. 1C). Flow cytometry analysis of leukocytes in tumors in the lung and skin and the normal lung showed that skin tumors contain a significantly higher frequency of CD4+Foxp3+ Treg cells than the lungs with tumors (Fig. 1D). This result is consistent with the observation that anti-CD25 treatment had no effect on tumor progression in the lung. It was also interesting to find that tumors in both tissues contained significant numbers of CD1d-restricted type I NKT cells. The majority of these NKT cells were a CD4−CD8− subset that has been suggested to be protective against lung metastasis of B16 melanoma.22 There was virtually no infiltation of CD8+ T cells in pre-necrotic tumors in the skin, and no increase in these cells in the tumor-bearing lung even though CT26 is known to express the immunogenic retrovirus-derived antigen, gp70. When investigating the dependence of subcutaneous CT26 tumor rejection on CD8+, CD4+ T or NK cells in vivo, we observed that if NK cells were depleted with anti-asialo GM1, tumors initially appeared and then regressed as seen with anti-CD25 treatment alone. However, if either CD4+ or CD8+ T cells were depleted, all mice developed tumors that were not rejected (Fig. 1E).
Figure 1.
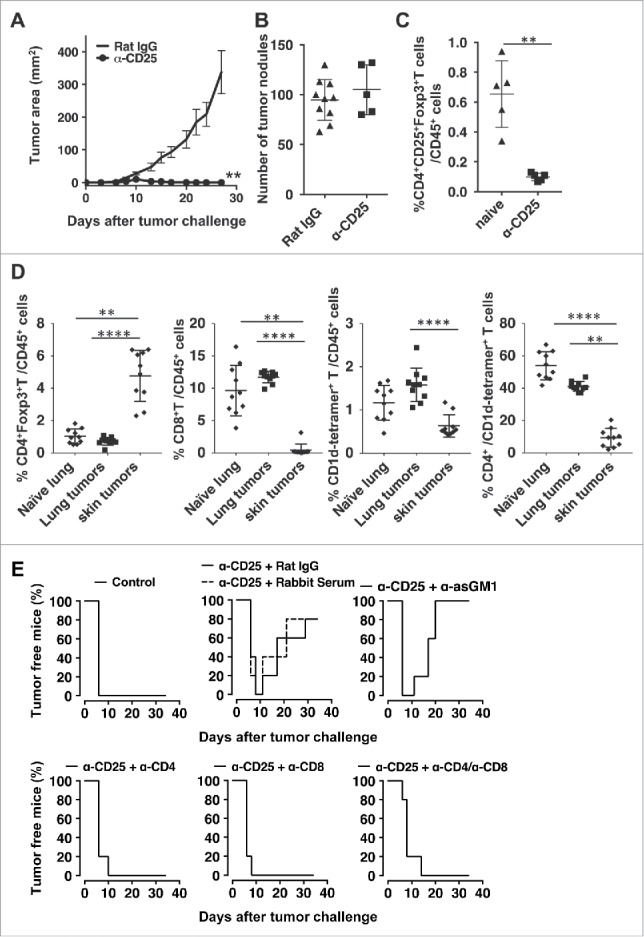
Depletion of CD25+ cells induces rejection of subcutaneous but not lung tumors. Mice were injected with 0.5 mg anti-CD25 (PC61) or Rat IgG i.v.. 5 days later, mice were challenged with CT26 (A and C) 50,000 cells s.c. or (B) 500,000 cells i.v. (A). s.c. tumor area was monitored at least three times weekly. Data are expressed as mean ± standard deviation (n = 5) Representative data from two independent experiments are shown. (Mann-Whitney test **p<0.01) (B) Mice challenged i.v. were sacrificed 14–16 days after tumor challenge, lungs were stained, and were tumors enumerated. Data are expressed as mean ± standard deviation (n = 10 for Rat IgG, n = 5 for anti-CD25). Representative data from two independent experiments are shown. (C) 1 day after anti-CD25 treatment, proportions of CD4+CD25+Foxp3+T cells of CD45+ cells in the lungs were enumerated. Data are expressed as mean ± standard deviation (n = 5). (Mann-Whitney test **p<0.01). (D) Flow cytometry analysis of leukocytes in skin tumors and lungs with/without tumors. On day 10 after tumor inoculation, subcutaneous tumors or lungs were harvested. After leukocyte preparation, cells were analyzed for infiltrating T cell subsets. Data are pooled from two independent experiments and expressed as mean ± standard deviation (n = 10). Kruskal-Wallis test with post hoc Dunn's test **p<0.01, ****p<0.0001 was performed for statistical analysis. (E) Depletion antibodies were administered the day before tumor challenge, the day of tumor challenge, and 5 days after tumor challenge. s.c. tumor area was measured, and tumor-free survival data plots are shown as representatives of two independent experiments (n = 5).
Anti-tumor immunity in skin uncovered by depletion of CD25+cells confers protection in the lung
We next determined if protection against s.c. tumors resulted in cross-protection against CT26 tumors at distant sites and tissues. Mice were treated with anti-CD25 and challenged with tumor as described above; however some mice were challenged simultaneously with both s.c. and i.v. CT26. While CD25 depletion by itself had no effect on lung tumor growth, when anti-CD25-pretreated mice challenged with s.c. CT26 were simultaneously inoculated i.v. with CT26, they rejected tumors in both the skin and the lung (Fig. 2A). Inoculation of dead CT26 s.c. along with anti-CD25 treatment had no effect on lung tumor growth, suggesting that an active immune response against live tumor cells was required for the cross-protection.
Figure 2.
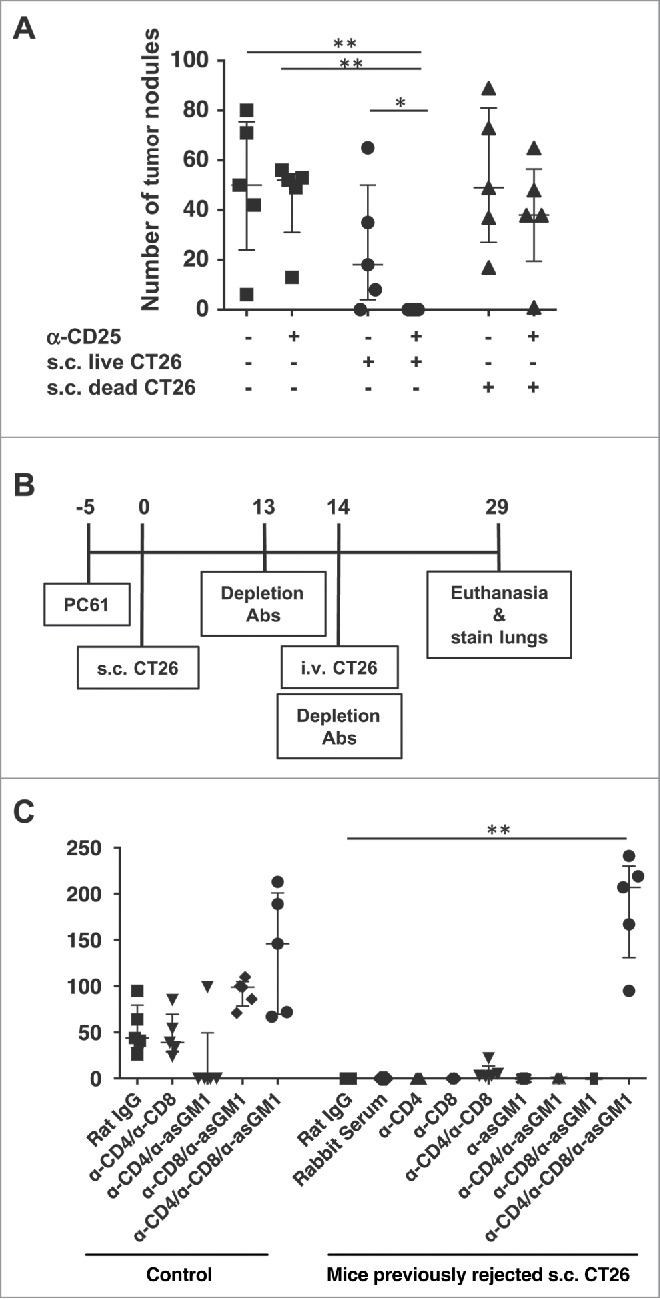
The protection against s.c. CT26 tumors induced by depletion of CD25+ cells confers protection against CT26 lung tumors. (A) Mice were injected with 0.5 mg anti-CD25 (PC61) or Rat IgG i.v.. 5 days later, mice were challenged with 50,000 CT26 cells s.c. and/or 200,000 CT26 cells i.v. Mice were sacrificed approximately 3 weeks later and lungs were stained and fixed, and tumors were enumerated. (B) Experimental scheme. (C) As shown in the experimental scheme in (B), after regression of the s.c. tumors in anti-CD25-treated mice, animals were challenged with 500,000 CT26 cells i.v., and depletion antibodies were administered as described. Mice were euthanized and lungs were stained approximately 2 weeks after i.v. CT26 challenge. Data are expressed as mean ± standard deviation (n = 5). Representative data from two independent experiments are shown. Statistically significant differences were determined by Mann-Whitney test *p<0.05 **p<0.01.
In order to investigate the anti-tumor immune responses that occur in the absence of CD25+ regulatory cells, CD25-depleted mice were inoculated with CT26 s.c. After two weeks, mice were challenged i.v. with CT26, and CD4+, CD8+ and/or NK cells were depleted to assess mechanism of protection (Fig. 2B). In naïve mice, depletion of CD4+ and CD8+ T cells had no impact on the growth of lung tumors. However tumor growth was inhibited in mice depleted of both CD4+ and NK cells, consistent with our previous observation that CD4+ type II NKT cells (that would be depleted by this anti-CD4 treatment) suppress tumor immunity in lungs.19 Depletion of CD8+ and NK cells or all three cell types enhanced lung tumor growth. However in mice pretreated with anti-CD25 during the time of s.c. CT26 challenge (which had rejected the s.c. tumor), significant lung tumor growth occurred only in mice depleted of CD4+, CD8+ and NK cells. Depletion of CD4+ and CD8+ T cells allowed for a small number of tumor nodules, but all other groups had sterilizing immunity to CT26 tumor cells. As the i.v. tumor challenge was performed two weeks following s.c. inoculation, the ability of the anti-tumor response against s.c. tumors to protect against a subsequent lung tumor challenge suggested that a memory response was induced that cross-protected in the lung, and that this response was dependent on all 3 effector cells, CD4+ T, CD8+ T and NK cells.
Depletion of CD25+ cells allows for development of long-term memory anti-tumor response
To further assess the immune memory and determine whether long-term memory was induced, mice were depleted of CD25+ cells and challenged s.c. with CT26. Three months after tumors were rejected, mice were re-challenged with CT26 tumor cells either s.c. on the same or opposite flank as the original tumor challenge, or i.v.. Regardless of the site of the second tumor challenge, no mice that had previously rejected s.c. CT26 had any tumor growth upon second challenge (Fig. 3A). Either CD4+ or CD8+ T cells were sufficient for the anti-tumor memory response, as it occurred if either only CD4+ or only CD8+ T cells were depleted, whereas tumors grew if both CD4+ and CD8+ T cells were depleted during the re-challenge (Fig. 3B). To further confirm the presence of a memory response, splenic T cells were isolated from mice that had rejected s.c. tumors and were transferred into RAG1−/− mice (Fig. 3C). Mice receiving T cells from wild-type mice that had rejected tumors were completely protected from a subsequent s.c. CT26 challenge, and this protection was abrogated if either CD4+ or CD8+ T cells were depleted from the inoculum. The small differences in relative roles of CD4+ and CD8+ T cells between the in vivo depletion and ex vivo depletion experiments may be due to differences in the depletion method in vivo vs in spleen cell preparations in vitro (although in both cases full depletion was verified by flow cytometry) and to the fact that the cells in Fig. 3C were adoptively transferred into RAG1−/− mice that have no T or B cells of their own, a very different environment from the intact mouse. Nevertheless, the same overall finding that both CD4+ and CD8+ T cells are involved in the protection was true in both models.
Figure 3.
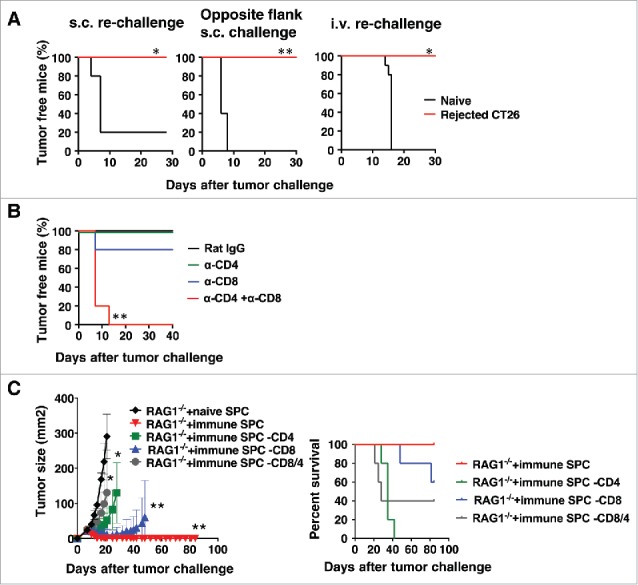
Rejection of s.c. tumors following CD25-depletion induces a memory response that protects from subsequent tumor rechallenge. (A) Mice were injected with 0.5 mg anti-CD25 (PC61) i.v., and 5 days later, mice were challenged with 50,000 CT26 cells s.c.. At 3 months after tumors were rejected, the mice were re-challenged with CT26 cells either s.c. on the same flank as the original challenge or on the opposite flank or i.v.. Survival data plots are shown as representatives of two independent experiments (n = 5). Statistical significance was determined by Log-rank test *p<0.05, **p<0.01. (B) Depletion antibodies were given the day before, the day of, and 5 days after the second tumor challenge. S.c. tumor area was measured, and tumor-free survival data plots are shown as representatives of two independent experiments (n = 5) (left panel), and survival data plots are shown (right panel). (C) 1 × 107 T cells from CD25-depleted mice that had rejected s.c. tumors were transferred into each RAG1−/− mouse. One day after the T cell transfer, mice were challenged with CT26 s.c. Data are expressed as mean ± standard deviation (n = 5). Representative data from two independent experiments are shown. Statistical significance was determined by 2-way ANOVA with Tukey post hoc adjustment *p<0.05, **p<0.01.
Protection from lung tumors in NKT cell-deficient mice does not confer protection against tumors in the skin
We next examined the converse question, whether protection from CT26 tumor growth in the lungs conferred cross-protection against s.c. CT26. CD1d−/− mice, which lack NKT cells, are protected from growth of lung tumors (Fig. 4A). While s.c. CT26 tumors grow slightly more slowly in CD1d−/− mice, there were no significant differences in s.c. tumor growth in either WT or CD1d−/− mice when mice were simultaneously challenged i.v. with CT26 (Fig. 4B). There was also no impact on s.c. tumor growth if CD1d−/− mice were challenged with s.c. CT26 one week after i.v. tumor challenge (Fig. 4C). This result suggests that there is no cross-protection by the immunity induced against lung tumors to subcutaneous tumors. To confirm this result, T cells from naïve or immunized CD1d−/− mice were transferred into RAG1−/− mice, and recipient mice were challenged either i.v. or s.c. with CT26. There was no effect on growth of s.c. CT26 tumor cells even though the transferred cells from immunized mice protected against lung tumor growth (Fig. 4D and 4E). This was in contrast to T cells from mice treated with anti-CD25 followed by s.c. CT26 challenge, which rejected s.c. tumors (Fig. 3C). These data indicated that the protection induced against CT26 cells in the lungs of CD1d−/− mice does not cross-protect against tumors in skin. Thus, cross-protection between two sites of the same tumor in the same mouse was not symmetrical.
Figure 4.
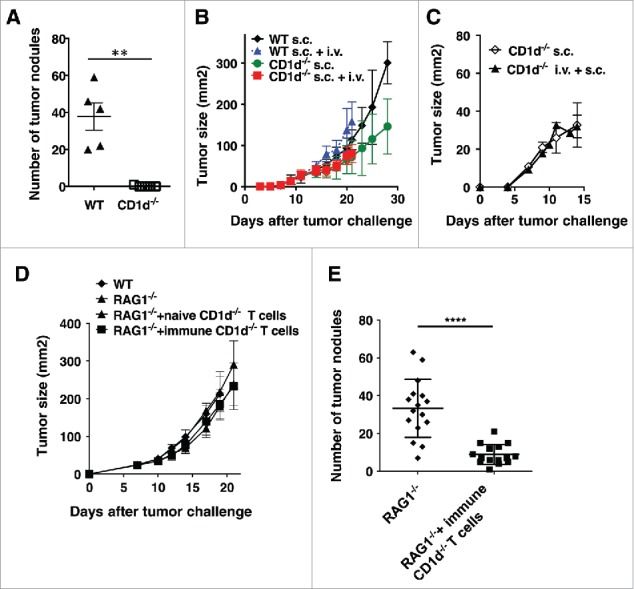
Protection from lung tumors in CD1d−/− mice does not confer protection against s.c. tumors. (A and B) WT or CD1d−/− mice were challenged with CT26 cells s.c. and/or i.v on the same day. S.c. tumors were measured until mice had to be euthanized due to lung tumors. Lungs from mice receiving i.v. tumor challenge were stained and tumors were enumerated. (C) Mice were challenged with CT26 cells i.v. One week later, mice were challenged with CT26 cells s.c and s.c. tumor growth monitored. (D and E) CD1d−/− mice were challenged with CT26 cells i.v. One week later, T cells from these mice were transferred into RAG1−/− mice. One day after T cell transfer, mice were challenged with CT26 s.c. (D) or i.v. (E). S.c. growth was monitored three times a week. Data are expressed as mean ± standard deviation (B-D, n = 5). Lungs from mice receiving i.v. tumor challenge were stained and tumors were enumerated on day 22. The numbers of lung metastases are expressed as mean ± standard deviation (A, n = 5 E, n = 15 for RAG1−/− group and n = 16 for RAG1−/− + immune CD1d−/− T cells group). Representative data from at least two independent experiments are shown. Statistically significant differences (**p<0.01,****p<0.0001) were determined by the Mann-Whitney test.
Discussion
Here, we investigated how tumor immunity is regulated in the skin and lungs, and how immunity at one site affects tumor growth at the other. We demonstrated that CD25+ cells, most likely Tregs, suppressed anti-tumor immunity in the skin. In contrast, anti-CD25 treatment did not induce protection in lungs whereas absence of NKT cells, presumably regulatory type II NKT cells, protected in the lungs. These observations clearly indicated that mechanisms of immune suppression induced by the same tumor are different in the two tissues. In addition to tissue microenvironment, immunological status of tumor bearing individuals23 as well as nature of cancer cells24,25 can also play roles in determination of predominant mechanisms of suppression of tumor immunity. Recent success of checkpoint inhibitors, especially antagonists of PD-1/PD-L1 interaction in cancer treatment, highlighted significant difference in the efficacy of the treatment among different cancer types. Many factors have been implicated to play roles to determine anti-tumor efficacy including the nature of the cancer cells (e.g. genomic instability) and expression of PD-L1 in tumor tissue. However, the data presented here suggest that tissue microenvironment should be considered as a factor which plays an important role determining the efficacy of immune checkpoint targeted therapy.
Skin and lungs not only utilize distinct mechanisms to suppress tumor immunosurveillance but also induce memory T cells with different quality even when immunosuppression is removed. Depletion of CD25+ cells allows the generation of memory T cells that can clear tumor cells both in the skin at local and distant sites as well as in the lungs. The memory responses were mostly T-cell mediated, as depletion of CD4+ and CD8+ cells allowed tumors to grow, and this protection could be achieved by adoptive transfer of T cells from immune mice into RAG1−/− recipients. In contrast, memory T cells from tumor-challenged CD1d−/− mice lacking NKT cells inhibited tumor growth in the lungs but not in the skin of the recipents of the T cells.
These immunological differences in the two tissues may be attributed to the different microenvironment provided by each tissue. Efficacy of immunotherapy against orthotopic (visceral) and subcutaneous tumors has been shown to be significantly different because of distinct macrophage populations attracted to the tumor sites.26 In other studies, it has been reported that induction of tissue resident T cells by tissue parenchymal DCs may explain dissociated clinical responses between orthotopic (mucosal) and subcutaneous tumors.27,28 Although in this study, we did not investigate contribution of tissue parenchymal cells, this will be further explored in the future because it may provide explanation for mixed responses to immunotherapy in humans.29
Generation of the memory anti-tumor immune response capable of clearing lung tumors may also be supported by NK cells, as depletion of NK cells along with CD4+ and CD8+ cells resulted in a complete loss of memory response to subsequent i.v. CT26 challenge, while CD4+ and CD8+ cells depletion alone had only a modest effect on reversing the protection (Fig. 2C). There are multiple mechanisms by which NK cells may be involved in this memory response. NK cells are most widely associated with their contribution to innate immunity; however, there is recent evidence of memory NK cells. While NK cell memory is most closely associated with its role in viral infection,30 a recent report has demonstrated that priming of NK cells with exposure to tumor cells causes phenotypic changes in NK cells, including increased cytotoxicity, perforin synthesis and survival.31 NK cells can also serve as secondary effector cells downstream of (activated by) memory CD4+ T cells without having memory themselves. The adoptive transfer experiments clearly show that T cells are sufficient for mediating the antitumor response; however, a role for NK cells in the development of the memory response cannot be discounted. NK cells can promote anti-tumor T cell responses in many ways, including activation of DCs that enhance Th1 polarization and CTL ability of T cells.32 NK cells also play a helper role in the re-activation of tumor-specific T cells.33 The increase in tumor burden upon depletion of NK cells is likely due to a reduction in tumor immunosurveillance that results from the cooperation of NK cells, DCs and T cells.
A hurdle for successful immunotherapy is the lack of understanding of how memory T cell responses are differentially regulated based on the tumor site. Tumor environment likely plays roles in both the generation of anti-tumor T cell responses, and the ability of those T cells to act at the tumor site to induce regression. It has been demonstrated that there are many subsets of tissue-resident or tissue-specific immune cells such as innate lymphoid cells (ILCs), macrophages, and memory T cells that are important for determining the type of immune response that is induced. One potential candidate cell type involved in the determination of the quality of induced memory cells is the population of antigen presenting cells including dendritic cells and macrophages in that tissue. It has been shown that T cells primed by APCs preexposed to factors, such as retinoic acid, involved in gut homeostasis express homing receptor for gut mucosa, α4β7.34,35 Our data suggests that memory T cells induced against tumors in the skin are able to circulate, as they could protect against skin tumors at distant sites as well as in the lungs. This is also supported by the fact that transfer of splenic T cells from these mice was sufficient to induce tumor rejection in recipient mice. While the memory CD8+ T cells that mediate protection against CT26 tumors in this study are not tissue resident, it is possible that other tissue resident cells are involved in the induction or trafficking of the memory T cells. In contrast to the memory T cell induction induced by the clearance of s.c. CT26 tumors, the memory T cell responses in CD1d−/− mice were able to protect only against lung tumors, and the adoptive transfer of splenic T cells from these mice did not significantly protect recipient mice against subcutaneous tumors, although they did protect recipients against lung tumors. The latter finding indicates that the protective cells from the lung circulated at least as far as the spleen. It is possible that the responses induced by lung-specific tumor clearance are more local and reside in the lung or lung-associated lymph nodes and do not circulate broadly systemically beyond, or at least have more restricted homing capacity to lung and spleen but not to the skin. It is not currently known how the tumor environment impacts homing of memory T cells to sites of metastasis. It is likely that both tumor type and site of metastasis impact the generation of memory T cell responses against tumor antigens as well as the ability of these cells to traffic to the tumor site.
While cancer immunotherapy has displayed significant promise, it remains hindered by a lack of understanding of how the tumor environment impacts immune cell function and how effector mechanisms are regulated. Further studies are needed to uncover markers of which regulatory cells are allowing for tumor growth and to determine which immunotherapy is most likely to have success under those conditions. As shown here, suppression of the regulatory mechanisms that predominate at a tumor site can result in the induction of memory T cells capable of inducing sterilizing immunity against the same tumor at distant sites, or can result in tissue-specific memory that does not protect at certain distinct tissue sites. These studies highlight the importance of characterization of the mechanisms of tissue-specific regulation of tumor immunity to choose the proper immunotherapy that can prevent recurrence of both the primary tumor and distant metastases.
Funding Statement
This work was supported by the Gui Foundation This work was supported by the Intramural Research Program of the Center for Cancer Research, National Cancer Institute, National Institutes of Health (Z01-C-004020) and by the Gui Foundation.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Acknowledgments
We thanks Amy James for assistance with mouse breeding, the NIH tetramer facility, the Vaccine Branch FACS core facility, the NCI animal facility and Lisa Smith for secretarial help.
References
- 1.Beyer M, Kochanek M, Darabi K, Popov A, Jensen M, Endl E, Knolle PA, Thomas RK, von Bergwelt-Baildon M, Debey S, et al. Reduced frequencies and suppressive function of CD4+CD25hi regulatory T cells in patients with chronic lymphocytic leukemia after therapy with fludarabine. Blood 2005;106:2018–25. doi: 10.1182/blood-2005-02-0642. PMID:15914560. [DOI] [PubMed] [Google Scholar]
- 2.Ghiringhelli F, Larmonier N, Schmitt E, Parcellier A, Cathelin D, Garrido C, Chauffert B, Solary E, Bonnotte B, Martin F. CD4+CD25+ regulatory T cells suppress tumor immunity but are sensitive to cyclophosphamide which allows immunotherapy of established tumors to be curative. Eur J Immunol 2004;34:336–44. doi: 10.1002/eji.200324181. PMID:14768038. [DOI] [PubMed] [Google Scholar]
- 3.Golden EB, Chhabra A, Chachoua A, Adams S, Donach M, Fenton-Kerimian M, Friedman K, Ponzo F, Babb JS, Goldberg J, et al. Local radiotherapy and granulocyte-macrophage colony-stimulating factor to generate abscopal responses in patients with metastatic solid tumours: a proof-of-principle trial. Lancet Oncol 2015;16:795–803. doi: 10.1016/S1470-2045(15)00054-6. PMID:26095785. [DOI] [PubMed] [Google Scholar]
- 4.Rosenberg SA. Raising the bar: the curative potential of human cancer immunotherapy. Sci Transl Med 2012;4:127ps8. doi: 10.1126/scitranslmed.3003634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Restifo NP, Dudley ME, Rosenberg SA. Adoptive immunotherapy for cancer: harnessing the T cell response. Nat Rev Immunol 2012;12:269–81. doi: 10.1038/nri3191. PMID:22437939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Topalian SL, Hodi FS, Brahmer JR, Gettinger SN, Smith DC, McDermott DF, Powderly JD, Carvajal RD, Sosman JA, Atkins MB, et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N Engl J Med 2012;366:2443–54. doi: 10.1056/NEJMoa1200690. PMID:22658127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tumeh PC, Harview CL, Yearley JH, Shintaku IP, Taylor EJ, Robert L, Chmielowski B, Spasic M, Henry G, Ciobanu V, et al. PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature 2014;515:568–71. doi: 10.1038/nature13954. PMID:25428505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Shevach EM. CD4+ CD25+ suppressor T cells: more questions than answers. Nat Rev Immunol 2002;2:389–400. doi: 10.1038/nri821. PMID:12093005. [DOI] [PubMed] [Google Scholar]
- 9.Thornton AM, Shevach EM. Suppressor effector function of CD4+CD25+ immunoregulatory T cells is antigen nonspecific. J Immunol 2000;164:183–90. doi: 10.4049/jimmunol.164.1.183. PMID:10605010. [DOI] [PubMed] [Google Scholar]
- 10.Onizuka S, Tawara I, Shimizu J, Sakaguchi S, Fujita T, Nakayama E. Tumor rejection by in vivo administration of anti-CD25 (interleukin-2 receptor alpha) monoclonal antibody. Cancer Res 1999;59:3128–33. PMID:10397255. [PubMed] [Google Scholar]
- 11.Knutson KL, Dang Y, Lu H, Lukas J, Almand B, Gad E, Azeke E, Disis ML. IL-2 immunotoxin therapy modulates tumor-associated regulatory T cells and leads to lasting immune-mediated rejection of breast cancers in neu-transgenic mice. J Immunol 2006;177:84–91. doi: 10.4049/jimmunol.177.1.84. PMID:16785502. [DOI] [PubMed] [Google Scholar]
- 12.Gritzapis AD, Voutsas IF, Baxevanis CN. Ontak reduces the immunosuppressive tumor environment and enhances successful therapeutic vaccination in HER-2/neu-tolerant mice. Cancer Immunol Immunother 2012;61:397–407. doi: 10.1007/s00262-011-1113-4. PMID:21928125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Litzinger MT, Fernando R, Curiel TJ, Grosenbach DW, Schlom J, Palena C. IL-2 immunotoxin denileukin diftitox reduces regulatory T cells and enhances vaccine-mediated T-cell immunity. Blood 2007;110:3192–201. doi: 10.1182/blood-2007-06-094615. PMID:17616639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tanaka A, Sakaguchi S. Regulatory T cells in cancer immunotherapy. Cell Res 2017;27:109–18. doi: 10.1038/cr.2016.151. PMID:27995907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bos PD, Plitas G, Rudra D, Lee SY, Rudensky AY. Transient regulatory T cell ablation deters oncogene-driven breast cancer and enhances radiotherapy. J Exp Med 2013;210:2435–66. doi: 10.1084/jem.20130762. PMID:24127486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Godfrey DI, Hammond KJ, Poulton LD, Smyth MJ, Baxter AG. NKT cells: facts, functions and fallacies. Immunol Today 2000;21:573–83. doi: 10.1016/S0167-5699(00)01735-7. PMID:11094262. [DOI] [PubMed] [Google Scholar]
- 17.Izhak L, Ambrosino E, Kato S, Parish ST, O'Konek JJ, Weber H, Xia Z, Venzon D, Berzofsky JA, Terabe M. Delicate balance among three types of T cells in concurrent regulation of tumor immunity. Cancer Res 2013;73:1514–23. doi: 10.1158/0008-5472.CAN-12-2567. PMID:23319803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Terabe M, Matsui S, Noben-Trauth N, Chen H, Watson C, Donaldson DD, Carbone DP, Paul WE, Berzofsky JA. NKT cell-mediated repression of tumor immunosurveillance by IL-13 and the IL-4R-STAT6 pathway. Nat Immunol 2000;1:515–20. doi: 10.1038/82771. PMID:11101874. [DOI] [PubMed] [Google Scholar]
- 19.Park JM, Terabe M, van den Broeke LT, Donaldson DD, Berzofsky JA. Unmasking immunosurveillance against a syngeneic colon cancer by elimination of CD4+ NKT regulatory cells and IL-13. Int J Cancer 2005;114:80–7. doi: 10.1002/ijc.20669. PMID:15523692. [DOI] [PubMed] [Google Scholar]
- 20.Terabe M, Swann J, Ambrosino E, Sinha P, Takaku S, Hayakawa Y, Godfrey DI, Ostrand-Rosenberg S, Smyth MJ, Berzofsky JA. A nonclassical non-Valpha14Jalpha18 CD1d-restricted (type II) NKT cell is sufficient for down-regulation of tumor immunosurveillance. J Exp Med 2005;202:1627–33. doi: 10.1084/jem.20051381. PMID:16365146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Terabe M, Robertson FC, Clark K, De Ravin E, Bloom A, Venzon D, Kato S, Mirza A, Berzofsky JA. Blockade of only TGF-β 1 and 2 is sufficient to enhance the efficacy of vaccine and PD-1 checkpoint blockade immunotherapy. OncoImmunology 2017;6:e1308616. doi: 10.1080/2162402X.2017.1308616. PMID:28638730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Crowe NY, Coquet JM, Berzins SP, Kyparissoudis K, Keating R, Pellicci DG, Hayakawa Y, Godfrey DI, Smyth MJ. Differential antitumor immunity mediated by NKT cell subsets in vivo. J Exp Med 2005;202:1279–88. doi: 10.1084/jem.20050953. PMID:16275765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Izhak L, Ambrosino E, Kato S, Parish ST, O'Konek JJ, Weber H, Xia Z, Venzon D, Berzofsky JA, Terabe M. Delicate balance among three types of T cells in concurrent regulation of tumor immunity. Cancer Res 2013;73:1514–23. doi: 10.1158/0008-5472.CAN-12-2567. PMID:23319803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mosely SI, Prime JE, Sainson RC, Koopmann JO, Wang DY, Greenawalt DM, Ahdesmaki MJ, Leyland R, Mullins S, Pacelli L. Rational selection of syngeneic preclinical tumor models for immunotherapeutic drug discovery. Cancer Immunol Res 2017;5:29–41. doi: 10.1158/2326-6066.CIR-16-0114. PMID:27923825. [DOI] [PubMed] [Google Scholar]
- 25.Llosa NJ, Cruise M, Tam A, Wicks EC, Hechenbleikner EM, Taube JM, Blosser RL, Fan H, Wang H, Luber BS, The vigorous immune microenvironment of microsatellite instable colon cancer is balanced by multiple counter-inhibitory checkpoints. Cancer Discov 2015;5:43–51. doi: 10.1158/2159-8290.CD-14-0863. PMID:25358689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Devaud C, Westwood JA, John LB, Flynn JK, Paquet-Fifield S, Duong CP, Yong CS, Pegram HJ, Stacker SA, Achen MG, et al. Tissues in different anatomical sites can sculpt and vary the tumor microenvironment to affect responses to therapy. Mol Ther 2014;22:18–27. doi: 10.1038/mt.2013.219. PMID:24048441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sandoval F, Terme M, Nizard M, Badoual C, Bureau MF, Freyburger L, Clement O, Marcheteau E, Gey A, Fraisse G, et al. Mucosal imprinting of vaccine-induced CD8(+) T cells is crucial to inhibit the growth of mucosal tumors. Sci Transl Med 2013;5:172ra20. doi: 10.1126/scitranslmed.3004888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nizard M, Roussel H, Diniz MO, Karaki S, Tran T, Voron T, Dransart E, Sandoval F, Riquet M, Rance B, et al. Induction of resident memory T cells enhances the efficacy of cancer vaccine. Nat Commun 2017;8:15221. doi: 10.1038/ncomms15221. PMID:28537262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.de Velasco G, Miao D, Voss MH, Hakimi AA, Hsieh JJ, Tannir NM, Tamboli P, Appleman LJ, Rathmell WK, Van Allen EM, et al. Tumor mutational load and immune parameters across metastatic renal cell carcinoma risk groups. Cancer Immunol Res 2016;4:820–2. doi: 10.1158/2326-6066.CIR-16-0110. PMID:27538576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lam VC, Lanier LL. NK cells in host responses to viral infections. Curr Opin Immunol 2017;44:43–51. doi: 10.1016/j.coi.2016.11.003. PMID:27984782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pal M, Schwab L, Yermakova A, Mace EM, Claus R, Krahl AC, Woiterski J, Hartwig UF, Orange JS, Handgretinger R, et al. Tumor-priming converts NK cells to memory-like NK cells. Oncoimmunology 2017;6:e1317411. doi: 10.1080/2162402X.2017.1317411. PMID:28680749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mailliard RB, Son YI, Redlinger R, Coates PT, Giermasz A, Morel PA, Storkus WJ, Kalinski P. Dendritic cells mediate NK cell help for Th1 and CTL responses: two-signal requirement for the induction of NK cell helper function. J Immunol 2003;171:2366–73. doi: 10.4049/jimmunol.171.5.2366. PMID:12928383. [DOI] [PubMed] [Google Scholar]
- 33.Bouwer AL, Saunderson SC, Caldwell FJ, Damani TT, Pelham SJ, Dunn AC, Jack RW, Stoitzner P, McLellan AD. NK cells are required for dendritic cell-based immunotherapy at the time of tumor challenge. J Immunol 2014;192:2514–21. doi: 10.4049/jimmunol.1202797. PMID:24477907. [DOI] [PubMed] [Google Scholar]
- 34.Bakdash G, Vogelpoel LT, van Capel TM, Kapsenberg ML, de Jong EC. Retinoic acid primes human dendritic cells to induce gut-homing, IL-10-producing regulatory T cells. Mucosal Immunol 2015;8:265–78. doi: 10.1038/mi.2014.64. PMID:25027601. [DOI] [PubMed] [Google Scholar]
- 35.Dzutsev A, Hogg A, Sui Y, Solaymani–Mohammadi S, Yu H, Frey B, Wang Y, Berzofsky JA. 2017. Differential T cell homing to colon vs small intestine is imprinted by local CD11c+ antigen presenting cells that determine homing receptors. J. Leuk. Biol 2017;102:1381–1388. doi: 10.1189/jlb.1A1116-463RR. [DOI] [PMC free article] [PubMed] [Google Scholar]