

Abstract
The Type 2 L-serine dehydratase from Legionella pneumophila (lpLSD) contains a [4Fe-4S]2+ cluster that acts as a Lewis acid to extract the hydroxyl group of L-serine during the dehydration reaction. Surprisingly, the crystal structure showed that all four of the iron atoms in the cluster were coordinated with protein cysteinyl residues and that the cluster was buried and not exposed to solvent. If the crystal structure of lpLSD accurately reflects the structure in solution, then substantial rearrangement at the active site is necessary for the substrate to enter. Furthermore, repair of the oxidized protein when the cluster has degraded would presumably entail exposure of the buried cysteine ligands. Thus, the conformation required for substrate to enter may be similar to those required for a new cluster to enter the active site. To address this, hydrogen-deuterium exchange combined with mass spectrometry (HDX MS) was used to probe the conformational changes that occur upon oxidative degradation of the Fe-S cluster. The regions that show the most significant differential HDX are either adjacent to the cluster location in the holo enzyme or connect regions that are adjacent to the cluster. The observed decrease in flexibility upon cluster binding provides direct evidence that the “tail in mouth” conformation observed in the crystal structure also occurs in solution and that the C-terminal peptide is coordinated to the [4Fe-4S] cluster in a pre-catalytic conformation. This observation is consistent with the requirement of an activation step prior to catalysis and with the unusually high level of resistance to oxygen-induced cluster degradation. Furthermore, peptide mapping of the apo form under non-reducing conditions revealed the formation of disulfide bonds between C396 and C485 and possibly between C343 and C385. These observations provide a picture of how the cluster loci are stabilized and poised to receive the cluster in the apo form and the requirement for an oxidation step in cluster formation.
Introduction
Protein-bound iron-sulfur (Fe-S) clusters have been found in all forms of life, including archaea, bacteria, and eukaryotes.1–4 They are among the oldest and most versatile inorganic cofactors and participate in such varied biological processes as respiration, photosynthesis, DNA replication and repair, gene regulation, and central metabolism.1–4 Furthermore, they are used by a large number of proteins. A larger number have been found in bacteria than in eukaryotes. E. coli, for example, contains well over 100 different iron-sulfur proteins. The electron-transfer systems account for approximately 50% of them, and as many as 17% function in non-redox roles such as dehydratases.1 The most common clusters are [2Fe-2S], [3Fe-4S], and [4Fe-4S]; the latter represents approximately 90% of the total.1 From the confirmed E. coli Fe-S proteins, more than 30 different cluster-binding motifs have been identified, and a genome search for these motifs indicates that iron-sulfur proteins may represent as much as 5% of the total bacterial proteins.
Although Fe-S clusters can be reconstituted in vitro by supplying iron and sulfide salts under reducing conditions5, living organisms have developed complex multi-protein in vivo systems for assembly and transfer of Fe-S clusters to the final acceptor protein. Two of the most common multi-protein complexes are the ISC (Iron Sulfur Cluster) and SUF (Sulfur Mobilization) systems that are found throughout living organisms.1–4 Fe-S proteins can be damaged by oxidative stress, and organisms have developed additional systems to repair the damage.2, 6 Some identified physiological oxidants are superoxide, hydrogen peroxide, and peroxynitrites.6 It is well known, however, that isolated Fe-S proteins can experience cluster degradation in vitro simply upon exposure to air.6, 7 In general, the half-life of Fe-S proteins in E. coli is approximately 40 min whereas the half-time of cluster repair is approximately 5 min.6 Thus, there appears to be a balance between oxidation and repair of these proteins during the normal aerobic growth of organisms.
In addition to Fe-S proteins with redox functions, there is a large class of non-redox enzymes called dehydratases where the clusters serve as Lewis acids to ligate a leaving hydroxyl group of the substrate. Aconitase is the archetypical member of this group and perhaps the most intensely studied.7–12 Aconitase contains a [4Fe-4S]2+ cluster, as do many of the non-redox dehydratases. To bind substrate, the clusters must be exposed to solvent, where they can also be attacked by oxidative agents. Initially, oxidation converts the cluster to an unstable [4Fe-4S]3+ form that rapidly degrades to a [3Fe-4S]1+ form. In vitro studies of this form have shown that it degrades further.13–15 Optical studies of aconitase have shown that most of the intermediate cluster forms retain some charge-transfer absorbance8, but that eventually all absorbance due to charge transfer of an Fe-S cluster is lost. These observations have also led to the suggestion that the cluster may degrade to an apoenzyme form in vivo, and that repair in this case may utilize the same pathways that originally assembled the cluster in newly synthesized proteins.6
Structures of aconitase11, 12 and other [4Fe-4S] dehydratases show that three of the four iron atoms in the cluster are bound to protein cysteine residues, and the fourth iron is not bound to the protein but exposed to solvent and free to accept the leaving group of a substrate. It came as a surprise when the crystal structure of an L-serine dehydratase from Legionella pneumophila (lpLSD) revealed that its [4Fe-4S]2+ cluster had all four iron atoms bound to protein cysteine residues.16 This is consistent, however, with this particular dehydratase exhibiting an increased resistance to oxygen inactivation17, 18 and with kinetic data that suggest it requires an activation step prior to catalysis.19 The crystal structure revealed that the fourth cysteine ligand is the C-terminal residue of the protein, and that it occupied the active site in much the same orientation as was expected for the substrate, L-serine. In addition, the structure also showed that the Fe-S cluster was completely buried inside the protein and not exposed to solvent.
Although a great deal of effort has been expended to understand how these in vivo systems work, little is known how the final acceptor proteins are recognized by the Fe-S carrier proteins or what characteristics of the acceptor proteins are necessary to accept the assembled cluster.a–f The crystal structure of lpLSD shows that the Fe-S cluster is buried within the structure and not directly exposed to solvent (Figure 1).16 A prominent feature of this structure shows the C-terminal cysteine residue forming a fourth coordination with an iron molecule of the cluster in what has been termed a “tail-in-mouth” configuration because the C-terminal portion of the protein is inserted into the active site. If the crystal structure of lpLSD accurately reflects the structure in solution, then substantial rearrangement at the active site is necessary for the substrate to enter. Furthermore, repair of the oxidized protein when the cluster has degraded would presumably entail exposure of the buried cysteine ligands. The conformational rearrangements required for a new cluster to enter the active site may be similar to those needed to open the active site for entry of substrate because both occur at nearby loci that are shielded from solvent by the same polypeptide segments.
Figure 1. Ribbon diagram and space filling models of the holo-lpLSD structure.

Top Left: The “tail-in-mouth” configuration of lpLSD is shown with the C-terminal segment of the protein in blue (tail) inserted into the active-site cleft (mouth). The Fe-S cluster is colored orange and yellow, respectively. Top Right; The same structure shown as a space-filling model. Lower Left and Right: The same structures as above with their tops rotated 90 degrees out from the plane of the page. Notice that the Fe-S cluster is not visible in the space-filling models.
To address these issues and characterize the L-serine dehydratase-FeS cluster topology in solution, we used hydrogen-deuterium exchange combined with mass spectrometry (HDX MS) to probe the conformational changes that occur upon oxidative degradation of the Fe-S cluster. One focus is the extent of HDX of peptide segments that enclose the active site and whether their HDX correlates with the loss of the cluster. Also of interest are the oxidative states adopted by the active-site cysteine ligands upon cluster degradation.
Materials and Methods
Materials
The protein was expressed and purified as previously reported.16 The native protein was maintained in an anaerobic environment to preserve the Fe-S cluster until just before hydrogen-deuterium exchange was initiated. The Fe-S cluster was degraded by removing the protein from the anaerobic chamber and exposing it to air until the charge-transfer absorbance at 400 nm was reduced to less than 10% of that prior to exposure, and the protein retained less than 5% of native activity. Any loss in volume that occurred during air exposure was corrected by adding back an equal volume of fresh buffer at several times during the course of cluster oxidation.
Guanidine HCl (8 M) was purchased from Thermo Scientific Pierce (Thermo Scientific Pierce, Grand Island, NY). All other reagents were purchased from Sigma-Aldrich (St. Louis, MO) unless otherwise specified.
HDX MS protocol
Stock solutions of both native protein containing the Fe-S cluster (holo) and inactive protein devoid of the cluster (apo) were prepared at 10 μM in 1×PBS. Continuous labeling with deuterium was initiated by diluting 5 μL of the stock solution into 20 μL of 1×PBS D2O buffer. HDX control samples (non-deuterated) were prepared in the same way with 1×PBS H2O buffer. Quenching was performed under reducing conditions with 1×PBS, 4 M guanidine HCl, 0.5 M tris-[2-carboxyethyl] phosphine (TCEP); pH adjusted to 2.5 with sodium hydroxide.
HDX reactions were conducted at 25 °C, and the extent of HD measured at 10 s, 30 s, 1 min, 2 min, 15 min, 1 h and 4 h. The half-life of the lpLSD Fe-S cluster is approximately 70 h,18 so little, if any degradation of the cluster would result during the exchange times. The exchange reaction was stopped by adding 1:1 (v/v) ice-cold reducing quench buffer. The quenched sample was then passed over a custom-packed pepsin column (2×20 mm) at 200 μL/min. Prior to LC/MS analysis, the digested peptides were captured on a 2×15 mm C8 trap column (Agilent, Santa Clara, CA, USA) and desalted with a 3 min flow at 200 μL/min of H2O containing 0.1% TFA. The peptic peptides were then separated by using a 2.1×50 mm reversed-phase C18 column (1.9 μm Hypersil Gold; Thermo Fisher Scientific, Waltham, MA, USA) with an 5.5 min linear gradient of 4%–40% CH3CN in 0.1% TFA at 50 μL/min with a nanoACQUITY UPLC (Waters, Milford, MA, USA). Protein digestion and peptide separation were carried out in water-ice bath to minimize back exchange. MS detection was performed on an LTQ-FT (Thermo Fisher Scientific, Santa Clara, CA, USA) using the following instrument parameters: spray voltage 5 kV, capillary temperature 275 °C, capillary voltage 38 V, and tube lens 185 V. Data were collected at a mass resolving power of 100,000 at m/z 400. Each experiment was carried out in duplicate.
HDX MS data analysis
To identify the peptic peptides generated from the protein and to provide a list of peptides to be followed during HDX, separate experiments without deuterium labeling were run, and product-ion mass spectra were acquired in a data-dependent mode, with the six most abundant ions from each scan selected for MS/MS. The MS/MS *.raw files were then converted to mzXML files and submitted to MassMatrix for peptide identification (20). This search was also performed against a decoy (reversed) sequence, and ambiguous identifications were discarded.
The created peptide list from three parallel runs and the *.raw files from non-deuterated runs were analyzed by using HDX Workbench (gift from the Griffin Laboratory, Scripps FL)21 to generate a peptide pool containing peptides with sufficient signal intensity and confidence to be reliably used for deuterium labeling analysis. The *.raw files for all HDX runs were then input into HDX Workbench to calculate the centroid masses of isotopic envelopes (m) and deuterium level (D%) for the peptide pool. As described previously (22), D% = {[m(P) − m(N)]/[m(F) − m(N)]} × 100%, where m(P), m(N) and m(F) are the centroid values of partially deuterated peptide, nondeuterated peptide, and fully deuterated peptide, respectively. The retention time window used for calculation of m of each peptide in each sample was manually inspected and adjusted for accurate calculation of D%, and peptides that showed interference by isotopic peaks from other peptides were discarded. The deuterium level was mathematically adjusted because the exchange media had 80% deuterium content. The data were not corrected for back exchange because two states were compared. Eventually, the D% values were exported from HDX workbench and input into Prism for generating HDX kinetic curves for both protein states.
Peptide mapping under non-reducing conditions
Non-reduced peptide mapping was performed to identify disulfide linkages on both cluster-containing (holo) and cluster-missing (apo) states of the L-serine dehydratase. The digestion was performed by mixing 4 μL of the protein stock solution with 16 μL of 1×PBS H2O buffer, which was then denatured by adding 30 μL of the non-reducing quench buffer composed of 3 M urea and 1% trifluoroacetic acid (TFA). The mixture was then submitted to online pepsin digestion, desalting, and 5.5-min gradient elution (same as the procedure described above for quenched HDX mixtures in “HDX MS protocol”). Data-dependent MS/MS scan (same as described above in “HDX MS data analysis”) was turned on during LC/MS acquisition to generate product-ion information for peptide assignment. The data was searched in MassMatrix using the same parameters as in reduced peptide mapping for peptide identification; disulfide-linked peptides were manually examined based on accurate masses and MS/MS fragmentation patterns.
Results and Discussion
Fe-S cluster binding induced protein conformational changes in L-serine dehydratase
To characterize the interaction between L-serine dehydratase and the [4Fe-4S] cluster in the protein active state, we applied comprehensive differential HDX analysis of the active, cluster-containing (holo) and inactive, cluster-missing (apo) states of the protein. In this approach, we compared the HDX kinetics of all peptic peptides (covering 95% of the protein sequence, Figure 2) derived from the apo and holo L-serine dehydratase (percentage-of-deuterium-incorporated, D%, versus time (log scale) plots were made for all peptides). To evaluate the significance of the differences in D% (ΔD%) observed between the apo and holo states, we calculated the average ΔD% for the duplicate analysis at the seven exchange time points we used at the peptide level for all peptic peptides and listed the results in the protein sequence coverage map in Figure 2, (Purple: ΔD% > 40%; Blue: 40% > ΔD% > 20%; Green: 20% > ΔD% > 5%; Gray: > 5%).
Figure 2. Sequence coverage map of peptic digestion of L-serine dehydratase.

Each bar indicates a peptide identified by mass spectrometry. The color and inside numbers represent the average ΔD% for the duplicate analysis of seven exchange time points between [4Fe-4S] free and bound states (Purple: ΔD% > 40%; Blue: 40% > ΔD% > 20%; Green: 20% > ΔD% > 5%). Positive values of average ΔD% indicate less D% in the holo enzyme state.
As expected, most regions in the protein have insignificant average ΔD% values (less than 5; colored in grey in Figure 2). For example, Figure 3a–c demonstrates three representative HDX kinetic plots of regions unaffected by [4Fe-4S] cluster binding in L-serine dehydratase, showing overlapping kinetic curves from both the [4Fe-4S] bound and unbound states. Region 41–50 is well structured and buried; therefore, it shows consistently low HDX extent at all time points. On the contrary, region 330–340 is highly flexible/disordered and undergoes extensive HDX, even at short times. Region 360–367 is intermediate, suggesting low flexibility or compromise behavior possibly due to constituent regions that contain both fast and slow exchangers.
Figure 3. Peptide-level HDX kinetics of L-serine dehydratse.

A comparison between [4Fe-4S] cluster-free (black) and bound (red) enzyme shows no differences in HDX at peptide regions shown in a, b, and c, and significant changes in HDX at the C-terminal region near C458 (d), the region near C396 (e), and the regions near C385 (f)(g).
In contrast, some regions in the protein show significant average ΔD% values (colored in green, blue and purple in Figure 2), and they all exhibit the same trend of having less D uptake after the [4Fe-4S] cluster binds to the protein. These regions are likely located at or near the binding site of the [4Fe-4S] cluster, and, therefore, undergo protection from HDX upon cluster binding. The observed differences in these regions are considered significant because all peptides covering the same region showed consistent HDX kinetics, smooth time-dependent curves, and similar ΔD% values. The significance of the structural perturbation in these regions is then evaluated by ranking by their average ΔD% values, as indicated by different colors in Figure 2 (Purple: ΔD% > 40%; Blue: 40% > ΔD% > 20%; Green: 20% > ΔD% > 5%). Representative HDX kinetic curves from these regions are shown in Figures 3d–g.
To compare the HDX mapping result of the Fe-S cluster binding interface to that determined by crystallography, the regions with significant HDX differences are mapped onto the crystal structure of the cluster containing L-serine dehydratase with the same color code (Figure 4, PDB:4RQO, monomer extracted). As expected, the most significant differences (colored in blue and purple) occur at regions near the binding site of the [Fe-S] cluster. Two of these regions, peptide 392–409 (Figure 3e) and peptide 452–458 (Figure 3d), contain two of the four cluster-coordinating cysteines, C396 and C458, respectively. Another short region with a large HDX difference, peptide 387–391 (Figure 3g), adjoins another binding cysteine, C385, although peptide 375–386 (Figure 3f) only has a relatively small average ΔD%. The peptide 387–391 contains a leucine residue (L389) that is in van der Waals contact with another leucine residue (L236) that is found in a loop between two helices that form a stretch of polypeptide from residue 215–249 (Figure 4) that shows an average HDX difference between 5 and 20%. The van der Waals contacts between these leucine residues appear to stabilize the loop structures, which themselves appear to form a cap over a possible entrance into the cluster binding site in the interior of the protein.
Figure 4. Structural analysis of L-serine dehydratase HDX kinetics.


Exchange rates are mapped onto the serine dehydratase crystal structure of the holo enzyme (PDB 4RQO). Regions exhibiting the largest difference (ΔD% > 40%) are colored purple; regions exhibiting a medium difference (40% > ΔD% > 20%) are colored blue; and regions exhibiting a small but significant difference (20% > ΔD% > 5%) are colored in green, as in Figure 1. The four cluster-coordinating cysteine residues are colored red and labeled. The two leucine residues (L236 and L389) forming van der Walls contacts between two loop structures that display reduced deuterium exchange in the apo enzyme form are shown in stick representation and colored in orange.
The last cysteine involved in the [4Fe-4S] cluster binding, C343, was not found in the pepsin digestion possibly owing to poor LC retention of short peptides containing this residue; therefore, HDX information is not available for the region 341–353.
Interestingly, the greatest difference in HDX before and after the [4Fe-4S] cluster binding occurs at the C-terminus peptide 452–458 (Purple in Figure 4), which has the average ΔD% of 53%. The HDX kinetics for C-terminal peptide 452–458 (Figure 3d) indicate it is flexible and solvent-accessible in the apo state of L-serine dehydratase because it becomes fully deuterated after 10 s exchange (black curve in Figure 3d); however, upon [4Fe-4S] cluster binding, this region undergoes much slower HDX (red curve in Figure 3d), indicating considerably more protection in the C-terminus in the holo form. The HDX result strongly suggests that the C-terminus folds to bury the [4Fe-4S] cluster upon its binding. Moreover, this folding is also consistent with the unusual extended half-life of this Fe-S protein.
Another area that shows significant deuterium exchange in the absence of the cluster is peptide 145–152 (Figure 4). This peptide forms a loop with three glycine residues immediately adjacent to S16-S18 that form hydrogen bonds with the carbonyl oxygens of the C-terminal cysteine residue (C458). Enhanced deuterium exchange in this region upon the [4Fe-4S] cluster release from the protein may result from increased flexibility of the S16-S18 region when the hydrogen bonds to the terminal cysteine are broken as C458 repositions to form a disulfide linkage to C396 (see below).
Disulfide bond formation in the inactive protein
To understand more completely the L-serine dehydratase apo/holo state transitions, we examined the oxidation states of the four local cysteine residues at the binding core for both [4Fe-4S]-bound and unbound states. In this case, we applied non-reducing digestion in the peptide mapping to preserve any disulfide bonds present. As expected, digestion of the cluster-containing L-serine dehydratase sample without disulfide reduction gives the same sequence coverage as that under reducing conditions, confirming there are no disulfide bonds in this form of the protein. Peptide mapping of non-reduced L-serine dehydratase apo protein, however, showed less sequence coverage under the same database search criteria (not considering disulfide bond linked peptides), missing the overlapping peptides containing the four binding cysteines.
A careful search for disulfide-bond-linked peptides in the non-reducing peptide mapping of apo L-serine dehydratase revealed disulfide bond formation among the coordinating cysteines in the binding core of the apo state. The four peptides, 375–386, 392–409/392–410, and 452–458, that we originally found in the peptide-mapping experiment under reducing conditions, contained three of the four cysteines. We examined them for any cross disulfide-bond formation and found, by using accurate mass measurements and MS/MS, that disulfide linkages occurred between peptides 452–458 and 392–409 as well as 452–458 to 392–410, indicating that C396 forms a disulfide bond with C458 in apo L-serine dehydratase (see Figure 5). We could not find any linkage involving peptide 375–386 with either 452–458 or 392–409/392–410. A possible explanation is that C385 forms a disulfide bond with C343 in the apo form, but can’t be detected with peptide mapping under non-reducing conditions because the coverage of C343, as mentioned above, is incomplete. Furthermore, tryptic peptide mapping of the same region did not give useful coverage because there are no trypsin cleavages sites (K, R) between G333 and E398.
Figure 5. Disulfide bond linkage of the four cluster coordinating cysteines in the apo enzyme form.
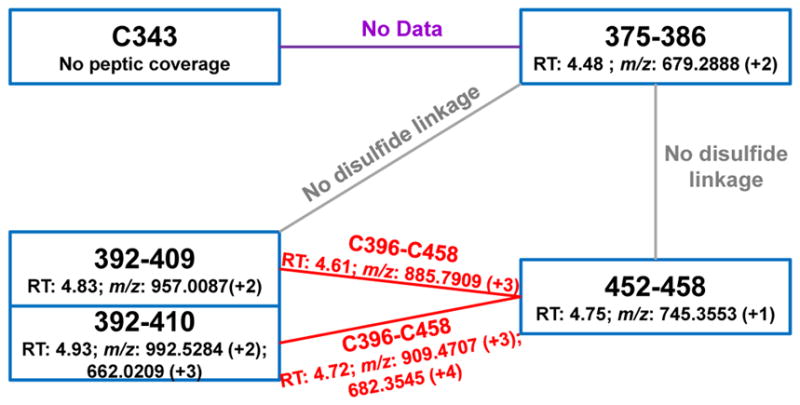
The state of disulfide linkage of C343, C385, C396, and C458 in the apo form as revealed by peptide mapping under non-reducing conditions is depicted.
Conclusion
Based on homology with other non-redox dehydratases containing a [4Fe-4S] cluster, we expected three of the four iron atoms to be coordinated with three protein cysteine residues. However, an X-ray crystallography study of lpLSD revealed a structure in which all four iron atoms of the [4Fe-4S] cluster are coordinated with protein cysteine residues. To investigate this observation further and to explore the transitions that occur upon cluster binding, we conducted peptide-level HDX MS under reducing conditions and peptide mapping under non-reducing conditions. The regions that show the most significant differential HDX are either adjacent to the cluster location in the holo enzyme or in connect regions that are adjacent to the cluster. The crystal structure shows that the C-terminal residues, 454–458, are inserted into the active site cleft and protected from solvent. Therefore, the observed decrease in flexibility upon cluster binding of this peptide segment provides direct evidence that the “tail in mouth” conformation (Figure 1) observed in the crystal structure is not a crystallization artifact and that the C-terminal peptide is coordinated to the [4Fe-4S] cluster in a pre-catalytic conformation. This observation is also consistent with the increased resistance to oxygen-induced cluster degradation compared to bacterial Fe-S proteins in general and for the requirement of an activation step prior to catalysis. Furthermore, characterization of the four binding cysteines after [4Fe-4S] cluster degradation with non-reduced peptide mapping revealed the formation of disulfide bonds between C396 and C485 and possibly between C343 and C385. Although the latter requires further evaluation, these observations provide a picture of how the cluster loci are stabilized and poised to receive the cluster in the apo form and the requirement for an oxidation step in cluster formation.
Supplementary Material
Acknowledgments
Funding: This research was supported by National Institute of General Medical Sciences (8 P41 GM103422) of the National Institutes of Health.
Abbreviations
- Fe-S
Iron-Sulfur
- LSD
L-serine dehydratase
- lpLSD
LSD from Legionella pneumophila
- HDX
hydrogen-deuterium exchange
- TCEP
0.5 M Tris[2-carboxyethyl] phosphine
- PBS
phosphate buffered saline
- holo
enzyme containing the Fe-S cluster
- apo
enzyme without the Fe-S cluster
Footnotes
Supporting Information. Figure S1 is included as supporting information. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Fontecave M. Iron-sulfur clusters: ever-expanding roles. Nature Chemical Biology. 2006;2:171–174. doi: 10.1038/nchembio0406-171. [DOI] [PubMed] [Google Scholar]
- 2.Py B, Barras F. Building Fe-S proteins: bacterial strategies. Nature Reviews/Microbiology. 2010;8:436–446. doi: 10.1038/nrmicro2356. [DOI] [PubMed] [Google Scholar]
- 3.Roche B, Aussel L, Ezraty B, Mandin P, Py B, Barras F. Iron/Sulfur proteins biogenesis in prokaryotes: Formation, regulation and diversity. Biochim Biophys Acta. 2013;1827:923–937. doi: 10.1016/j.bbabio.2013.05.001. [DOI] [PubMed] [Google Scholar]
- 4.Py B, Mareau PL, Barras F. Fe-S clusters, fragile sentinels of the cell. Curr Opin Microbiol. 2011;14:218–223. doi: 10.1016/j.mib.2011.01.004. [DOI] [PubMed] [Google Scholar]
- 5.Malkin R, Rabinowitz JC. The reconstitution of clostridial ferredoxin. Biochem Biophy Res Commun. 1966;23:822–827. doi: 10.1016/0006-291x(66)90561-4. [DOI] [PubMed] [Google Scholar]
- 6.Djaman O, Outten FW, Imlay JA. Repair of oxidized iron-sulfur clusters in Escherichia coli. J Biol Chem. 2004;279:44590–44599. doi: 10.1074/jbc.M406487200. [DOI] [PubMed] [Google Scholar]
- 7.Flint DH, Allen RM. Iron-Sulfur Proteins with Nonredox Functions. Chem Rev. 1996;96:2315–2334. doi: 10.1021/cr950041r. [DOI] [PubMed] [Google Scholar]
- 8.Emptage MH, Dreyer JL, Kennedy MC, Beinert H. Optical and EPR characterization of different species of active and inactive aconitase. J Biol Chem. 1983;258:11106–11111. [PubMed] [Google Scholar]
- 9.Beinert H, Kennedy MC, Stout CD. Aconitase as Iron-Sulfur Protein, Enzyme, and Iron-Regulatory Protein. Chem Rev. 1996;96:2335–2373. doi: 10.1021/cr950040z. [DOI] [PubMed] [Google Scholar]
- 10.Emptage MH, Kent TA, Kennedy MC, Beinert H, Munck E. Mössbauer and EPR studies of activated aconitase: development of a localized valence state at a subsite of the [4Fe-4S] cluster on binding of citrate. Proc Natl Acad Sci. 1983;80:4674–4678. doi: 10.1073/pnas.80.15.4674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lauble H, Kennedy MC, Beinert H, Stout CD. Crystal structures of aconitase with isocitrate and nitroisocitrate bound. Biochemistry. 1992;31:2735–2748. doi: 10.1021/bi00125a014. [DOI] [PubMed] [Google Scholar]
- 12.Lloyd SJ, Lauble L, Prasad GS, Stout CD. The Mechanism of Aconitase: 1.8 A Resolution Crystal Structure of the S642A:Citrate Complex. Prot Sci. 1999;8:2655–2662. doi: 10.1110/ps.8.12.2655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Flint DH, Smyk-Randall E, Tuminello JF, Draczynska-Lusiak B, Brown OR. The inactivation of dihydroxy-acid dehydratase in Escherichia coli treated with hyperbaric oxygen occurs because of the destruction of its Fe-S cluster, but the enzyme remains in the cell in a form that can be reactivated. J Biol Chem. 1993;268:25547–25552. [PubMed] [Google Scholar]
- 14.Varghese S, Tang Y, Imlay JA. Contrasting sensitivities of Escherichia coli aconitases A and B to oxidation and iron depletion. J Bacteriol. 2003;185:221–230. doi: 10.1128/JB.185.1.221-230.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Flint DH, Tuminello JF, Emptage MH. The inactivation of Fe-S cluster containing hydro-lyases by superoxide. J Biol Chem. 1993;268:22369–22376. [PubMed] [Google Scholar]
- 16.Thoden JB, Holden HM, Grant GA. Structure of L-serine dehydratase from Legionella pneumophila: Novel use of the C-terminal cysteine as an intrinsic competitive inhibitor. Biochemistry. 2014;53:7615–7624. doi: 10.1021/bi501253w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chen S, Xu XL, Grant GA. Allosteric activation and contrasting properties of L-serine dehydratase types 1 and 2. Biochemistry. 2012;51:5320–5328. doi: 10.1021/bi300523p. [DOI] [PubMed] [Google Scholar]
- 18.Xu XL, Grant GA. Identification and characterization of two new types of bacterial l-serine dehydratases and assessment of the function of the ACT domain. Arch Biochem Biophys. 2013;540:62–69. doi: 10.1016/j.abb.2013.10.009. [DOI] [PubMed] [Google Scholar]
- 19.Grant GA. Kinetic evidence of a noncatalytic substrate binding site that regulates activity in Legionella pneumophila L-serine dehydratase. Biochemistry. 2012;51:6961–6967. doi: 10.1021/bi3008774. [DOI] [PubMed] [Google Scholar]
- 20.Xu H, Freitas MA. MassMatrix: a database search program for rapid characterization of proteins and peptides from tandem mass spectrometry data. Proteomics. 2009;9:1548–1555. doi: 10.1002/pmic.200700322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pascal BD, Willis S, Lauer JL, Landgraf RR, West GM, Marciano D, Novick S, Goswami D, Chalmers MJ, Griffin PR. HDX workbench: software for the analysis of H/D exchange MS data. J Am Soc Mass Spect. 2012;23:1512–1521. doi: 10.1007/s13361-012-0419-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhang Z, Smith DL. Determination of amide hydrogen exchange by mass spectrometry: a new tool for protein structure elucidation. Prot Sci. 1993;2:522–53. doi: 10.1002/pro.5560020404. [DOI] [PMC free article] [PubMed] [Google Scholar]
References
- a.Boyd ES, Thomas KM, Dai Y, Boyd JM, Outten FW. Interplay between oxygen and Fe-S cluster biogenesis: Insights from the SUF pathway. Biochemistry. 2014;53:5834–5847. doi: 10.1021/bi500488r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- b.Fox NG, Chakrabarti M, McCormick SP, Lindahl PA, Barondeau DP. Biochemistry. 2015;54:3871–3879. doi: 10.1021/bi5014485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- c.Netz DJA, Mascarenhas J, Stehling O, Pierik AJ, Lill R. Maturation of cytosolic and nuclear iron-sulfur proteins. Trends in Cell Biol. 2014;24:303–312. doi: 10.1016/j.tcb.2013.11.005. [DOI] [PubMed] [Google Scholar]
- d.Rouault TA. Mammalian iron-sulfur proteins: Novel insights into biogenesis and function. Nature Rev Mol Cell Biol. 2015;16:45–55. doi: 10.1038/nrm3909. [DOI] [PubMed] [Google Scholar]
- e.Maio N, Rouault TA. Iron-sulfur cluster biogenesis in mammalian cells: New insights into the molecular mechanisms of cluster delivery. Biochim Biophys Acta. 2015;1853:1493–1512. doi: 10.1016/j.bbamcr.2014.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- f.Stehling O, Wilbrecht C, Lill R. Mitochondrial iron-sulfur protein biogenesis and human disease. Biochimie. 2014;100:61–77. doi: 10.1016/j.biochi.2014.01.010. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.