

Abstract
Enzymes are attractive as immunotherapeutics because they can catalyze shifts in the local availability of immunostimulatory and immunosuppressive signals. Clinical success of enzyme immunotherapeutics frequently hinges upon achieving sustained biocatalysis over relevant timescales. The timescale and location of biocatalysis are often dictated by the location of the substrate. For example, therapeutic enzymes that convert substrates distributed systemically are typically designed to have a long half-life in circulation, whereas enzymes that convert substrates localized to a specific tissue or cell population can be more effective when designed to accumulate at the target site. This Topical Review surveys approaches to improve enzyme immunotherapeutic efficacy via chemical modification, encapsulation, and immobilization that increases enzyme accumulation at target sites or extends enzyme half-life in circulation. Examples provided illustrate “replacement therapies” to restore deficient enzyme function, as well as “enhancement therapies” that augment native enzyme function via supraphysiologic doses. Existing FDA-approved enzyme immunotherapies are highlighted, followed by discussion of emerging experimental strategies such as those designed to enhance anti-tumor immunity or resolve inflammation.
Graphical abstract
Enzymes drive key processes in immunology
Enzymes are proteins that rapidly convert many copies of substrate molecules into products with fine specificity. Nearly all reactions within living systems are catalyzed by enzymes. Within the innate immune system, for example, neutrophils secrete granules containing various enzymes that can synthesize anti-bacterial compounds, as well as degrade the host extracellular matrix to enable cell migration through damaged or diseased tissue.1–2 Likewise, classically-activated macrophages produce pro-inflammatory nitric oxide via upregulated expression of inducible nitric oxide synthase, and secrete matrix metalloproteinases (e.g., MMP-1, -2, -7, -9, and -12) that degrade various extracellular matrix components, including collagen, elastin, and fibronectin.3 Enzymes also mediate resolution of inflammation. For example, arginase 1 expressed by alternatively-activated macrophages depletes arginine supplies required for nitric oxide synthesis, and also contributes to polyamine and proline syntheses that are important for cell proliferation and tissue repair.4 Tissue transglutaminase secreted by alternatively-activated macrophages stabilizes the extracellular matrix during wound healing by covalently crosslinking matrix components.5 Additionally, CD39 and CD73 work in concert to resolve inflammation by dephosphorylating adenosine triphosphate, a pro-inflammatory signal, to adenosine, an anti-inflammatory signal.6–7 As a final example, immunological tolerance at the fetal-maternal interface and within tumor microenvironments is established in part by the enzyme indoleamine 2,3-dioxygenase, which catalyzes the breakdown of tryptophan into kynurenines, where both tryptophan depletion and kynurenine production act in concert to produce immunosuppressive effects.8–9
Enzymes as immunotherapeutics
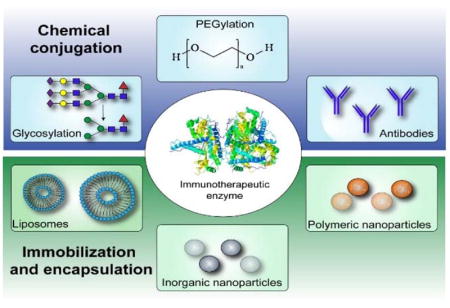
The diverse, yet highly specific activities of enzymes have fostered interest in their use as therapeutics since the early 1960s. At present, approximately 15% of all FDA-approved proteins are enzymes for use in a wide variety of therapeutic applications ranging from cancer treatment, to clot lysis for ischemic stroke, and digestion.10–11 A smaller subset of these are used to treat immune-related disorders, either by replacing enzymes that are deficient or to supply supra-physiologic enzyme doses to enhance activity. Surveyed herein are both established and emerging examples of enzymes as immunotherapeutics, with emphasis placed on strategies to improve enzyme stability, bioavailability, and targeting to specific cell or tissue sites via chemical modification, immobilization, or encapsulation (Figure 1). In particular, the following sections illustrate examples of enzyme therapies to restore or enhance immune system function via glycosylation or PEGylation, the use of enzyme-antibody conjugation for cancer immunotherapy and to treat neuroinflammation, and the use of nanoparticle vehicles for localized enzyme delivery to treat acute and chronic inflammation in different areas of the body.
Figure 1.

Enzyme therapy delivery strategies to restore or enhance immune system function.
1. FDA-approved enzyme immunotherapeutics
Deficiencies in enzyme expression or catalytic activity underlie numerous pathologies. For example, inadequate lysosomal enzyme function underpins the family of lysosomal storage diseases (LSDs) characterized by substrate accumulation within lysosomes leading to cell dysfunction or death.12 Numerous LSDs are linked to immune system irregularities.13 Likewise, adenosine deaminase-severe combined immunodeficiency disorder (ADA-SCID) is characterized by lymphocyte dysfunction due to a deficiency in adenosine deaminase. Enzyme replacement therapy (ERT) is a clinical modality to restore deficient enzyme function in which patients receive regular parenteral enzyme infusions.14 Despite active investigation since the early 1960s, however, only a handful of enzymes have proven successful for ERT, while many more have failed.15
In contrast, enzyme-based cancer therapy is an adjunctive clinical modality to degrade a substrate into a product that renders tumor cells more susceptible to chemotherapy or immunotherapy. At present, there is only one FDA-approved enzyme for cancer treatment, L-asparaginase, for acute lymphoblastic leukemia.10 The success of both ERT and enzyme-based cancer therapy is limited primarily due to issues of rapid enzyme clearance from the body, enzyme degradation via proteases, enzyme immunogenicity, or the inability to accumulate enzymes at target tissue sites or within specific cell populations.16–18 Glycosylation and PEGylation are two chemical modification strategies to address these issues that have enabled successful clinical translation of immunotherapeutic enzymes for both ERT and enzyme-based cancer therapy, as discussed in more detail below.
A. Glycosylation to improve the efficacy of immunotherapeutic enzyme replacement therapy
Glycosylation of β-glucocerebrosidase for treatment of Gaucher disease
Increased lysosomal levels of glucosylceramide, a type of sphingolipid, can lead to onset of Gaucher disease, a lysosomal storage disease characterized by anemia, thrombocytopenia, bone disease, hepatomegaly, and splenomegaly.14, 19–20 β-glucocerebrosidase is a lysosomal enzyme that catalyzes the hydrolysis of glucosylceramide to glucose and ceramide. β-glucocerebrosidase deficiency leads to glucosylceramide accumulation within lysosomes, particularly the lysosomes of reticular endothelial system (RES) macrophages. ERT using placenta-derived β-glucocerebrosidase has poor efficacy because only small amounts of enzyme are taken up into lysosomes of RES macrophages, with the vast majority instead taken up by hepatocytes.21 Targeting the macrophage mannose receptor (MMR; CD206) and mannose-6-phosphate receptor (M6PR) by modifying native glycans on β-glucocerebrosidase with mannose can increase recognition and uptake by RES macrophages, and also facilitate receptor-mediated endocytosis into lysosomal compartments,22 thereby improving ERT efficacy. For example, mannosylated placenta-derived β-glucocerebrosidase (Ceradase®) significantly increases pediatric patient hemoglobin levels and platelet counts, decreases hepatosplenomegaly, and improves skeletal symptoms.23–25 To address limited availability of donated placental tissue, a recombinant form (Cerezyme®) has since been developed with mannose-terminated N-glycans. Notably, Ceredase® and Cerezyme® are the first protein therapeutics for which carbohydrate engineering was used to improve efficacy by targeting a specific cell type.25 Building from this success, glycoconjugation has been used to improve the efficacy of various enzyme and other protein drugs. For more on this topic, we direct the reader to an excellent recent review.26
Glycosylation of α-galactosidase A for treatment of Fabry disease
Given the success of mannosylated β-glucocerebrosidase to treat Gaucher disease, glycoconjugate targeting for ERT has been expanded to other lysosomal storage diseases. Fabry disease for example, is caused by deficiency of α-galactosidase A which digests globotriaosylceramide and other related glycolipids. Excess globotriaosylceramide accumulates primarily within lysosomes of endothelial cells in the microvasculature of the kidneys, heart, and brain,27 and is linked to chronic pro-inflammatory Toll-like receptor activation.28 Recombinant α-galactosidase A (Fabrazyme®) has been modified with a terminal mannose to target MMR and M6PR. However, this glycoconjugate also targets enzyme uptake via the asialoglycoprotein receptor in the liver, as well as uptake in the spleen. For example, 10–100 times more α-galactosidase A activity was observed in the liver and spleen of mice after intravenous infusion, compared to the kidney and heart.29 Because the liver and spleen are not severely affected in Fabry disease, this significant off-site uptake reduces the effective dose of Fabrazyme reaching the kidneys, heart, and brain where it is most needed. Thus, treatment of Fabry disease via ERT will likely benefit from improved targeting approaches.
B. PEGylation to improve the efficacy of immunotherapeutic enzyme replacement therapy
PEGylation of adenosine deaminase for treatment of SCID
Adenosine deaminase is an enzyme that converts adenosine and deoxyadenosine to inosine and deoxyinosine, respectively. Adenosine deaminase deficiency (ADA) accounts for 20% of SCID cases, and is an autosomal recessive immune system deficiency which results in diminished or absent T, B, and NK cells. Readers interested in ADA and the mechanisms that lead to its pathological presentation are directed to an excellent recent review.30 The hallmark of ADA-SCID is recurrent infections due to impaired adaptive immunity as a result of progressive lymphopenia after birth.31 A treatment for ADA-SCID is infusion of adenosine deaminase to reduce intra- and extracellular deoxyadenosine levels that are toxic to developing lymphocytes,32 as well as extracellular adenosine levels that can inhibit T cell activation and expansion.33–34
Native adenosine deaminase has a short in vivo half-life, with activity lasting only a few hours after injection.35 Covalent conjugation of polyethylene glycol (i.e., “PEGylation”) to lysine residues of adenosine deaminase significantly increases its hydrodynamic radius, decreases its renal excretion, and shields it from proteolytic cleavage, which together increase its half-life in circulation from hours to days.36 A commercial formulation of PEG-adenosine deaminase, known as Adagen®, has been approved in the United States since 1990. For more on the history and use of Adagen®, we direct the reader to a recent review.32
C. PEGylation to improve the efficacy of immunotherapeutic enzymes for cancer treatment
PEGylation of L-asparaginase for treatment of acute lymphoblastic leukemia
Lymphoma is a group of cancers found in the blood, which originate from aberrant lymphocytes. Acute lymphoblastic leukemia (ALL) occurs specifically in lymphocyte precursors that become arrested during development and replace normal bone marrow components.37 Lymphoblastic leukemia cells have reduced capacity for L-asparagine biosynthesis, which renders them more susceptible to chemotherapeutic-induced death in asparagine-deficient plasma. Infusion of supraphysiologic doses of L-asparaginase, an enzyme that degrades L-asparagine, is FDA-approved for treatment of ALL. However, recombinant L-asparaginase has a half-life in blood of only ~12 hours. PEGylation can extend L-asparaginase half-life to approximately 6 days,38 and PEGylated asparaginase administered five-fold less frequently than the non-PEGylated formulation had comparable efficacy for event-free survival three years after a safety study.39 This serves as another example of how systemic enzyme infusions can have remarkable therapeutic efficacy, if the challenges of proteolytic degradation and renal clearance can be avoided via simple PEG modification.
Challenges of PEG-protein conjugates
Although PEGylation has shown promise in the past and has led to approval of approximately 10 protein-based therapies, there is increasing concern regarding its potential immunogenicity. Anti-PEG antibodies can diminish efficacy of the PEG therapeutic or result in adverse reactions, such as anaphylaxis.40 A number of pre-clinical and clinical reports indicate the rise of anti-PEG antibodies in both patients and animals.41 Reports by Armstrong and colleagues suggest that more than 25% of blood donors are positive for anti-PEG IgG antibodies.42–43 However, contrasting reports that claim only 1 in 500 (0.2%) donors have anti-PEG antibodies.44 In light of this ongoing debate, alternatives to PEG should be investigated, especially for enzymes that are delivered systemically and repeatedly.
2. Emerging opportunities in enzyme immunotherapeutics
A. Targeted immunotherapeutic enzyme delivery via antibody conjugation
By the mid-1970s, monoclonal antibodies were emerging as a new a class of targeted therapeutics. The concurrent advent of recombinant DNA technology enabled development of antibody-enzyme fusion proteins, in which therapeutic enzymes are endowed with targeting properties by joining their gene with that of an antibody or antibody fragment. In 1987, for example, fusion proteins of chemotherapeutic prodrug converting enzymes and tumor targeting antibodies emerged as an experimental cancer treatment known as, “antibody-directed enzyme prodrug therapy (ADEPT)”.45,46 Inspired by this concept, a plethora of different antibody-enzyme fusion proteins emerged over the last several decades. Alternatively, covalent conjugation or cross-linking of enzymes onto antibodies provides a simple approach to circumvent difficulties with recombinant antibody-enzyme fusion protein production. Here, we highlight pre-clinical examples of antibody-mediated enzyme targeting for cancer immunotherapy and anti-inflammatory ERT.
Antibody-enzyme conjugates to amplify tumor immune recognition
Natural killer (NK) cells play a vital role in eliminating cancer cells and restricting tumor growth. For example, NK cells can detect tumor cells via the natural killer group 2D (NKG2D) receptor that recognizes abnormal glycans.47–48 However, surface glycans on tumor cells that become modified with excess sialyl groups (i.e., “hypersialylated”) are preferentially recognized by Siglec receptors expressed by NK cells instead of NKG2D, which serves as an immune escape mechanism by inhibiting NK-mediated tumor cell lysis.48 Desialylating tumor cell glycans with enzymes, such as sialidase, can therefore increase tumor immunogenicity by concurrently preventing Siglec binding and promoting NKG2D binding.49 Recently, Bertozzi and colleagues developed a novel strategy to increase tumor cell susceptibility to antibody-dependent cell-mediated cytotoxicity (ADCC) by conjugating sialidase to trastuzumab, a HER2-specific antibody (Figure 2).50 The trastuzumab-sialidase fusion protein desialylated cancer cell surface glycans, which increased NK cell activation by promoting NKG2D binding and preventing Siglec binding in vitro. Thus, using antibody-enzyme conjugates to edit the cancer cell surface glycocalyx provides a promising new approach to improve cancer immunotherapy.
Figure 2.

Antibody-sialidase conjugates (T-Sia) that improve cancer immunotherapy by targeting the sialic acid axis of immune modulation. (A) Hypersialylated glycans on cancer cells bind to NK inhibitory receptors (Siglecs) and block interactions with NK-activating receptors (NKG2Ds). (B) Sialidase fused to trastuzumab (Tras) is localized to HER2+ cancer cells. Sialidase desialylates cancer cell surface glycans, which concurrently prevents Siglec binding and promotes NKG2D binding, thereby increasing tumor cell susceptibility to NK cell-mediated ADCC or “antibody dependent cell-mediated cytotoxicity”. (C) Cytotoxic activity of NK cells against different HER2-expressing cancer cells in the presence of either Tras or T-Sia in vitro. Reprinted with permission from ref 50. Copyright 2016 National Academy of Sciences.
Antibody-enzyme conjugates to treat neuroinflammation resulting from lysosomal storage diseases in the central nervous system
Many lysosomal storage diseases are associated with altered inflammatory cytokine levels that result in neuroinflammation.51 Several mucopolysaccharidosis (MPS) metabolic disorders, for example, are characterized by a deficiency of enzymes that catalyze glycosaminoglycan degradation, which leads to neuroinflammation and neurodegeneration. Conventional ERT approaches often fail for lysosomal storage diseases within the brain because the enzymes are too large to cross the blood-brain barrier (BBB). Antibody-enzyme conjugates can address this challenge by engaging insulin receptor-mediated IgG antibody transport across the BBB.52 For example, IgG fused to iduronate 2-sulfatase penetrated the brain and treated mice with MPS Type II after intravenous administration.53 Similarly, IgG fused to arylsulfatase A for treatment of metachromatic leukodystrophy and IgG fused to N-sulfoglucosamine sulfohydrolase for treatment of MPS type IIIA rapidly penetrated the brain of Rhesus monkeys following intravenous delivery.54–55
B. Nanocarriers for sustained delivery of anti-inflammatory enzymes
Immobilizing anti-inflammatory enzymes on or within nano-scale carriers is emerging as a useful approach to accumulate enzyme within a target tissue. Below we survey recent pre-clinical examples of PEG-modified (i.e., “PEGylated”) liposomes, as well as organic and inorganic nanoparticles, for sustained local delivery of enzymes to treat acute and chronic inflammation.
Liposome-encapsulated enzymes to treat acute and chronic inflammation
Superoxide dismutase (SOD) is an enzyme that generates molecular oxygen or hydrogen peroxide from superoxide produced by pro-inflammatory cytokine signaling, and extracellular SOD (EC-SOD) can inhibit inflammation.56–57 Likewise, catalase is an enzyme that regulates the signaling activity of hydrogen peroxide, such as that generated by SOD, by converting it into water and oxygen. Encapsulating SOD in PEGylated liposomes can decrease inflammation in a rat model of rheumatoid arthritis by increasing enzyme intra-articular retention time and improving localization.58,59 SOD-loaded PEGylated liposomes also reduced levels of TNF-α and oxidative species more effectively than free SOD in a rat model of peritonitis established via intraperitoneal injection of lipopolysaccharide (LPS), a pro-inflammatory bacterial endotoxin.60 SOD and catalase encapsulated within liposomes also reduced skin inflammation in a murine ear edema model,61 as well as periodontal inflammation in a canine model.62
Nanoparticle encapsulated enzymes to treat acute and chronic inflammation
In recent years, various enzyme-nanoparticle formulations have been developed to treat acute and chronic inflammation in different areas of the body. For example, protective antioxidant carriers for endothelial targeting (“PACkET”), are enzyme-loaded nanoparticles engineered to bind pulmonary vasculature endothelium (Figure 3).63 PACkET nanoparticles are formulated with oleate, oleate-coated magnetite, calcium cations, and a biotinylated Pluronic F-127 copolymer that is used to immobilize streptavidin-conjugated mouse anti-PECAM antibodies on the particle surface. Antioxidant enzymes (SOD or catalase) loaded into PACkET nanoparticles were efficiently delivered to the lung after intravenous injection in LPS-challenged mice. Catalase PACkET alleviated endothelial damage and inflammation by mitigating the pathogenic accumulation of bronchoalveolar lavage protein in the alveolar compartment in LPS-challenged mice. Likewise, catalase PACkET inhibited leukocyte transmigration at inflammatory sites more effectively than nanoparticles containing the same enzyme but coated with isotype control IgG. Alternatively, in LPS-challenged mice, SOD PACkET effectively inhibited endothelial activation by pro-inflammatory cytokines and significantly reduced the pulmonary level of macrophage inflammatory protein-2 (MIP-2) and TNF when compared to particles coated with isotype control IgG.
Figure 3.

Protective Antioxidant Carriers for Endothelial Targeting (PACkET). (A) Antioxidant enzymes are encapsulated within protective antioxidant carriers (PACs), while anti-PECAM monoclonal antibodies (Ab) are immobilized on the PAC surface. (B) Protection of endothelial cells exposed to H2O2 with catalase (CAT)-loaded Ab or IgG PACs. (C) Cytokine (MIP2 and TNF) expression levels in mice receiving superoxide dismutase (SOD)-loaded Ab or IgG PACs with and without LPS for 24 h. Reprinted with permission from ref 63. Copyright 2014 Elsevier.
Along this line, SOD encapsulated in nanoparticles of poly(lactic-co-glycolic acid) (PLGA), a polymer widely used in FDA-approved devices and formulations, protected human neurons from oxidative stress ex vivo more effectively than native SOD and PEG-SOD.64 Likewise, SOD-nanoparticles constructed from polybutylcyanoacrylate and PLGA conjugated to antibodies reduced neuroinflammation and improved behavior in a mouse model of cerebral ischemia and reperfusion injury.65 Furthermore, SOD encapsulated within poly(L-lysine-PEG) nanoparticles reduced aortic inflammation associated with hypertension in a mouse model of diet-induced obesity.66 Finally, a copolymer system of SOD conjugated to divinyl ether and maleic anhydride reduced inflammation associated with fibrosis of rat liver via uptake by Kupffer and liver endothelial cells.67
Nanocarriers have also been used to deliver enzymes other than SOD and catalase. For example, serratiopeptidase is an enzyme that is widely used to reduce pain and inflammation associated with arthritis, chronic bronchitis, and atherosclerosis.68 Local delivery of serratiopeptidase via magnetic nanoparticles enhanced its anti-inflammatory effects in a rat paw edema model.69 Additionally, nanoparticles comprising an anti-inflammatory Salmonella acetyltransferase enzyme (AvrA) and crosslinked green fluorescent protein effectively treated acute inflammation characteristic of inflammatory bowel disease using a murine colitis model.70
Enzyme-nanoparticle formulations are also being investigated to treat autoimmune diseases, such as rheumatoid arthritis. Rheumatoid arthritis is characterized by chronic joint inflammation due to overexpression of various pro-inflammatory cytokines, such as TNF-α.71 Treating rheumatoid arthritis with TNF-α inhibitors remains the clinical standard,72–73 despite growing evidence that systemic delivery of TNF-α antagonists over prolonged periods can adversely affect normal TNF-α function, or establish TNF-α reservoirs.74 Alternatively, Champion and colleagues recently reported self-assembling hybrid supraparticles functionalized with arginine-specific gingipain A (RgpA), a TNF-α-degrading enzyme (Figure 4).75 Specifically, a recombinant ZE peptide-RgpA fusion protein was non-covalently immobilized within porous calcium supraparticles modified with ZR peptide via heterodimeric ZR:ZE leucine zipper assembly. RgpA supraparticles significantly reduced TNF-α concentration in solution, and increased L929 murine fibrosarcoma cell viability upon TNF-α challenge in vitro more effectively than soluble enzyme. Thus, local delivery of enzymes that degrade pro-inflammatory cytokines may eventually provide a useful alternative to cytokine inhibitors currently used to treat rheumatoid arthritis and other autoimmune diseases.
Figure 4.

Supraparticles that proteolytically degrade TNF-α. (A) Schematic representation of self-assembled porous hybrid supraparticles with immobilized pRgpACAT, a TNF-α-degrading enzyme. (B) Survival of L929 mouse fibroblasts treated with TNF-α (“Controls”), compared to TNF-α in the presence of soluble enzyme (black bars) or enzyme supraparticles (red bars). Reprinted with permission from ref 75. Copyright 2016 The Royal Society of Chemistry.
Conclusions
To date, as few as 20 therapeutic enzymes are FDA-approved.76–77 Enzymes have typically failed to reach the clinic due to immunogenicity, decreased localized catalytic activity, and rapid clearance from the body.78 Approaches to encapsulate, immobilize, or append enzymes with chemical moieties can greatly improve therapeutic efficacy by extending in vivo half-life or targeting enzymes to specific cell and tissue sites. This Topical Review illustrates FDA-approved therapies and emerging technologies that use these approaches to treat immune diseases that manifest both systemically and locally. The increasing number of clinical and pre-clinical successes in using immunotherapeutic enzymes to replace or augment native enzyme activity highlights their enormous promise to treat diverse immune-related pathologies.
Acknowledgments
The authors gratefully acknowledge support by the following grants: R01 AI133623, R01 DE027301, and R01 DK098589 to BGK, as well as R03 EB019684 and R21 EB024762 to GAH. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
References
- 1.Kjeldsen L, Sengelov H, Lollike K, Nielsen MH, Borregaard N. Isolation and characterization of gelatinase granules from human neutrophils. Blood. 1994;83:1640–1649. [PubMed] [Google Scholar]
- 2.Faurschou M, Sørensen OE, Johnsen AH, Askaa J, Borregaard N. Defensin-rich granules of human neutrophils: characterization of secretory properties. Biochim Biophys Acta, Mol Cell Res. 2002;1591:29–35. doi: 10.1016/s0167-4889(02)00243-4. [DOI] [PubMed] [Google Scholar]
- 3.Jones JA, McNally AK, Chang DT, Qin LA, Meyerson H, Colton E, Kwon ILK, Matsuda T, Anderson JM. Matrix metalloproteinases and their inhibitors in the foreign body reaction on biomaterials. J Biomed Mater Res, Part A. 2008;84A:158–166. doi: 10.1002/jbm.a.31220. [DOI] [PubMed] [Google Scholar]
- 4.Modolell M, Corraliza IM, Link F, Soler G, Eichmann K. Reciprocal regulation of the nitric oxide synthase/arginase balance in mouse bone marrow-derived macrophages by TH 1 and TH 2 cytokines. Eur J Immunol. 1995;25:1101–1104. doi: 10.1002/eji.1830250436. [DOI] [PubMed] [Google Scholar]
- 5.Haroon ZA, Hettasch JM, Lai TS, Dewhirst MW, Greenberg CS. Tissue transglutaminase is expressed, active, and directly involved in rat dermal wound healing and angiogenesis. FASEB J. 1999;13:1787–1795. doi: 10.1096/fasebj.13.13.1787. [DOI] [PubMed] [Google Scholar]
- 6.Deaglio S, et al. Adenosine generation catalyzed by CD39 and CD73 expressed on regulatory T cells mediates immune suppression. J Exp Med. 2007;204:1257–1265. doi: 10.1084/jem.20062512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Beavis PA, Stagg J, Darcy PK, Smyth MJ. CD73: a potent suppressor of antitumor immune responses. Trends Immunol. 2012;33:231–237. doi: 10.1016/j.it.2012.02.009. [DOI] [PubMed] [Google Scholar]
- 8.Munn DH, Zhou M, Attwood JT, Bondarev I, Conway SJ, Marshall B, Brown C, Mellor AL. Prevention of Allogeneic Fetal Rejection by Tryptophan Catabolism. Science. 1998;281:1191–1193. doi: 10.1126/science.281.5380.1191. [DOI] [PubMed] [Google Scholar]
- 9.Munn DH, Mellor AL. IDO in the Tumor Microenvironment: Inflammation, Counter-regulation and Tolerance. Trends Immunol. 2016;37:193–207. doi: 10.1016/j.it.2016.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lagassé HAD, Alexaki A, Simhadri VL, Katagiri NH, Jankowski W, Sauna ZE, Kimchi-Sarfaty C. Recent advances in (therapeutic protein) drug development. F1000Research. 2017;6:113. doi: 10.12688/f1000research.9970.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Vellard M. The enzyme as drug: application of enzymes as pharmaceuticals. Curr Opin Biotechnol. 2003;14:444–450. doi: 10.1016/s0958-1669(03)00092-2. [DOI] [PubMed] [Google Scholar]
- 12.Schultz ML, Tecedor L, Chang M, Davidson BL. Clarifying lysosomal storage diseases. Trends Neurosci. 2011;34:401–410. doi: 10.1016/j.tins.2011.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Castaneda JA, Lim MJ, Cooper JD, Pearce DA. Immune system irregularities in lysosomal storage disorders. Acta Neuropathol. 2008;115:159–174. doi: 10.1007/s00401-007-0296-4. [DOI] [PubMed] [Google Scholar]
- 14.Lachmann RH. Enzyme replacement therapy for lysosomal storage diseases. Curr Opin Pediatr. 2011;23:588–93. doi: 10.1097/MOP.0b013e32834c20d9. [DOI] [PubMed] [Google Scholar]
- 15.Brady RO. Enzyme Replacement for Lysosomal Diseases. Annu Rev Med. 2006;57:283–296. doi: 10.1146/annurev.med.57.110104.115650. [DOI] [PubMed] [Google Scholar]
- 16.Wraith JE. Limitations of enzyme replacement therapy: Current and future. J Inherited Metab Dis. 2006;29:442–447. doi: 10.1007/s10545-006-0239-6. [DOI] [PubMed] [Google Scholar]
- 17.Harmatz P. Enzyme Replacement Therapies and Immunogenicity in Lysosomal Storage Diseases: Is There a Pattern? Clin Ther. 2015;37:2130–2134. doi: 10.1016/j.clinthera.2015.06.004. [DOI] [PubMed] [Google Scholar]
- 18.Chuang HY, Suen CS, Hwang MJ, Roffler SR. Toward reducing immunogenicity of enzyme replacement therapy: altering the specificity of human β-glucuronidase to compensate for α-iduronidase deficiency. Protein Eng, Des Sel. 2015;28:519–530. doi: 10.1093/protein/gzv041. [DOI] [PubMed] [Google Scholar]
- 19.Barton NW, et al. Replacement Therapy for Inherited Enzyme Deficiency — Macrophage-Targeted Glucocerebrosidase for Gaucher’s Disease. N Engl J Med. 1991;324:1464–1470. doi: 10.1056/NEJM199105233242104. [DOI] [PubMed] [Google Scholar]
- 20.Rosenbloom BE, Weinreb NJ. Gaucher disease: a comprehensive review. Crit Rev Oncog. 2013;18:163–175. doi: 10.1615/critrevoncog.2013006060. [DOI] [PubMed] [Google Scholar]
- 21.Furbish FS, Steer CJ, Barranger JA, Jones EA, Brady RO. The uptake of native and desialylated glucocerebrosidase by rat hepatocytes and Kupffer cells. Biochem Biophys Res Commun. 1978;81:1047–1053. doi: 10.1016/0006-291x(78)91456-0. [DOI] [PubMed] [Google Scholar]
- 22.Gary-Bobo M, Nirdé P, Jeanjean A, Morère A, Garcia M. Mannose 6-phosphate receptor targeting and its applications in human diseases. Curr Med Chem. 2007;14:2945–2953. doi: 10.2174/092986707782794005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wade AA, Rosenthal DI. Gaucher’s Disease. In: Weissman BN, editor. Imaging of Arthritis and Metabolic Bone Disease. Imaging of Degenerative and Traumatic Conditions. Chapter 27. W.B. Saunders; Philadelphia: 2009. pp. 520–528. [Google Scholar]
- 24.Syed Haneef SA, George Priya Doss C. Personalized Pharmacoperones for Lysosomal Storage Disorder: Approach for Next-Generation Treatment. In: Donev R, editor. Advances in Protein Chemistry and Structural Biology. Volume 102: Personalized Medicine. Chapter 8. Academic Press; New York: 2016. pp. 225–265. [DOI] [PubMed] [Google Scholar]
- 25.Brady RO, Murray GJ, Barton NW. Modifying exogenous glucocerebrosidase for effective replacement therapy in Gaucher disease. J Inherited Metab Dis. 1994;17:510–519. doi: 10.1007/BF00711365. [DOI] [PubMed] [Google Scholar]
- 26.Lingg N, Zhang P, Song Z, Bardor M. The sweet tooth of biopharmaceuticals: importance of recombinant protein glycosylation analysis. Biotechnol J. 2012;7:1462–1472. doi: 10.1002/biot.201200078. [DOI] [PubMed] [Google Scholar]
- 27.Rombach SM, Smid BE, Bouwman MG, Linthorst GE, Dijkgraaf MGW, Hollak CEM. Long term enzyme replacement therapy for Fabry disease: effectiveness on kidney, heart and brain. Orphanet J Rare Dis. 2013;8:47. doi: 10.1186/1750-1172-8-47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rozenfeld P, Feriozzi S. Contribution of inflammatory pathways to Fabry disease pathogenesis. Mol Genet Metab. 2017;122:19–27. doi: 10.1016/j.ymgme.2017.09.004. [DOI] [PubMed] [Google Scholar]
- 29.Murray GJ, Anver MR, Kennedy MA, Quirk JM, Schiffmann R. Cellular and tissue distribution of intravenously administered agalsidase alfa. Mol Genet Metab. 2007;90:307–312. doi: 10.1016/j.ymgme.2006.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Whitmore KV, Gaspar HB. Adenosine Deaminase Deficiency – More Than Just an Immunodeficiency. Front Immunol. 2016;7:314. doi: 10.3389/fimmu.2016.00314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sauer A, Brigida I, Carriglio N, Aiuti A. Autoimmune Dysregulation and Purine Metabolism in Adenosine Deaminase Deficiency. Front Immunol. 2012;3:265. doi: 10.3389/fimmu.2012.00265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Booth C, Gaspar HB. Pegademase bovine (PEG-ADA) for the treatment of infants and children with severe combined immunodeficiency (SCID) Biol: Targets Ther. 2009;3:349–358. [PMC free article] [PubMed] [Google Scholar]
- 33.Kizaki H, Suzuki K, Tadakuma T, Ishimura Y. Adenosine receptor-mediated accumulation of cyclic AMP-induced T-lymphocyte death through internucleosomal DNA cleavage. J Biol Chem. 1990;265:5280–5284. [PubMed] [Google Scholar]
- 34.Huang S, Apasov S, Koshiba M, Sitkovsky M. Role of A2a Extracellular Adenosine Receptor-Mediated Signaling in Adenosine-Mediated Inhibition of T-Cell Activation and Expansion. Blood. 1997;90:1600–1610. [PubMed] [Google Scholar]
- 35.Davis S, Abuchowski A, Park YK, Davis FF. Alteration of the circulating life and antigenic properties of bovine adenosine deaminase in mice by attachment of polyethylene glycol. Clin Exp Immunol. 1981;46:649–652. [PMC free article] [PubMed] [Google Scholar]
- 36.Molineux G. Pegylation: engineering improved pharmaceuticals for enhanced therapy. Cancer Treat Rev. 2002;28(Suppl A):13–6. doi: 10.1016/s0305-7372(02)80004-4. [DOI] [PubMed] [Google Scholar]
- 37.Pui CH, Robison LL, Look AT. Acute lymphoblastic leukaemia. The Lancet. 2008;371:1030–1043. doi: 10.1016/S0140-6736(08)60457-2. [DOI] [PubMed] [Google Scholar]
- 38.Asselin BL, Whitin JC, Coppola DJ, Rupp IP, Sallan SE, Cohen HJ. Comparative pharmacokinetic studies of three asparaginase preparations. J Clin Oncol. 1993;11:1780–1786. doi: 10.1200/JCO.1993.11.9.1780. [DOI] [PubMed] [Google Scholar]
- 39.Dinndorf PA, Gootenberg J, Cohen MH, Keegan P, Pazdur R. FDA drug approval summary: pegaspargase (oncaspar) for the first-line treatment of children with acute lymphoblastic leukemia (ALL) Oncologist. 2007;12:991–998. doi: 10.1634/theoncologist.12-8-991. [DOI] [PubMed] [Google Scholar]
- 40.Wylon K, Dölle S, Worm M. Polyethylene glycol as a cause of anaphylaxis. Allergy, Asthma, Clin Immunol. 2016;12:67. doi: 10.1186/s13223-016-0172-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zhang P, Sun F, Liu S, Jiang S. Anti-PEG antibodies in the clinic: Current issues and beyond PEGylation. J Controlled Release. 2016;244B:184–193. doi: 10.1016/j.jconrel.2016.06.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Armstrong JK, Hempel G, Koling S, Chan LS, Fisher T, Meiselman HJ, Garratty G. Antibody against poly(ethylene glycol) adversely affects PEG-asparaginase therapy in acute lymphoblastic leukemia patients. Cancer. 2007;110:103–111. doi: 10.1002/cncr.22739. [DOI] [PubMed] [Google Scholar]
- 43.Garay RP, El-Gewely R, Armstrong JK, Garratty G, Richette P. Antibodies against polyethylene glycol in healthy subjects and in patients treated with PEG-conjugated agents. Expert Opin Drug Delivery. 2012;9:1319–1323. doi: 10.1517/17425247.2012.720969. [DOI] [PubMed] [Google Scholar]
- 44.Richter AW, Akerblom E. Polyethylene glycol reactive antibodies in man: titer distribution in allergic patients treated with monomethoxy polyethylene glycol modified allergens or placebo, and in healthy blood donors. Int Arch Allergy Appl Immunol. 1984;74:36–39. doi: 10.1159/000233512. [DOI] [PubMed] [Google Scholar]
- 45.Bagshawe KD. Antibody directed enzymes revive anti-cancer prodrugs concept. Br J Cancer. 1987;56:531–532. doi: 10.1038/bjc.1987.237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sharma SK, Bagshawe KD. Antibody Directed Enzyme Prodrug Therapy (ADEPT): Trials and tribulations. Adv Drug Delivery Rev. 2017;118:2–7. doi: 10.1016/j.addr.2017.09.009. [DOI] [PubMed] [Google Scholar]
- 47.Nausch N, Cerwenka A. NKG2D ligands in tumor immunity. Oncogene. 2008;27:5944–5958. doi: 10.1038/onc.2008.272. [DOI] [PubMed] [Google Scholar]
- 48.Hudak JE, Canham SM, Bertozzi CR. Glycocalyx Engineering Reveals a Siglec-Based Mechanism for NK Cell Immunoevasion. Nat Chem Biol. 2014;10:69–75. doi: 10.1038/nchembio.1388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Cohen M, Elkabets M, Perlmutter M, Porgador A, Voronov E, Apte RN, Lichtenstein RG. Sialylation of 3-methylcholanthrene-induced fibrosarcoma determines antitumor immune responses during immunoediting. J Immunol. 2010;185:5869–5878. doi: 10.4049/jimmunol.1001635. [DOI] [PubMed] [Google Scholar]
- 50.Xiao H, Woods EC, Vukojicic P, Bertozzi CR. Precision glycocalyx editing as a strategy for cancer immunotherapy. Proc Natl Acad Sci U S A. 2016;113:10304–10309. doi: 10.1073/pnas.1608069113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Simonaro CM. Lysosomes, Lysosomal Storage Diseases, and Inflammation. J Inborn Errors Metab Screening. 2016;4:1–8. [Google Scholar]
- 52.Pardridge WM, Boado RJ. Reengineering Biopharmaceuticals for Targeted Delivery Across the Blood–Brain Barrier. In: Wittrup KD, Verdine GL, editors. Methods in Enzymology. Volume 503: Protein Engineering for Therapeutics B. Chapter 11. Academic Press; New York: 2012. pp. 269–292. [DOI] [PubMed] [Google Scholar]
- 53.Zhou QH, Boado RJ, Lu JZ, Hui EK, Pardridge WM. Brain-penetrating IgG-iduronate 2-sulfatase fusion protein for the mouse. Drug Metab Dispos. 2012;40:329–335. doi: 10.1124/dmd.111.042903. [DOI] [PubMed] [Google Scholar]
- 54.Boado RJ, Lu JZ, Hui EK, Sumbria RK, Pardridge WM. Pharmacokinetics and brain uptake in the rhesus monkey of a fusion protein of arylsulfatase a and a monoclonal antibody against the human insulin receptor. Biotechnol Bioeng. 2013;110:1456–1465. doi: 10.1002/bit.24795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Boado RJ, Lu JZ, Hui EKW, Pardridge WM. Insulin Receptor Antibody–Sulfamidase Fusion Protein Penetrates the Primate Blood–Brain Barrier and Reduces Glycosoaminoglycans in Sanfilippo Type A Cells. Mol Pharmaceutics. 2014;11:2928–2934. doi: 10.1021/mp500258p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Gao F, Koenitzer JR, Tobolewski JM, Jiang D, Liang J, Noble PW, Oury TD. Extracellular Superoxide Dismutase Inhibits Inflammation by Preventing Oxidative Fragmentation of Hyaluronan. J Biol Chem. 2008;283:6058–6066. doi: 10.1074/jbc.M709273200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Yu DH, et al. Over-expression of extracellular superoxide dismutase in mouse synovial tissue attenuates the inflammatory arthritis. Exp Mol Med. 2012;44:529–535. doi: 10.3858/emm.2012.44.9.060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Eugénia M, Cruz M, Manuela Gaspar M, Bárbara M, Martins F, Luísa Corvo M. Liposomal Superoxide Dismutases and Their Use in the Treatment of Experimental Arthritis. In: Duzgunes N, editor. Methods in Enzymology. Volume 391: Liposomes. Chapter 22. Academic Press; New York: 2005. pp. 395–413. [DOI] [PubMed] [Google Scholar]
- 59.Luisa Corvo M, Jorge JCS, van’t Hof R, Cruz MEM, Crommelin DJA, Storm G. Superoxide dismutase entrapped in long-circulating liposomes: formulation design and therapeutic activity in rat adjuvant arthritis. Biochim Biophys Acta, Biomembr. 2002;1564:227–236. doi: 10.1016/s0005-2736(02)00457-1. [DOI] [PubMed] [Google Scholar]
- 60.Porfire AS, Leucuţa SE, Kiss B, Loghin F, Pârvu AE. Investigation into the role of Cu/Zn-SOD delivery system on its antioxidant and antiinflammatory activity in rat model of peritonitis. Pharmacol Rep. 2014;66:670–676. doi: 10.1016/j.pharep.2014.03.011. [DOI] [PubMed] [Google Scholar]
- 61.Simões S, Marques C, Cruz ME, Figueira Martins MB. Anti-inflammatory effects of locally applied enzyme-loaded ultradeformable vesicles on an acute cutaneous model. J Microencapsulation. 2009;26:649–658. doi: 10.3109/02652040802630403. [DOI] [PubMed] [Google Scholar]
- 62.Petelin M, Pavlica Z, Ivanuša T, Šentjurc M, Skalerič U. Local delivery of liposome-encapsulated superoxide dismutase and catalase suppress periodontal inflammation in beagles. J Clin Periodontol. 2000;27:918–925. doi: 10.1034/j.1600-051x.2000.027012918.x. [DOI] [PubMed] [Google Scholar]
- 63.Hood ED, Chorny M, Greineder CF, Alferiev I, Levy RJ, Muzykantov VR. Endothelial targeting of nanocarriers loaded with antioxidant enzymes for protection against vascular oxidative stress and inflammation. Biomaterials. 2014;35:3708–3715. doi: 10.1016/j.biomaterials.2014.01.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Reddy MK, Wu L, Kou W, Ghorpade A, Labhasetwar V. Superoxide dismutase-loaded PLGA nanoparticles protect cultured human neurons under oxidative stress. Appl Biochem Biotechnol. 2008;151:565–577. doi: 10.1007/s12010-008-8232-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Yun X, Maximov VD, Yu J, Zhu H, Vertegel AA, Kindy MS. Nanoparticles for targeted delivery of antioxidant enzymes to the brain after cerebral ischemia and reperfusion injury. J Cereb Blood Flow Metab. 2013;33:583–592. doi: 10.1038/jcbfm.2012.209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Saraswathi V, Ganesan M, Perriotte-Olson C, Manickam DS, Westwood RA, Zimmerman MC, Ahmad IM, Desouza CV, Kabanov AV. Nanoformulated copper/zinc superoxide dismutase attenuates vascular cell activation and aortic inflammation in obesity. Biochem Biophys Res Commun. 2016;469:495–500. doi: 10.1016/j.bbrc.2015.12.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Swart PJ, Hirano T, Kuipers ME, Ito Y, Smit C, Hashida M, Nishikawa M, Beljaars L, Meijer DKF, Poelstra K. Targeting of superoxide dismutase to the liver results in anti-inflammatory effects in rats with fibrotic livers. J Hepatol. 1999;31:1034–1043. doi: 10.1016/s0168-8278(99)80316-x. [DOI] [PubMed] [Google Scholar]
- 68.Tiwari M. The role of serratiopeptidase in the resolution of inflammation. Asian J Pharm Sci. 2017;12:209–215. doi: 10.1016/j.ajps.2017.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Kumar S, Jana AK, Dhamija I, Singla Y, Maiti M. Preparation, characterization and targeted delivery of serratiopeptidase immobilized on amino-functionalized magnetic nanoparticles. Eur J Pharm Biopharm. 2013;85(3):413–426. doi: 10.1016/j.ejpb.2013.06.019. [DOI] [PubMed] [Google Scholar]
- 70.Herrera Estrada L, Wu H, Ling K, Zhang G, Sumagin R, Parkos CA, Jones RM, Champion JA, Neish AS. Bioengineering Bacterially Derived Immunomodulants: A Therapeutic Approach to Inflammatory Bowel Disease. ACS Nano. 2017;11:9650–9662. doi: 10.1021/acsnano.7b03239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Bek S, Bojesen AB, Nielsen JV, Sode J, Bank S, Vogel U, Andersen V. Systematic review and meta-analysis: pharmacogenetics of anti-TNF treatment response in rheumatoid arthritis. Pharmacogenomics J. 2017;17:403–411. doi: 10.1038/tpj.2017.26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Croft M, Siegel RM. Beyond TNF: TNF superfamily cytokines as targets for the treatment of rheumatic diseases. Nat Rev Rheumatol. 2017;13:217–233. doi: 10.1038/nrrheum.2017.22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Siebert S, Tsoukas A, Robertson J, McInnes I. Cytokines as therapeutic targets in rheumatoid arthritis and other inflammatory diseases. Pharmacol Rev. 2015;67:280–309. doi: 10.1124/pr.114.009639. [DOI] [PubMed] [Google Scholar]
- 74.Chan JMK, Villarreal G, Jin WW, Stepan T, Burstein H, Wahl SM. Intraarticular Gene Transfer of TNFR:Fc Suppresses Experimental Arthritis with Reduced Systemic Distribution of the Gene Product. Mol Ther. 2002;6:727–736. doi: 10.1006/mthe.2002.0808. [DOI] [PubMed] [Google Scholar]
- 75.Park WM, Yee CM, Champion JA. Self-assembled hybrid supraparticles that proteolytically degrade tumor necrosis factor-α. J Mater Chem B. 2016;4:1633–1639. doi: 10.1039/c5tb01647a. [DOI] [PubMed] [Google Scholar]
- 76.Maximov V, Reukov V, Vertegel AA. Targeted delivery of therapeutic enzymes. J Drug Delivery Sci Technol. 2009;19:311–320. [Google Scholar]
- 77.Baldo BA. Enzymes approved for human therapy: indications, mechanisms and adverse effects. BioDrugs. 2015;29:31–55. doi: 10.1007/s40259-015-0116-7. [DOI] [PubMed] [Google Scholar]
- 78.Kontermann RE. Strategies for extended serum half-life of protein therapeutics. Curr Opin Biotechnol. 2011;22:868–876. doi: 10.1016/j.copbio.2011.06.012. [DOI] [PubMed] [Google Scholar]