

Abstract
A recent study reports on five different mutations as sources of Dopamine transporter (DAT) deficiency syndrome (DTDS). One of these mutations, R445C, is believed to be located on the intracellular side of DAT distant to the primary (S1) or secondary (S2) sites to which substrate binding is understood to occur. Thus, the molecular mechanism by which the R445C mutation results in DAT transport deficiency has eluded explanation. However, the recently reported X-ray structures of the endogenous amine transporters for dDAT and hSERT revealed the presence of a putative salt bridge between R445 and E428 suggesting a possible mechanism. To evaluate whether the R445C effect is a result of a salt bridge interaction, the mutants R445E, E428R, and the double mutant E428R/R445E were generated. The single mutants R445E and E428R displayed loss of binding and transport properties of the substrate [3H]DA and inhibitor [3H]CFT at the cell surface while the double mutant E428R/R445E, although nonfunctional, restored [3H]DA and [3H]CFT binding affinity to that of WT. Structure based analyses of these results led to a model wherein R445 plays a dual role in normal DAT function. R445 acts as a component of a latch in its formation of a salt bridge with E428 which holds the primary substrate binding site (S1) in place and helps enforce the inward closed protein state. When this salt bridge is broken, R445 acts as a trigger which disrupts a local polar network and leads to the release of the N-terminus from its position inducing the inward closed state to one allowing the inward open state. In this manner, both the loss of binding and transport properties of the R445C variant are explained.
Graphical abstract
Introduction
A key protein in the control of optimal dopamine levels, the dopamine transporter protein (DAT), operates through reuptake of extracellular dopamine. DAT is a Na+/Cl− dependent neurotransmitter sodium symporter comprised of 12 transmembrane spanning helices connected by extracellular and intracellular (ECL and ICL) loops. The primary binding site of the substrate is centrally located. This primary binding site (S1) was detailed by the first X-ray structures of the bacterial homolog of DAT, LeuT1, 2. Computational studies of the migration of substrate through LeuT suggested a number of more weakly bound “stopover” sites on the EC side of the protein3, 4. One of these sites more proximal to S1, the S2 site, was found to be occupied by tricyclic antidepressants in other X-ray structures of LeuT2, 4. These models have now been validated by an X-ray structure of hSERT which showed that two molecules of the SSRI S-citalopram (SCIT) were bound to the protein, specifically, one at each of the S1 and S2 sites. Computational studies using the X-ray structure of hSERT supported a similar pathway for the endogenous substrate 5-HT migration through SERT5 including the presence of the S2 stopover site, also referred to as the vestibule. Based on these findings and others, the emerging model for the overall protein conformations associated with substrate migration considers 3 protein conformational states; 1) outward open (inward closed) 2) outward open and ligand occluded (inward closed) and 3) outward closed (inward open)1, 4, 6–13.
Defects in the binding and transport of DA have been shown to have severe effects as evidenced by patients diagnosed with dopamine transporter deficiency syndrome (DTDS)14–16. The DTDS symptomology is characterized by hyperkinetic movement disorder which progresses to parkinsonism-dystonia. In our recent study, we reported on a cohort of patients whose onset of DTDS ranges from infancy to adolescence and whose DAT deficiency is attributed to various identified mutations in DAT16. In order to better understand the molecular basis of DTDS, we had embarked on structural investigations of DAT and related transporter proteins. Using comparative modeling methods based on the X-ray crystal structure of LeuT14–16, we examined the location of five independent mutations16 heretofore ascribed to DTDS patients. Four of these mutations were in the extracellular vestibule (S2) region of DAT suggesting an adverse effect on initial substrate binding at this site. A fifth mutation, R445C, was located well towards the intracellular side of the protein distal to the S1 primary and S2 secondary substrate binding sites and thus confounding rationalization of its role. Subsequent X-ray structures of dDAT and hSERT17–20 indicated a possible mechanism which led us to investigate the molecular basis of R445C DAT deficiency as well as to reassess the four previously examined mutations.
Results and Discussion
Mutations in the extracellular a.k.a. vestibule region of DAT
In the Ng et al. study16, we used a homology model for DAT based on the X-ray structure of LeuT to examine the 4 extracellular region mutants associated heretofore with DTDS, i.e., A314V, G386R, Y470S and R85L. Here we reassessed these variants using the subsequently published X-ray structures of dDAT and hSERT which inherently provide more relevant structures. Figure 1 shows an X-ray structure of dDAT (PDB entry 4XP120 highlighting the various residues associated with DTDS occurrence. For visual reference purposes we show the position of the SCIT molecule occupying the S2 position in SERT (Figure 1B, D) based on a superposition of the X-ray structures of dDAT and hSERT. In support of our previous findings, the mutant variants are located in the upper, vestibule region near the S2 site suggesting interference with substrate binding. For instance, A314 sits on the outer surface of dDAT pointing away from the S2 pocket which is consistent with the relatively smaller effect of the A314V mutant on DA function and, consequently, the extent of DTDS phenotype. Conversely, R85, which is fairly close to A314 points directly into the vestibule and overlaps with the position at which SCIT binds to SERT. This is consistent with the more deleterious effect of the R85L mutation on DA binding (vida infra) and thus the severity of disease.
Figure 1.
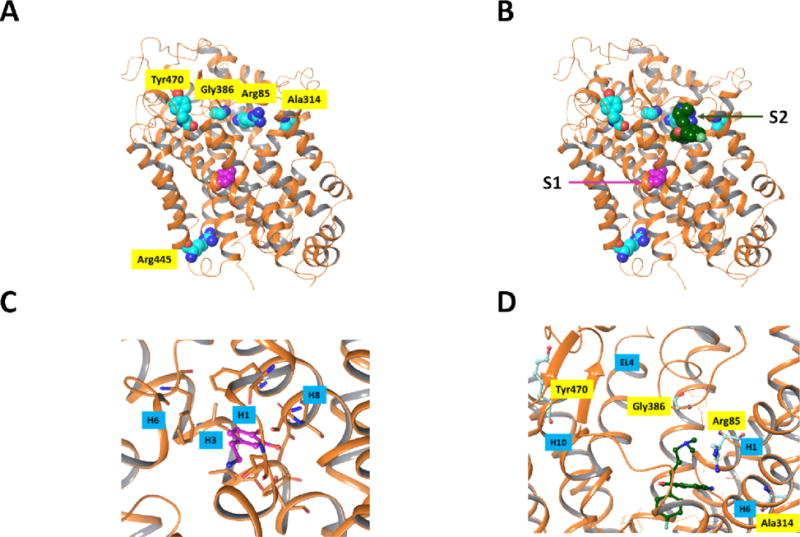
Positions of residues mutated in previous DTDS studies. The protein is shown in ribbon rendering and atoms which are displayed are rendered as space filling structures. Protein carbon atoms are aqua colored and dopamine carbon atoms are depicted in magenta. The X-ray structure is that of dDAT20 [PDB entry 4XP1] and the residues which were mutated are indicated using the hDAT numbering. A) All mutated residues, with the exception of R445, are in the upper region of the protein. None of the mutated residues are in close contact with the dopamine substrate in the primary binding site. B) Based on a superposition of dDAT and the recently published X-ray structure of SERT17 with SCIT occupying both the primary binding site (S1) as well as the vestibule binding site on the intracellular entryway to SERT (S2) it can be seen that the 4 mutated residues in the upper portion of the protein are in the vestibule region and could potentially interfere with the substrate binding at this stopover site. Only the SCIT ligand of the hSERT X-ray structure is shown. C) Closeup view of the primary substrate binding site in dDAT showing the residues close to the DA substrate in stick models. Nearby helices H1, H3, H6 and H8 are indicated. D) Closeup view of the “vestibule” region in dDAT in A). For reference, the residues of dDAT corresponding to the dDAT mutants observed in DTDS are shown as aqua ball and stick structures. Nearby helices H1, H6 and H10 and extracellular loop EL4 are indicated. Molecular graphics were generated with Maestro21.
The R445C mutation
In agreement with our earlier report, R445 is located on the intracellular side of TM9 distant from S2 and S1 sites (Figure 1). It has thus been unclear as to how the R445C mutation linked to cases of DTDS can have the reported adverse effects on DA binding and uptake. Inspection of the dDAT X-ray structure reveals that R445 forms a salt bridge with E428 on TM8. While E428 is not in contact with DA at S1, we note that in the EC direction, TM8 is one of the helices surrounding DA at S1 (Figure 1C). We therefore postulated that this salt bridge between R445 and E428 holds TM8 in a position optimal for DA binding at S1 and may therefore play a role in DA binding and/or transport, thereby explaining the effect of the R445C mutation. This would suggest that mutation at E428 which precludes the salt bridge would also have a similar effect as the R445C mutant. Furthermore, replacing the E428-R445 salt bridge with a similar salt bridge at these sites, e.g., by interchanging the residues at these positions, may restore DA binding and/or transport. To test these hypotheses we have made the following mutants R445E, E428R and the double mutant E428R/R445E, predicted to re-establish the salt bridge interaction, and report herein the effects of these mutations on substrate binding as compared to the native WT protein.
Substrate binding and transport
Transient expression of the DAT constructs was somewhat suppressed compared with WT, except for that of E428R, both in total protein and in cell surface protein (Figure 2 left panel). In stably expressing LLCK-PK1 cells, total and surface DAT expression appeared somewhat decreased for the double mutant (Figure 2 right panel). The latter pattern was mimicked by the apparent average Bmax values of cocaine analog [3H]CFT binding in stable cell lines (Table 1), although the only statistically significant Bmax effect was an increase in E428R. More important than the modest changes in DAT expression along with changes in [3H]CFT Bmax for the constructs were the observed changes in CFT and DA recognition (Table 1). The Kd value for [3H]CFT binding was increased 3- to 5-fold in the single mutants R445E and E428R, and restored to WT level in the double mutant R445E/E428R. The same phenomenon occurred for DA binding as measured via its inhibitory effect on [3H]CFT binding. DA Ki was increased (i.e., affinity decreased) ~ 6-fold in the single mutants and restored back to normal in the double mutant. The fact that both the CFT Kd and DA Ki are restored in the double mutant, suggests the residues E and R can be in either position (428 or 445) for enabling the formation of the salt bridge. If DA affinity (Ki) would be the only property that is changing in the single mutants, a 6-fold increase in DA Ki would be expected to cause a ~ 6-fold decrease in DA uptake measured at [DA] far below its Km. Indeed, the ability of the single constructs to take up [3H]DA was virtually collapsed with uptake levels at only 2 -4 % of WT uptake (Figure 3). This indicates that additional factors, other than DA recognition, underlie the greatly reduced DA uptake of the single mutants. In support, while the R445E/E428R double mutant exhibited restored CFT and DA affinity/recognition, DAT function/DA uptake was still notably debilitated. Thus, the specific residues are likely critical for the conformational transitions in DAT that allow for DA transport. Figure 2. Immunoblotting studies of mutant hDAT. LLC-PK1 cells transiently (left panel) or stably (right panel) transfected with indicated hDAT constructs were subjected to cell surface biotinylation. Biotinylated proteins (“Surface”) and whole cell lysates (“Total”) were analyzed with antibodies against DAT and actin as indicated. Equal amounts of total lysate protein were loaded for each mutated hDAT as for wild-type (WT).
Figure 2.
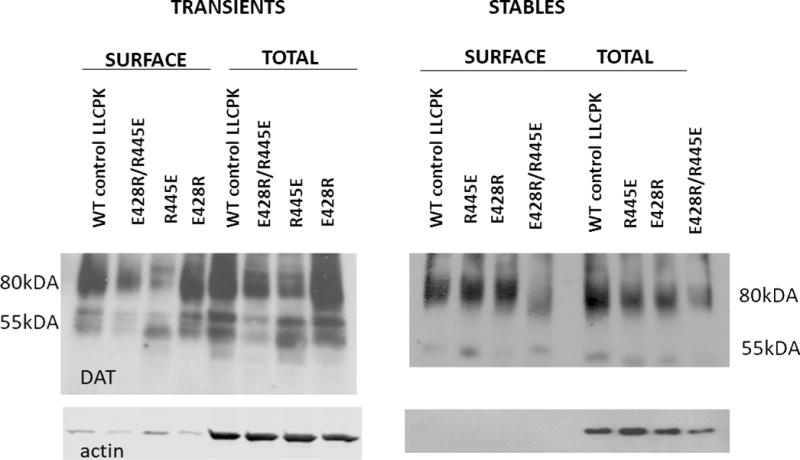
Immunoblotting studies of mutant hDAT. LLC-PK1 cells transiently (left panel) or stably (right panel) transfected with indicated hDAT constructs were subjected to cell surface biotinylation. Biotinylated proteins (“Surface”) and whole cell lysates (“Total”) were analyzed with antibodies against DAT and actin as indicated. Equal amounts of total lysate protein were loaded for each mutated hDAT as for wild-type (WT).
Table 1.
[3]CFT binding to WT, R445E, E428R, and R445E-E428R DAT and its inhibition by DA
| [3H]CFT binding | WT | R445E | E428R | R445E-E428R |
|---|---|---|---|---|
| Kd (nM) | 13.1 ± 2.62 | 67.4 ± 7.26* | 35.2 ± 2.14* | 4.83 ± 0.83 |
| Bmax (pmol/mg) | 1.04 ± 0.14 | 3.27 ± 0.63 | 2.50 ± 0.30* | 0.56 ± 0.11 |
| DA Ki (μM) | 3.83 ± 0.579 | 22.4 ± 4.7** | 21.8 ± 10.2** | 3.66 ± 0.53 |
Values are mean ± SE; n = 4 for [3H]CFT Kd and Bmax values and n = 5–6 for DA Ki values.
P<0.05,
P<0.01 compared with wild-type (WT) (one-way ANOVA followed by Dunnett multiple comparisons with WT).
Figure 3.
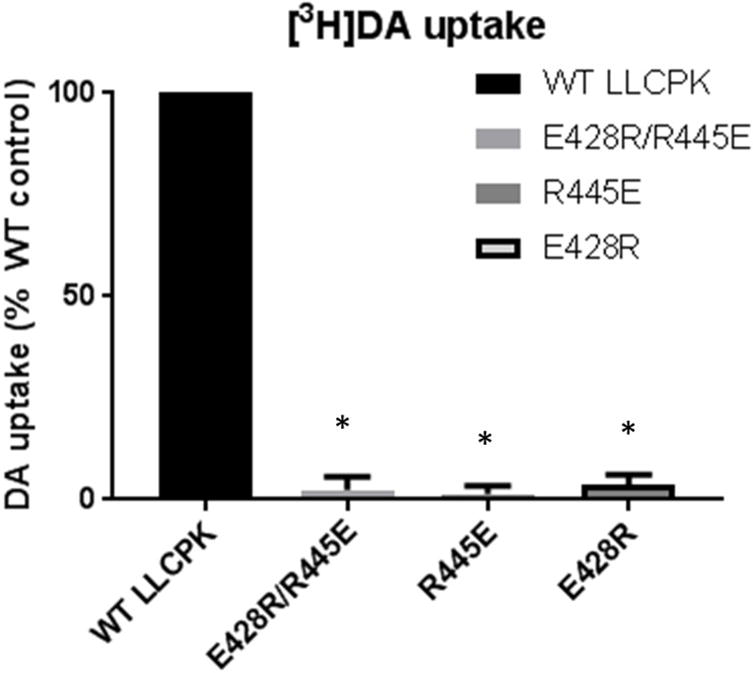
Dopamine transport by wild-type (WT) and mutant hDAT. Uptake was measured with [3H]dopamine in the assay. Control [3H]dopamine uptake activity was ~ 0.11 pmol/mg protein/min. Results shown are mean with error bar depicting SE. * P < 0.001 compared with wild-type (WT) (one-sample Student’s t-test, with WT set at 100%, Bonferroni-corrected for multiple comparisons; n = 5 for each expression condition).
Structural basis for binding and transport effects of R445 and ancillary mutations
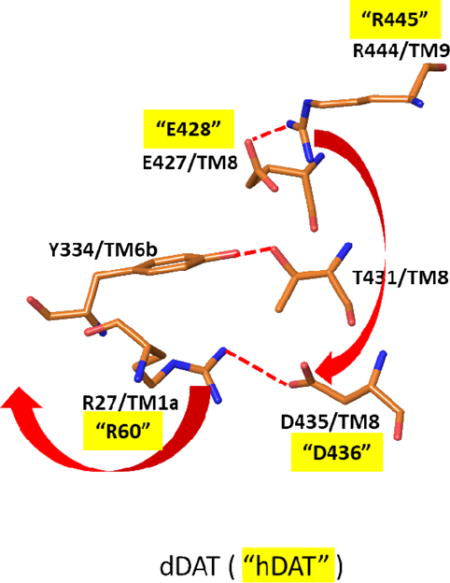
Our previous studies have shown that the R445C mutation linked to DTDS adversely affects both binding and transport of DA when compared with WT. These findings mirror the quantitative effects observed with the R445E mutant as presented above which is consistent with the hypothesis that the intact E428-R445 salt bridge is needed for efficacious substrate binding and transport. Similarly, the E428R mutant undergoes an analogous loss of DA binding and transport. Thus, while R445 is distal from the substrate binding site, it’s role in forming the salt bridge with E428 on TM8 which lines the DA S1 binding site can be important in maintaining the structure of the S1 site required for effective DA binding. This would explain the loss of DA affinity, and in turn transport, upon disruption of this salt bridge by any of the mutations above. Indeed, evidence to support a role of the native salt bridge in maintaining the correct structure at S1 was shown with the finding that restoration of DA and CFT binding occurred with the E428R/R445E double mutant. However, the inability of the double mutant to maintain substrate transport suggests more complex mechanisms. Based on available structural and mechanistic information, binding and transport of substrate involves dynamic shifts between inward and outward facing states of DAT. Precluding substrate egress from DAT, the transporter is in the outward open or outward open occluded state while the intracellular gates are closed (inward closed). To achieve final substrate egress and concomitant transport into the intracellular milieu, DAT must reconfigure to an outward closed and inward open protein conformation. To date, one such outward closed (inward open) X-ray structure has been reported. This structure is for LeuT and is shown in Figure 4 in comparison with an outward open (inward closed) X-ray structure of LeuT. As viewed from the IC side of the proteins, there is an opening of the IC region of the protein which is most evident by the dramatic outward shift of TM1a in the inward open (magenta) versus closed (green) structures (Figures 4a and 4b). This shift is connected to a release of the N-terminus. This N-terminus in the inward closed form is held in its position largely by a network of hydrogen bonds and polar interactions involving residues on TM8. The N-terminus is thus covering the exit of the substrate and inducing a more closed form of the protein in that region. Closer inspection of this region in the inward closed state reveals a network of polar interactions which involve the N-terminal residues (Figure 4c and 4d). Disruption of these interactions would accompany dissociation of the N-terminus when adopting the inward open conformation. While no X-ray structure is available for an inward open state of any of the endogenous amine transporters, the dDAT X-ray structures of the inward closed form indicate a similar mechanism is at play. As a shown in the example in Figure 5, the inward closed form of dDAT has a similar tertiary conformation to that of LeuT in the IC region, including a similar positioning of the N-terminus and TM1a. This conformation utilizes a similar polar interaction network as described for LeuT. Indeed, the X-ray structures of hSERT (inward closed forms) have such a network as well. We note that the salt bridge in dDAT between R27 on TM1b and D435 on the N-terminus is associated with this closed conformation (R27 and D435 in dDAT are equivalent to R60 and D436, respectively, in hDAT). Disruption of the R444-E427 salt bridge (equivalent to R445-E428 in hDAT) would allow R444 to associate with D435 thereby replacing the D435 interaction with R27; R27 and the N-terminus would then be free to swing out in order to adopt the inward open conformation (see red arrows in Figure 5b). These results are consistent with earlier analyses of the polar network in X-ray structures and through examination via molecular dynamics studies22. This mechanistic model provides an explanation for the findings herein for the various mutations as follows. For the DTDS associated R445C (or R445E) hDAT mutation, disruption of the E428-R445 salt bridge disrupts the local structure around the S1 site as described earlier. However, the Cys residue now at position 445 cannot form the salt bridge with D436 necessary for initiating the N-terminus release. Thus, both binding and transport of the substrate are hindered. (Of course, lack of S1 binding alone would preclude transport.) The double mutant reversal of the E428-R445 members of the salt bridge holds the S1 binding site intact, but when this salt bridge is disrupted there is no longer an Arg at position 445 to form a salt bridge with D436 as it has been replaced by a Glu and therefore cannot initiate the N-terminus release. The E428R single mutant would be similar to the natural R445C mutant in its deleterious effect on the structure at S1 and therefore on substrate binding. Thus, the availability of an Arg at position 445 could lead to the inward open form but would be rendered ineffective by the diminishing of the prerequisite substrate binding at S1. The mechanistic model described herein is closely related to earlier reports including molecular dynamics simulations on DAT models. The role of the R60-D436 (corresponding to R27-D435 in dDAT as described above) salt bridge in stabilizing the inward closed conformation has been pointed out22–26. It has been proposed that the breaking of the R60-D436 salt bridge can be compensated by the formation of an alternate E61-K260 salt bridge26. The coordinated movement of R445 from proximity to E428 towards D436 along with the separation of R60 from D436 was shown by MD simulations of DAT22. Along with changes a hydrogen bond between E428 and Y335 was also broken.
Figure 4.
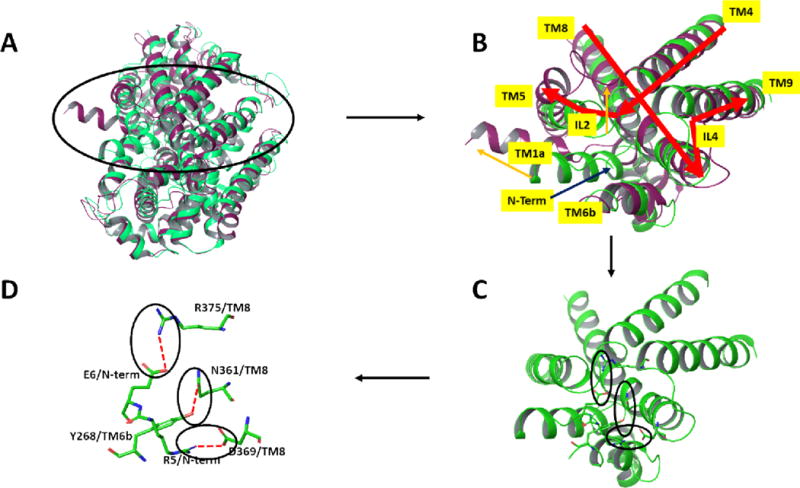
Comparison via superposition of the outward open (in green, PDB entry 3TT1) and inward open (magenta, PDB entry 3TT3) X-ray structures of LeuT12 viewed from the intracellular side of the proteins. A) Ribbon representation of the full proteins. B) Close-up view of the region encircled in A. Major differences between the 2 protein structures are indicated by orange arrows. Most notably, the N-terminus of the LeuT which is held near the core of the protein through multiple polar interactions in the outward open form (green) is, in the inward-open form, removed from that region (not resolved in the X-ray structure) and TM1a concomitantly swings outward. C) and D) depict the key polar residues comprising the network in the outward open form, in ribbon and stick figures respectively. Molecular graphics were generated with Maestro21.
Figure 5.
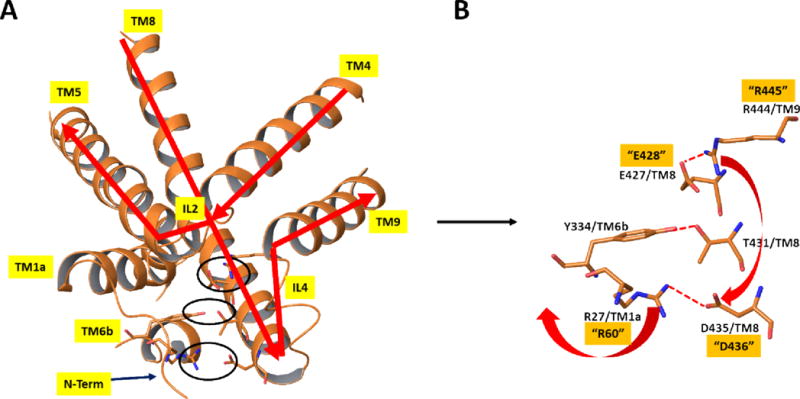
X-ray structure of dDAT19 [PDB entry 4XP1] in the same orientation and from the same view as in Figure 4. A) Close-up view of the protein in the region discussed using ribbon representation of the protein and depicting key polar interactions in stick figures. B) Stick figure representation of key polar interactions in the extracellular side responsible for maintaining the closed-inward (open-outward) protein form in the region shown. Numbering of residues is based on dDAT, as in the X-ray structure. The corresponding hDAT numbering for the residues mutated in the current study are depicted with quotes. Red arrows indicate the proposed structural rearrangements which would occur if the salt bridge between E427 and R444 were broken. Specifically, R444 could alternatively form a salt bridge with D435, thereby disrupting the salt bridge between D435 and R27 and, in turn releasing the N-terminus to form the inward open structure. Molecular graphics were generated with Maestro21.
Conclusions
The R445C mutation in DAT has been found to be responsible for cases of DTDS. The location of the R445 residue on the intracellular region, far removed from both the primary and secondary DA binding sites have confounded earlier attempts at explaining the molecular basis for the deleterious effects of the R445C mutation. The studies herein suggest that R445 has a dual role in DA transport by DAT. R445 forms a salt bridge with E428 which is part of a network of polar interactions on the intracellular side of DAT. As part of the intact salt bridge with E428, R445 serves as a latch holding TM8 in place thereby maintaining the structure needed for DA binding at S1 as well as the closed state of DAT. Upon release of R445 from the salt bridge, it then serves as a trigger which has a cascading effect through the local polar network leading to the disruption of the E428-R445 salt bridge freeing the N-terminal and opening the IC region to allow DA egress. The DTDS condition characterized by the R445C mutation is thus a consequence of the loss of these two (latch and trigger) capabilities at the 445 position of DAT. From the perspective of therapeutic possibilities, the understanding provided herein suggests that approaches aimed at restoration of the deficient DAT functioning of the R445C mutant, such as gene therapy to restore residue 445 to the native Arg be sought. Finally, it is noteworthy that the 5 mutants hitherto identified as associated with DTDS operate via 2 different apparent molecular mechanisms. R445C as described herein results in a loss of function of DAT residing in a region not directly involved with substrate binding whereas the other four mutations appear to play a role in diminishing substrate binding at the vestibule entranceway site.
Materials and Methods
Site-directed mutagenesis and heterologous expression of hDAT mutant constructs
The mutants were generated (WT-hDAT cDNA is from Dr. Jonathan Javitch at Columbia University) with the Quick-change site-directed mutagenesis kit (Agilent Technologies) according to the manufacturer’s protocol and confirmed by DNA sequencing. The following primers were used: R445E sense 5′- GCTCCTGCACCGGCATGAGGAGCTGTTCAC-3′ and antisense 5′- GTGAACAGCTCCTCATGCCGGTGCAGGAGC-3′ and E428R sense 5′-GCTATGGGAGGCATGAGGAGCGTCATCACCGG-3′ and antisense 5′-CCGGTGATGACGCTCCTCATGCCTCCCATAGC-3′.
E428R plasmid was used as template for the generation of E428R/R445E double mutant.Culturing and stable transfection of Lewis Lung Carcinoma-Porcine Kidney (LLC-PK1) cells (gift from Dr. Roxanne Vaughan at University of North Dakota) with hDAT constructs by Lipofectamine 2000 (Invitrogen) was carried out as in our previous work27.
[3H]CFT binding assay and [3H]dopamine uptake assay
Binding of [3H]CFT27 and [3H]DA uptake assays16, 28 to stably transfected LLC-PK1 cells were studied as described previously. Briefly, intact cell suspensions were incubated with [3H]CFT (4 nM final concentration) and varying concentrations of non-labelled CFT spanning its IC50 value for 20 minutes at 21°C. For uptake measurements with intact adhering cells, 3H]DA (10 nM final concentration) was added into 24-well cell culture plates and incubation continued for 5 min at 21°C as described by us previously28.
Immunoblotting studies
Surface DAT was assessed with sulfo-NHS-SS-biotin as described by us previously16. Total lysates and biotinylated proteins were separated by 7.5% SDS-polyacrylamide gel electrophoresis and transferred to a nitrocellulose membrane. Membranes were probed using goat-anti DAT (Santa Cruz, sc-1433; 1:500) and anti-rabbit β-actin antibody (Sigma; 1:5000). Secondary antibodies conjugated to horseradish peroxidase were obtained from Pierce (Rockford, IL; 1:5000). Immunoblots were developed with Supersignal West Pico Chemiluminescent Substrate (Thermo Scientific) using a film developer.
Acknowledgments
M. Reith received funding from a grant from National Institutes of Health, National Institute on Drug Abuse R01 DA019676
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Reference list
- 1.Yamashita A, Singh SK, Kawate T, Jin Y, Gouaux E. Crystal structure of a bacterial homologue of Na+/Cl−-dependent neurotransmitter transporters. Nature. 2005;437:215–223. doi: 10.1038/nature03978. [DOI] [PubMed] [Google Scholar]
- 2.Zhou Z, Zhen J, Karpowich NK, et al. LeuT-desipramine structure reveals how antidepressants block neurotransmitter reuptake. Science. 2007;317:1390–1393. doi: 10.1126/science.1147614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Jorgensen AM, Topiol S. Driving forces for ligand migration in the leucine transporter. Chem Biol Drug Des. 2008;72:265–272. doi: 10.1111/j.1747-0285.2008.00707.x. [DOI] [PubMed] [Google Scholar]
- 4.Singh SK, Yamashita A, Gouaux E. Antidepressant binding site in a bacterial homologue of neurotransmitter transporters. Nature. 2007;448:952–956. doi: 10.1038/nature06038. [DOI] [PubMed] [Google Scholar]
- 5.Topiol S, Bang-Andersen B, Sanchez C, Bogeso KP. Exploration of insights, opportunities and caveats provided by the X-ray structures of hSERT. Bioorg Med Chem Lett. 2016;26:5058–5064. doi: 10.1016/j.bmcl.2016.08.087. [DOI] [PubMed] [Google Scholar]
- 6.Piscitelli CL, Gouaux E. Insights into transport mechanism from LeuT engineered to transport tryptophan. EMBO J. 2012;31:228–235. doi: 10.1038/emboj.2011.353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Piscitelli CL, Krishnamurthy H, Gouaux E. Neurotransmitter/sodium symporter orthologue LeuT has a single high-affinity substrate site. Nature. 2010;468:1129–1132. doi: 10.1038/nature09581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Shi L, Quick M, Zhao Y, Weinstein H, Javitch JA. The mechanism of a neurotransmitter:sodium symporter–inward release of Na+ and substrate is triggered by substrate in a second binding site. Mol Cell. 2008;30:667–677. doi: 10.1016/j.molcel.2008.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Singh SK, Piscitelli CL, Yamashita A, Gouaux E. A competitive inhibitor traps LeuT in an open-to-out conformation. Science. 2008;322:1655–1661. doi: 10.1126/science.1166777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wang H, Elferich J, Gouaux E. Structures of LeuT in bicelles define conformation and substrate binding in a membrane-like context. Nat Struct Mol Biol. 2012;19:212–219. doi: 10.1038/nsmb.2215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wang H, Gouaux E. Substrate binds in the S1 site of the F253A mutant of LeuT, a neurotransmitter sodium symporter homologue. EMBO Rep. 2012;13:861–866. doi: 10.1038/embor.2012.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Krishnamurthy H, Gouaux E. X-ray structures of LeuT in substrate-free outward-open and apo inward-open states. Nature. 2012;481:469–474. doi: 10.1038/nature10737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Shan J, Javitch JA, Shi L, Weinstein H. The substrate-driven transition to an inward-facing conformation in the functional mechanism of the dopamine transporter. PLoS ONE. 2011;6:e16350. doi: 10.1371/journal.pone.0016350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kurian MA, Li Y, Zhen J, et al. Clinical and molecular characterisation of hereditary dopamine transporter deficiency syndrome: an observational cohort and experimental study. Lancet Neurol. 2011;10:54–62. doi: 10.1016/S1474-4422(10)70269-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kurian MA, Zhen J, Cheng SY, et al. Homozygous loss-of-function mutations in the gene encoding the dopamine transporter are associated with infantile parkinsonism-dystonia. J Clin Invest. 2009;119:1595–1603. doi: 10.1172/JCI39060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ng J, Zhen J, Meyer E, et al. Dopamine transporter deficiency syndrome: phenotypic spectrum from infancy to adulthood. Brain. 2014;137:1107–1119. doi: 10.1093/brain/awu022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Coleman JA, Green EM, Gouaux E. X-ray structures and mechanism of the human serotonin transporter. Nature. 2016;532:334–339. doi: 10.1038/nature17629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Penmatsa A, Wang KH, Gouaux E. X-ray structure of dopamine transporter elucidates antidepressant mechanism. Nature. 2013;503:85–90. doi: 10.1038/nature12533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Penmatsa A, Wang KH, Gouaux E. X-ray structures of Drosophila dopamine transporter in complex with nisoxetine and reboxetine. Nat Struct Mol Biol. 2015;22:506–508. doi: 10.1038/nsmb.3029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wang KH, Penmatsa A, Gouaux E. Neurotransmitter and psychostimulant recognition by the dopamine transporter. Nature. 2015;521:322–327. doi: 10.1038/nature14431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Maestro. Schrodinger Release 2016-1: Maestro. Schrodinger, LLC; New York, NY: 2016. version 10.5. [Google Scholar]
- 22.Khelashvili G, Stanley N, Sahai MA, et al. Spontaneous inward opening of the dopamine transporter is triggered by PIP2-regulated dynamics of the N-terminus. ACS Chem Neurosci. 2015;6:1825–1837. doi: 10.1021/acschemneuro.5b00179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cheng MH, Bahar I. Complete mapping of substrate translocation highlights the role of LeuT N-terminal segment in regulating transport cycle. PLoS Comput Biol. 2014;10:e1003879. doi: 10.1371/journal.pcbi.1003879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cheng MH, Bahar I. Molecular Mechanism of Dopamine Transport by Human Dopamine Transporter. Structure. 2015;23:2171–2181. doi: 10.1016/j.str.2015.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Loland CJ, Granas C, Javitch JA, Gether U. Identification of intracellular residues in the dopamine transporter critical for regulation of transporter conformation and cocaine binding. J Biol Chem. 2004;279:3228–3238. doi: 10.1074/jbc.M304755200. [DOI] [PubMed] [Google Scholar]
- 26.Ma S, Cheng MH, Guthrie DA, Newman AH, Bahar I, Sorkin A. Targeting of dopamine transporter to filopodia requires an outward-facing conformation of the transporter. Sci Rep. 2017;7:5399. doi: 10.1038/s41598-017-05637-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chen N, Vaughan RA, Reith ME. The role of conserved tryptophan and acidic residues in the human dopamine transporter as characterized by site-directed mutagenesis. J Neurochem. 2001;77:1116–1127. doi: 10.1046/j.1471-4159.2001.00312.x. [DOI] [PubMed] [Google Scholar]
- 28.Stouffer MA, Ali S, Reith ME, et al. SKF-83566, a D(1) -dopamine receptor antagonist, inhibits the dopamine transporter. J Neurochem. 2011;118:714–720. doi: 10.1111/j.1471-4159.2011.07357.x. [DOI] [PMC free article] [PubMed] [Google Scholar]